

Abstract
Objective
Folate is involved in the one-carbon metabolism that plays an essential role in the synthesis, repair and methylation of DNA. We examined whether child’s germline genetic variation in the folate pathway is associated with childhood acute lymphoblastic leukemia (ALL), and whether periconception maternal folate and alcohol intake modify the risk.
Methods
Seventy-six single nucleotide polymorphisms (SNPs), including 66 haplotype-tagging SNPs in 10 genes (CBS, DHFR, FOLH1, MTHFD1, MTHFR, MTR, MTRR, SHMT1, SLC19A1, and TYMS) were genotyped in 377 ALL cases and 448 controls. Log-additive associations between genotypes and ALL risk were adjusted for age, sex, Hispanic ethnicity (when appropriate), and maternal race.
Results
Single and haplotype SNPs analyses showed statistically significant associations between SNPs located in (or adjacent to) CBS, MTRR, TYMS/ENOFS and childhood ALL. Many regions of CBS were associated with childhood ALL in Hispanics and non-Hispanics (P <0.01). Levels of maternal folate intake modified associations with SNPs in CBS, MTRR, and TYMS.
Conclusion
Our data suggest the importance of genetic variability in the folate pathway and childhood ALL risk.
Keywords: Case-control study, Children, DNA methylation, Folate, Genetic polymorphisms, Leukemia
Introduction
Leukemia is the most common cancer worldwide in children (ages 0-14 years), with more than 2600 cases diagnosed annually in the USA (1). The main histological type is acute lymphoblastic leukemia (ALL) representing 80% of all cases in the USA (2). The causes of childhood leukemia remain largely unknown and, as for many cancers, genetic susceptibility combined with environmental exposures are likely to play a significant role in the mechanisms of childhood leukemia development (3).
Folate is a water-soluble B vitamin involved in one-carbon metabolism that plays an essential role in the synthesis, repair and methylation of DNA. Folate metabolism provides one-carbon units necessary for the synthesis of nucleic acid bases, and enables the conversion of methionine into S-adenosylmethionine (SAM), via its ability to methylate homocysteine. SAM is the universal methyl group donor in the majority of biochemical reactions including DNA methylation (4, 5). Low folate intake or defects in folate metabolism may lead to DNA strand breaks, reduced DNA repair, and aberrant DNA methylation. Low levels of folate in utero have been hypothesized to increase the risk of cancer, including childhood leukemia (5-10).
Although there is growing evidence that genetic variants of methylenetetrahydrofolate reductase (MTHFR), a key enzyme in the regulation of folate metabolism, interact with folate bioavailability in the association with cancer risk (11), few studies on childhood leukemia have investigated gene-environment interactions, and those that have included limited information on folate intake and yielded conflicting results (9, 12, 13). Moreover, these previous reports mainly focused on MTHFR polymorphisms (7-9, 12-19), and few studies have examined other genes in the folate pathway (10, 20-26).
Our objectives were to determine whether common variants in 10 genes that encode enzymes involved in the folate pathway (Table 1) are associated with the risk of childhood ALL, and to assess whether maternal daily folate intake in the peri-conception period act as effect modifier. We will also examine the effect modification of alcohol consumption, known to reduce folate bioavailability (27).
Table 1. Description of genes and SNPs included in the analyses (the Northern California Childhood Leukemia Study, 1996-2002).
|
GENE, HUGO nomenclature [aliases, where appropriate] |
Function of the protein encoded by the gene | Chr. | N. of SNPs selected for genotyping |
SNPs excluded from analyses |
|---|---|---|---|---|
|
CBS: cystathionine-beta-synthase [HIP4] |
Catalyzes homocysteine to cystationine (first step of the transsulfuration pathway). |
21 | 17 | rs11203172* rs1672123* rs2124461* rs234709* rs8132811† |
| DHFR: dihydrofolate reductase | Converts dihydrofolate into tetrahydrofolate (methyl group shuttle required for the de novo synthesis of purines, thymidylic acid, and certain amino acids). |
5 | 4 | None |
|
FOLH1: folate hydrolase (prostate-specific membrane antigen) 1 [PSM, FGCP, FOLH, GCP2, PSMA, Mgcp, GCPII, NAALAD1, NAALAdase] |
Acts as a glutamate carboxypeptidase on different alternative substrates, including the nutrient folate; dysfunction may be associated with impaired intestinal absorption of dietary folates, resulting in hyperhomocysteinemia. |
11 | 4 | rs7113251‡ |
|
MTHFD1: methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1, methenyltetrahydrofolate cyclohydrolase, formyltetrahydrofolate synthetase [MTHFC, MTHFD] |
Catalyzes three sequential reactions in the interconversion of 1.3 carbon derivatives of tetrahydrofolate (substrates for methionine, thymidylate, and de novo purine syntheses). |
14 | 10 | rs2236225† |
|
MTHFR: 5,10-methylenetetrahydrofolate reductase (NADPH) |
Catalyzes the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate (cosubstrate for homocysteine remethylation to methionine). |
1 | 10 | rs4846040* rs7538516* |
|
MTR: 5-methyltetrahydrofolate-homocysteine methyltransferase [MS, FLJ33168, FLJ43216, FLJ45386] |
Catalyzes the final step in methionine biosynthesis. | 1 | 4 | None |
|
MTRR: 5-methyltetrahydrofolate- homocysteine methyltransferase reductase [MSR, MGC129643] |
Regenerates a functional 5-methyltetrahydrofolate- homocysteine methyltransferase via reductive methylation (which becomes inactive due to the oxidation of its cob(I)alamin cofactor). |
5 | 18 | rs1046012* |
|
SHMT1: serine hydroxymethyltransferase 1 (soluble) [SHMT, CSHMT, MGC15229, MGC24556] |
Catalyzes the reversible conversion of serine and tetrahydrofolate to glycine and 5,10-methylene tetrahydrofolate (this reaction provides one carbon units for synthesis of methionine, thymidylate, and purines in the cytoplasm). |
17 | 5 | None |
|
SLC19A1: solute carrier family 19 (folate transporter), member 1 [CHMD, FOLT, IFC1, REFC, RFC1] |
Main entry point of folate into cell; plays a role in maintaining intracellular concentrations of folate. |
21 | 6 | rs1050351* rs2838950* rs2838951* |
|
TYMS: thymidylate synthetase [TS, TMS, Tsase, HsT422, MGC88736] |
Catalyzes the methylation of deoxyuridylate to deoxythymidylate using 5,10-methylenetetrahydrofolate (methylene-THF) as a cofactor (this function maintains the dTMP (thymidine-5-prime monophosphate) pool critical for DNA replication and repair). |
18 | 11 | None |
Genotyping failure
Hardy-Weinberg disequilibrium in both Hispanic and non-Hispanic controls (p<0.01)
Minor allele frequency <5%
Material and Methods
This study was approved by the University of California, Berkeley Committee for the Protection of Human Subjects, the California Health and Human Services Agency Committee for the Protection of Human Subjects, and the Institutional Review Boards of the nine participating hospitals. Written informed consent was obtained prior to interview from the responding parent of each participating child.
Study population
The present analyses were based on incident childhood ALL cases and controls born between 1982 and 2001 and recruited between April 1996 and December 2002 in the NCCLS, a population-based case-control study. A total of nine hospitals from 35 counties in the San Francisco Bay Area and California Central Valley participated in the ultra-rapid case recruitment. Comparison to the California Cancer Registry shows that leukemia cases ascertained through the NCCLS protocol covered approximately 76% of the cases diagnosed in all participating and non-participating hospitals in the 35 study counties. Cases were eligible if they were under 15 years of age at diagnosis, had no prior cancer diagnosis, lived in the study area, and if their biological parents spoke either English or Spanish for the purpose of completing the interview. Eighty six percent of cases determined to be eligible consented to participate. Control selection has been described in detail in previous reports (28-30). In brief, one or two healthy controls who resided within the study area were randomly selected from birth certificates supplied by the California Department of Public Health and were individually matched to cases on child’s date of birth, sex, Hispanic ethnicity (a child was considered Hispanic if either parent was Hispanic), and maternal race. During the study period of 1995-2002, 80% of potential controls identified were located and deemed to be eligible, and of these, 84% agreed to participate (29). Table 2 describes general characteristics of cases and controls included in our analyses.
