

Summary
Prion diseases are fatal neurodegenerative diseases characterised by the accumulation of misfolded prion protein (PrPSc) in the brain. They are caused by the templated misfolding of normal cellular protein, PrPC, by PrPSc. We have recently generated a unique cell system in which epitope-tagged PrPC competent to produce bona fide PrPSc is expressed in neuroblastoma cells. Using this system we demonstrated that PrPSc forms on the cell surface within minutes of prion exposure. Here, we describe the intracellular trafficking of newly formed PrPSc. After formation in GM1-enriched lipid microdomains at the plasma membrane, PrPSc is rapidly internalised to early endosomes containing transferrin and cholera toxin B subunit. Following endocytosis, PrPSc intracellular trafficking diverges: some is recycled to the plasma membrane via Rab11-labelled recycling endosomes; the remaining PrPSc is subject to retromer-mediated retrograde transport to the Golgi. This pathway leads to lysosomal degradation, and we show that this is the dominant PrPSc degradative mechanism in the early stages of prion infection.
Key words: Prions, Intracellular trafficking, Retrograde transport, Lysosomes
Introduction
Many neurodegenerative diseases result from the aggregation of misfolded proteins. Indeed, their shared mechanism provides the basis for a new class of pathologies: the protein misfolding disorders. These include Alzheimer's, Huntington's and Parkinson's diseases, as well as prion diseases. Prion diseases are the prototypical protein misfolding disorders, as their pathogenesis is associated solely with aberrant misfolding of a host cellular protein, the normal cellular PrP (PrPC) to abnormal conformers (PrPSc), in agreement with the ‘protein-only’ hypothesis (Prusiner, 1982). PrPC is a glycophosphatidylinositol (GPI)-linked peripheral membrane protein found principally at the plasma membrane. It is monomeric and rich in α-helical structure whereas the disease-associated PrPSc is characterised by an increase in β-sheets, detergent insolubility and partial resistance to proteolysis. Neuropathologically, prion diseases are associated with severe neuronal loss, marked gliosis, spongiform change and accumulation of PrPSc in the brain. A major gap exists in our understanding of how conversion of PrPC to PrPSc causes neuronal dysfunction and death. Various mechanisms have been proposed including synaptic impairment (Senatore et al., 2012), ubiquitin–proteasome system (UPS) dysfunction (Kristiansen et al., 2007), endoplasmic reticulum (ER) stress (Hetz et al., 2003; Rane et al., 2008), toxicity due to the presence of PrP species in the cytosol (Chakrabarti and Hegde, 2009; Kristiansen et al., 2005; Ma et al., 2002), and unfolded protein response induction and translational arrest (Moreno et al., 2012). To assess the contribution of these mechanisms to prion-induced toxicity, it is essential to know the exact cellular trafficking pathways of PrPSc. That is, where is it formed and what cellular compartments does it traffic to within the prion-infected cell.
Numerous intracellular compartments have been proposed as the initial site of prion conversion (i.e. PrPSc formation): the endolysosomal system (Borchelt et al., 1992; Magalhães et al., 2005), the endosomal recycling compartment (ERC) (Marijanovic et al., 2009) and the trans-Golgi network (TGN)/Golgi/ER (Béranger et al., 2002). Critically, these studies relied on the use of chronically infected cell lines, which require serial sub-passage to ensure inocula-derived PrPSc had been eliminated. These studies are further complicated by the lack of PrPSc-specific antibodies, making fixation and processing to remove PrPC prior to immunostaining necessary (Kristiansen et al., 2005; Veith et al., 2009). As a result, details of the initial stages of prion conversion and early trafficking events remained elusive. We recently generated a unique cell system in which epitope-tagged PrPC is expressed in PrP knockdown neuroblastoma cells. The PrP-224AlaMYC chimera supports prion replication and results in the production of a bona fide epitope-tagged PrPSc (Goold et al., 2011). Using anti-MYC antibodies in conjunction with formic acid treatment to remove PrPC (Goold et al., 2011; Kristiansen et al., 2005), we were able to distinguish newly formed PrPSc generated in the recipient cell from both cellular PrPC and inocula-derived PrPSc species (Goold et al., 2011; Kristiansen et al., 2005). Our initial studies have unequivocally demonstrated that prion infection of cells is extremely rapid, occurring within one minute of prion exposure, and that the plasma membrane is the initial site of prion conversion. Further, de novo produced PrPSc formed at the plasma membrane is rapidly endocytosed and trafficked to a perinuclear compartment. Cells fixed shortly after prion exposure host PrPSc in a diffuse cellular pattern, reflecting its transition through an early endosomal compartment. A short time later, the cells have assumed a characteristic phenotype with PrPSc found primarily at the plasma membrane and in the perinuclear region, which is densely packed with organelles, including early endosomes, recycling endosomes, the TGN and Golgi. This steady-state distribution is maintained thereafter as the cells continue to stably propagate PrPSc.
Here, we extend our previous work by taking advantage of the PrP-224AlaMYC cell system to map the intracellular trafficking of PrPSc following its initial formation at the plasma membrane. We show that newly formed PrPSc co-localises with cholera toxin B subunit (CTB), a well characterised marker of GM1-enriched membrane microdomains, at and near the cell surface. PrPSc is endocytosed to early endosome-associated protein 1 (EEA1), transferrin (Tf) and CTB-labelled organelles. PrPSc is then segregated into two pathways: it can be recycled back to the plasma membrane via a Rab11-positive recycling compartment or rapidly sorted to the TGN and the Golgi apparatus. We show that the retromer complex mediates PrPSc trafficking to the TGN. Further, we provide evidence that PrPSc reaching the Golgi is rapidly transferred to lysosomes and degraded, suggesting that this pathway is the major degradative mechanism in the early stages of prion infection.
Results
PrPSc co-localises with transferrin and cholera toxin B following endocytosis
To analyse PrPSc intracellular transport in detail, we compared its distribution shortly after its formation with the distribution of three well-defined trafficking cargoes – Tf, CTB and dextran. These fluorescently labelled molecules are predominantly found in the ERC (Tf), in GM1-enriched membrane microdomains and along the retrograde pathway (CTB), and within the endolysosomal system (dextran) (Baravalle et al., 2005; Lencer and Tsai, 2003). PrP-224AlaMYC cells were labelled with the individual cargoes and exposed to RML prions for 2 min prior to fixation. Cells were then treated with formic acid and immunostained with anti-MYC antibodies, a process that allows specific visualisation of de novo produced PrPSc by confocal microscopy. After 2 min exposure to prions, co-localisation was observed with all three cargoes, but most notably with CTB (Fig. 1). PrPSc/CTB co-localisation was observed at and near the plasma membrane and also in more perinuclear compartments. PrPSc also co-distributed with Tf in this compartment (Fig. 1).
Fig. 1.
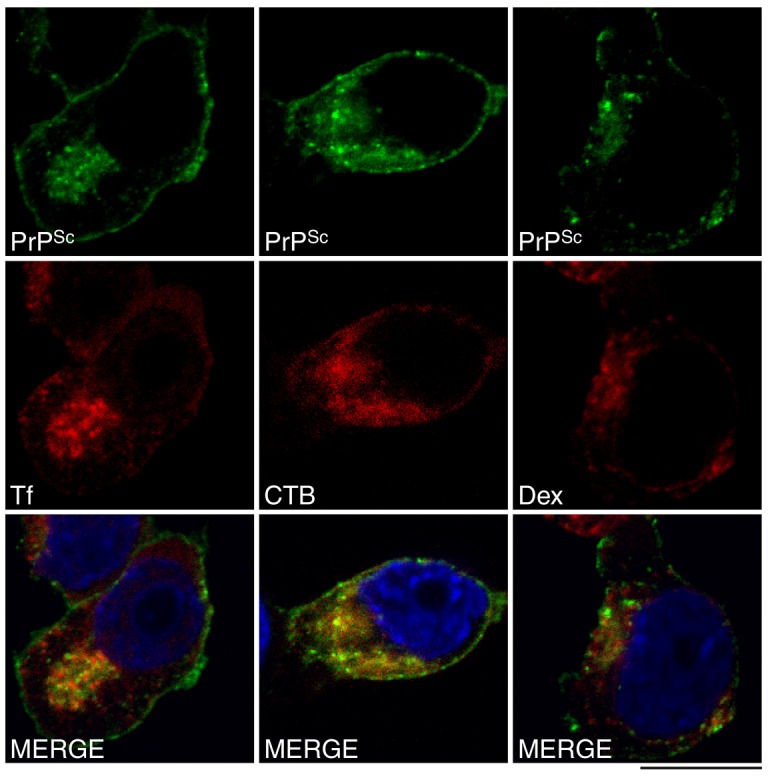
Newly formed PrPSc co-localises with cholera toxin B and transferrin in prion-infected cells. PrP-224AlaMYC cells were incubated with labelled cholera toxin B (CTB), transferrin (Tf) and dextran (Dex) and then exposed to prions for 2 min. Cells were fixed and processed to reveal PrPSc and imaged by confocal microscopy. Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. PrPSc (green) co-localises with all cargoes (red) but most notably with CTB. Scale bar: 10 µm.
