

Abstract
The properties of stem cells can be induced during the epithelial to mesenchymal transition (EMT). The responsible molecular mechanisms, however, remain largely undefined. Here we report the identification of the microRNA-146a (miR-146a) as a common target of Krüppel-like factor 8 (KLF8) and TGF-β, both of which are known EMT-inducers. Upon KLF8 overexpression or TGF-β treatment, a significant portion of the MCF-10A cells gained stem cell traits as demonstrated by an increased expression of CD44high/CD24low, activity of aldehyde dehydrogenase (ALDH), mammosphere formation and chemoresistance. Along with this change, the expression of miR-146a was highly upregulated in the cells. Importantly, we found that miR-146a was aberrantly co-overexpressed with KLF8 in a panel of invasive human breast cancer cell lines. Ectopic expression of KLF8 failed to induce the stem cell traits in the MCF-10A cells if the cells were pre-treated with miR-146a inhibitor, whereas overexpression of miR-146a in the MCF-10A cells alone was sufficient to induce the stem cell traits. Co-staining and luciferase reporter analyses indicated that miR-146a targets the 3’-UTR of the Notch signaling inhibitor NUMB for translational inhibition. Overexpression of KLF8 dramatically potentiated the tumorigenecity of MCF-10A cells expressing the H-Ras oncogene, which was accompanied by a loss of NUMB expression in the tumors. Taken together, this study identifies a novel role and mechanism for KLF8 in inducing pro-tumorigenic mammary stem cells via miR-146a potentially by activating Notch signaling. This mechanism could be exploited as a therapeutic target against drug resistance of breast cancer.
Keywords: KLF8, miR-146a, EMT, mammary stem cells, tumorigenesis
Introduction
EMT plays a key role in normal embryogenesis and wound healing [1]. A large body of evidence suggests a role of aberrantly activated EMT in tumor initiation and malignant progression [1,2]. Cancer stem cells (CSCs) are a rare subpopulation of cancer cells that are able to self-renew, differentiate and are thought to be responsible for tumor initiation, growth, invasion, metastasis, recurrence, and resistance to various therapies [3]. Recent studies have linked EMT to induction or maintenance of mammary stem cells associated with malignant transformation [4,5]. MicroRNAs (miRs), a family of short non-coding regulatory RNAs targeting mRNA 3’-untranslated region (3’-UTR) to regulate gene expression, have recently emerged as critical regulators of tumor progression and CSCs and are thus tightly regulated in the cell [6,7]. However, how miRs regulate EMT-associated CSC properties remain in its infancy of studies.
KLF8 is among a few master EMT-promoting transcriptional factors [8,9]. In addition to repressing E-cadherin to promote EMT [9,10], KLF8 also targets other important genes such as cyclin D1 [9,11-14] and MMP9 [15] that are associated with tumor progression of various cancer types including breast cancer [8-10,15-25]. KLF8 has also been reported to regulate the transcription of KLF4 [13] and Wnt signaling [26] both of which are important regulators of stem cells and cancer [27,28]. Given these important roles of KLF8 as a dual transcription factor [13-15,29,30], its expression is tightly regulated in the cells at various levels including transcription [14,16,31,32], post-translational modifications [11-13,33] and subcellular localization [33,34]. Unlike other regulators of EMT such as Snail, TWIST and ZEBs which target a consensus E-box(es) at their target gene promoters [1], KLF8 represents a distinct family of transcription factors (i.e., KLFs) and targets a consensus GT-box(es) to regulate its target gene expression [8]. This difference suggests that KLF8 may regulate a distinct group of genes or miRs associated with EMT or stem cells during cancer progression.
In this study, we sought to find out the first miRs that are regulated by KLF8 and identified miR-146a as one of primary target miRs of KLF8 that links mammary stem cell induction to oncogenic transformation.
Material and methods
Cell culture, reagents and plasmid construction
MCF-10A, MCF-7, T47D, MDA-MB-231, Hs578T and BT-549 cell lines [9,17] and the MCF-10A line that expresses inducible KLF8 (10A-iK8) and the MDA-MB-231 line that expresses inducible KLF8 short hairpin RNA (231-K8ikd) [15,35] were described previously. The overexpression or knockdown of KLF8 was induced by doxycycline included in the culture medium. The Cy3 dye-labeled negative control miR inhibitor and miR-146a inhibitor were purchased from Dharmacon (Lafayette, CO, USA). Their transfection was done using Oligofectamine (Invitrogen, Grand Island, NY, USA). To construct pIS1-NUMB-3’-UTR luciferase reporter, we amplified the NUMB 3’-UTR fragment (nucleotides 3-1253) by RT-PCR and ligated it into pIS1 reporter vector (Addgene ID: 12179, Cambridge, MA, USA) [36] between the AgeI and XbaI sites. Primers used for the cloning were 5’-ATG CAC CGG TAA TCA TTA TGG CTA TGT ATC TTG TC (forward) and 5’-ATG CTC TAG AAG ATG AGC TCT CTT ATT GTT ATC C (reverse). To construct the miR-146a-binding defective mutant reporter, the miR-146a binding site in the 3’-UTR was disabled by site-directed mutagenesis PCR using the pIS1-NUMB-3’-UTR luciferase reporter as template and the primers of 5’-ata gac tac aga tat taa gaa g (forward) and 5’-tct gta gtc tat tta aaa tat tc (reverse). To construct pBabe-mCherry-miR-146a sponge, we cloned mCherry into pBabe-puro vector [9] between the BamHI and EcoRI sites. We then cloned the annealed oligonucleotides for miR-146a sponge into pBabe-puro-mCherry vector between the EcoRI and SalI sites. The sequences of the sponge oligos consisting of 4 tandem miR-146a binding sites with 4-nucleotide spacers at the bulged sites were 5’-AAT TCA ACC CAT GGA TGC AGT TCT CAA GTA ACC CAT GGA TGC AGT TCT CAT CAA ACC CAT GGA TGC AGT TCT CACT GAA CCC ATG GAT GCA GTT CTC AA (sense) and 5’-CCG GTT GAG AAC TGC ATC CAT GGG TTC AGT GAG AAC TGC ATC CAT GGG TTT GAT GAG AAC TGC ATC CAT GGG TTA CTT GAG AAC TGC ATC CAT GGG TTG (antisense). Retroviruses derived from the pBabe-puro-mCherry-miR-146a-sponge or pBabe-puro-mCherry vector were used to infect the 10A-iK8 cells. The respective 10A-iK8-miR-146a-sponge cell line and 10A-iK8-miR-sponge mock cell line were established by puromycin selection. To stably overexpress miR-146a, we transferred the pre-miR-146a template from pcDNA3-miR-146a vector (Addgene ID: 15092, Cambridge, MA, USA) [37] between BamHI and SalI sites into pBabe-neo vector (Addgene ID: 1767, Cambridge, MA, USA) between BamH I and XhoI sites to form the pBabe-neo-miR-146a retroviral vector. Viruses derived from this vector or pBabe-neo control vector were used to infect MCF-10A cells. The respective MCF-10A-miR-146a and MCF-10A-miR-mock cell lines were established by G418 selection. 10A-iK8-V12 and 10A-iK8-GFP control cell lines were generated by retroviral infection using the retroviral vector MSCV H-Ras V12 IRES GFP (Addgene, Plasmid 18780, Cambridge, MA, USA) and control vector pMIG (Addgene: Plasmid 9044, Cambridge, MA, USA), respectively, followed by GFP-based 96-well plate cloning. A 3’-UTR-less human NUMB-GFP fusion was used to replace the GFP in the pMIG vector to generate 10A-iK8-NUMB cell line similarly.