Table 2. Selected characteristics of participating cases and controls (the Northern California Childhood Leukemia Study, 1996-2002).
| Characteristics | Cases | Controls |
|---|---|---|
|
|
|
|
| N (%) | N (%) | |
| Total | 377 | 448 |
| Sex | ||
| Male | 200 (53.1) | 237 (52.9) |
| Female | 177 (46.9) | 211 (47.1) |
| Ethnicity | ||
| Hispanic | 156 (41.4) | 179 (40.0) |
| Non-Hispanic White | 162 (43.0) | 198 (44.2) |
| Non-Hispanic Other | 59 (15.6) | 71 (15.8) |
| Morphological subtypes | ||
| B cell lineage: c-ALL | 189 (50.1) | — |
| B-cell lineage: other | 153 (40.6) | — |
| T cell lineage | 31 (8.2) | — |
| Undefined | 4 (1.1) | — |
| Age at diagnosis/reference date* (years) | ||
| 0.0–1.9 | 37 (9.8) | 63 (14.1) |
| 2.0–3.9 | 125 (33.2) | 127 (28.3) |
| 4.0–5.9 | 93 (24.7) | 111 (24.8) |
| 6.0–7.9 | 41 (10.9) | 45 (10.0) |
| 8.0–9.9 | 30 (8.0) | 37 (8.3) |
| At least 10 | 51 (13.5) | 65 (14.5) |
| Down syndrome | ||
| Yes | 12 (3.2) | 1 (0.2) |
| No | 364 (96.8) | 447 (99.8) |
| Maternal alcohol consumption in the 3 months before pregnancy |
||
| Less than 1 drink per month | 198 (52.5) | 223 (49.8) |
| 1–3 drink(s) per month | 45 (11.9) | 35 (7.8) |
| 1 drink per week | 53 (14.1) | 76 (17.0) |
| 2–4 drinks per week | 41 (10.9) | 54 (12.1) |
| 5–6 drinks per week | 10 (2.7) | 23 (5.1) |
| At least 1 drink per day | 27 (7.2) | 29 (6.5) |
| Missing† | 3 (0.8) | 8 (1.8) |
| Maternal alcohol consumption during pregnancy |
||
| Less than 1 drink per month | 351 (93.1) | 414 (92.4) |
| At least 1 drink per month | 16 (4.2) | 16 (5.9) |
| Missing | 10 (2.7) | 12 (2.7) |
| Maternal folate intake from food and supplements 3 months before pregnancy (mcg DFEs/day) |
||
| 63–279 | 77 (20.4) | 105 (23.4) |
| 280–429 | 78 (20.7) | 105 (23.4) |
| 430–764 | 122 (32.4) | 105 (23.4) |
| 765–2,163 | 92 (24.4) | 104 (23.2) |
| Missing | 23 (6.1) | 29 (6.5) |
| Mean (s.d.) | 570 (347) | 556 (362) |
| Household annual income (USD) | ||
| <15,000 | 53 (14.1) | 39 (8.7) |
| 15,000-29,999 | 70 (18.6) | 62 (13.8) |
| 30,000-44,999 | 54 (14.3) | 55 (12.3) |
| 45,000-59,999 | 65 (17.2) | 69 (15.4) |
| 60,000-74,999 | 40 (10.6) | 62 (13.8) |
| 75,000+ | 95 (25.2) | 161 (35.9) |
Reference date for controls = date at diagnosis of matched case.
Three mothers (one from case and two from control groups) reported to be drinkers but quantity was missing.
Biospecimen collection and DNA processing
Buccal cells were collected as the primary DNA source for case and control children. Buccal cytobrushes were collected at the time of interview by trained interviewers. DNA from cytobrush samples was extracted by heating (98-100°C) in the presence of NaOH, followed by neutralization with Tris-HCl buffer, and whole-genome amplification (WGA) using GenomePlex reagents (Sigma Aldrich, St. Luis, MO). In the event that buccal cell DNA was insufficient for genotyping (26.6% of subjects), archived newborn blood (ANB) specimens were used as a secondary DNA source. ANB specimens are collected at birth on a paper card for each child born in California and archived at −20°C by the California Department of Public Health. The NCCLS receives one spot containing approximately 60 microliters of blood, per child. A small piece of the bloodspot was excised and DNA was extracted using the QIAamp DNA mini-extraction kit. Isolated ANB DNA was whole-genome amplified using REPLI-g reagents (Qiagen, Hilden, Germany). WGA products were tested for minimum acceptable amplifiable human DNA content using an ALUq real time PCR method published elsewhere.(31) When analyzed using multiplexed GoldenGate genotyping (Illumina, San Diego, CA), whole-genome amplified DNA from both buccal cells and ANB specimens yielded genotypes that were highly concordant with those from genomic DNA from peripheral blood (31, 32).
SNP selection and genotyping
Within the context of a study of candidate pathways, we selected 89 SNPs in 10 folate metabolism genes (CBS, DHFR, FOLH1, MTHFD1, MTHFR, MTR, MTRR, SHMT1, SLC19A1, and TYMS) (Table 1) on the basis of two criteria: (i) SNPs that result in amino acid changes; and (ii) SNPs that tag haplotype blocks, or groups of linked SNPs, within regions encompassing the target genes and 10,000 base pairs up- and down-stream of the gene. Using HaploView (33), in conjunction with SNP data from both the 30 Caucasian trios in the HapMap project (34) (Release 19, Build 34) and the 23 Hispanics in the SNP500Cancer project (35), we applied the method of Gabriel et al (36) to select haplotype-tagging SNPs (htSNPs) that captured at least 80% of the haplotype diversity for common haplotypes (>5% frequency) in the Caucasian or Hispanic populations.
In addition, 82 ancestry informative markers (AIMs) for defining European versus African versus Native American ancestral origin were selected for genotyping. Genetic ancestry was estimated using a maximum likelihood approach as described by Chakraborty et al (37) and Hanis et al (38). Ancestral population AIM allele frequencies were input along with the genotypes of the admixed participants to improve ancestry estimation. Genotyping of 385 cases and 456 controls was performed using a custom Illumina GoldenGate genotyping panel (1536 SNPs). Across this panel, we used a GenTrain cutoff of 0.25, a SNP-wise call rate threshold of 90% and a subject-wise call rate threshold of 95%. Quality of genotyping was verified by comparing duplicate samples: (i) 59 samples were run in duplicate after processing with the same WGA method and typed on the same plate; these showed a 0.88% discordance of genotype; (ii) DNA specimens extracted from both buccal cell and ANB spots were genotyped for 9 subjects and showed a 1.05% discordance of genotype. In addition, we estimated the Mendelian errors in 10 trios from the HapMap Centre d’Etude du Polymorphisme Humain (CEPH) and only 28 pedigree errors in 25 markers were found (overall Mendelian error rate = 0.2%).
Out of the 79 SNPs that satisfied the call rate threshold described above, two were excluded because they departed from Hardy-Weinberg equilibrium (p<0.01) in both Hispanic and non-Hispanic controls, and one SNP was excluded because the minor allele frequency was less than 5% in both Hispanic and non-Hispanic controls (Table 1). In addition, eight cases and eight controls were below the subject-wise call rate threshold, leaving a total of 76 SNPs in 377 ALL cases and 448 controls available for analysis.
Diet and alcohol intake assessment
Details of data collection have been published elsewhere (39). In brief, maternal folate intake estimates were assessed using a modified version of the Block Food Frequency Questionnaire (FFQ) administered during the in-home interviews to assess the pre-pregnancy diet of the mother. This period was chosen rather than diet during pregnancy because it represents the probable state of nutritional adequacy at the time of conception and during early pregnancy. Daily folate intake in micrograms per day of dietary folate equivalents (mcg DFEs) was calculated as a composite variable of vitamin/supplement use and dietary intake (folate from natural source and from fortification when it was in effect), taking into account all food items, the reported portion sizes, the folate content per 100 grams, and the bioavailability corresponding to the source of folate (USDA SR 16-1, 2004) (Table 2).
Alcohol consumption was also measured through the FFQ. Consumption of different types of alcohol (beer, wine or wine coolers, and hard liquor or mixed drinks) was assessed in frequency before and during pregnancy and combined variables were derived for total alcohol consumption (number of drinks per month, week, or day) (Table 2).