Newly formed PrPSc traffics through early endosomes, recycling endosomes, the TGN and the Golgi apparatus
Co-localisation with CTB and Tf suggests PrPSc undergoes retrograde transport and may be trafficked to recycling endosomes. To explore this further we measured the extent of PrPSc co-localisation with well-defined organelle markers. PrP-224AlaMYC cells were exposed to RML prions and fixed at serial time-points up to 16 min. Cells were then processed to reveal PrPSc and the different organelle markers for confocal microscopy. The proportion of total PrPSc that co-localised with each organelle marker was determined. Co-localisation of PrPSc with the early endosomal marker EEA1 and the recycling endosomal marker Rab11 was observed at early time points (Fig. 2A,B). Later, PrPSc co-localisation with the TGN marker TGN46 and the Golgi marker GM130 was observed (Fig. 2A,B). Little co-localisation with the ER marker protein disulphide isomerase (PDI) was observed (Fig. 2C,D; supplementary material Fig. S1). This apparent co-distribution between PDI and PrPSc is likely to be due to our inability to distinguish juxtaposed signals in the crowded perinuclear region of the cell. This non-specific co-localisation accounted for less than 5% of that measured for EEA1 after 16 min. A low, yet consistent, level of co-localisation of PrPSc with LAMP1, a late endosome and lysosomal marker, was also observed (Fig. 2C,D). Overall, our data suggests that PrPSc moves from its site of formation at the plasma membrane through the cell periphery to the ERC, TGN and Golgi apparatus. This implies at least two trafficking routes are followed by PrPSc; that is, the recycling and the retrograde pathways.
Fig. 2.

PrPSc co-localises with markers for early endosomes, recycling endosomes, the trans Golgi network and Golgi. (A) PrP-224AlaMYC cells were fixed after 2 and 16 min of exposure to prions and processed to reveal PrPSc and organelle markers EEA1 (early endosomes), Rab11 (recycling endosomes), TGN46 (TGN) and GM130 (Golgi). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. After 2 min, PrPSc (green) is found at the plasma membrane, the cell periphery and in the perinuclear region and co-localises with EEA1 and Rab11 and less well with TGN46 and GM130 (red). After 16 min of exposure to prions, intracellular PrPSc accumulates close to the nucleus where it co-localises with EEA1, Rab11, TGN46 and GM130. Scale bar: 10 µm. (B) The proportion of PrPSc that co-localises with the indicated organelle markers was quantified at various time points following prion addition (mean ± s.e.m., n = 4). A transition from early/recycling endosomes to the TGN and Golgi is revealed. (C) PrP-224AlaMYC cells were fixed after a 4 min exposure to prions and processed to reveal PrPSc (green) and organelle markers protein disulphide isomerase (ER; red) and LAMP1 (late endosomes and lysosomes; red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. Little co-localisation with the ER was observed but a low level of PrPSc was consistently found in lysosomes. Scale bar: 10 µm. (D) The proportion of PrPSc that co-localises with protein disulphide isomerase and LAMP1 after a 4 min exposure to prions was quantified (mean ± s.e.m., n = 4).
PrPSc is a cargo for retrograde transport
Close association with CTB and high co-localisation with TGN46 and GM130 markers suggests that PrPSc undergoes retrograde transport to the TGN and Golgi. However, the close juxtaposition of several types of organelles in the perinuclear region of PrP-224AlaMYC cells makes a precise localisation difficult. Therefore, we added brefeldin A (BFA) to chronically prion-infected PrP-224AlaMYC cells to break up the Golgi apparatus into dispersed vesicles termed ‘ministacks’ (Alcalde et al., 1992). BFA treatment enabled us to visualise the degree of co-localisation of PrPSc with GM130 with a higher spatial resolution. Distinct vesicles labelled with both PrPSc and GM130 were clearly visible (Fig. 3A). In an independent experiment, we added BFA prior to RML prion exposure and examined the effect on PrPSc distribution. A gross disruption in PrPSc distribution was observed, with a more diffuse localisation pattern, indicating delivery to the perinuclear region was inhibited (supplementary material Fig. S2A,B). This effect occurred within 16 min of prion exposure, well before any reduction in PrPSc levels induced by BFA were observed (supplementary material Fig. S2C). BFA also inhibited accumulation of CTB in the perinuclear region indicating that retrograde trafficking of this cargo was inhibited (supplementary material Fig. S3A). This suggests that the trafficking of PrPSc to the perinuclear region is dependent on an intact Golgi and/or ER and is consistent with the retrograde trafficking of PrPSc. To test this hypothesis we used Retro-2, a small molecule inhibitor of retrograde transport (Stechmann et al., 2010). Consistent with published data, we found no detectable changes in EEA1, Rab11, TGN46 or GM130 distribution in PrP-224AlaMYC cells following Retro-2 treatment (data not shown). However, co-localisation of CTB with a Golgi marker was reduced indicating that retrograde trafficking of this cargo was inhibited (supplementary material Fig. S3A). Retro-2 treatment of PrP-224AlaMYC cells prior to prion exposure did not affect prion infection or change the gross distribution of PrPSc in the infected cells. However, PrPSc co-localisation with GM130 was reduced in treated cells relative to controls, indicating that transport to the Golgi was inhibited (Fig. 3B,C). This suggests that PrPSc is trafficked via the retrograde pathway from endosomes to the Golgi, a process which has been shown to be mediated by the retromer complex (Bonifacino and Hurley, 2008).
Fig. 3.

PrPSc is a substrate for retrograde transport. (A) Chronically infected PrP-224AlaMYC cells were treated with brefeldin A (BFA) for 15 min prior to fixation and processed to reveal PrPSc (green) and GM130 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. BFA treatment induces the formation of dispersed Golgi ministacks, some of which contain PrPSc, which appear yellow in the merged image (arrow). Scale bar: 10 µm. (B) PrP-224AlaMYC cells were treated with vehicle or Retro-2 prior to exposure to prions for 4 min and fixation. Cells were processed to reveal PrPSc (green) and GM130 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. No gross effects on PrPSc or GM130 distribution were observed. Scale bar: 10 µm. (C) The proportion of PrPSc that co-localises with GM130 in the presence and absence of Retro-2 was quantified at various time points following prion addition (mean ± s.e.m., n = 4). Retro-2 inhibits PrPSc transfer to the Golgi. (D) PrP-224AlaMYC cells were mock transfected or transfected with siRNA oligos directed against Vps35. Cells were exposed to prions for 4 min, fixed and processed to reveal PrPSc (green), VPS35 or TGN46 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. RNAi treatment reduced VPS35 expression but had no gross effects on PrPSc or TGN46 distribution. Scale bars: 10 µm. (E) The proportion of PrPSc that co-localises with TGN46 after mock or VPS35 RNAi transfection was quantified at various time points following prion addition (mean ± s.e.m., n = 3). VPS35 knockdown retarded PrPSc delivery to the TGN.
To confirm this hypothesis, we targeted retromer function with RNAi directed against one of its core subunits, VPS35. This treatment reduced VPS35 expression in PrP-224AlaMYC cells by ∼70%, as determined by western blot (supplementary material Fig. S3B) and confirmed by immunofluorescence staining (Fig. 3D). We tested its functional role in PrPSc trafficking by quantifying the extent of co-localisation with TGN46, in VPS35 RNAi and mock-treated PrP-224AlaMYC cells in the early minutes of prion exposure. Gross PrPSc and TGN46 distribution were unchanged by this treatment (Fig. 3D). However, we found that co-localisation between PrPSc and TGN46 was significantly reduced by the downregulation of VPS35 relative to control after 4 min prion exposure (Fig. 3E). By 16 min, no difference in co-localisation was detected. This partial effect is likely to be due to the incomplete VPS35 knockdown, which in turn affects the kinetics of transport.