Fluorescence activated cell-sorting (FACS) analysis
For quantification of cells expressing the human breast stem cell marker proteins CD44 and CD24, cells were trypsinized and washed with PBS containing 2% of FBS. The cell surface marker proteins were detected by incubating the cells with anti-CD44-FITC (clone G44-26) and anti-CD24-PE (clone ML5) antibodies (BD Bioscience, Franklin Lakes, NJ, USA) in the manufacturer-recommended dilutions at 4°C for 30 minutes. After washing three times, analysis was performed using a flow cytometer (FACScalibur, BD Biosciences, Franklin Lakes, NJ, USA). Ten thousand events were acquired for each sample and analyzed with FlowJo software.
ALDEFLUOR assay
The ALDEFLUOR kit (StemCell Technologies, Vancouver, BC, Canada) was used to isolate the aldehyde dehydrogenase (ALDH) enzymatic activity-positive cell population according to the manufacturer’s instruction. Briefly, cells were detached with 0.25% trypsin/EDTA, washed with PBS supplemented with 2% FBS and suspended in ALDEFLUOR assay buffer containing ALDH substrate (BAAA, 1 μmol/L for 1×106 cells) and incubated at 37°C for 40 minutes. As a negative control, an equivalent cell aliquot was treated with 50 μmol/L of diethylaminobenzaldehyde (DEAB), a specific ALDH inhibitor. The gates for FACS were established using the ALDEFLUOR-stained cells treated with DEAB to define the baseline fluorescence of the cells (R1) and the ALDEFLUOR-positive region (R2).
MicroRNA expression profiling analysis
The 10A-iK8 cells were grown under uninduced (U) or induced (I) conditions for 7 days. The total RNA was prepared from the cells using Trizol (Invitrogen, Carlsbad, CA, USA). Forty μg of the total RNA was used to isolate total miroRNA using the small RNA isolation kit-RT²-qPCR Grade miRNA Isolation Kit (MA-01, SABioscienc, Valencia, CA, USA). Then, 100 ng of the small RNA sample was used to synthesize the first strand cDNAs for the microRNAs using RT2 miRNA first strand kit (MA-03, SABioscienc, Valencia, CA, USA). The cDNAs were then used as templates to screen the whole human genome miRNA PCR array (MAH-100A, SABioscienc, Valencia, CA, USA) using quantitative real-time reverse transcription-PCR (qRT-PCR) for the profiling by following the manufacturer’s instruction. In some experiments, individual microRNAs were examined similarly to determine their expression in MCF-10A cells that were treated with TGF-β (2.5 ng/ml) for 7 days or in a panel of human breast cancer cell lines.
Promoter reporter assays
Luciferase reporter assays were performed essentially as described previously [9]. Briefly, HEK293 cells were co-transfected with the wild-type or mutant pIS1-NUMB-3’-UTR luciferase reporter pIS1 and pBABE-neo-miR-146a or pBABE-neo control vector along with the normalizing control vector pIS0 encoding Firefly luciferase (Addgene ID: 12178, Cambridge, MA, USA) [36]. After 36 hours, luciferase activity was determined using the dual luciferase reporter assay system (Promega, Fitchburg, WI, USA) and 20/20n luminometer (Turner BioSystems, Sunnyvale, CA, USA) according to the manufacturers’ instructions.
Mammosphere formation assay
Mammosphere culture was performed as described [38]. Cells (3000 to 104 per well) were plated in the ultra-low adherent 6-well plates and cultured in the MammoCultTM basal medium with proliferation supplements (STEMCELL Technologies, Vancouver, BC, Canada) for 7-10 days prior to sphere counting and photography. Mammospheres greater than 75 μm in diameter were counted as positive spheres.
Co-staining of cellular miR-146a and NUMB
The fluorescent in situ hybridization coupled with tyramide-signal amplification (FISH-TSA) was performed as previously described [39]. Briefly, cells grown on glass coverslips were fixed, permeabilized and prehybridized. Cells were then hybridized with 20 μM of miR-146a probes (EXIQON, Woburn, MA, USA) for overnight at 52°C, washed and blocked with blocking buffer. After incubated with 0.05 units/ml of peroxidase-conjugated sheep anti-DIG antibody (Roche, Indianapolis, IN), cover slips were further incubated with biotin-tyramide for TSA in a TSA-PLUS system (Perkin-Elmer, Boston, MA, USA) followed by incubation with SA-Fluorescein. After the FISH-TSA, the cellular NUMB protein was stained with anti-NUMB antibody (Cell Signaling, Danvers, MA, USA) and Texas-red conjugated secondary antibody. Cell imaging was processed and analyzed with a fluorescent confocal microscope.
Western blotting (WB) and immunohistochemical (IHC) staining
WB and IHC staining were carried out as previously described [15,23]. The antibodies used in IHC include rabbit anti-human KLF8 antibody [9] and rabbit anti-human NUMB monoclonal antibody (C29G1, Cell Signaling, Danvers, MA, USA).
Mammary tumorigenecity in immunocompromised mice
Four to 5-week-old NOD/SCID mice (12 mice per cell line. Taconic, Germantown, NY, USA) were injected with the 10A-iK8, 10A-iK8-V12 or 10A-iK8-GFP cells (2 x 106 in 100 μl mixture of PBS and Matrigel mix (1:1)) into the right hind mammary fat pad. The mice were fed with the Dox Diet (3888) (Bio Servs, Frenchtown, NJ, USA) supplemented with doxycycline (200 mg/kg) to induce the expression of KLF8 in the tumor cells in vivo or with the Control Diet (S4207) not containing doxycycline. Tumors formed by the cells were monitored for 7 months before the tumors were photographed and dissected. Tumor length (L) and width (W) were measured using a Vernier caliper, and mean tumor volume was calculated by the formula of V = 0.52*L*W2 and compared between groups of mice by box-plot analysis. The mice were housed and maintained in specific pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the United States Department of Agriculture, United States Department of Health and Human Services, and the National Institute of Health. Animal care and use was approved by the Institutional Animal Care and Use Committee. Human care of the mice was thoroughly considered.
Statistical analysis
All the data was summarized and presented as mean +/- the standard deviation (SD) with a minimum of three observations per group. Unpaired, paired or single sample Student’s t-test with the Bonferroni correction for the multiple comparisons or Qi-Square test was applied as appropriate. Significance was determined by the alpha level of 0.05.
Results
KLF8 induces EMT-associated mammary stem cell traits in MCF-10A cells
Our previous findings showed that stable expression of KLF8 could induce EMT in breast epithelial cells [9]. An intriguing recent finding is that breast epithelial cells gain the properties of stem cells during EMT [4,5]. To determine whether during KLF8-induced EMT, the cells obtain the properties of stem cells, we used our recently generated MCF-10A cell line that expresses tetracycline-regulated inducible expression of KLF8 (10A-iK8) [15]. Effective induction of EMT by the induced expression of KLF8 in the cells, using TGF-b-induced MET in the parental MCF-10A cells as a positive control, was verified by analysis of the changes in the cell morphology and expression switch between epithelial markers and mesenchymal markers proteins (data now shown). Changes in the human mammary stem cell surface markers CD44high/CD24low were first examined by flow cytometry. We found that more than 90% of the cells acquired a CD44high/CD24low expression pattern when the expression of KLF8 was induced (10A-iK8/I) compared to only 33% of the uninduced cells (10A-iK8/U) (Figure 1A and 1B). To verify this result, an independent human mammary stem cell marker aldehyde dehydrogenase (ALDH) was analyzed. We found that KLF8-expressing cells (10A-iK8/I) gained 3 times more ALDH-positive cells (5.76%) than the cells not expressing KLF8 (10A-iK8/U) did (1.34%) (Figure 1C and 1D). Similar results were obtained in the cells treated with TGF-β (Figures 1A, 1B and 2). Taken together, these results clearly demonstrated that like TGF-β, KLF8 could induce the mammary stem cell traits via EMT.
Figure 1.