Statistical analyses
We used unconditional logistic regression models to estimate the odds ratios (ORs) associated between childhood ALL and genetic polymorphisms, maternal folate intake and alcohol consumption in the year preceding the pregnancy, adjusting for child’s age at diagnosis/reference date, sex, race and Hispanic ethnicity (for analyses including all subjects). These models employed the following variables of interest: none, one or two copies of the variant allele for SNPs; folate intake using continuous or categorical variables (below or above the median of 430 mcg DFE per day in the control population for folate intake); alcohol consumption using ordinal categories (<1 drink per month, 1-3 drink(s) per month, 1 drink per week, 2-4 drinks per week, 5-6 drinks per week, or at least 1 drink per day) or two categories (<3 drinks per month, at least 3 drinks per month). Analyses involving folate or alcohol consumption were adjusted for household income. No statistically significant (p<0.05) main effects were detected between the risk of ALL and maternal folate intake (ORcontinuous=1.01 per 100 DFE unit, p=0.65; and ORcategorical=1.29 for high vs. low folate level, p=0.13) and alcohol consumption before pregnancy (ORordinal=0.98, p=0.63; and OR1+ drink per week vs. less=0.90, p=0.56), or alcohol consumption during pregnancy (OR1+ drink per month vs. less=0.86, p=0.66). No statistically significant interaction was reported between folate (as a continuous variable) and alcohol intake (ever/never) variables before (p=0.21) or during (p=0.41) pregnancy. These environmental factors were only used to test for interaction with genetic factors thereafter, using the Wald chi-square test. Interactions between SNPs and folate intake were estimated after adjustment for alcohol consumption, and interaction with alcohol consumption was estimated after adjustment for folate intake.
Nominal log-additive p-values were calculated for all 76 SNPs for all subjects together and separately for Hispanic and non-Hispanic subjects. To test for heterogeneity between ethnic groups, a p-value for the multiplicative interaction term between the genotype and Hispanic ethnicity was calculated for each SNP, using the Wald chi-square test.
We used a “sliding window” approach to conduct systematic analyses of haplotypes, with a “window” defined as 2 to 5 contiguous SNPs (40). For each possible window, haplotype frequencies were computed and a global score statistic (Obs) was calculated. We then calculated 1,000 simulated global score statistics (Simi) by randomly permuting case/control status. The p-value for the window was defined as the number of times Simi was greater than Obs, divided by 1,000. These analyses were conducted separately in Hispanics and non-Hispanics when there was a statistically significant interaction between at least one SNP within the gene and Hispanic ethnicity (p <0.05).
Haplotype blocks with the lowest p-value across all sliding windows (including single SNP windows) were selected for additional haplotype analyses. Analyses to examine specific haplotypes in the significant genomic regions were performed with haplotype trend regression techniques to calculate ORs associated with each copy of a specific haplotype using the most frequent haplotype as the referent group (41).
Adjustment for genetic ancestry derived from AIMs, in addition to maternal race, did not change the risk estimates associated with SNPs by more than 10% and was therefore not included in the final models for any SNP or haplotype.
Thirteen children with Down syndrome were excluded from all the analyses of SNPs located on chromosome 21 (gene CBS and SLC19A1). Analyses were implemented using SAS 9.1.3 and R 2.4.1 (Haplo.stats and SNPassoc packages). Graphical representations of the sliding window results were constructed using GrASP (42).
Results
The results for log-additive effects of individual SNPs are shown in Table 3. Overall, statistically significant associations (nominal p-values <0.05) were found between childhood ALL and SNPs genotyped in CBS (rs400660 and rs11909493, downstream from the gene region), MTHFR (rs1537515), and TYMS/ENOFS1 (rs1059393) and ENOFS1 (rs2612092) – a neighboring gene with overlapping transcripts, that regulates the expression of TYMS. Several SNPs in CBS, FOLH1, and MTRR showed statistically significant (p<0.05) heterogeneity in ALL risk between the two ethnic groups. Of these, rs6586281 and rs234705 in CBS, and rs162031 and rs10380 in MTRR showed significant associations in Hispanic children but not in non-Hispanic children or the population as a whole. The association observed for SNP rs400660 downstream of CBS appears to be limited to non-Hispanic children (p-value for test of heterogeneity =0.059). No significant association was detected overall for the eight non-synonymous SNPs and two synonymous SNPs located in MTHFD1, MTHFR, MTR, MTRR, and SLC19A1 (Table 4). However, a significantly increased risk of childhood ALL was observed in Hispanic children heterozygous for MTRR rs2287779 (OR=1.92, 95% CI: 1.14-3.25, Table 4).
Table 3. Association between the risk of childhood acute lymphoblastic leukemia and SNPs in the folate pathway genes, overall and by Hispanic ethnicity (the Northern California Childhood Leukemia Study, 1996-2002).
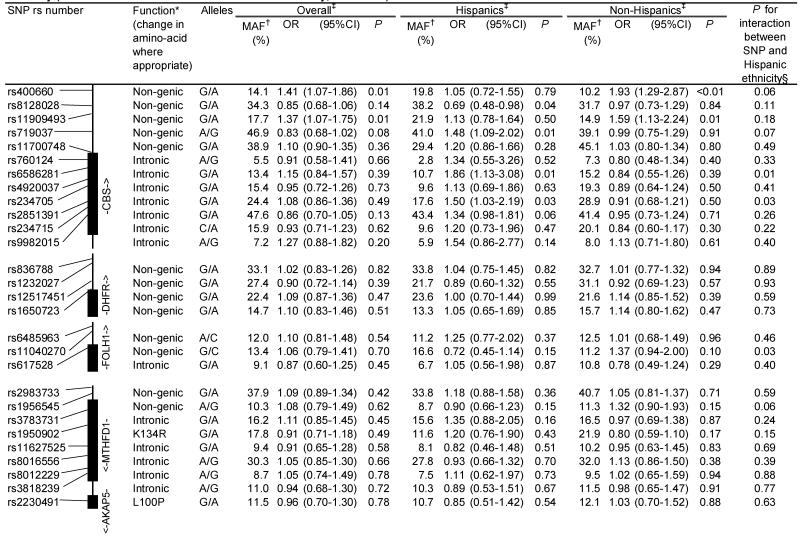
|
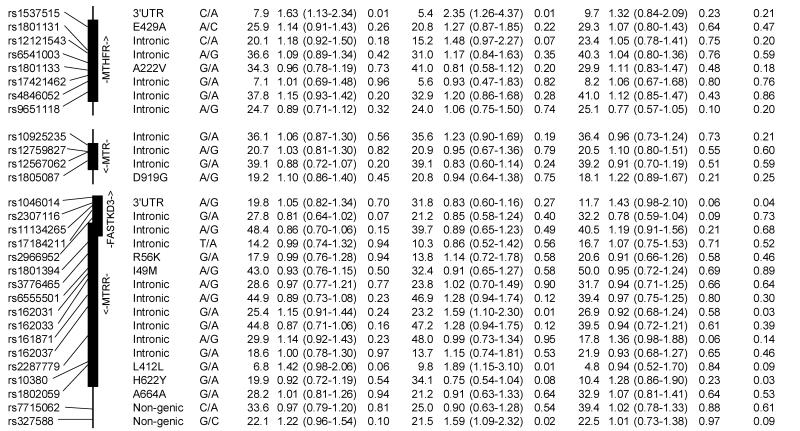
|
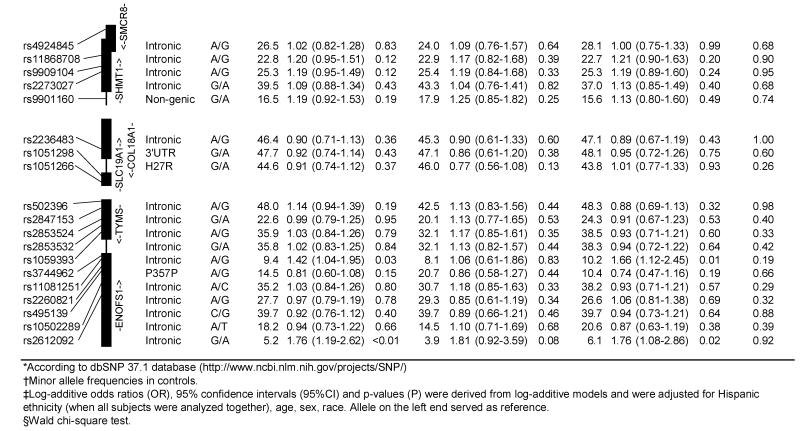
|
Table 4. Odds ratio estimates* for SNPs located in coding regions of genes (the Northern California Childhood Leukemia Study, 1996-2002).