A fraction of PrPSc is recycled to the plasma membrane
If our hypothesis that the plasma membrane is a major site of prion conversion is correct, then prion propagation (i.e. PrPSc formation in the absence of exogenous inocula) would require recycling of at least a portion of the internalised PrPSc to the cell surface. To test this hypothesis, we used phosphoinositol-specific phospholipase C (PI-PLC), an enzyme that cleaves GPI-linked proteins from their membrane anchor (Enari et al., 2001). Chronically infected PrP-224AlaMYC cells were treated with PI-PLC at 4°C for 30 min to cleave surface PrP (PrPC and PrPSc) from cells. Since this temperature shift inhibits vesicular trafficking (including recycling to the plasma membrane), intracellular PrP remains largely unaffected. Cells were washed with cold media, shifted to 37°C and the reappearance of cell surface PrPSc monitored. Nearly all of the control cells incubated at 4°C in the absence of PI-PLC showed plasma membrane PrPSc (Fig. 4A,B). As expected, PI-PLC effectively removed most of the PrPSc from the surface of the cells whilst leaving PrPSc in the perinuclear region (Fig. 4A,B). Interestingly, after 10 min at 37°C more cells showed membrane PrPSc and by 30 min at 37°C, the proportion had nearly reached control levels (Fig. 4A,B). Overall levels of PrPSc were reduced by PI-PLC treatment even after 30 min incubation at 37°C. This is probably due both to the initial reduction in PrPSc cleaved from the plasma membrane and also due to the reduction of the overall level of PrPC at the cell surface, the substrate for further PrPSc formation, which will not have returned to steady state in the timespan of the assay (Nunziante et al., 2003).
Fig. 4.
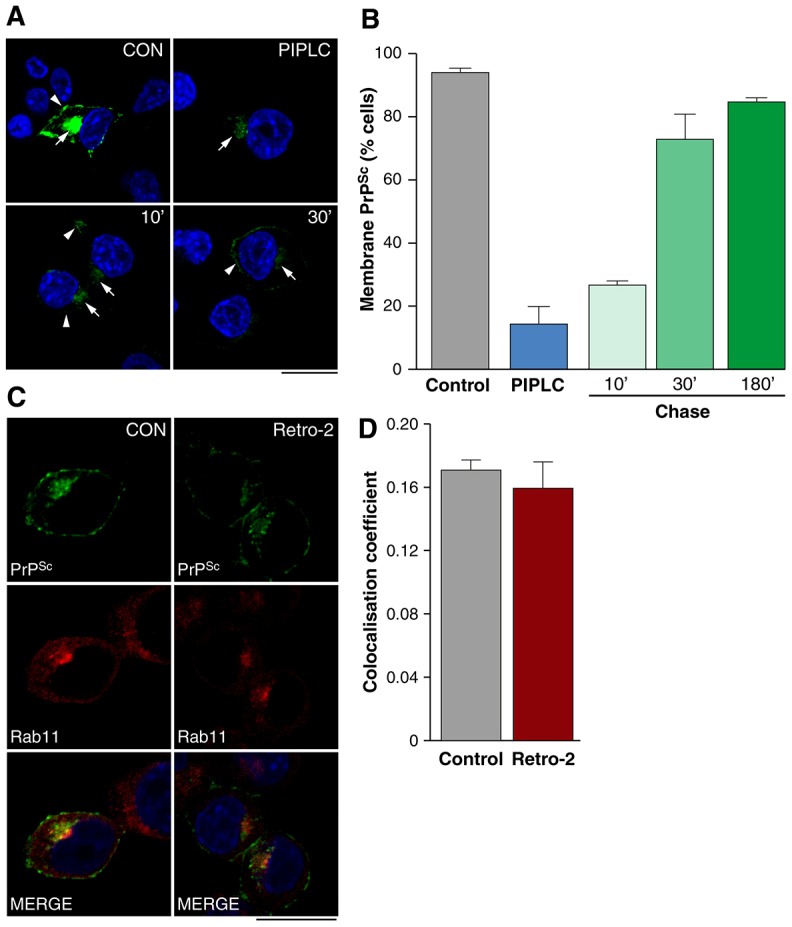
PrPSc is recycled to the plasma membrane. (A) Chronically infected PrP-224AlaMYC cells were incubated for 30 min at 4°C in control conditions or in the presence of PIPLC then fixed immediately or transferred to 37°C for the indicated times. Cells were processed to reveal PrPSc (green) and nuclei were visualised with DAPI (blue). Merged confocal images are shown. After incubation at 4°C, PrPSc was found at the plasma membrane (arrowhead) and in the perinuclear region (arrow). PIPLC effectively removes PrPSc from the cell surface under these conditions but does not affect intracellular PrPSc (arrow). Incubation at 37°C results in the recycling of internal PrPSc to the cell surface (arrowheads). Scale bar: 10 µm. (B) Quantification of the proportion of cells with membrane PrPSc (mean ± s.e.m., n = 3). (C) PrP-224AlaMYC cells were treated with vehicle or Retro-2 prior to exposure to prions for 4 min and fixation. The cells were processed to reveal PrPSc (green) and Rab11 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. Scale bar: 10 µm. (D) The proportion of PrPSc that co-localises with Rab11 in the presence and absence of Retro-2 was quantified 4 min following prion addition (mean ± s.e.m., n = 4). Retro-2 did not affect PrPSc trafficking through the recycling endosome.
This data confirms the recycling of PrPSc and supports our hypothesis that the plasma membrane is a major site of prion conversion. We used Retro-2 to assess if passage through the TGN is necessary for PrPSc recycling via the ERC. Control PrP-224AlaMYC cells or cells pretreated with Retro-2 were exposed to prions and fixed after 4 min when the effect of Retro-2 is significant (Fig. 3C). Cells were then processed to reveal PrPSc and Rab11 and the proportion that co-localised was determined. No significant difference in the co-localisation was detected (Fig. 4C,D). This indicates that the presence of PrPSc in the ERC was not sensitive to Retro-2 and is therefore not dependent on retrograde transport of PrPSc.
Newly formed PrPSc is targeted for lysosomal degradation
We previously showed that prion infected cells assume a steady state PrPSc distribution within minutes of prion infection suggesting that PrPSc is formed at the plasma membrane and internalised continuously. In addition, we observed that PrPSc levels remained relatively unchanged within 2–24 h from prion infection (Goold et al., 2011) indicating that PrPSc formation and degradation kinetics are balanced. This implies that PrPSc degradation is rapid and continuous in the early stages of prion infection. At steady state, a low level of PrPSc is found co-localised with LAMP1 (Fig. 2C,D), suggesting that PrPSc is delivered to lysosomes for degradation. To explore this further, we investigated the effects of lysosomal protease inhibitors (leupeptin, E64d and pepstatin A) on PrPSc distribution. PrP-224AlaMYC cells were pre-treated with lysosomal protease inhibitors for 15 min, then exposed to RML prions for 180 min, fixed and processed to reveal PrPSc. In agreement with previously published data (Luhr et al., 2004), treatment with the cysteine protease inhibitors leupeptin and E64d caused an increase in PrPSc levels, whereas treatment with pepstatin A did not (supplementary material Fig. S4).
In view of these findings, we probed routes that PrPSc may follow to reach the late endosome/lysosomal system. Delivery via the endolysosomal system akin to the trafficking of the epidermal growth factor receptor (Dikic, 2003) would provide the most direct route. However, when we compared the co-localisation of PrPSc with TGN46 and Rab7, it showed an increased accumulation of PrPSc within the TGN relative to the late endosome at early time points, indicating that the trafficking through the TGN represents the major transport route for PrPSc (Fig. 5A,B). To explore this further we inhibited lysosomal PrPSc degradation using leupeptin and E64d, combining the two reagents to maximise their effects. Leupeptin and E64d were added to PrP-224AlaMYC cells prior to prion infection and the distribution of PrPSc was analysed. Under control conditions, PrPSc is found principally in the perinuclear region and at the plasma membrane by 4 min, and this steady-state distribution is maintained thereafter (Fig. 5D). A similar PrPSc distribution is observed under leupeptin and E64d treatment at 4 min, with most of the intracellular PrPSc found close to the nucleus (Fig. 5C). However, by 16 min, PrPSc shows a more widespread distribution that overlaps closely with LAMP-1 (Fig. 5C,D). Quantifying the co-localisation of PrPSc with LAMP1 supports this observation: co-localisation is relatively low (similar to control conditions) up to 4 min following RML prion addition in the presence of leupeptin and E64d (Fig. 5E); at 16 min post infection, this co-localisation is significantly increased relative to controls. This suggests that PrPSc is delivered to lysosomes after transit through the perinuclear region (i.e. endosomes and the TGN). In support of this, BFA treatment, known to inhibit retrograde transport (supplementary material Figs S2, S3), prevents the lysosomal accumulation of PrPSc caused by combined leupeptin and E64d treatment (supplementary material Fig. S5).
Fig. 5.