KLF8 induces stem cell traits in MCF-10A cells associated with EMT. A and B: Induction of an increase in CD44high/CD24low cells by KLF8 and TGF-β. The 10A-iK8 cells were grown under uninduced (U) or induced (I) conditions, or the parental MCF-10A cells were grown untreated (-TGF-β) or treated (+TGF-β) with recombinant TGF-β (2.5 ng/ml) for two weeks. The cells were then prepared for FACS analysis of expression of the cell-surface markers CD44 and CD24 as described in the Materials and Methods to identify CD44high/CD24low cell population (% shown in red). C and D: Induction of an increase in ALDH+ cells by KLF8 and TGF-β. The 10A-iK8 cells or parental MCF-10A cells were grown and treated as described above, followed by FACS analysis of ALDH expression in the cells using the ALDEFLIOR assay as described in the Materials and Methods to identify the ALDH+ cell population. Representative FACS results are shown C where percentage of ALDH+ cell populations are indicated in red in the absence of DEAB, normalized to those in the presence of DEAB. Fold changes in the ALDH+ cell populations caused by KLF8 expression or TGF-β treatment was summarized in D. *P < 0.01 compared to the uninduced or untreated cells.
Figure 2.
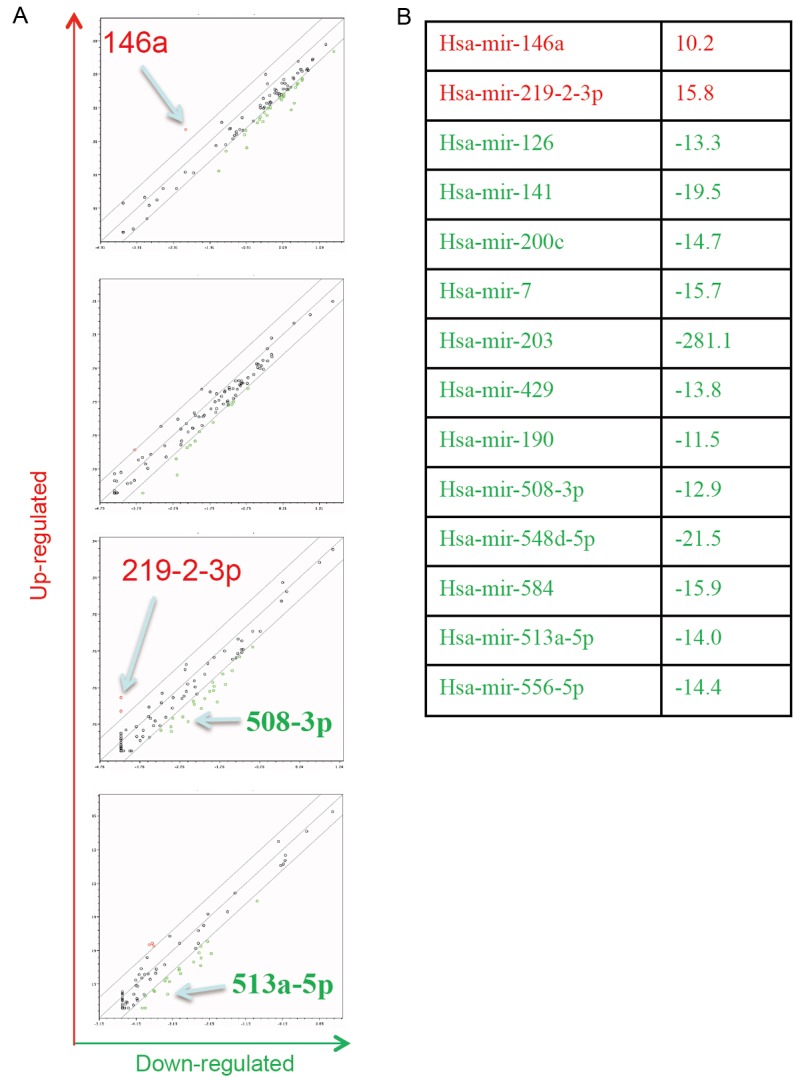
MicroRNA profile in 10A-iK8 cells. A: The whole genomic miR PCR array (consisting of 4 consecutive sets) was screened for KLF8-regulated miRs in the 10A-iK8 induced cells compared to the uninduced cells. Assay and analysis were described in the Materials and Methods. B: Shown are miRs whose expression levels were increased or decreased by > 10 times in the induced cells compared to the uninduced cells.
KLF8-upregulated expression of miR-146a is responsible for the induction of stem cell traits in MCF-10A cells
MiRs have been implicated in the regulation of both EMT and stem cells [40-43]. We sought to examine the change in the expression profile of miRs to identify target miRs of KLF8 responsible for the stem cell trait induction during EMT. Among the 14 miRs whose levels were changed ≥ 10 times by the induction of KLF8 expression (Figure 2), four were similarly regulated by TGF-β treatment (Figure 3A). Among these 4 miRs, the miR-146a was the most significantly upregulated in the invasive breast cancer cells (MDA-MB-231, Hs578T and BT-549), but not in the non-invasive breast cancer cells (MCF-7 and T47-D) (Figure 3B and 3C). Given that the invasive and non-invasive cells represent the mesenchymal and epithelial phenotypes, respectively, during EMT and KLF8 is aberrantly overexpressed in these invasive cancer cells [17], we decided to focus firstly on the miR-146a for the subsequent experiments.
Figure 3.
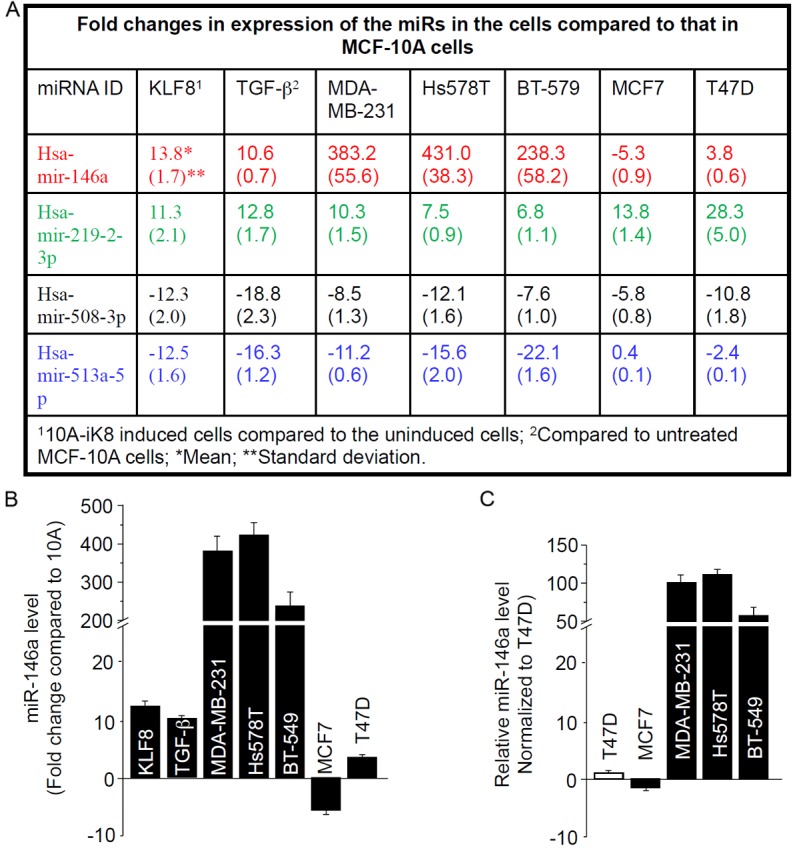
MiR-146a is upregulated by KLF8 and TGF-β in MCF-10A cells and is highly overexpressed in invasive breast cancer cells. A: Expression of the 14 miRs shown in Figure 2 was further compared between TGF-β treated and untreated MCF-10A cells to identify target miRs (Hsa-mir-146a, -219-2-3p, -508-3p, and -513a-5p) common to KLF8 and TGF-β. The expression of these 4 miRs was then examined in the indicated human breast cancer cell lines. B: MiR-146a fold difference from that in the MCF-10A cells. C: The expression of MiR-146a was compared between the cancer cells.