| GENE | SNPs | Overall | Hispanics | Non-Hispanics | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| Numbers |
OR (95% CI) | Numbers |
OR (95% CI) | Numbers |
OR (95% CI) | |||||
| Ca | Co | Ca | Co | Ca | Co | |||||
| MTHFD1 | rs1950902 (K134R) | |||||||||
| R/R | 263 | 302 | 1.00 | 112 | 140 | 1.00 | 151 | 162 | 1.00 | |
| R/K | 98 | 124 | 0.92 (0.67-1.26) | 40 | 31 | 1.62 (0.95-2.75) | 58 | 93 | 0.67 (0.45-1.00) | |
| K/K | 12 | 17 | 0.82 (0.38-1.77) | 1 | 5 | 0.25 (0.03-2.17) | 11 | 12 | 0.99 (0.42-2.32) | |
| MTHFR | rs1801131 (E429A) | |||||||||
| E/E | 190 | 238 | 1.00 | 86 | 110 | 1.00 | 104 | 129 | 1.00 | |
| E/A | 159 | 184 | 1.10 (0.82-1.47) | 60 | 62 | 1.24 (0.79-1.95) | 99 | 121 | 1.02 (0.70-1.49) | |
| A/A | 26 | 24 | 1.37 (0.76-2.48) | 8 | 6 | 1.72 (0.57-5.19) | 18 | 18 | 1.24 (0.61-2.51) | |
| rs1801133 (A222V) | ||||||||||
| A/A | 169 | 186 | 1.00 | 62 | 59 | 1.00 | 107 | 127 | 1.00 | |
| A/V | 159 | 214 | 0.80 (0.60-1.08) | 72 | 91 | 0.74 (0.46-1.20) | 87 | 123 | 0.84 (0.57-1.24) | |
| V/V | 47 | 46 | 1.10 (0.69-1.75) | 20 | 27 | 0.69 (0.35-1.38) | 27 | 19 | 1.72 (0.89-3.31) | |
| MTR | rs1805087 (D919G) | |||||||||
| G/G | 237 | 292 | 1.00 | 99 | 11 | 1.00 | 138 | 181 | 1.00 | |
| G/D | 123 | 137 | 1.11 (0.82-1.49) | 51 | 60 | 0.97 (0.61-1.55) | 72 | 77 | 1.22 (0.83-1.81) | |
| D/D | 16 | 18 | 1.11 (0.55-2.22) | 5 | 7 | 0.78 (0.24-2.56) | 11 | 10 | 1.46 (0.60-3.56) | |
| MTRR | rs2966952 (R56K) | |||||||||
| R/R | 253 | 301 | 1.00 | 109 | 132 | 1.00 | 144 | 169 | 1.00 | |
| R/K | 114 | 127 | 1.08 (0.80-1.47) | 46 | 41 | 1.36 (0.83-2.23) | 68 | 86 | 0.93 (0.63-1.38) | |
| K/K | 9 | 16 | 0.69 (0.30-1.60) | 1 | 4 | 0.29 (0.03-2.72) | 8 | 12 | 0.79 (0.31-2.00) | |
| rs1801394 (I49M) | ||||||||||
| I/I | 133 | 145 | 1.00 | 74 | 84 | 1.00 | 59 | 61 | 1.00 | |
| I/M | 178 | 220 | 0.88 (0.64-1.21) | 69 | 74 | 1.05 (0.67-1.66) | 109 | 146 | 0.77 (0.49-1.20) | |
| M/M | 66 | 82 | 0.88 (0.58-1.35) | 13 | 21 | 0.69 (0.32-1.49) | 53 | 61 | 0.90 (0.53-1.53) | |
| rs2287779 (L412L) | ||||||||||
| G/G | 310 | 389 | 1.00 | 108 | 145 | 1.00 | 202 | 244 | 1.00 | |
| G/A | 64 | 57 | 1.43 (0.96-2.13) | 46 | 33 | 1.92 (1.14-3.25) | 18 | 24 | 0.91 (0.48-1.74) | |
| A/A | 3 | 2 | 1.92 (0.32-11.7) | 2 | 1 | 2.69 (0.24-30.2) | 1 | 1 | 1.18 (0.07-19.8) | |
| rs10380 (H622Y) | ||||||||||
| H/H | 252 | 293 | 1.00 | 82 | 79 | 1.00 | 170 | 213 | 1.00 | |
| H/Y | 103 | 131 | 0.88 (0.63-1.22) | 59 | 78 | 0.73 (0.46-1.15) | 44 | 54 | 1.00 (0.63-1.58) | |
| Y/Y | 20 | 23 | 0.97 (0.51-1.84) | 13 | 22 | 0.57 (0.27-1.22) | 7 | 1 | 8.69 (1.06-71.6) | |
| rs1802059 (A664A) | ||||||||||
| G/G | 189 | 238 | 1.00 | 98 | 117 | 1.00 | 91 | 121 | 1.00 | |
| G/C | 161 | 167 | 1.23 (0.91-1.65) | 53 | 48 | 1.33 (0.82-2.15) | 108 | 119 | 1.20 (0.82-1.76) | |
| C/C | 26 | 43 | 0.77 (0.45-1.30) | 4 | 14 | 0.34 (0.11-1.07) | 22 | 29 | 1.00 (0.54-1.88) | |
| SLC19A1 | rs1051266 (H27R) | |||||||||
| R/R | 106 | 132 | 1.00 | 49 | 49 | 1.00 | 58 | 83 | 1.00 | |
| R/H | 188 | 205 | 1.12 (0.81-1.56) | 74 | 89 | 0.83 (0.50-1.37) | 115 | 122 | 1.34 (0.87-2.05) | |
| H/H | 54 | 85 | 0.76 (0.49-1.18) | 21 | 35 | 0.59 (0.30-1.17) | 34 | 51 | 0.94 (0.53-1.65) | |
Adjustment for Hispanic ethnicity (when all subjects were analyzed together), age, sex, race.
Figures 1a and 1b show haplotype “sliding window” results for genes in which the p-value for at least one window was less than 0.10. Results are shown separately for Hispanics and non-Hispanics when at least one SNP within a given gene had a p-value for interaction by Hispanic ethnicity <0.10, as listed in Table 3. Single SNP associations observed for CBS, MTRR (in Hispanics) and TYMS/ENOFS1 persisted through increasingly larger haplotype windows. The haplotype block (rs400660 to rs719037) outside CBS appeared to be associated with childhood ALL, in both Hispanic children (minimum p<0.01 for the 4 SNP-window), and to some extent in non-Hispanics (minimum p<0.05 for a nested 3 SNP-window). In particular, the GAGG haplotype was associated with a decreased risk of childhood ALL in Hispanic children (Table 5). In contrast, the GAGCA and GGGCG haplotypes in the CBS region tagged by rs4920037 to rs9982015 were associated with an increased risk of childhood ALL.
Figure 1.


a. “Sliding window” haplotype analyses for all subjects and by Hispanic ethnicity: Genes CBS, MTHFD1 and MTHFR
Position of single nucleotide polymorphisms on chromosomes was based on dbSNP 37.1 database (http://www.ncbi.nlm.nih.gov/projects/SNP/). Results are presented for genes in which the p-value for at least one window was less than 0.10, and are shown separately for Hispanics and non-Hispanics when at least one single nucleotide polymorphisms within a given gene had a p-value for interaction by Hispanic ethnicity <0.10. Results are adjusted for child’s race, Hispanic ethnicity (when all children analyzed together), sex and age.
b. “Sliding window” haplotype analyses for all subjects and by Hispanic ethnicity: Genes MTRR and TYMS
Position of single nucleotide polymorphisms on chromosomes was based on dbSNP 37.1 database (http://www.ncbi.nlm.nih.gov/projects/SNP/). Results are presented for genes in which the p-value for at least one window was less than 0.10, and are shown separately for Hispanics and non-Hispanics when at least one single nucleotide polymorphisms within a given gene had a p-value for interaction by Hispanic ethnicity <0.10. Results are adjusted for child’s race, Hispanic ethnicity (when all children analyzed together), sex and age.