Newly formed PrPSc is trafficked to lysosomes following retrograde transport. (A) PrP-224AlaMYC cells were fixed after 2, 4 and 16 min of exposure to prions and processed to reveal PrPSc (green) and Rab7 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. Intracellular PrPSc localisation shows a typical perinuclear accumulation. Rab7-labelled late endosomes show a more widespread distribution. Scale bar: 10 µm. (B) The proportion of PrPSc that co-localises with Rab7 and TGN46 was quantified after various times following prion exposure (mean ± s.e.m., n = 4). More PrPSc accumulates in the TGN at early time points. (C) PrP-224AlaMYC cells were treated with leupeptin (Leu) and E64d for 15 min then exposed to prions for 2, 4 and 16 min prior to fixation and processing to reveal PrPSc (green) and LAMP1 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. After 4 min, PrPSc is observed concentrated in the perinuclear region (arrow). After 16 min, PrPSc is shifted to the cell periphery in puncta that often coincide with lysosomes, appearing yellow in the merged image. No major effects on LAMP1 distribution were observed. Scale bar: 10 µm. (D) PrP-224AlaMYC cells were treated with vehicle or leupeptin and E64d as in C. The proportion of cells showing a pronounced perinuclear PrPSc distribution was quantified (mean ± s.e.m., n = 4). (E) PrP-224AlaMYC cells were treated as in D and the proportion of PrPSc that co-localises with LAMP1 was quantified after various times following prion exposure (mean ± s.e.m., n = 4). In the presence of leupeptin and E64d, PrPSc builds up firstly in the perinuclear region and shows relatively low co-localisation with LAMP1 before assuming a more peripheral distribution that shows a higher level of co-localisation with LAMP1.
To further test the role of retrograde transport of PrPSc in its lysosomal delivery and degradation, we assayed PrPSc levels in acutely infected PrP-224AlaMYC cells after treatment with Retro-2. PrP-224AlaMYC cells were pre-treated with Retro-2 then exposed to prions for 180 min, fixed and processed to reveal PrPSc and EEA1 or LAMP1. This treatment resulted in the accumulation of PrPSc in the perinuclear region of the cell, coincident with EEA1 immunostaining (Fig. 6A). The intensity of PrPSc immunostaining was then quantified from confocal images obtained from three independent experiments. This analysis showed a significant increase in pixel intensity relative to control cells (Fig. 6D). Interestingly, the increased PrPSc levels did not result in an increase in its co-localisation with LAMP1 (Fig. 6E). This data suggests Retro-2 treatment retards PrPSc delivery to the lysosome and thereby inhibits its degradation (Fig. 6A,D). A similar paradigm was used to directly analyse the role of lysosomes in PrPSc degradation. We assayed PrPSc levels in acutely infected PrP-224AlaMYC cells after combined treatment with leupeptin and E64d. This analysis showed a robust increase in pixel intensity relative to control cells indicating an accumulation of PrPSc under these conditions (Fig. 6B,D). PrPSc distribution was altered such that it was more widely spread through the cell. An increase in co-localisation with LAMP1 was also noted, indicating that PrPSc builds up in lysosomes (Fig. 6E).
Fig. 6.
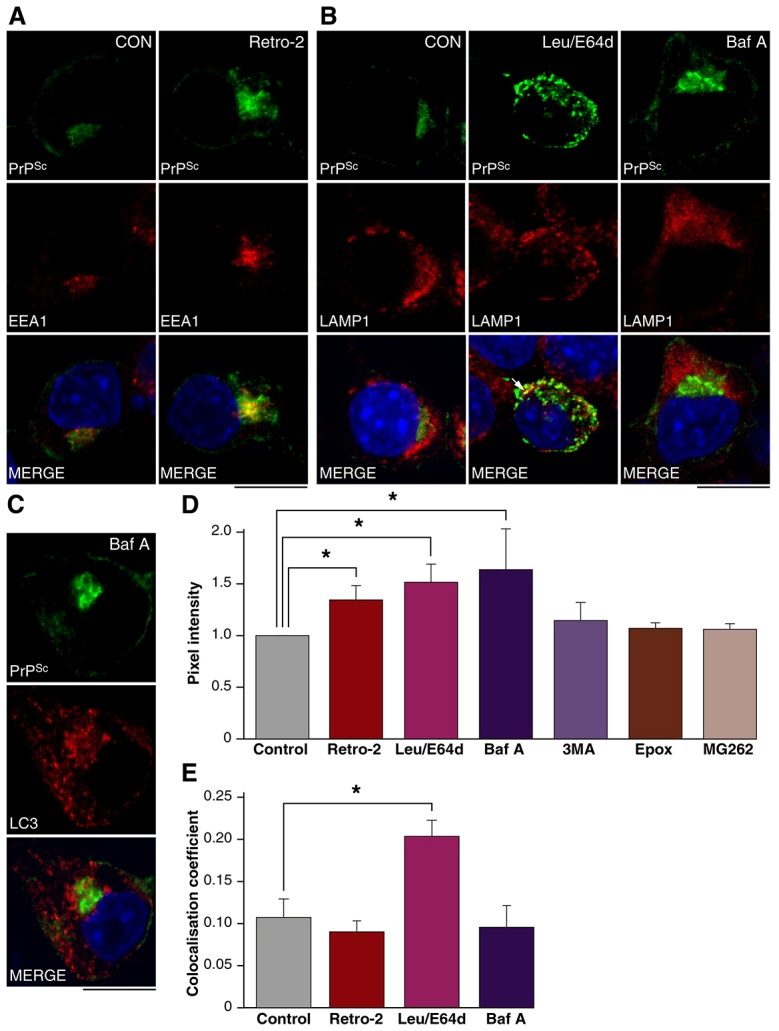
Newly formed PrPSc is degraded in lysosomes. (A) PrP-224AlaMYC cells were treated with vehicle or Retro-2 for 15 min then exposed to prions for 180 min prior to fixation and processing to reveal PrPSc (green) and EEA1 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. Retro-2 induces PrPSc accumulation in the perinuclear region. Scale bar: 10 µm. (B) PrP-224AlaMYC cells were treated with vehicle, leupeptin (Leu) and E64d or Baf A for 15 min then exposed to prions for 180 min prior to fixation and processing to reveal PrPSc (green) and LAMP1 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. Leupeptin and E64d treatment increases the intensity and changes the distribution of PrPSc immunostaining, causing a build-up in the cell periphery, coincident with LAMP1 (arrow). Baf A treatment also increases PrPSc levels but an accumulation in the perinuclear region, distinct from LAMP1, is induced. Scale bar: 10 µm. (C) PrP-224AlaMYC cells were treated with Baf A for 15 min then exposed to prions for 180 min prior to fixation and processing to reveal PrPSc (green) and LC3 (red). Nuclei were visualised with DAPI (blue). Individual channels and merged confocal images are shown. Baf A treatment induces the formation of distinct LC3-labelled puncta and causes PrPSc accumulation near the nucleus. Scale bar: 10 µm. (D) Quantification of the pixel intensity per cell after vehicle (control), Retro-2, leupeptin and E64d, Baf A, 3-methyladenine (3MA), epoxomicin (Epox), or MG262 treatment. Data are normalised to control levels (mean ± s.e.m., n = 4). (E) Quantification of the co-localisation of PrPSc with LAMP1 after treatment with vehicle, Retro-2, leupeptin and E64d or Baf A (mean ± s.e.m., n = 4).
To assess the contribution of autophagy and the UPS to PrPSc breakdown we used Bafilomycin A (Baf A) and 3-methyladenine (3-MA), two reagents commonly used to inhibit autophagy (Klionsky et al., 2008) and the proteasome inhibitors epoxomicin and MG262. PrP-224AlaMYC cells were pre-treated with the inhibitors then exposed to prions for 180 min, fixed and processed to reveal PrPSc, which was then quantified from confocal images. Baf A treatment caused a significant increase PrPSc levels whereas 3-MA, epoxomicin and MG262 had little effect (Fig. 6D). The efficacy of proteasomal inhibition was confirmed by using the proteasome activity probe MV151 (Verdoes et al., 2006). Little probe was incorporated into the proteasome in the presence of epoxomicin or MG262 (supplementary material Fig. S6A). No changes in PrPC or 20S levels were detectable (supplementary material Fig. S6B). Baf A treatment caused a redistribution of LC3, a protein marker of the autophagosomal membrane, into discrete puncta indicating a block in autophagic flux (Fig. 6C). A pronounced increase in PrPSc levels in the perinuclear region was also induced, which was quite distinct from the distribution observed after leupeptin/E64d treatment (Fig. 6B,C). No increase in lysosomal co-localisation was observed under these conditions (Fig. 6B,E) and little co-localisation with LC3 was detected (Fig. 6C).
PrPSc is targeted for lysosomal degradation in chronically infected cells
We were interested to compare PrPSc degradation in cells acutely infected with prions to that observed in chronically infected cells that have been propagating PrPSc for several passages. Therefore, we assayed PrPSc levels in chronically infected PK1 (ScPK1) cells treated for 180 min with the lysosomal, autophagy and UPS inhibitors used above. Western blot analysis of proteinase K (PK) digested lysates showed a significant increase in PrPSc levels following treatment with leupeptin and E64d, Baf A, 3-MA and epoxomicin (supplementary material Fig. S7A–C). MG262 treatment resulted in a small but non statistically significant increase in PrPSc levels (supplementary material Fig. S7B,C). None of the treatments increased PrPC levels in the time course of our experiments (supplementary material Fig. S7A,B). The effects of 3-MA and epoxomicin indicate that autophagy and the proteasome play additional roles in PrPSc degradation in ScPK1 cells.