To test if the upregulation of miR-146a is required for the induction of stem cell traits by KLF8, we blocked the function of miR-146a in the 10A-iK8 cells. We found that when the cells were treated with the miR-146a inhibitor, the numbers of both ALDH positive cells and mammospheres were reduced by approximately 50% regardless of the induction of KLF8 expression (Figure 4A and 4B). This result suggested that miR-146a plays a mediating role downstream of KLF8 for the induction of the stem cell traits in the cells.
Figure 4.
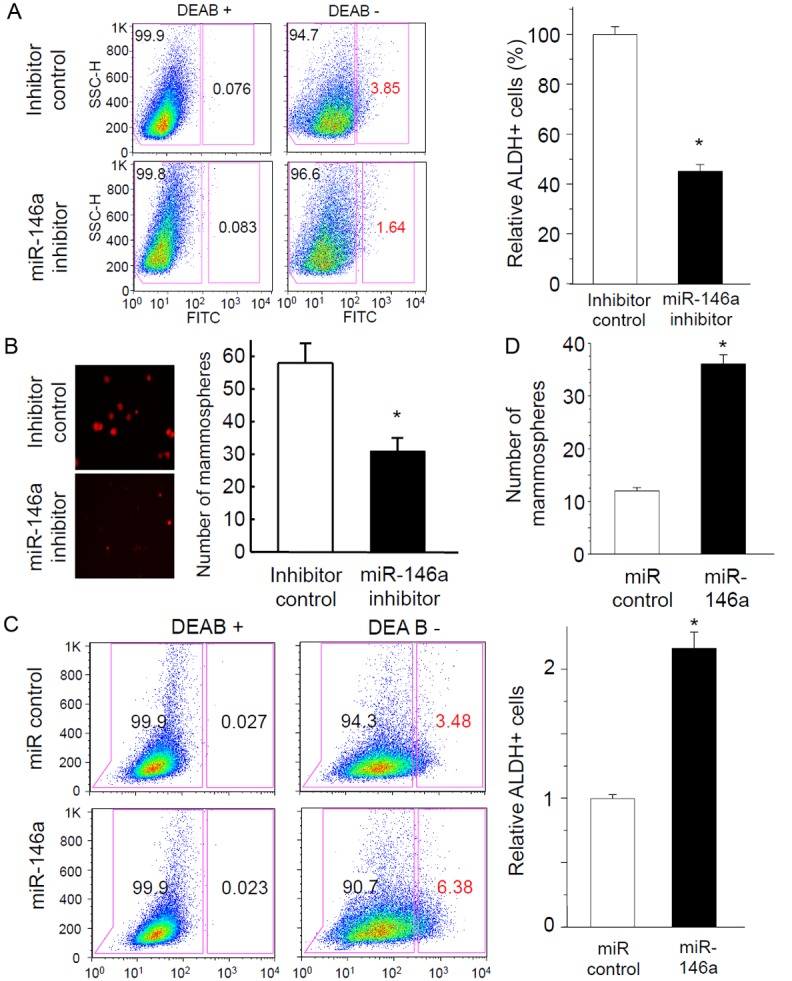
KLF8 upregulated expression of miR-146a is responsible for the induction of stem cell traits in MCF-10A cells. A and B: Inhibition of miR-146a suppresses the KLF8-dependent expression of ALDH and mammosphere formation. The 10A-iK8 cells were transfected with a Cy3 dye-labeled miR-146a inhibitor or an inhibitor control. After 3 days, the expression of KLF8 was induced in the cells and the cells were cultured for 7-10 days either in regular culture plates for analysis of ALDH+ population (% shown in red) by FACS in the absence or presence of the ALDH inhibitor DEAB (A) or in mammosphere culture dishes for mammosphere formation (B). Representative FACS results and mammosphere images were shown in the left panels. C and D: Overexpression of miR-146a is sufficient to induce stem cell traits in MCF-10A cells. MCF-10A cells were infected with retroviral viruses expressing pre-miR-146a or pre-miR control and selected with G418. The cells were then used for analysis of ALDH+ population (% shown in red) by FACS (C) and mammosphere formation (D) similarly as described in A and B. The data represent the mean ± S.E. of at least three independent experiments in triplicate. *P < 0.05.
To determine if overexpression of miR-146a alone is sufficient to induce the stem cell traits, we overexpressed miR-146a in the parental MCF-10A cells. We found that the cells contained more than 2 times of ALDH positive cells and mammosphere forming capability compared to the mock control cells (Figure 4C and 4D). This result suggested that the miR-146a plays a critical role in inducing stem cell traits in the cells.
MiR-146a-mediated induction of the stem cell traits by KLF8 is correlated with drug resistance
Drug resistance is one of the hallmarks for stem cells. To test if KLF8 grants the 10A-iK8 cells with the capability of resisting the cell killing effect of therapeutic drugs and whether miR-146a plays such a role downstream of KLF8 in the cells, we first generated the 10A-iK8-sponge cell line where miR-146a was constitutively blocked by its inhibiting sponge (10A-iK8-146a-sponge) and a sponge mock control cell line (10A-iK8-mock-sponge). After the inhibition of the stem cell traits under the induced conditions by the miR-146a sponge but not the mock sponge was confirmed (Figure 5B and 5C), the response of the cells to the killing effect of paclitaxel was determined. We found that ~90% of the uninduced cells were killed after treated for 6 days regardless of the expression of the miR-146a-sponge or mock-sponge (Figure 5D, columns 3 and 4, compare days 6 to 1). In sharp contrast, induction of KLF8 expression kept ~80% of the mock-sponge cells viable (Figure 5D, columns 1, compare days 6 to 1). However, only ~30% of the miR-146a-sponge cells survived the drug treatment regardless of the KLF8 expression (Figure 5D, columns 2, compare days 6 to 1). These results strongly suggested that KLF8-induced miR-146a-dependent stem cell traits are a major contribution to the resistance of the cells to the cytotoxic effect of the drug.
Figure 5.

MiR-146a-mediated induction of stem cell traits by KLF8 is correlated with drug resistance. A-C: Constitutive blocking of miR-146a function inhibits the induction of stem cell traits by KLF8. Retroviral viruses expressing the miR-146a sponge (A) were used to infect the 10A-iK8 cells. After selected with puromycin, the 10A-iK8-146a-sponge and mock-sponge control cells were cultured under uninduced (U) or induced (I) conditions for 7-10 days prior to ALDH expression based FACS analysis (B) and mammosphere formation (C). *P < 0.05 compared to I-Mock group. D: KLF8 induces the drug resistance in a miR-146a-dependeant manner. The 10A-iK8-sponge and mock control cells were grown under uninduced (U) or induced (I) conditions for 6 days. The cells were then treated with paclitaxel (10 nM) for another 6 days. At the indicated time points, the cell viability was examined using the Promega CellTiter-Glo® Luminescent Cell Viability Assay. Relative viability was normalized to the mock control cells (I-Mock) a day after treatment was started. *P < 0.05 compared to I-Mock group at the same time points. The data represent the mean ± S.E. of at least three independent experiments in triplicate.
MiR-146a mediates KLF8 induced stem cell traits by inhibiting NUMB expression
Potential molecular targets of miR-146a were predicted by using several software programs including TargetScan 5.1, miRanda, miRbase and PITA. Among the potential targets, we focused on NUMB given its well conserved 3’-UTR binding site for miR-146a and the inhibitory role in regulating Notch signaling, an important stem cell regulating pathway [44]. To test if miR-146a directly targets NUMB transcript to reduce its translation, we first performed NUMB 3’-UTR luciferase reporter analysis. We found that the presence of the 3’-UTR caused approximately 40% reduction in the luciferase activity when miR-146a was co-transfected (Figure 6B, compare columns 2 to 1). The inhibitory effect of miR-146a was lost when the miR-146a binding site in the 3’-UTR was disabled by mutagenesis (Figure 6B, compare columns 4 to 3). This result suggested that NUMB is likely a specific inhibitory target of the miR-146a in the cells. As described above (Figure 3), miR-146a expression is differentially expressed between invasive and non-invasive breast cancer cells by more than 100 times. To test if the miR-146a expression is inversely correlated with the expression of NUMB protein in the cancer cells, we compared the NUMB protein expression between the indicated invasive and non-invasive breast cancer cells by western blotting (Figure 6C) followed by co-staining miR-146a and NUMB protein in the cells (Figure 6D). The western blotting results showed a highly inverse correlation of expression between miR-146a and NUMB protein in the cancer cells (Figures 3 and 4C). Similarly, the co-staining assay showed that miR-146a was abundantly expressed in the invasive Hs578T cells in which there was only little expression of NUMB protein, whereas in the non-invasive T47D cells the expression of NUMB was high and localized at the cell-cell junctions where miR-146a was absent (Figure 6D). Taken together, these data suggested that miR-146a can be induced by KLF8 and bind to NUMB 3’-UTR to suppress NUMB expression in the invasive breast cancer cells.