Table 5. Haplotype trend regression results (the Northern California Childhood Leukemia Study, 1996-2002).
| Genes/Haplotypes | Case % | Control % | OR (95% CI)* | P |
|---|---|---|---|---|
|
CBS (rs400660, rs8128028, rs11909493, rs719037) - Hispanics |
||||
| GGGA | 30.6 | 24.0 | Reference | |
| AGAG | 18.6 | 19.1 | 0.62 (0.24-1.65) | 0.33 |
| GAGA | 16.5 | 15.2 | 0.72 (0.23-2.27) | 0.58 |
| GAGG | 14.2 | 22.8 | 0.22 (0.08-0.60) | <0.01 |
| GGGG | 12.2 | 14.7 | 0.46 (0.13-1.60) | 0.22 |
| Rare haplotypes† | 7.8 | 4.3 | 2.66 (0.58-12.25) | 0.21 |
| Global P: 0.01 | ||||
|
CBS (rs400660, rs8128028, rs11909493, rs719037) - Non-Hispanics |
||||
| GAGA | 29.2 | 29.3 | Reference | |
| GGGA | 26.4 | 27.5 | 0.97 (0.48-1.97) | 0.93 |
| GGGG | 20.2 | 24.6 | 0.65 (0.30-1.38) | 0.26 |
| AGAG | 14.2 | 9.1 | 2.67 (1.04-6.81) | 0.04 |
| Rare haplotypes† | 10.1 | 9.5 | 1.24 (0.46-3.33) | 0.67 |
| Global P: 0.04 | ||||
|
CBS (rs4920037, rs234705, rs2851391, rs234715, rs9982015) - Hispanics |
||||
| GGACA | 38.3 | 47.8 | Reference | |
| GGGCA | 27.9 | 28.6 | 1.31 (0.61-2.79) | 0.49 |
| GAGCA | 14.8 | 8.2 | 4.17 (1.42-12.21) | <0.01 |
| AAAAA | 11.0 | 9.0 | 1.89 (0.67-5.35) | 0.23 |
| GGGCG | 8.7 | 5.6 | 3.11 (0.92-10.48) | 0.07 |
| Rare haplotypes† | 0.3 | 0.8 | 0.27 (0.01-26.48) | 0.58 |
| Global P: 0.06 | ||||
|
MTHFD (rs3783731, rs1950902, rs11627525) - Hispanics |
||||
| GGG | 70.4 | 74.6 | Reference | |
| AGG | 13.6 | 12.2 | 1.40 (0.52-3.77) | 0.51 |
| GAA | 4.3 | 6.6 | 0.45 (0.11-1.83) | 0.27 |
| Rare haplotypes† | 11.7 | 6.6 | 4.38 (1.35-14.14) | 0.01 |
| Global P: 0.04 | ||||
|
MTHFR (rs4846052, rs9651118) - Both ethnic groups |
||||
| AA | 40.4 | 37.8 | Reference | |
| GA | 37.2 | 37.6 | 0.90 (0.55-1.45) | 0.66 |
| GG | 21.5 | 24.5 | 0.66 (0.39-1.11) | 0.12 |
| AG | 0.81 | 0.17 | does not converge | 0.25 |
| Global P: 0.02 | ||||
|
MTRR (rs162037, rs2287779, rs10380) - Hispanics |
||||
| GGG | 40.9 | 42.5 | Reference | |
| GGA | 27.7 | 34.1 | 0.72 (0.35-1.48) | 0.37 |
| AGG | 15.4 | 13.7 | 1.40 (0.53-3.74) | 0.50 |
| GAG | 16.0 | 9.8 | 3.39 (1.20-9.57) | 0.02 |
| Global P: 0.04 | ||||
|
TYMS (rs3744962, rs11081251, rs2260821, rs495139) - Both ethnic groups |
||||
| ACAC | 33.6 | 33.4 | Reference | |
| AAGG | 21.1 | 22.4 | 0.83 (0.49-1.42) | 0.50 |
| AAAG | 13.9 | 15.2 | 0.77 (0.41-1.47) | 0.43 |
| AAAC | 15.5 | 11.2 | 1.93 (1.0-3.74) | 0.05 |
| GAAC | 9.5 | 12.4 | 0.53 (0.26-1.08) | 0.08 |
| Rare haplotypes† | 6.4 | 5.4 | 1.57 (0.64-3.83) | 0.33 |
| Global P: 0.04 | ||||
Adjustment for Hispanic ethnicity (when all subjects were analyzed together), age, sex, race.
Haplotypes with an estimate frequency of less than 5% overall were grouped together.
Other gene regions, which did not include significant single SNPs, were associated with childhood ALL (minimum p-value <0.05) as shown in Figures 1a and 1b (“sliding window” analyses) and Table 5 (haplotype trend regression analyses). These gene regions included (i) the haplotype block tagged by rs3783731 to rs11627525 in MTHFD1 among Hispanics, where associations were mainly driven by the rare haplotypes; (ii) the haplotype block tagged by rs4846052 and rs9651118 in MTHFR, where the GG haplotype appears to be slightly more prevalent in controls compared to cases (p=0.12); (iii) the haplotype block tagged by rs162037 to rs10380 in MTRR in Hispanics, where the GAG haplotype was associated with an increased risk of childhood ALL; and (iv) the haplotype block tagged by rs3744962 to rs495139 in gene TYMS/ENOFS1, where the AAAC haplotype conferred a two-fold increased risk of childhood ALL.
The associations of individual SNPs located in CBS, MTRR, and TYMS/ENOFS1 varied by level of maternal folate intake at the time of conception (P for interaction <0.05). These selected results are shown in Table 6, after adjustment for maternal alcohol intake during pregnancy.
Table 6. Interactions between child's SNPs in folate pathway genes and maternal folate intake (the Northern California Childhood Leukemia Study, 1996-2002).
| Gene | SNP rs number | Alleles | Odds ratios* (95% CI) |
P for interaction† |
|
|---|---|---|---|---|---|
|
| |||||
| Maternal folate intake less than 430 mcg DFEs per day (N=152 ca. and 204 co.) |
Maternal folate intake at least 430 mcg DFEs per day (N=196 ca. and 208 co.) |
||||
| CBS | rs6586281 | G/A | 0.56 (0.33-0.94) | 1.60 (1.02-2.53) | <0.01 |
| rs2851391 | G/A | 1.09 (0.58-2.03) | 0.66 (0.48-0.89) | 0.01 | |
| rs9982015 | A/G | 0.63 (0.34-1.17) | 1.57 (0.92-2.67) | 0.04 | |
| MTRR | rs162031 | G/A | 1.62 (1.12-2.32) | 0.74 (0.53-1.04) | <0.01 |
| rs162037 | G/A | 1.42 (0.94-2.15) | 0.70 (0.48-1.01) | 0.05 | |
| rs327588 | G/C | 1.59 (1.09-2.31) | 0.88 (0.62-1.26) | 0.04 | |
| TYMS | rs3744962 | A/G | 1.19 (0.76-1.89) | 0.51 (0.32-0.80) | 0.01 |
Log-additive odds ratios, adjusted for Hispanic ethnicity, age, sex, race, household income, and maternal alcohol intake during pregnancy. Allele on the left end served as reference.
Wald chi-square test.
Discussion
We examined the relationship between 76 SNPs in 10 genes involved in the folate pathway (i.e., CBS, DHFR, FOLH1, MTHFD1, MTHFR, MTR, MTRR, SHMT1, SLC19A1, and TYMS) and the development of childhood ALL. Our single SNP analyses did not provide evidence for an association with the most commonly studied MTHFR polymorphisms, i.e., 1298A>C (rs1801131) and 677C>T (rs1801133). We observed associations between childhood ALL and single SNPs in CBS and TYMS. Haplotype blocks in CBS, MTHFD1, MTRR, and MTHFR, as well as blocks just outside CBS and TYMS, were associated with ALL risk in children. Analyses conducted separately for Hispanic and non-Hispanic children showed differences in risk, especially for CBS and MTRR. In a few instances, associations with SNPs in CBS, MTRR, and TYMS – but not MTHFR – vary by level of maternal folate intake around the time of conception/early pregnancy.
Previous studies examining the association between childhood leukemia and MTHFR variants 1298A>C or 677C>T, and self-reported maternal folate intake have shown positive, negative, or null associations (7-10, 12-16, 18-24). The lack of associations with 1298A>C and 677C>T variants in our series is consistent with results from a comprehensive meta-analysis where the pooled OR for 677C>T variant was 0.88 (95% CI: 0.73-1.06; p=0.18) and the pooled OR for 1298A>C variant was 0.80 (95% CI: 0.56-1.16; p=0.24), suggesting that these variants are not likely to contribute to childhood ALL risk.(15) It is well-established that the 677C>T variant modulates folate and homocysteine levels, while the biological significance of 1298A>C polymorphism has been questioned as it does not appear to influence folate and homocysteine levels in populations mildly deficient in folate (43). Alternatively, 1298A>C polymorphism may only have an effect among subjects with low folate level (9, 23). Our null findings for the 1298A>C and 677C>T variants, as well as the lack of interaction between MTHFR variants and maternal folate intake, may be due to the increase in folate serum levels among women of childbearing age since the early 1990’s in the US (44). A case-only study in Australia (13) found no evidence of a multiplicative interaction between MTHFR variants 1298A>C and 677C>T and self-reported maternal folate supplementation during pregnancy, although this finding was based on small numbers. Similarly, one study in Germany (12) reported no association between childhood ALL with these MTHFR variants overall and after taking into account calendar years when folate supplementation during pregnancy was implemented. These observations contrasted with positive findings reported earlier in a Canadian study (9).