To quantify the proportion of plasma membrane PrPSc that is targeted for degradation we performed surface labelling experiments. ScPK1 cells were treated with cell impermeable NHS-sulpho-biotin at 4°C for 30 min, a procedure that specifically labels cell surface proteins (Gottardi et al., 1995). Cells were analysed immediately or returned to 37°C for a chase period of up to 6 h. Lysates from surface-labelled cells were digested with PK and detergent insoluble material was recovered by centrifugation. Immunoblotting showed the PrPSc was recovered in the insoluble material (supplementary material Fig. S8). Probing the membranes with NeutrAvidin-HRP revealed biotinylated bands that migrated with the same apparent molecular weight as PK-resistant PrP (supplementary material Fig. S8). Uninfected cells did not contain these bands nor did unbiotinylated ScPK1 cells (supplementary material Fig. S8; Fig. 7A). We were also able to detect PK-resistant PrP in NeutrAvidin affinity chromatography eluates, albeit with low efficiency because of the difficulty in solubilising PrPSc (data not shown). Probing similar eluates not treated with PK showed the Tf receptor was labelled and captured efficiently but that ERK1/2 was not (data not shown). Together, our data shows we were able to effectively label and detect surface PrPSc.
Fig. 7.
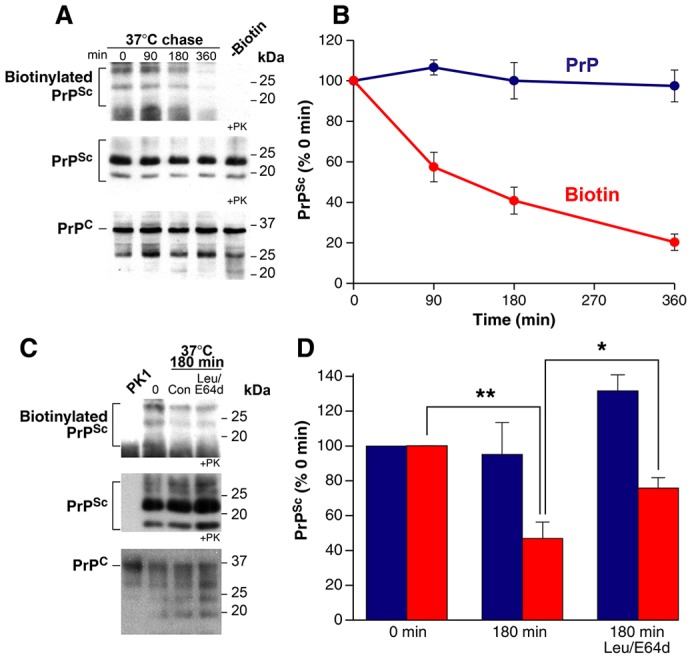
Surface PrPSc is rapidly degraded. (A) ScPK1 cells were surface labelled with NHS-sulpho-biotin at 4°C for 30 min and cells were harvested immediately or returned to 37°C for a chase period of up to 6 h. Cell lysates were analysed directly or after limited PK digestion and centrifugation to enrich for PrPSc (+PK). ScPK1 cells not exposed to NHS-sulpho-biotin but processed in parallel were included as a control (-Biotin). Western blots were probed with anti-PrP antibodies or with NeutrAvidin-HRP to visualise biotinylated proteins. (B) Western blots similar to those in A were quantified to determine total PrPSc and biotinylated PrPSc levels (mean ± s.e.m., n = 3). Biotinylated PrPSc levels are rapidly reduced. Total PrPSc levels are unchanged. (C) ScPK1 cells were surface labelled with NHS-sulpho-biotin at 4°C for 30 min and cells were harvested immediately or returned to 37°C for a chase period of 180 min in control media (Con) or in media containing leupeptin (Leu)/E64d before analysis. Samples were analysed as in A. PK1 cells processed in parallel were included as a control. (D) Western blots similar to those in C were quantified to determine total PrPSc and biotinylated PrPSc levels (mean ± s.e.m., n = 4). Inclusion of leupeptin and E64d in the chase media reduced but did not completely abolish the loss of biotinylated PrPSc. Total levels of PrPSc were increased by this treatment.
We then analysed the levels of labelled PrPSc in our pulse–chase experiments (Fig. 7). Little change was seen in the levels of total PrP or PK-resistant PrP (PrPSc) over a 6 h time course (Fig. 7A). However, the levels of biotinylated PrPSc decrease appreciably in the same time frame (Fig. 7B). Addition of leupeptin and E64d in the chase media reduced but did not completely abolish the loss of biotinylated PrPSc relative to control conditions (Fig. 7C,D). As expected, total levels of PrPSc were increased by leupeptin and E64d treatment (Fig. 7C,D). Our experiments show 60% of surface PrPSc is degraded in 3 h, with a large proportion processed in the lysosome. The rest is presumably en route to this organelle, sorted through the recycling pathway or becomes resistant to degradation. However, this quantification may be an underestimate of the true level of degradation because surface-labelled PrPC may well be a substrate for prion conversion during the chase period, as previously observed for surface-iodinated PrP (Caughey and Raymond, 1991).
Discussion
To gain a better understanding of how PrPSc formation causes cytotoxicity, it is essential to know where PrPSc is formed and to what cellular compartments it traffics to within prion-infected cells. In earlier work, we used our PrP-224AlaMYC cell system to demonstrate that PrPSc is formed at the cell surface and rapidly endocytosed to a perinuclear region (Goold et al., 2011). Here, we extend this work by characterising in more detail the trafficking pathways utilised by newly formed PrPSc. We have performed a precise spatio-temporal analysis of PrPSc intracellular distribution following prion infection using established methods to visualise de novo PrPSc (Goold et al., 2011). In the absence of PrPSc-specific antibodies, we employed immunodetection of formic acid-resistant MYC-tagged PrP to produce a detailed trafficking itinerary of newly formed PrPSc and elaborate further on the kinetics of prion propagation (Borchelt et al., 1992; Marijanovic et al., 2009; Veith et al., 2009). Data presented here suggests that PrPSc forms at the plasma membrane and is then endocytosed to early endosomes containing CTB and Tf. Following endocytosis, a proportion of PrPSc is recycled to the plasma membrane via the ERC, whilst the remainder undergoes retrograde transport to the TGN and the Golgi apparatus. Recycling then provides further PrPSc at the plasma membrane, for template-seeded PrPC conversion, whereas retrograde transport leads to lysosomal delivery and PrPSc degradation. The PrPSc trafficking kinetics described here in cells support previously observed immunohistochemistry analyses, where PrPSc has been detected on the plasma membrane (Jeffrey et al., 1994), in the Golgi (Barmada and Harris, 2005) and endolysosomal system (Arnold et al., 1995) in the prion-infected mouse brain.
Shortly after prion infection, we observed extensive co-localisation of newly formed PrPSc with CTB, at and near the plasma membrane (Fig. 1), suggesting that PrPSc may enter the cell in GM1-enriched membrane microdomains (Lencer and Tsai, 2003). This conclusion is in agreement with previous studies of PrPSc endocytosis. Pharmacological manipulation of the lipid content and organisation of membrane microdomains has been shown to disrupt PrPSc trafficking and propagation (Marella et al., 2002). In addition, PrPSc is known to segregate with membrane microdomain fractions (Naslavsky et al., 1997; Pimpinelli et al., 2005). It is, however, important to acknowledge the possibility of cell type-specific differences in PrPSc trafficking. For example, the endocytosis of PrPC and PrPSc fibrils in sensory neurons appears to be dependent upon clathrin-coated pits (Jen et al., 2010; Parkyn et al., 2008). This apparent heterogeneity in PrPSc trafficking in different cell types could also be explained by differences in the uptake of newly formed PrPSc versus the more mature PrPSc fibrils purified from end-stage prion-infected mouse brain that have been previously used to study endocytosis.
Following endocytosis, internalised PrPSc is mainly segregated by two intracellular trafficking pathways: recycling to the cell surface via the ERC and retrograde transport to the TGN/Golgi. We have previously observed co-localisation of PrPSc with Tf in perinuclear regions (Goold et al., 2011) and detailed kinetic analysis demonstrated co-localisation with Rab11 early in the course of prion infection (Fig. 2). These findings suggest that PrPSc is rapidly transferred to recycling endosomes. In support of this hypothesis, we observed that a proportion of PrPSc is recycled to the plasma membrane from an internal compartment (Fig. 4A,B). Evidence of rapid PrPSc recycling has important implications for sustained prion propagation as it replenishes plasma membrane PrPSc and thus facilitates further seeded-misfolding of PrPC at the cell surface. This hypothesis is consistent with the work of Marijanovic et al. (Marijanovic et al., 2009), who demonstrated the importance of the ERC to PrPSc formation. However, our data does not rule out further PrPC prion conversion in the ERC as both PrPC and PrPSc traverse this compartment. Our data does not differentiate between PrPSc formed at the cell surface from that formed within the recycling pathway. Indeed, the rapid return of plasma membrane PrPSc to control levels suggests PrPSc may form within intracellular compartments during recycling and supports the hypothesis that the ERC plays an important part in prion conversion.