Figure 6.
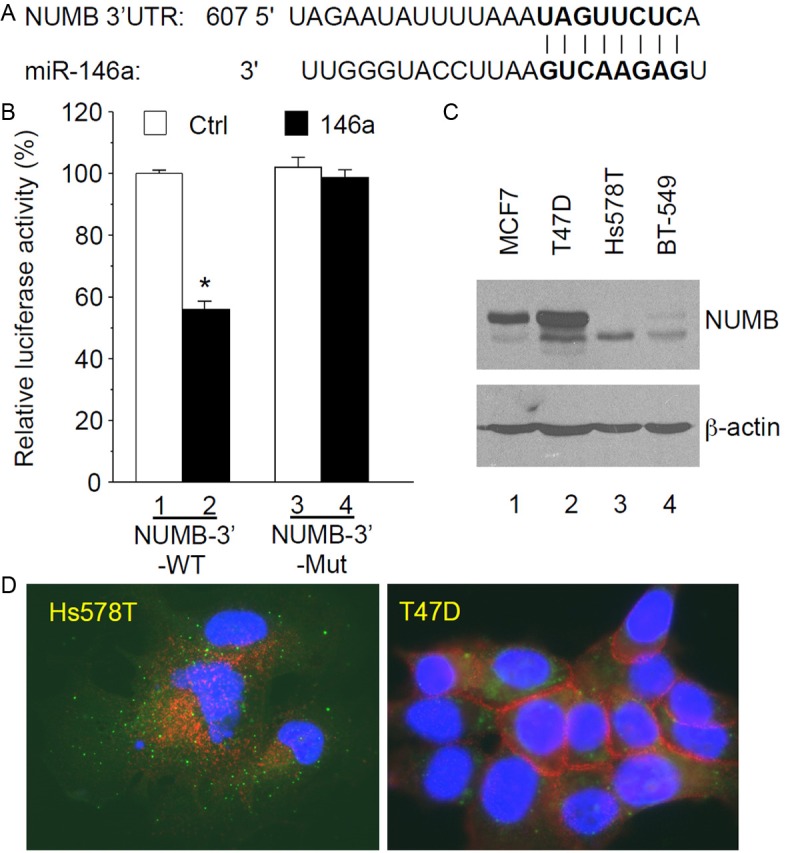
MiR-146a targets the Notch signaling inhibitor NUMB. A and B: miR-146a binds the 3’-UTR of NUMB transcript to inhibit NUMB translation. The miR-146a binding site in NUMB 3’-UTR was aligned with the miR-146a sequence (A). The wild type (WT) NUMB 3’-UTR or its mutant defective in miR-146a binding (MUT) was cloned into a luciferase reporter vector. The reporter plasmids were transfected into the MCF-10A cells stably expressing miR-146a or miR control (described in Figure 4C and 4D) for luciferase activity analysis (B). *P < 0.05 compared to column 1. C and D: NUMB expression is inversely correlated with miR-146a expression and invasiveness in breast cancer cell lines. Whole cell lysates were used for anti-NUMB blotting with β-actin as a loading control (C). The expression of the endogenous miR-146a (green dots) and NUMB protein (red) in the indicated breast cancer cell lines was determined by FISH-TSA as described in the Materials and Methods (D). The data represent the mean ± S.E. of at least three independent experiments in triplicate.
We then tested if overexpression of NUMB can interfere with the induction of the stem cell traits by KLF8. We infected the 10A-iK8 cells with lentiviruses expressing a NUMB-GFP fusion protein that does not contain the 3’-UTR of NUMB. Lentivirus expressing only GFP was used as a control. The cells were grown in mammosphere culture medium for 10 days in the presence of KLF8 expression. We found that the NUMB-GFP expressing cells formed 80% less mammospheres compared to the GFP expressing cells (Figure 7). This result suggested that KLF8 induces the stem cell traits by inhibiting NUMB expression via likely the induction of the miR-146a expression.
Figure 7.
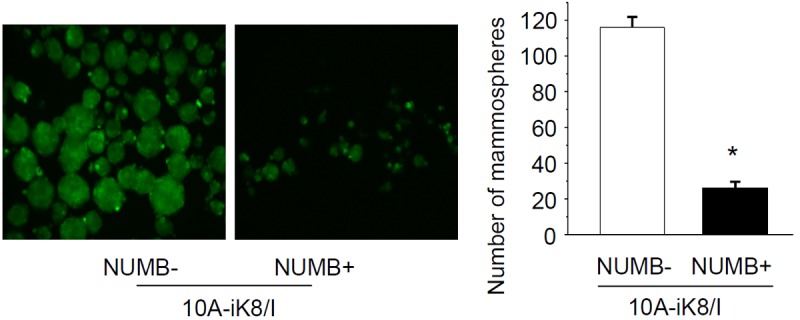
Overexpression of NUMB inhibits the induction of the stem cell traits by KLF8. 10A-iK8 cells expressing NUMB-GFP or GFP control were generated as described in Materials and Methods. The NUMB cDNA did not contain the 3’-UTR template. The cells were grown in stem cell culture plate under the induced conditions for 10 days to form mammospheres. The mammospheres were imaged and counted. The data represent the mean ± S.E. of at least three independent experiments in triplicate. *P < 0.05 compared to NUMB-group.
KLF8 potentiates oncogenic transformation of MCF-10A cells
To test if KLF8 plays a role in transforming the MCF-10A cells, we introduced the H-RasV12 oncogene into the 10A-iK8 cells. The tumorigenecity of the cells were determined in nude mice after mammary fat pad injection. By the end of month 7, no detectable tumors were formed by the parental MCF-10A cells or the 10A-iK8 cells regardless of the induction of KLF8 expression (data not shown). In the absence of KLF8 induction, the 10A-iK8-RasV12 cells formed extremely small tumors of < 5 mm3 (Figure 8A and 8B, U). In sharp contrast, when KLF8 expression in the cells was induced, the tumors grew up to 1.3 cm3 with a median size of approximately 0.5 cm3 (Figure 8A and 8B, I). This result suggested that KLF8 makes a critical contribution to the cell transformation presumably by inducing the stem cell traits. To confirm the induced expression of KLF8 and determine the expression pattern of NUMB protein in the tumors, we performed IHC co-staining experiments. We observed a high level of NUMB protein expression in the absence of KLF8 expression and a loss of NUMB expression in the tumors that expressed KLF8 (Figure 8C). Taken together, these results support that inhibition of NUMB expression and thus induction of stem cell traits by KLF8 (likely via miR-146a) is an important mechanism responsible for the cell transformation.
Figure 8.