To our knowledge, no studies of childhood leukemia to date have examined SNPs in the MTHFR other than the 1298A>C (rs1801131) and 677C>T (rs1801133) variants. We examined eight tagging SNPs in MTHFR, and report an association between childhood ALL and single SNP rs1537515 (p=0.008) located in the 3′UTR regulatory region of the gene. This association, however, did not persist through increasingly larger haplotype windows. The haplotype analyses revealed that the block composed of rs4846052 and rs9651118 was also associated with childhood ALL risk (minimum p-value <0.05), although no specific haplotype could be identified as responsible for this association.
With the exception of MTHFR, few studies have examined the role of other key genes in the folate pathways (10, 20-23, 25). We report associations between childhood ALL and single and haplotype tagging SNPs, inside and outside the boundaries of CBS localized on chromosome 21 (minimum p<0.01). While no conclusions in terms of causality can be drawn from observations derived from non-synonymous SNPs, our data provide useful information for future validation studies. Several CBS gene variants have been previously associated with adult cancers at various sites, i.e., brain (45), digestive tract (46, 47), and lung (48). The CBS enzyme is involved in the trans-sulfuration pathway, inducing irreversible catalysis of homocysteine to cystathionine. As a result, overexpression of CBS decreases levels of homocysteine inducing functional folate deficiency (“folate trap”). Because homocysteine is at the intersection of the DNA synthesis and repair pathway and the DNA methylation pathway, overexpression of CBS may lead to alteration in all these functions (5). CBS is over-expressed in children with trisomy 21 (Down syndrome) (49), and it has been postulated that the cellular lesions caused by the “folate trap” may contribute to leukemia risk in children with Down syndrome. There were only 13 children with DS in our series; these were excluded from our analyses of genes localized on the chromosome 21. It will be of interest in future studies to consider possible interactions of CBS SNPs with risk for cytogenetic subtypes of ALL containing additional copies of chromosome 21. A clue to the possible functional properties of CBS SNPs might be finding that specific CBS alleles are overrepresented among the additional chromosome 21 within hyperdiploid leukemias, thus suggesting a selection for CBS SNP variants during leukemogenesis. In addition to comparing germline and leukemic CBS genotypes, future studies in our group are underway to examine the correlation between CBS gene variants, folate intake, cytogenetic markers, and methylation profiles.
The SNPs in SLC19A1, a folate transport gene located on chromosome 21 and close to CBS, were not associated with childhood leukemia. However, coverage of that gene was limited. The enzyme regulated by MTRR maintains the MTR enzyme in its active form, which is essential for maintaining an adequate level of methionine, a precursor of the universal methyl group donor SAM. We observed a statistically significant increased risk of childhood ALL in children carrying the A variant of SNP rs2287779 (L412L) in exon 9 of MTRR, where the association appears to be limited to Hispanic children. In contrast to recent studies (10, 20, 21), we did not report association with rs1801394 (I49M) in MTRR.
We reported associations between childhood ALL and intronic SNPs in ENOFS1, a gene regulating the expression of TYMS in the DNA synthesis pathway. One childhood leukemia study examined two polymorphisms (2R>3R and 1494del6) of TYMS and found no difference in haplotype distribution between ALL cases and controls (10). Lower risks of childhood ALL were reported in Malay carriers of both the 3′-TYMS 1494- 6p/−6p genotype and SLC19A1 80G>A (rs1051266) (24). No statistically significant associations were seen between the TYMS 6bp and 2R>3R polymorphisms and adult ALL, while a decreased risk of adult AML was reported for the TYMS 6bp polymorphism (50). Although single SNPs in MTHFD1 were not associated with childhood ALL in our series, haplotype analyses suggest an association with the gene region tagged by rs3783731 to rs11627525. No single SNP or haplotype in the SHMT1 and MTR genes was associated with childhood ALL risk in our study. Null findings were also previously reported for MTHFD1 1958 G>A (rs2236225) (10), MTHFD1 401 G>A (rs1950902) (10), TYMS 28-bp,(23) SHMT1 1420 C>T (rs1979277) (10, 20, 23), and MTR 2756A>G (rs1805087) (10, 20) polymorphisms. In contrast, a large study in the UK reported an association between MTR 2756A>G and ALL and AML in children, specifically in the presence of MLL chromosomal abnormality (23). We reported that associations between several folate pathway genes and risk of childhood ALL varied by levels of maternal folate and alcohol intake. Although similar findings have been observed for various solid cancers in adults (51-54), no previous studies of childhood leukemia has comprehensively examined gene-environment interaction.
The current study presents several strengths and limitations. Haplotype analyses using tagging SNPs provide useful information on gene regions possibly involved in the etiology of childhood leukemia, but are not sufficient to establish causality. Although coverage of the folate genes in our study is not complete, it is more comprehensive than most investigations of this pathway in childhood ALL studies to date. Our analyses were initiated based on a priori hypotheses about the role of genes in folate metabolism pathways. This candidate gene approach, however, still suffers from the possibility of false positive reporting as none of the observed single SNP associations remained statistically significant after strict multiple testing correction using the Bonferroni method (results not shown). In contrast to single SNP analyses, the “sliding window” haplotype analyses has the advantage of providing additional power for mapping disease genes by examining larger sets of neighboring SNPs (55). Most individual epidemiologic studies of childhood leukemia are hampered by low statistical power. To overcome this limitation, the Childhood Leukemia International Consortium (http://clic.berkeley.edu) was recently established to conduct pooled and replication studies of genetic and environmental factors with greater sample size within the Consortium.
Our study population is unique in that it includes approximately 40% Hispanics. We observed several differences by Hispanic ethnicity in the association between several SNPs and childhood ALL, but the underlying reasons remain unclear. The analyses of ancestry informative markers in our series showed that potential confounding by population stratification was very low, likely due to close matching on race and ethnicity. We used all available information to select haplotype-tagging SNPs based on ethnic groups, but this was not possible for all SNPs. As a result, coverage of genetic variation by the selected SNPs might be unequal in the two populations, limiting our ability to interpret the observed ethnic differences. Haplotype-tagging SNPs have been mostly defined in US residents from northern and western European ancestry, such as in the HapMap project, and any given haplotype may be more or less common depending on the population. Finally, findings by ethnic group may be due to chance alone due to low statistical power for stratified analyses, and therefore need to be replicated.
In our study, annual household income, a potential confounder of alcohol and/or folate intake, was lower in cases compared to controls. We previously assessed whether this observation may be due to selection bias (28), using information available on California birth certificates to compare characteristics such as parental age and education and maternal reproductive history between participating controls (i.e., whose location was successfully traced and provided consent) and “ideal” controls from the birth registry (with no tracing effort). We found little difference in socio-demographic characteristics between the two (28), suggesting that participating controls are representative of the study population base, therefore reducing the potential for selection bias. In the current study, the amount of maternal alcohol consumption before and during pregnancy increases with the level of household income (p<0.01). Although we adjusted for this possible confounder, we cannot rule out residual confounding by socio-economic status. In contrast, folate intake was not associated with household income (p=0.82).
Estimates of maternal folate intake were derived from a detailed food frequency questionnaire with information on dietary and supplemental source three months prior to conception, as a surrogate for “folate environment” of the fetus during early pregnancy. No data on folate supplementation during the entire pregnancy were available for this analysis. Inherent to the case-control design, collection of self-reported information may suffer from recall bias. The public knowledge of “healthy” diets and lifestyles and their impacts on disease prevention may have led to differential recall between case and control mothers. However, the direction of a possible bias is difficult to evaluate since both over- and under-reporting may have occurred. The lack of association between maternal intake of alcohol and folate and childhood leukemia in our series may be due to non-differential misclassification of diet between case and control mothers, leading to risk estimates biased toward the null (56). However, the data collection methods and estimation of maternal folate level were similar between cases and controls. An alternative explanation is that folate levels are high in the California population (57) so that lack of variation in exposure may have affected our ability to identify possible associations or interactions.
In conclusion, our data do not support associations between childhood ALL and SNPs or haplotypes in the MTHFR gene. The strongest associations were found for the CBS gene, with SNPs located in or adjacent to the gene region. There were also suggestions of associations with haplotype blocks in genes MTRR, and TYMS/ENOFS1.