Detailed spatio-temporal analysis revealed a sequential build-up of PrPSc in the endosomal system, followed by the TGN and Golgi, consistent with trafficking along the retrograde transport route. However, we found that the presence of PrPSc in recycling endosomes was independent of retrograde transport (Fig. 4C,D). This suggests that the PrPSc trafficking pathway diverges at an early point into a recycling/propagative pathway and a retrograde/degradative pathway. We confirmed that PrPSc reaches the Golgi using BFA, a reagent that destabilises the Golgi/ER interface to generate Golgi ministacks (Alcalde et al., 1992). BFA is known to exhibit pleiotropic effects within the cell. For instance, BFA-induced disruption of the secretory pathway has previously been shown to inhibit PrP delivery to the cell surface and hence reduce prion propagation (Taraboulos et al., 1992). BFA has also been shown to inhibit retrograde transport of two well-defined cargos: shiga toxin (Mallard et al., 1998) and CTB (supplementary material Fig. S3A). We exploited the latter effect to show that transfer of newly formed PrPSc to the perinuclear region is dependent upon an intact Golgi and/or ER. To confirm this finding, we pre-treated cells with Retro-2, a small molecule inhibitor of retrograde transport (Stechmann et al., 2010), and observed a block of PrPSc and CTB trafficking to the Golgi (Fig. 3B,C; supplementary material Fig. S3A). Further, inhibition of retromer function by RNAi-mediated knockdown of one of its core subunits (VPS35) delayed PrPSc transfer to the TGN (Fig. 3D,E). This data indicates that the retrograde transfer of PrPSc to the TGN is mediated at least in part by the retromer complex. Taken together, this data demonstrates that PrPSc is rapidly trafficked to the Golgi via retrograde transport, a route also followed by other GPI-linked proteins (Nichols et al., 2001).
The ultimate destination for many retrograde transport cargos is the ER, and indeed PrPSc has previously been shown to build up in this compartment, albeit in a cell system overexpressing Rab6A (Béranger et al., 2002). In our system, we find little evidence for PrPSc accumulation in the ER, at least in the timespan analysed in our experiments (Fig. 2C,D; supplementary material Fig. S1). Rather than transfer from the Golgi to the ER, our data suggests that PrPSc is instead routed to the lysosome for degradation: (1) more PrPSc accumulates in the TGN relative to the late endosome at early time points (Fig. 5A,B); (2) leupeptin and E64d treatment resulted in an early accumulation of PrPSc in the perinuclear region, followed by a more peripheral distribution with a high degree of LAMP1 co-localisation (Fig. 5C–E); (3) BFA treatment abrogates lysosomal accumulation of PrPSc (supplementary material Fig. S5); (4) Retro-2 treatment produces a significant increase in PrPSc levels, causing an accumulation in the perinuclear region and not in lysosomes (Fig. 6A,E). Although our data cannot rule out the trafficking of a proportion of PrPSc through the endolysosomal system, taken together our observations indicate that retrograde transport plays an important role in the lysosomal delivery and degradation of PrPSc, and are consistent with previous work in sensory neurons, where purified PrPSc was shown to accumulate in a perinuclear compartment prior to trafficking to the lysosome (Jen et al., 2010).
The absence of detectable PrPSc in the ER and evidence of PrPSc delivery to the lysosome from the Golgi apparatus suggests that PrPSc may be a substrate of Golgi quality control. This mechanism clears misfolded or aggregated protein from the Golgi and transfers them to the lysosome for degradation (Anelli and Sitia, 2008; Arvan et al., 2002). This pathway has previously been implicated in the clearance of misfolded PrP isoforms generated by inherited prion disease mutations or pharmacological manipulation of PrP trafficking in neuronal cells (Ashok and Hegde, 2009; Gilch et al., 2001). The results presented here extend these findings to infectious PrP isoforms, where PrPSc formed following inocula exposure at the cell surface enters the Golgi via the retrograde pathway before being targeted for degradation. We propose that Golgi quality control is necessary to maintain PrPSc at a constant level in the acute phase of prion infection. To support this we showed a robust increase in PrPSc levels in as little as three hours of leupeptin/E64d treatment following acute prion infection. Interestingly, Baf A treatment also caused an increase in PrPSc levels after three hours (Fig. 6D). However, the lack of PrPSc in LC3-labelled puncta suggests little PrPSc is targeted for macroautophagy at this stage of prion infection (Fig. 6C). Consistent with this, 3-MA, an inhibitor of autophagosome sequestration (Klionsky et al., 2008), had little effect on PrPSc levels under these conditions (Fig. 6D). These findings indicate the primary effect of Baf A under these conditions is to block PrPSc progression to the lysosome, probably trapping it in an early endosomal compartment (Baravalle et al., 2005). Inhibition of the UPS in this paradigm did not result in PrPSc accumulation (Fig. 6D). Taken together, these data indicate the Golgi/lysosome route represents the primary pathway for PrPSc breakdown during the acute phase of prion infection.
As cells advance towards a state of chronic infection, the appearance of cytosolic or large aggregated forms of PrPSc may stimulate other degradative pathways, including the UPS and macroautophagy, which have previously been shown to regulate PrPSc levels (Aguib et al., 2009; Nunziante et al., 2011). Our demonstration that treatment of ScPK1 cells with inhibitors of autophagy and the proteasome results in PrPSc accumulation is consistent with this hypothesis (supplementary material Fig. S7). The increase in PrPSc levels after as little as three hours in the presence of lysosomal, autophagy and proteasome inhibitors emphasises the dynamic nature of its metabolism. It also demonstrates the delicate equilibrium that exists between PrPSc formation and degradation in these cells, which is clearly visualised by our surface labelling experiments (Fig. 7). Our data are consistent with previously published work showing the degradation of surface PrPSc (Caughey and Raymond, 1991). However, not all surface PrPSc was degraded in both sets of experiments. Our data suggests some of the PrPSc is recycled but it is possible a proportion of the PrPSc is resistant to degradation, as indicated by metabolic labelling experiments (Borchelt et al., 1990; Caughey and Raymond, 1991). This may be due to alternative PrPSc trafficking pathways not detected in our experiments using acutely infected cells. In chronically infected cells PrPSc that escapes degradation may be routed to compartments, such as aggresomes, known to accumulate aggregated proteins (Kristiansen et al., 2005). Such differences in the trafficking and metabolism of PrPSc between acute and chronic phases of prion infection may also be inferred by the differential contributions made by the lysosome, macroautophagy and the UPS to PrPSc degradation (Fig. 6; supplementary material Fig. S7). Overall, our data suggests a large proportion of the PrPSc formed at the cell surface is endocytosed, recycled or targeted for degradation in a few minutes.
Elucidation of the PrPSc trafficking pathway has relevance to other proteinopathies associated with the endocytosis of misfolded proteins that access the cytosol such as amyloid-beta (Aβ), superoxide dismutase 1 and α-synuclein (Münch et al., 2011). Indeed, striking similarities exist between some aspects of Aβ metabolism and that of PrPSc. In particular, the retrograde trafficking route and involvement of lysosomes in Aβ degradation and toxicity have been widely reported (Muhammad et al., 2008; Small and Gandy, 2006). It has been suggested that destabilisation of the lysosomal limiting membrane in response to luminal accumulation of aggregated Aβ may contribute to cytotoxicity. A similar mechanism has also been postulated in prion diseases (Kovács et al., 2007; Zhang et al., 2003). Accumulation of PrPSc in lysosomes may lead to membrane destabilisation and the leakage of luminal contents. This mechanism could provide a route through which PrPSc gains access to the cytosol, a likely prerequisite for reported sources of cytotoxicity, including proteasomal inhibition and aberrant interactions with other cytosolic proteins (Chakrabarti and Hegde, 2009; Kristiansen et al., 2005; Kristiansen et al., 2007). Whilst PrP-224AlaMYC and related neuroblastoma cell lines have proved invaluable in studying prion infection and trafficking, they are limited in their application to studies of cytotoxicity as they stably propagate prions without noticeable effect on cell viability (Weissmann, 2004). Indeed, previous attempts to study prion-associated toxicity in neuroblastoma subclones, have required the addition of exogenous stressors (Kristiansen et al., 2005; Nunziante et al., 2011). Identification of the trafficking compartments at the interface between prion infection and prion cytotoxicity demands the use of a post-mitotic cell line expressing PrP-224AlaMYC, which is currently under development.