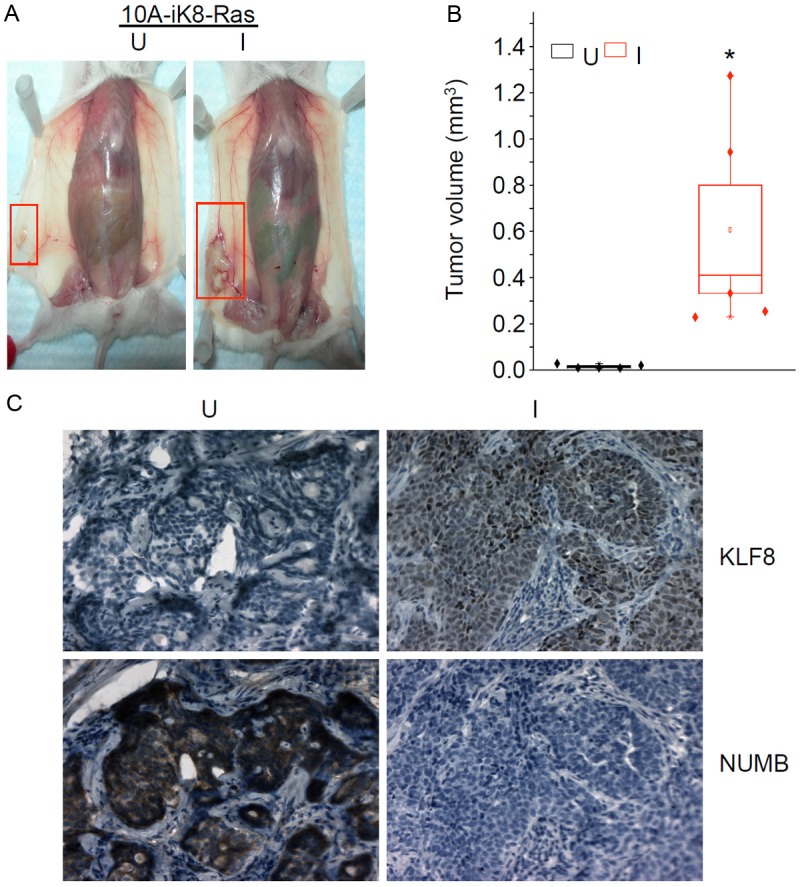
KLF8 potentiates Ras-induced transformation of MCF-10A cells. A and B: KLF8 enhances tumorigenecity of MCF-10A cells induced by H-Ras-V12. The 10A-iK8-V12 cells were generated as described in Materials and Methods. Cells of 1 x 106 were injected into the fat pad of nude mice. The mice were fed with the Dox Diet to induce (I) the expression of KLF8 in the tumor cells in vivo. Uninduced (U) KLF8 expression was maintained by feeding the mice with the Control Diet not supplemented with doxycyclin. Tumors formed by the cells were monitored for 7 months before the tumor images were taken (A) and tumor volumes were analyzed by box plot (B). P < 0.01 compared to U group. C: Induction of KLF8 expression led to a decrease in NUMB expression in the tumors. Specimens of the tumors from A & B were prepared for immunohistochemical staining of the expression of KLF8 and NUMB proteins.
Discussion
Using immortalized human non-tumorigenic mammary epithelial cells expressing inducible KLF8 or treated with TGF-β, a panel of human breast cancer cell lines representing epithelial or mesenchymal features, a combination of multiple markers for mammary stem cells, array screening and genetic modifications of miRs in the cells, we demonstrate that the miR-146a plays an important role in mediating the induction and maintenance of breast CSCs during EMT potentially by repressing the expression of the Notch signaling inhibitor NUMB. This finding provides new insights into the mechanisms for breast cancer progression and developing novel therapeutic strategies.
Our results are consistent with the recent reports supporting cancer-promoting role of miR-146a [45]. For example, in cisplatin-resistant MCF-7 lines that are characteristic of mesenchymal invasive cancer cell phenotype miR-146a is the most highly upregulated among all miRs compared to the cisplatin-sensitive, parental epithelial non-invasive MCF-7 cells [46]. MiR-146a was also found to be the most highly upregulated miR in triple negative sporadic breast tumors and basal-like mammary tumor epithelial cell lines and target the tumor suppressor BRCA1 to promote proliferation [47]. Evidence is also accumulating that supports a similar role of miR-146a in promoting EMT, malignant transformation or therapeutic resistance of various other types of cancer including bronchial [48], gastric [49], liver [50], thyroid [51], and pancreatic cancer [52], leukemia [53,54] and lymphoma [55,56]. On the other hand, some reports have indicated a tumor-inhibiting role of miR146a [57-59]. It remains to be answered whether this discrepancy was due to the difference in the experimental conditions, the stages of tumors or cancer types studied, or even an undiscovered double-faced role of miR-146a in cancer. Nevertheless, given that KLF8 supports tumor progression of multiple cancer types [8], miR-146a may contribute to the role for KLF8 in regulating the cancer types other than breast cancer as well.
Several miR-146a regulators have recently been reported including NF-κB [51,53,55,56], TGF-β [48] and breast cancer metastasis suppressor 1 [58]. On the other hand, transcripts of several genes have been identified as miR-146a targets such as NF-κB [57,59], BRCA1 [47], and SMAD4 [49,50,54]. It will be interesting to determine if KLF8 activates miR-146a promoter directly or indirectly by targeting these or other miR-146a regulating genes and whether other miR-146a target transcripts besides NUMB [21] are also regulated by KLF8 via miR-146a in the regulation of EMT and/or CSCs.
It should be noted that in addition to miR-146a, several other KLF8-regulated miRs also respond to EMT including miR-219-2-3p, miR-508-3p, and miR-513a-5p (see Figure 3). Interestingly, expression of some of these miRs is highly altered in either invasive or non-invasive breast cancer cells, yet others are modestly underexpressed in both invasive and non-invasive breast cancer cells. These results suggest that these KLF8 target/effector miRs may contribute differentially to the EMT-dependent induction of breast stem cells responsible for the development and growth of primary tumors, the initiation of tumor invasion and metastasis, the colonization and growth of the secondary tumors at the metastatic sites, or throughout the entire process of tumor progression. Besides these four miRs, other potential KLF8 target miRs (see Figure 2) may also play important roles in EMT-associated CSC induction. Studies have shown that miR-200 family and associated miRs regulate EMT and play an important role in tumor progression [60,61]. These miRs are similarly regulated in the cells expressing high levels of KLF8 (Figures 2 and 3), cisplatin-resistant mesenchymal breast cancer cells [46], and invasive prostate cancer cells [62]. Interestingly, miR-200 family miRs and the ZEBs mutually down-regulate each other, which has been shown to play an important role in EMT, CSCs and metastatic progression of cancer [60,63-69]. These results indicate that KLF8 and ZEBs may cooperate in regulating their common target miRs and also suggest an interesting possibility of cross-talk between KLF8 and ZEBs and potentially other EMT-regulating transcription factors through the miRs-mediated feedback regulation [60].
Resistance to cancer therapies is a major hurdle of clinical intervention of cancer and one of the hallmarks of CSCs [3]. Several recent studies have shown that miR-146a is an important positive regulator of drug resistance [46,50,54]. The other potential KLF8-downregulated miRs such as miR-200 family have been reported to inhibit resistance to therapeutic drugs such as microtubule-interfering chemotherapeutic agents [70-72]. We have recently demonstrated that KLF8 promotes cell resistance against the cytotoxic effects of DNA damaging agents via PARP-1-dependent DNA damage responses [23]. Interestingly, PARP-1 has been reported to contribute to resistance of hepatocellular carcinoma CSCs against multiple therapeutic drug types [73]. These results suggest that in addition to promoting tumorigenesis and metastasis, KLF8 could also play a role in maintaining or enhancing therapeutic resistance of CSCs by more than one mechanism and miRs could play a role downstream of KLF8 in the regulation of DNA damage responses. Lastly, the cytotoxic resistance could be one of the potential mechanisms provided by KLF8 that help the H-RasV12 expressing cells form tumors more easily and quickly. Experiments are in progress to test these interesting possibilities.
In summary, this report demonstrates a novel mechanism of breast cancer progression by which KLF8 induces CSCs to promote tumorigenesis and drug resistance. This finding provides new insights into developing novel therapeutic strategies to inhibit CSCs and sensitize tumor cells to the otherwise ineffective conventional therapies.