Acknowledgements
This research could not have been conducted without the strong support from our clinical collaborators and participating hospitals which include: University of California Davis Medical Center (Dr. Jonathan Ducore), University of California San Francisco (Dr. Mignon Loh and Dr Katherine Matthay), Children’s Hospital of Central California (Dr. Vonda Crouse), Lucile Packard Children’s Hospital (Dr. Gary Dahl), Children’s Hospital Oakland (Dr. James Feusner), Kaiser Permanente Sacramento (Dr. Vincent Kiley), Kaiser Permanente Santa Clara (Dr. Carolyn Russo and Dr. Alan Wong), Kaiser Permanente San Francisco (Dr. Kenneth Leung), and Kaiser Permanente Oakland (Dr. Stacy Month), and the families of the study participants. We also acknowledge our collaborators at the California Department of Public Health, and the entire Northern California Childhood Leukemia Study staff for their effort and dedication.
Financial support: Children With Leukaemia, UK, grants 2005/027 and 2006/051; National Institute of Environmental Health Sciences, grants P42-ES04705 and R01 ES09137. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences.
Footnotes
Authorship
Contributions: C.M., A.C., and P.A.B. designed the study and directed its implementation; G.S. performed statistical analyses; G.S. and C.M. equally analyzed and interpreted the data, and prepared the manuscript; L.F.B., J.W., J.L.W., M.C.A, J.S.C., N.G., K.Y.U., and G.B. contributed to the study design; G.B. assessed the maternal folate intake and alcohol consumption; V.K. contributed to data collection; H.M.H performed laboratory work for genotyping data; all authors provided critical review of the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Ferlay J, Bray F, Pisani P, Parkin DM. Globocan 2002 – Cancer incidence, mortality and prevalence worldwide (IARC CancerBase No. 5, version 2.0) 2004 [Google Scholar]
- 2.Parkin DM, Whelan SL, Ferlay J, Storm H. Cancer in five continents, Vol. I to VIII (IARC CancerBase No. 7) 2005 [Google Scholar]
- 3.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6:193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 4.Friso S, Choi SW. Gene-nutrient interactions in one-carbon metabolism. Curr Drug Metab. 2005;6:37–46. doi: 10.2174/1389200052997339. [DOI] [PubMed] [Google Scholar]
- 5.Duthie SJ. Folic acid deficiency and cancer: mechanisms of DNA instability. Br Med Bull. 1999;55:578–592. doi: 10.1258/0007142991902646. [DOI] [PubMed] [Google Scholar]
- 6.Thompson JR, Gerald PF, Willoughby ML, Armstrong BK. Maternal folate supplementation in pregnancy and protection against acute lymphoblastic leukaemia in childhood: a case-control study. Lancet. 2001;358:1935–1940. doi: 10.1016/S0140-6736(01)06959-8. [DOI] [PubMed] [Google Scholar]
- 7.Wiemels JL, Smith RN, Taylor GM, Eden OB, Alexander FE, Greaves MF. Methylenetetrahydrofolate reductase (MTHFR) polymorphisms and risk of molecularly defined subtypes of childhood acute leukemia. Proc Natl Acad Sci U S A. 2001;98:4004–4009. doi: 10.1073/pnas.061408298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franco RF, Simoes BP, Tone LG, Gabellini SM, Zago MA, Falcao RP. The methylenetetrahydrofolate reductase C677T gene polymorphism decreases the risk of childhood acute lymphocytic leukaemia. Br J Haematol. 2001;115:616–618. doi: 10.1046/j.1365-2141.2001.03140.x. [DOI] [PubMed] [Google Scholar]
- 9.Krajinovic M, Lamothe S, Labuda D, et al. Role of MTHFR genetic polymorphisms in the susceptibility to childhood acute lymphoblastic leukemia. Blood. 2004;103:252–257. doi: 10.1182/blood-2003-06-1794. [DOI] [PubMed] [Google Scholar]
- 10.Gast A, Bermejo JL, Flohr T, et al. Folate metabolic gene polymorphisms and childhood acute lymphoblastic leukemia: a case-control study. Leukemia. 2007;21:320–325. doi: 10.1038/sj.leu.2404474. [DOI] [PubMed] [Google Scholar]
- 11.Bailey LB. Folate, methyl-related nutrients, alcohol, and the MTHFR 677C-->T polymorphism affect cancer risk: intake recommendations. J Nutr. 2003;133:3748S–3753S. doi: 10.1093/jn/133.11.3748S. [DOI] [PubMed] [Google Scholar]
- 12.Thirumaran RK, Gast A, Flohr T, et al. MTHFR genetic polymorphisms and susceptibility to childhood acute lymphoblastic leukemia [letter] Blood. 2005;106:2590–2591. doi: 10.1182/blood-2005-04-1719. [DOI] [PubMed] [Google Scholar]
- 13.Milne E, de Klerk NH, van Bockxmeer F, et al. Is there a folate-related gene-environment interaction in the etiology of childhood acute lymphoblastic leukemia? Int J Cancer. 2006;119:229–232. doi: 10.1002/ijc.21803. [DOI] [PubMed] [Google Scholar]
- 14.Zintzaras E, Koufakis T, Ziakas PD, Rodopoulou P, Giannouli S, Voulgarelis M. A meta-analysis of genotypes and haplotypes of methylenetetrahydrofolate reductase gene polymorphisms in acute lymphoblastic leukemia. Eur J Epidemiol. 2006;21:501–510. doi: 10.1007/s10654-006-9027-8. [DOI] [PubMed] [Google Scholar]
- 15.Pereira TV, Rudnicki M, Pereira AC, Pombo-de-Oliveira MS, Franco RF. 5,10-Methylenetetrahydrofolate reductase polymorphisms and acute lymphoblastic leukemia risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:1956–1963. doi: 10.1158/1055-9965.EPI-06-0334. [DOI] [PubMed] [Google Scholar]
- 16.Alcasabas P, Ravindranath Y, Goyette G, et al. 5,10-methylenetetrahydrofolate reductase (MTHFR) polymorphisms and the risk of acute lymphoblastic leukemia (ALL) in Filipino children. Pediatr Blood Cancer. 2008;51:178–182. doi: 10.1002/pbc.21511. [DOI] [PubMed] [Google Scholar]
- 17.Amorim MR, Zanrosso CW, Magalhaes IQ, et al. MTHFR 677C-->T and 1298A-->C polymorphisms in children with Down syndrome and acute myeloid leukemia in Brazil. Pediatr Hematol Oncol. 2008;25:744–750. doi: 10.1080/08880010802435104. [DOI] [PubMed] [Google Scholar]
- 18.Schnakenberg E, Mehles A, Cario G, et al. Polymorphisms of methylenetetrahydrofolate reductase (MTHFR) and susceptibility to pediatric acute lymphoblastic leukemia in a German study population. BMC Med Genet. 2005;6:23. doi: 10.1186/1471-2350-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balta G, Yuksek N, Ozyurek E, et al. Characterization of MTHFR, GSTM1, GSTT1, GSTP1, and CYP1A1 genotypes in childhood acute leukemia. Am J Hematol. 2003;73:154–160. doi: 10.1002/ajh.10339. [DOI] [PubMed] [Google Scholar]
- 20.de Jonge R, Tissing WJ, Hooijberg JH, et al. Polymorphisms in folate-related genes and risk of pediatric acute lymphoblastic leukemia. Blood. 2009;113:2284–2289. doi: 10.1182/blood-2008-07-165928. [DOI] [PubMed] [Google Scholar]
- 21.Petra BG, Janez J, Vita D. Gene-gene interactions in the folate metabolic pathway influence the risk for acute lymphoblastic leukemia in children. Leuk Lymphoma. 2007;48:786–792. doi: 10.1080/10428190601187711. [DOI] [PubMed] [Google Scholar]
- 22.Kamel AM, Moussa HS, Ebid GT, Bu RR, Bhatia KG. Synergistic effect of methyltetrahydrofolate reductase (MTHFR) C677T and A1298C polymorphism as risk modifiers of pediatric acute lymphoblastic leukemia. J Egypt Natl Canc Inst. 2007;19:96–105. [PubMed] [Google Scholar]
- 23.Lightfoot TJ, Johnston WT, Painter D, et al. Genetic variation in the folate metabolic pathway and risk of childhood leukemia. Blood. 2010;115:3923–3929. doi: 10.1182/blood-2009-10-249722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeoh AE, Lu Y, Chan JY, et al. Genetic susceptibility to childhood acute lymphoblastic leukemia shows protection in Malay boys: results from the Malaysia-Singapore ALL Study Group. Leuk Res. 2010;34:276–283. doi: 10.1016/j.leukres.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Gra OA, Glotov AS, Kozhekbaeva Z, Makarova OV, Nasedkina TV. [Genetic polymorphism in GST, NAT2, and MTRR and susceptibility to childhood acute leukemia] Mol Biol (Mosk) 2008;42:214–225. Russian. [PubMed] [Google Scholar]