Materials and Methods
Cell culture and chemicals
PrP-224AlaMYC cells were generated as described previously (Goold et al., 2011) and routinely cultured in dual selection media (puromycin and G418) in OptiMEM, FCS (10%) and penicillin/streptomycin. ScPK1 cells were prepared as described (Klöhn et al., 2003). Tissue culture media, labelled cargo proteins and AlexaFluor-conjugated antibodies were purchased from Invitrogen. Retro-2 was obtained from Merck. Other chemicals were purchased from Sigma. The primary antibodies used in this study are detailed in supplementary material Table S1.
Cellular prion infection
Brain homogenates were prepared from end-stage RML prion-infected CD-1 mice as described (Klöhn et al., 2003). These were diluted to 0.1% in media, and added to cells grown to 70% confluence that were seeded on tissue culture plastic or poly-L-lysine-coated glass coverslips. Chronically prion-infected PrP-224AlaMYC cells were prepared by exposing cells grown on plastic to RML prions for 16 h then washing the cells three times with fresh media and passaging the cells for at least seven days in the absence of external prions prior to use. Cells were washed and fixed with pre-warmed formaldehyde (3.7% in PBS) for 13 min at 37°C. All animal experiments were performed according to approved guidelines.
Cell manipulations
Cells were treated with vehicle, BFA (10 µg/ml), Retro-2 (20 µM), leupeptin (200 µM), E64d (2 µM), pepstatin A (20 µM), epoxomicin (1 µM), MG262 (1 µM), bafilomycin A (20 nM) and 3-MA (10 mM). Cells were transfected with RNAi oligonucleotides using Dharmafect according to the manufacturer's instructions. A mixture of four RNAi oligonucleotides targeting VPS35 (Dharmacon ON-TARGETplus kit) was added at 100 nM for 72 h prior to use. PI-PLC (Sigma) treatment was carried out using 0.5 U/ml PI-PLC for 30 min at 4°C. The cells were fixed at 4°C for 10 min then transferred to 37°C for a further 10 min prior to washing or transferred to 37°C and fixed as described above. Texas Red-labelled transferrin (5 µg/ml), AlexaFluor555-CTB (10 µg/ml) or AlexaFluor555-Dextran (10,000 kDa, 2.5 mg/ml) was added to the media for 30 min prior to prion exposure. Proteasome activity probe MV151 (1 µM) was added to the cells for 60 min prior to fixation.
Immunofluorescence analysis
Cells were routinely prepared for immunofluorescence analysis with 5 min pre-treatment with 98% formic acid to remove host PrPC and expose PrPSc as described previously (Goold et al., 2011). Formic acid treated cells were permeabilised with cold methanol for 10 min at −20°C then washed with PBS, prior to immunostaining as described previously (Goold et al., 2011). Fluorescence images were acquired using a Zeiss LSM510 META confocal microscope with a plan-Apochromat 63×/1.40 oil DIC objective. Control uninfected cells were processed for each experiment and LSM settings were chosen to give minimal background staining. The same settings were used to examine all infected cells in that experiment. Co-localisation coefficients were calculated using the co-localisation tool in the Zeiss LSM software version 4.2. Background co-localisation levels were determined using uninfected cells and subtracted from values obtained from prion-infected cells. The intensity of intracellular MYC staining was determined using Volocity software (Improvision). At least 25 cells in four independent experiments were analysed per condition. Differential interference contrast images were obtained and merged with confocal images to allow an accurate determination of the proportion of cells that showed membrane staining.
Biochemical analysis
Cell lysates were prepared for biochemical analysis as described previously (Goold et al., 2011). Surface labelling was performed using NHS-sulpho-biotin (Thermo Scientific). Cells were washed twice in ice cold PBS with 0.5 mM MgCl2 and 1 mM CaCl2 then incubated on ice with 1 mg/ml NHS-sulpho-biotin in PBS with 0.5 mM MgCl2 and 1 mM CaCl2 for 30 min. The reaction was quenched with 50 mM glycine, 0.5% BSA in PBS, then cells were washed once with TBS and harvested immediately or treated with warmed media and returned to 37°C for the chase period. Harvested cells were washed with PBS then resuspended in lysis buffer (PBS, 0.5% Triton X-100, 0.5% sodium deoxycholate). The protein concentration of the lysates was adjusted to 1 mg/ml and aliquots were methanol precipitated for total PrP analysis or digested with PK (10 µg/ml, 90 min at 37°C) for the analysis of PrPSc. To enrich for detergent-insoluble proteins the PK digested samples were centrifuged at 20,000 g for 30 min at 4°C. The pellet was washed once with lysis buffer and once with methanol. The pellet was then air dried and resuspended in SDS sample buffer. Biotinylated proteins were detected with NeutrAvidin-HRP (Invitrogen) used at a 1∶1000 dilution. ScPK1 lysates were incubated with NeutrAvidin-agarose (Thermo Scientific) according to the manufacturer's instructions.
Statistical analysis
Data are expressed as mean ± s.e.m., unless otherwise stated. Data were compared by two-tailed t-tests and considered significantly different when P<0.05. Degree of significance is expressed as *P<0.05, **P<0.01, unless otherwise specified.
Supplementary Material
Footnotes
Author contributions
R.G. designed and performed experiments, analysed data and wrote the paper; C.M. wrote the paper; S.R. performed experiments; J.C. analysed data; G.S. designed experiments, analysed data and revised the manuscript; S.J.T. designed experiments, analysed data and wrote the paper.
Funding
This work was funded by the Medical Research Council [grant number G0700877 to S.J.T.]; Brain Research Trust [studentship number STU07103 to S.R.]; and Cancer Research UK [grant number LF4605 to G.S.]. Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.120477/-/DC1
References
- Aguib Y., Heiseke A., Gilch S., Riemer C., Baier M., Schätzl H. M., Ertmer A. (2009). Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 5, 361–369 10.4161/auto.5.3.7662 [DOI] [PubMed] [Google Scholar]
- Alcalde J., Bonay P., Roa A., Vilaro S., Sandoval I. V. (1992). Assembly and disassembly of the Golgi complex: two processes arranged in a cis-trans direction. J. Cell Biol. 116, 69–83 10.1083/jcb.116.1.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli T., Sitia R. (2008). Protein quality control in the early secretory pathway. EMBO J. 27, 315–327 10.1038/sj.emboj.7601974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J. E., Tipler C., Laszlo L., Hope J., Landon M., Mayer R. J. (1995). The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J. Pathol. 176, 403–411 10.1002/path.1711760412 [DOI] [PubMed] [Google Scholar]
- Arvan P., Zhao X., Ramos-Castaneda J., Chang A. (2002). Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic 3, 771–780 10.1034/j.1600-0854.2002.31102.x [DOI] [PubMed] [Google Scholar]
- Ashok A., Hegde R. S. (2009). Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 5, e1000479 10.1371/journal.ppat.1000479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baravalle G., Schober D., Huber M., Bayer N., Murphy R. F., Fuchs R. (2005). Transferrin recycling and dextran transport to lysosomes is differentially affected by bafilomycin, nocodazole, and low temperature. Cell Tissue Res. 320, 99–113 10.1007/s00441-004-1060-x [DOI] [PubMed] [Google Scholar]
- Barmada S. J., Harris D. A. (2005). Visualization of prion infection in transgenic mice expressing green fluorescent protein-tagged prion protein. J. Neurosci. 25, 5824–5832 10.1523/JNEUROSCI.1192-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béranger F., Mangé A., Goud B., Lehmann S. (2002). Stimulation of PrP(C) retrograde transport toward the endoplasmic reticulum increases accumulation of PrP(Sc) in prion-infected cells. J. Biol. Chem. 277, 38972–38977 10.1074/jbc.M205110200 [DOI] [PubMed] [Google Scholar]
- Bonifacino J. S., Hurley J. H. (2008). Retromer. Curr. Opin. Cell Biol. 20, 427–436 10.1016/j.ceb.2008.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt D. R., Scott M., Taraboulos A., Stahl N., Prusiner S. B. (1990). Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J. Cell Biol. 110, 743–752 10.1083/jcb.110.3.743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt D. R., Taraboulos A., Prusiner S. B. (1992). Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 267, 16188–16199 [PubMed] [Google Scholar]
- Caughey B., Raymond G. J. (1991). The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 266, 18217–18223 [PubMed] [Google Scholar]
- Chakrabarti O., Hegde R. S. (2009). Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell 137, 1136–1147 10.1016/j.cell.2009.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I. (2003). Mechanisms controlling EGF receptor endocytosis and degradation. Biochem. Soc. Trans. 31, 1178–1181 10.1042/BST0311178 [DOI] [PubMed] [Google Scholar]