Acknowledgements
This work was supported by grants from NCI (CA132977), Susan G. Komen for the Cure Breast Cancer Foundation (KG090444 and KG080616) and NY State Stem Cell Research Program (N08G-497) to JZ.
Disclosure of conflict of interest
The authors declare no conflicts of interest.
Abbreviations
- KLF8
Krüppel-like factor 8
- EMT
epithelial to mesenchymal transition
- miR-146a
microRNA-146a
- ALDH
aldehyde dehydrogenase
- CSC
cancer stem cell
- 10A-iK8
MCF-10A line that expresses inducible KLF8
- 10A-iK8-146a-sponge
the 10A-iK8-sponge cell line where miR-146a was constitutively blocked by its inhibiting sponge
- 10A-iK8-mock-sponge
a sponge mock control cell line
- 231-K8ikd
the MDA-MB-231 line that expresses inducible KLF8 short hairpin RNA
References
- 1.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Takebe N, Warren RQ, Ivy SP. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011;13:211. doi: 10.1186/bcr2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou N, Mo YY. Roles of microRNAs in cancer stem cells. Front Biosci (Schol Ed) 2012;4:810–818. doi: 10.2741/s301. [DOI] [PubMed] [Google Scholar]
- 7.Mo YY. MicroRNA regulatory networks and human disease. Cell Mol Life Sci. 2012;69:3529–3531. doi: 10.1007/s00018-012-1123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lahiri SK, Zhao J. Kruppel-like factor 8 emerges as an important regulator of cancer. Am J Transl Res. 2012;4:357–363. [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung MC, Zhao J. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007;67:7184–7193. doi: 10.1158/0008-5472.CAN-06-4729. [DOI] [PubMed] [Google Scholar]
- 10.Li JC, Yang XR, Sun HX, Xu Y, Zhou J, Qiu SJ, Ke AW, Cui YH, Wang ZJ, Wang WM, Liu KD, Fan J. Up-regulation of Kruppel-like factor 8 promotes tumor invasion and indicates poor prognosis for hepatocellular carcinoma. Gastroenterology. 2010;139:2146–2157. e2112. doi: 10.1053/j.gastro.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Urvalek AM, Lu H, Wang X, Li T, Yu L, Zhu J, Lin Q, Zhao J. Regulation of the oncoprotein KLF8 by a switch between acetylation and sumoylation. Am J Transl Res. 2011;3:121–132. [PMC free article] [PubMed] [Google Scholar]
- 12.Urvalek AM, Wang X, Lu H, Zhao J. KLF8 recruits the p300 and PCAF co-activators to its amino terminal activation domain to activate transcription. Cell Cycle. 2010;9:601–611. doi: 10.4161/cc.9.3.10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei H, Wang X, Gan B, Urvalek AM, Melkoumian ZK, Guan JL, Zhao J. Sumoylation delimits KLF8 transcriptional activity associated with the cell cycle regulation. J Biol Chem. 2006;281:16664–16671. doi: 10.1074/jbc.M513135200. [DOI] [PubMed] [Google Scholar]
- 14.Zhao J, Bian ZC, Yee K, Chen BP, Chien S, Guan JL. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol Cell. 2003;11:1503–1515. doi: 10.1016/s1097-2765(03)00179-5. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, DiPersio CM, Feustel PJ, Zhao J. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene. 2011;30:1901–1911. doi: 10.1038/onc.2010.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Urvalek AM, Liu J, Zhao J. Activation of KLF8 transcription by focal adhesion kinase in human ovarian epithelial and cancer cells. J Biol Chem. 2008;283:13934–13942. doi: 10.1074/jbc.M709300200. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Zhao J. KLF8 transcription factor participates in oncogenic transformation. Oncogene. 2007;26:456–461. doi: 10.1038/sj.onc.1209796. [DOI] [PubMed] [Google Scholar]
- 18.Fu WJ, Li JC, Wu XY, Yang ZB, Mo ZN, Huang JW, Xia GW, Ding Q, Liu KD, Zhu HG. Small interference RNA targeting Kruppel-like factor 8 inhibits the renal carcinoma 786-0 cells growth in vitro and in vivo. J Cancer Res Clin Oncol. 2010;136:1255–1265. doi: 10.1007/s00432-010-0776-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L, Liu N, Xu M, Liu Y, Min J, Pang H, Zhang N, Zhang H. Lentivirus-delivered Kruppel-like factor 8 small interfering RNA inhibits gastric cancer cell growth in vitro and in vivo. Tumour Biol. 2012;33:53–61. doi: 10.1007/s13277-011-0245-7. [DOI] [PubMed] [Google Scholar]
- 20.Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006;99:35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- 21.Schnell O, Romagna A, Jaehnert I, Albrecht V, Eigenbrod S, Juerchott K, Kretzschmar H, Tonn JC, Schichor C. Kruppel-like factor 8 (KLF8) is expressed in gliomas of different WHO grades and is essential for tumor cell proliferation. PLoS One. 2012;7:e30429. doi: 10.1371/journal.pone.0030429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan W, Zhu J, Sun X, Tang W. Small interfering RNA targeting Kruppel-like factor 8 inhibits U251 glioblastoma cell growth by inducing apoptosis. Mol Med Rep. 2012;5:347–350. doi: 10.3892/mmr.2011.669. [DOI] [PubMed] [Google Scholar]
- 23.Lu H, Hu L, Li T, Lahiri S, Shen C, Wason MS, Mukherjee D, Xie H, Yu L, Zhao J. A novel role of Kruppel-like factor 8 in DNA repair in breast cancer cells. J Biol Chem. 2012 Dec 21;287:43720–9. doi: 10.1074/jbc.M112.418053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT, Turkson J. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109:9623–9628. doi: 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu H, Wang X, Urvalek AM, Li T, Xie H, Yu L, Zhao J. Transformation of human ovarian surface epithelial cells by Krüppel-like factor 8. Oncogene. 2012 doi: 10.1038/onc.2012.545. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang T, Cai SY, Zhang J, Lu J, Lin C, Zhai J, Wu M, Shen F. Krüppel-Like Factor 8 Is a New Wnt/Beta-Catenin Signaling Target Gene and Regulator in Hepatocellular Carcinoma. PLoS One. 2012;7:e39668. doi: 10.1371/journal.pone.0039668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q, Gao Y, Jia Z, Mishra L, Guo K, Li Z, Le X, Wei D, Huang S, Xie K. Dysregulated Kruppel-like factor 4 and vitamin D receptor signaling contribute to progression of hepatocellular carcinoma. Gastroenterology. 2012;143:799–810. e791–792. doi: 10.1053/j.gastro.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nandan MO, Yang VW. The role of Kruppel-like factors in the reprogramming of somatic cells to induced pluripotent stem cells. Histol Histopathol. 2009;24:1343–1355. doi: 10.14670/hh-24.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Vliet J, Turner J, Crossley M. Human Kruppel-like factor 8: a CACCC-box binding protein that associates with CtBP and represses transcription. Nucleic Acids Res. 2000;28:1955–1962. doi: 10.1093/nar/28.9.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang P, Basu P, Redmond LC, Morris PE, Rupon JW, Ginder GD, Lloyd JA. A functional screen for Kruppel-like factors that regulate the human gamma-globin gene through the CACCC promoter element. Blood Cells Mol Dis. 2005;35:227–235. doi: 10.1016/j.bcmd.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Eaton SA, Funnell AP, Sue N, Nicholas H, Pearson RC, Crossley M. A network of Kruppel-like Factors (Klfs). Klf8 is repressed by Klf3 and activated by Klf1 in vivo. J Biol Chem. 2008;283:26937–26947. doi: 10.1074/jbc.M804831200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 33.Lu H, Wang X, Li T, Urvalek AM, Yu L, Li J, Zhu J, Lin Q, Peng X, Zhao J. Identification of poly (ADP-ribose) polymerase-1 (PARP-1) as a novel Kruppel-like factor 8-interacting and -regulating protein. J Biol Chem. 2011;286:20335–20344. doi: 10.1074/jbc.M110.215632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehta TS, Lu H, Wang X, Urvalek AM, Nguyen KH, Monzur F, Hammond JD, Ma JQ, Zhao J. A unique sequence in the N-terminal regulatory region controls the nuclear localization of KLF8 by cooperating with the C-terminal zinc-fingers. Cell Res. 2009;19:1098–1109. doi: 10.1038/cr.2009.64. [DOI] [PubMed] [Google Scholar]