- 26.Koppen IJ, Hermans FJ, Kaspers GJ. Folate related gene polymorphisms and susceptibility to develop childhood acute lymphoblastic leukaemia. Br J Haematol. 2010;148:3–14. doi: 10.1111/j.1365-2141.2009.07898.x. [DOI] [PubMed] [Google Scholar]
- 27.Mason JB, Choi SW. Effects of alcohol on folate metabolism: implications for carcinogenesis. Alcohol. 2005;35:235–241. doi: 10.1016/j.alcohol.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 28.Ma X, Buffler PA, Layefsky M, Does MB, Reynolds P. Control selection strategies in case-control studies of childhood diseases. Am J Epidemiol. 2004;159:915–21. doi: 10.1093/aje/kwh136. [DOI] [PubMed] [Google Scholar]
- 29.Kwan ML, Block G, Selvin S, Month S, Buffler PA. Food consumption by children and the risk of childhood acute leukemia. Am J Epidemiol. 2004;160:1098–107. doi: 10.1093/aje/kwh317. [DOI] [PubMed] [Google Scholar]
- 30.Bartley K, Metayer C, Selvin S, Ducore J, Buffler P. Diagnostic X-rays and risk of childhood leukaemia. Int J Epidemiol. 2010;39:1628–37. doi: 10.1093/ije/dyq162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen HM, Wiemels JL, Wrensch M, Wiencke JK. DNA quantification of whole genome amplified samples for genotyping on a multiplexed bead array platform. Cancer Epidemiol Biomarkers Prev. 2007;16:1686–1690. doi: 10.1158/1055-9965.EPI-06-1024. [DOI] [PubMed] [Google Scholar]
- 32.Paynter RA, Skibola DR, Skibola CF, Buffler PA, Wiemels JL, Smith MT. Accuracy of multiplexed Illumina platform-based single-nucleotide polymorphism genotyping compared between genomic and whole genome amplified DNA collected from multiple sources. Cancer Epidemiol Biomarkers Prev. 2006;15:2533–2536. doi: 10.1158/1055-9965.EPI-06-0219. [DOI] [PubMed] [Google Scholar]
- 33.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 34.The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 35.Packer BR, Yeager M, Burdett L, Koppen IJ, Hermans FJ, Kaspers GJ. SNP500Cancer: a public resource for sequence validation, assay development, and frequency analysis for genetic variation in candidate genes. Nucleic Acids Res. 2006;34:D617–D621. doi: 10.1093/nar/gkj151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 37.Chakraborty R, Weiss KM. Frequencies of complex diseases in hybrid populations. Am J Phys Anthropol. 1986;70:489–503. doi: 10.1002/ajpa.1330700408. [DOI] [PubMed] [Google Scholar]
- 38.Hanis CL, Chakraborty R, Ferrell RE, Schull WJ. Individual admixture estimates: disease associations and individual risk of diabetes and gallbladder disease among Mexican-Americans in Starr County, Texas. Am J Phys Anthropol. 1986;70:433–441. doi: 10.1002/ajpa.1330700404. [DOI] [PubMed] [Google Scholar]
- 39.Jensen CD, Block G, Buffler P, Ma X, Selvin S, Month S. Maternal dietary risk factors in childhood acute lymphoblastic leukemia (United States) Cancer Causes Control. 2004;15:559–570. doi: 10.1023/B:CACO.0000036161.98734.17. [DOI] [PubMed] [Google Scholar]
- 40.Yu Z, Schaid DJ. Sequential haplotype scan methods for association analysis. Genet Epidemiol. 2007;31:553–564. doi: 10.1002/gepi.20228. [DOI] [PubMed] [Google Scholar]
- 41.Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered. 2002;53:79–91. doi: 10.1159/000057986. [DOI] [PubMed] [Google Scholar]
- 42.Mathias RA, Gao P, Goldstein JL, et al. A graphical assessment of p-values from sliding window haplotype tests of association to identify asthma susceptibility loci on chromosome 11q. BMC Genet. 2006;7:38. doi: 10.1186/1471-2156-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pereira AC, Schettert IT, Morandini Filho AA, Guerra-Shinohara EM, Krieger JE. Methylenetetrahydrofolate reductase (MTHFR) c677t gene variant modulates the homocysteine folate correlation in a mild folate-deficient population. Clin Chim Acta. 2004;340:99–105. doi: 10.1016/j.cccn.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 44.McDowell MA, Lacher DA, Pfeiffer CM, et al. Blood folate levels: the latest NHANES results. NCHS Data Brief. 2008:1–8. [PubMed] [Google Scholar]
- 45.Semmler A, Simon M, Moskau S, Linnebank M. Polymorphisms of methionine metabolism and susceptibility to meningioma formation: laboratory investigation. J Neurosurg. 2008;108:999–1004. doi: 10.3171/JNS/2008/108/5/0999. [DOI] [PubMed] [Google Scholar]
- 46.Le Marchand L, Donlon T, Hankin JH, Kolonel LN, Wilkens LR, Seifried A. B-vitamin intake, metabolic genes, and colorectal cancer risk (United States) Cancer Causes Control. 2002;13:239–248. doi: 10.1023/a:1015057614870. [DOI] [PubMed] [Google Scholar]
- 47.Ott N, Geddert H, Sarbia M. Polymorphisms in methionine synthase (A2756G) and cystathionine beta-synthase (844ins68) and susceptibility to carcinomas of the upper gastrointestinal tract. J Cancer Res Clin Oncol. 2008;134:405–410. doi: 10.1007/s00432-007-0301-2. [DOI] [PubMed] [Google Scholar]
- 48.Shen M, Rothman N, Berndt SI, et al. Polymorphisms in folate metabolic genes and lung cancer risk in Xuan Wei, China. Lung Cancer. 2005;49:299–309. doi: 10.1016/j.lungcan.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Chadefaux B, Rethore MO, Raoul O, et al. Cystathionine beta synthase: gene dosage effect in trisomy 21. Biochem Biophys Res Commun. 1985;128:40–44. doi: 10.1016/0006-291x(85)91641-9. [DOI] [PubMed] [Google Scholar]
- 50.Kim HN, Kim YK, Lee IK, et al. Association between polymorphisms of folate-metabolizing enzymes and hematological malignancies. Leuk Res. 2009;33:82–87. doi: 10.1016/j.leukres.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 51.Murtaugh MA, Curtin K, Sweeney C, et al. Dietary intake of folate and co-factors in folate metabolism, MTHFR polymorphisms, and reduced rectal cancer. Cancer Causes Control. 2007;18:153–163. doi: 10.1007/s10552-006-0099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang FF, Terry MB, Hou L, et al. Genetic polymorphisms in folate metabolism and the risk of stomach cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:115–121. doi: 10.1158/1055-9965.EPI-06-0513. [DOI] [PubMed] [Google Scholar]
- 53.Arasaradnam RP, Commane DM, Bradburn D, Mathers JC. A review of dietary factors and its influence on DNA methylation in colorectal carcinogenesis. Epigenetics. 2008;3:193–198. doi: 10.4161/epi.3.4.6508. [DOI] [PubMed] [Google Scholar]
- 54.Hung RJ, Hashibe M, McKay J, et al. Folate-related genes and the risk of tobacco-related cancers in Central Europe. Carcinogenesis. 2007;28:1334–40. doi: 10.1093/carcin/bgm067. [DOI] [PubMed] [Google Scholar]
- 55.Liu N, Zhang K, Zhao H. Haplotype-association analysis. Adv Genet. 2008;60:335–405. doi: 10.1016/S0065-2660(07)00414-2. [DOI] [PubMed] [Google Scholar]
- 56.Kwan ML, Jensen CD, Block G, Hudes ML, Chu LW, Buffler PA. Maternal diet and risk of childhood acute lymphoblastic leukemia. Public Health Rep. 2009;124:503–514. doi: 10.1177/003335490912400407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caudill MA, Le T, Moonie SA, Esfahani ST, Cogger EA. Folate status in women of childbearing age residing in Southern California after folic acid fortification. J Am Coll Nutr. 2001;20:129–134. doi: 10.1080/07315724.2001.10719024. [DOI] [PubMed] [Google Scholar]