- Enari M., Flechsig E., Weissmann C. (2001). Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. USA 98, 9295–9299 10.1073/pnas.151242598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilch S., Winklhofer K. F., Groschup M. H., Nunziante M., Lucassen R., Spielhaupter C., Muranyi W., Riesner D., Tatzelt J., Schätzl H. M. (2001). Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. EMBO J. 20, 3957–3966 10.1093/emboj/20.15.3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold R., Rabbanian S., Sutton L., Andre R., Arora P., Moonga J., Clarke A. R., Schiavo G., Jat P., Collinge J. et al. (2011). Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun 2, 281 10.1038/ncomms1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottardi C. J., Dunbar L. A., Caplan M. J. (1995). Biotinylation and assessment of membrane polarity: caveats and methodological concerns. Am J Physiol 268, F285–F295 [DOI] [PubMed] [Google Scholar]
- Hetz C., Russelakis-Carneiro M., Maundrell K., Castilla J., Soto C. (2003). Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 22, 5435–5445 10.1093/emboj/cdg537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey M., Goodsir C. M., Bruce M., McBride P. A., Scott J. R., Halliday W. G. (1994). Correlative light and electron microscopy studies of PrP localisation in 87V scrapie. Brain Res. 656, 329–343 10.1016/0006-8993(94)91477-X [DOI] [PubMed] [Google Scholar]
- Jen A., Parkyn C. J., Mootoosamy R. C., Ford M. J., Warley A., Liu Q., Bu G. J., Baskakov I. V., Moestrup S., McGuinness L. et al. (2010). Neuronal low-density lipoprotein receptor-related protein 1 binds and endocytoses prion fibrils via receptor cluster 4. J. Cell Sci. 123, 246–255 10.1242/jcs.058099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J., Abeliovich H., Agostinis P., Agrawal D. K., Aliev G., Askew D. S., Baba M., Baehrecke E. H., Bahr B. A., Ballabio A. et al. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöhn P. C., Stoltze L., Flechsig E., Enari M., Weissmann C. (2003). A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. USA 100, 11666–11671 10.1073/pnas.1834432100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács G. G., Gelpi E., Ströbel T., Ricken G., Nyengaard J. R., Bernheimer H., Budka H. (2007). Involvement of the endosomal-lysosomal system correlates with regional pathology in Creutzfeldt-Jakob disease. J. Neuropathol. Exp. Neurol. 66, 628–636 10.1097/nen.0b013e318093ecc7 [DOI] [PubMed] [Google Scholar]
- Kristiansen M., Messenger M. J., Klöhn P. C., Brandner S., Wadsworth J. D., Collinge J., Tabrizi S. J. (2005). Disease-related prion protein forms aggresomes in neuronal cells leading to caspase activation and apoptosis. J. Biol. Chem. 280, 38851–38861 10.1074/jbc.M506600200 [DOI] [PubMed] [Google Scholar]
- Kristiansen M., Deriziotis P., Dimcheff D. E., Jackson G. S., Ovaa H., Naumann H., Clarke A. R., van Leeuwen F. W., Menéndez-Benito V., Dantuma N. P. et al. (2007). Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell 26, 175–188 10.1016/j.molcel.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Lencer W. I., Tsai B. (2003). The intracellular voyage of cholera toxin: going retro. Trends Biochem. Sci. 28, 639–645 10.1016/j.tibs.2003.10.002 [DOI] [PubMed] [Google Scholar]
- Luhr K. M., Nordström E. K., Löw P., Ljunggren H. G., Taraboulos A., Kristensson K. (2004). Scrapie protein degradation by cysteine proteases in CD11c+ dendritic cells and GT1-1 neuronal cells. J. Virol. 78, 4776–4782 10.1128/JVI.78.9.4776-4782.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Wollmann R., Lindquist S. (2002). Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298, 1781–1785 10.1126/science.1073725 [DOI] [PubMed] [Google Scholar]
- Magalhães A. C., Baron G. S., Lee K. S., Steele-Mortimer O., Dorward D., Prado M. A. M., Caughey B. (2005). Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 25, 5207–5216 10.1523/JNEUROSCI.0653-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallard F., Antony C., Tenza D., Salamero J., Goud B., Johannes L. (1998). Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. J. Cell Biol. 143, 973–990 10.1083/jcb.143.4.973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella M., Lehmann S., Grassi J., Chabry J. (2002). Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J. Biol. Chem. 277, 25457–25464 10.1074/jbc.M203248200 [DOI] [PubMed] [Google Scholar]
- Marijanovic Z., Caputo A., Campana V., Zurzolo C. (2009). Identification of an intracellular site of prion conversion. PLoS Pathog. 5, e1000426 10.1371/journal.ppat.1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno J. A., Radford H., Peretti D., Steinert J. R., Verity N., Martin M. G., Halliday M., Morgan J., Dinsdale D., Ortori C. A. et al. (2012). Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad A., Flores I., Zhang H., Yu R., Staniszewski A., Planel E., Herman M., Ho L., Kreber R., Honig L. S. et al. (2008). Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. USA 105, 7327–7332 10.1073/pnas.0802545105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch C., O'Brien J., Bertolotti A. (2011). Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 108, 3548–3553 10.1073/pnas.1017275108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N., Stein R., Yanai A., Friedlander G., Taraboulos A. (1997). Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J. Biol. Chem. 272, 6324–6331 10.1074/jbc.272.10.6324 [DOI] [PubMed] [Google Scholar]
- Nichols B. J., Kenworthy A. K., Polishchuk R. S., Lodge R., Roberts T. H., Hirschberg K., Phair R. D., Lippincott-Schwartz J. (2001). Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 153, 529–541 10.1083/jcb.153.3.529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunziante M., Gilch S., Schätzl H. M. (2003). Essential role of the prion protein N terminus in subcellular trafficking and half-life of cellular prion protein. J. Biol. Chem. 278, 3726–3734 10.1074/jbc.M206313200 [DOI] [PubMed] [Google Scholar]
- Nunziante M., Ackermann K., Dietrich K., Wolf H., Gädtke L., Gilch S., Vorberg I., Groschup M., Schätzl H. M. (2011). Proteasomal dysfunction and endoplasmic reticulum stress enhance trafficking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. J. Biol. Chem. 286, 33942–33953 10.1074/jbc.M111.272617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkyn C. J., Vermeulen E. G. M., Mootoosamy R. C., Sunyach C., Jacobsen C., Oxvig C., Moestrup S., Liu Q., Bu G. J., Jen A. et al. (2008). LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell Sci. 121, 773–783 10.1242/jcs.021816 [DOI] [PubMed] [Google Scholar]
- Pimpinelli F., Lehmann S., Maridonneau-Parini I. (2005). The scrapie prion protein is present in flotillin-1-positive vesicles in central- but not peripheral-derived neuronal cell lines. Eur. J. Neurosci. 21, 2063–2072 10.1111/j.1460-9568.2005.04049.x [DOI] [PubMed] [Google Scholar]
- Prusiner S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 10.1126/science.6801762 [DOI] [PubMed] [Google Scholar]
- Rane N. S., Kang S. W., Chakrabarti O., Feigenbaum L., Hegde R. S. (2008). Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev. Cell 15, 359–370 10.1016/j.devcel.2008.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatore A., Colleoni S., Verderio C., Restelli E., Morini R., Condliffe S. B., Bertani I., Mantovani S., Canovi M., Micotti E. et al. (2012). Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC α(2)δ-1 Subunit. Neuron 74, 300–313 10.1016/j.neuron.2012.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small S. A., Gandy S. (2006). Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron 52, 15–31 10.1016/j.neuron.2006.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stechmann B., Bai S. K., Gobbo E., Lopez R., Merer G., Pinchard S., Panigai L., Tenza D., Raposo G., Beaumelle B. et al. (2010). Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 141, 231–242 10.1016/j.cell.2010.01.043 [DOI] [PubMed] [Google Scholar]
- Taraboulos A., Raeber A. J., Borchelt D. R., Serban D., Prusiner S. B. (1992). Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 3, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veith N. M., Plattner H., Stuermer C. A., Schulz-Schaeffer W. J., Bürkle A. (2009). Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur. J. Cell Biol. 88, 45–63 10.1016/j.ejcb.2008.08.001 [DOI] [PubMed] [Google Scholar]
- Verdoes M., Florea B. I., Menendez-Benito V., Maynard C. J., Witte M. D., van der Linden W. A., van den Nieuwendijk A. M. C. H., Hofmann T., Berkers C. R., van Leeuwen F. W. B. et al. (2006). A fluorescent broad-spectrum proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem. Biol. 13, 1217–1226 10.1016/j.chembiol.2006.09.013 [DOI] [PubMed] [Google Scholar]
- Weissmann C. (2004). The state of the prion. Nat. Rev. Microbiol. 2, 861–871 10.1038/nrmicro1025 [DOI] [PubMed] [Google Scholar]
- Zhang Y. H., Spiess E., Groschup M. H., Bürkle A. (2003). Up-regulation of cathepsin B and cathepsin L activities in scrapie-infected mouse Neuro2a cells. J. Gen. Virol. 84, 2279–2283 10.1099/vir.0.19153-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.