- 35.Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36(Suppl 1):59–72. doi: 10.1046/j.1365-2184.36.s.1.6.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- 37.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mingle LA, Bonamy G, Barroso M, Liao G, Liu G. LPA-induced mutually exclusive subcellular localization of active RhoA and Arp2 mRNA revealed by sequential FRET and FISH. Histochem Cell Biol. 2009;132:47–58. doi: 10.1007/s00418-009-0589-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu C, Tang DG. MicroRNA regulation of cancer stem cells. Cancer Res. 2011;71:5950–5954. doi: 10.1158/0008-5472.CAN-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu S, Clouthier SG, Wicha MS. Role of microRNAs in the regulation of breast cancer stem cells. J Mammary Gland Biol Neoplasia. 2012;17:15–21. doi: 10.1007/s10911-012-9242-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wright JA, Richer JK, Goodall GJ. microRNAs and EMT in mammary cells and breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:213–223. doi: 10.1007/s10911-010-9183-z. [DOI] [PubMed] [Google Scholar]
- 43.Zimmerman AL, Wu S. MicroRNAs, cancer and cancer stem cells. Cancer Lett. 2011;300:10–19. doi: 10.1016/j.canlet.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009;279:8–12. doi: 10.1016/j.canlet.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elsarraj HS, Stecklein SR, Valdez K, Behbod F. Emerging functions of microRNA-146a/b in development and breast cancer: microRNA-146a/b in development and breast cancer. J Mammary Gland Biol Neoplasia. 2012;17:79–87. doi: 10.1007/s10911-012-9240-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pogribny IP, Filkowski JN, Tryndyak VP, Golubov A, Shpyleva SI, Kovalchuk O. Alterations of microRNAs and their targets are associated with acquired resistance of MCF-7 breast cancer cells to cisplatin. Int J Cancer. 2010;127:1785–1794. doi: 10.1002/ijc.25191. [DOI] [PubMed] [Google Scholar]
- 47.Garcia AI, Buisson M, Bertrand P, Rimokh R, Rouleau E, Lopez BS, Lidereau R, Mikaelian I, Mazoyer S. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol Med. 2011;3:279–290. doi: 10.1002/emmm.201100136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Nelson A, Wang X, Kanaji N, Kim M, Sato T, Nakanishi M, Li Y, Sun J, Michalski J, Patil A, Basma H, Rennard SI. MicroRNA-146a modulates human bronchial epithelial cell survival in response to the cytokine-induced apoptosis. Biochem Biophys Res Commun. 2009;380:177–182. doi: 10.1016/j.bbrc.2009.01.066. [DOI] [PubMed] [Google Scholar]
- 49.Xiao B, Zhu ED, Li N, Lu DS, Li W, Li BS, Zhao YL, Mao XH, Guo G, Yu PW, Zou QM. Increased miR-146a in gastric cancer directly targets SMAD4 and is involved in modulating cell proliferation and apoptosis. Oncol Rep. 2012;27:559–566. doi: 10.3892/or.2011.1514. [DOI] [PubMed] [Google Scholar]
- 50.Tomokuni A, Eguchi H, Tomimaru Y, Wada H, Kawamoto K, Kobayashi S, Marubashi S, Tanemura M, Nagano H, Mori M, Doki Y. miR-146a suppresses the sensitivity to interferon-alpha in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2011;414:675–680. doi: 10.1016/j.bbrc.2011.09.124. [DOI] [PubMed] [Google Scholar]
- 51.Pacifico F, Crescenzi E, Mellone S, Iannetti A, Porrino N, Liguoro D, Moscato F, Grieco M, Formisano S, Leonardi A. Nuclear factor-{kappa}B contributes to anaplastic thyroid carcinomas through up-regulation of miR-146a. J Clin Endocrinol Metab. 2010;95:1421–1430. doi: 10.1210/jc.2009-1128. [DOI] [PubMed] [Google Scholar]
- 52.Yu J, Li A, Hong SM, Hruban RH, Goggins M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin Cancer Res. 2012;18:981–992. doi: 10.1158/1078-0432.CCR-11-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pichler K, Schneider G, Grassmann R. MicroRNA miR-146a and further oncogenesis-related cellular microRNAs are dysregulated in HTLV-1-transformed T lymphocytes. Retrovirology. 2008;5:100. doi: 10.1186/1742-4690-5-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhong H, Wang HR, Yang S, Zhong JH, Wang T, Wang C, Chen FY. Targeting Smad4 links microRNA-146a to the TGF-beta pathway during retinoid acid induction in acute promyelocytic leukemia cell line. Int J Hematol. 2010;92:129–135. doi: 10.1007/s12185-010-0626-5. [DOI] [PubMed] [Google Scholar]
- 55.Cameron JE, Yin Q, Fewell C, Lacey M, McBride J, Wang X, Lin Z, Schaefer BC, Flemington EK. Epstein-Barr virus latent membrane protein 1 induces cellular MicroRNA miR-146a, a modulator of lymphocyte signaling pathways. J Virol. 2008;82:1946–1958. doi: 10.1128/JVI.02136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Motsch N, Pfuhl T, Mrazek J, Barth S, Grasser FA. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microRNA miR-146a. RNA Biol. 2007;4:131–137. doi: 10.4161/rna.4.3.5206. [DOI] [PubMed] [Google Scholar]
- 57.Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008;27:5643–5647. doi: 10.1038/onc.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hurst DR, Edmonds MD, Scott GK, Benz CC, Vaidya KS, Welch DR. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 2009;69:1279–1283. doi: 10.1158/0008-5472.CAN-08-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y, Vandenboom TG 2nd, Wang Z, Kong D, Ali S, Philip PA, Sarkar FH. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70:1486–1495. doi: 10.1158/0008-5472.CAN-09-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korpal M, Kang Y. The emerging role of miR-200 family of microRNAs in epithelial-mesenchymal transition and cancer metastasis. RNA Biol. 2008;5:115–119. doi: 10.4161/rna.5.3.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, Wei Y, Hu G, Garcia BA, Ragoussis J, Amadori D, Harris AL, Kang Y. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17:1101–1108. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu J, Ohuchida K, Mizumoto K, Sato N, Kayashima T, Fujita H, Nakata K, Tanaka M. MicroRNA, hsa-miR-200c, is an independent prognostic factor in pancreatic cancer and its upregulation inhibits pancreatic cancer invasion but increases cell proliferation. Mol Cancer. 2010;9:169. doi: 10.1186/1476-4598-9-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 65.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 66.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schuler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 69.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A, Kurie JM. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140–2151. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cochrane DR, Spoelstra NS, Howe EN, Nordeen SK, Richer JK. MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol Cancer Ther. 2009;8:1055–1066. doi: 10.1158/1535-7163.MCT-08-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y, VandenBoom TG 2nd, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adam L, Zhong M, Choi W, Qi W, Nicoloso M, Arora A, Calin G, Wang H, Siefker-Radtke A, McConkey D, Bar-Eli M, Dinney C. miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res. 2009;15:5060–5072. doi: 10.1158/1078-0432.CCR-08-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu XL, Xing BC, Han HB, Zhao W, Hu MH, Xu ZL, Li JY, Xie Y, Gu J, Wang Y, Zhang ZQ. The properties of tumor-initiating cells from a hepatocellular carcinoma patient’s primary and recurrent tumor. Carcinogenesis. 2010;31:167–174. doi: 10.1093/carcin/bgp232. [DOI] [PubMed] [Google Scholar]