

Abstract
Purpose
It is highly challenging to develop reliable diagnostic tests to predict patients’ responsiveness to anticancer treatments on clinical endpoints before commencing the definitive phase III randomized trial. Development and validation of genomic signatures in the randomized trial can be a promising solution. Such signatures are required to predict quantitatively the underlying heterogeneity in the magnitude of treatment effects.
Experimental Design
We propose a framework for developing and validating genomic signatures in randomized trials. Codevelopment of predictive and prognostic signatures can allow prediction of patient-level survival curves as basic diagnostic tools for treating individual patients.
Results
We applied our framework to gene-expression microarray data from a large-scale randomized trial to determine whether the addition of thalidomide improves survival for patients with multiple myeloma. The results indicated that approximately half of the patients were responsive to thalidomide, and the average improvement in survival for the responsive patients was statistically significant. Cross-validated patient-level survival curves were developed to predict survival distributions of individual future patients as a function of whether or not they are treated with thalidomide and with regard to their baseline prognostic and predictive signature indices.
Conclusion
The proposed framework represents an important step toward reliable predictive medicine. It provides an internally validated mechanism for using randomized clinical trials to assess treatment efficacy for a patient population in a manner that takes into consideration the heterogeneity in patients’ responsiveness to treatment. It also provides cross-validated patient-level survival curves that can be used for selecting treatments for future patients.
Introduction
Advances in our understanding of the biology of human cancers have revealed substantial molecular heterogeneity of most conventional histologic diagnoses. In treating patients with cancer, this implies that only a subset of treated patients is likely to benefit from a given therapy. This is particularly relevant for molecularly targeted drugs (1–3). As such, there is a substantial growing need for developing a diagnostic test to predict responsiveness of a given treatment for individual patients. To this end, basic research often suggests a panel of candidate predictive markers. Genomic technologies, such as microarrays, have provided powerful tools for developing genomic signatures (diagnostic tests) based on genome-wide screening. Ideally, the diagnostic test would be developed before initiating the definitive phase III trial that evaluates its effectiveness in distinguishing patients who benefit from a new treatment from those who do not with regard to clinical endpoints, such as survival or disease-free survival (4). However, this is difficult because of the complexity of signaling pathways and the inherent difficulty in developing reliable diagnostic tests for clinical endpoints using early phase II data. One approach to address this issue, in which a signature to identify responsive patients is unavailable, is to design and analyze the randomized phase III trial in such a way that both developing a genomic signature and testing treatment efficacy based on the developed signature is possible and conducted validly. The adaptive signature designs (ASD) have been proposed for this purpose (5, 6; see Appendix A).
A genomic signature is usually developed as a composite score integrating the status or values of multiple component genes. Such signatures are continuously valued and represent varying treatment effects among patients (7), reflecting the complexity of disease biology. For developing a diagnostic test, one conventional framework is to define responsive and nonresponsive patients by invoking a thresholding of the continuous score (i.e., patients whose scores are higher or smaller than a threshold are defined as responsive patients) as done in the ASDs (5, 6). However, in this framework, information about varying treatment effects among responsive patients would be lost. This can be a concern for plausible situations where treatment selection is affected by many factors, including adverse effects, cost of treatment, and patient’s preference, and thus the size of treatment effect that leads to a patient’s selecting the treatment can differ among patients. This suggests the need for another framework that presents the treatment effect as a function of the continuous genomic signature as a more relevant diagnostic tool for predictive medicine.
In this article, we consider this framework and propose a new methodology for developing and validating genomic signatures in randomized trials, through estimating the underlying profile of the variation of treatment effects as a function of continuous genomic signatures. On the basis of an estimate of this function, we can provide a cross-validation–based test of treatment efficacy for the patient population, and also provide cross-validated patient-level survival curves and treatment effects for use in treatment decisions for individual patients. The motivating example is a phase III trial for assessing whether the addition of thalidomide improves survival for patients with multiple myeloma undergoing high-dose therapy (8, 9). Figure 1 shows a small survival difference between the 2 treatment arms (thalidomide and no thalidomide) for the subset of patients with genomic data in the trial. This small average effect may reflect the heterogeneity in the effects of thalidomide. By applying our framework to the data of this trial, we can estimate the profile of treatment effects as a function of the genomic signature (with a normalized range from 0 to 1), as shown in Fig. 2. This allows us to predict the treatment effect for any individual patient. On the basis of the estimated treatment effects function, the treatment effect for the overall population and its heterogeneity can be assessed. Furthermore, our framework allows prediction of patient-level survival curves for each treatment for any subset of patients with various degrees of responsiveness to treatments and various degrees of baseline risks, represented by the predictive and prognostic signatures, as shown in Fig. 3. Such prediction curves can serve as a basic diagnostic tool for selecting appropriate treatments for future patients.
Figure 1.

Survival curves for all 351 patients with genomic data in the randomized trial for multiple myeloma.
Figure 2.
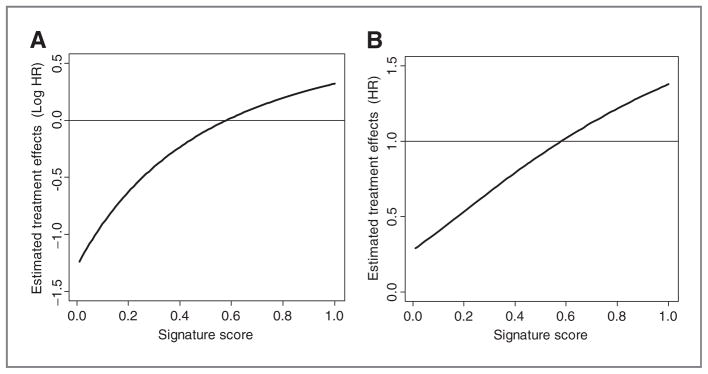
The estimated treatment effects functions for the predicted signature score, S, in terms of logarithm of HR, that is, Ψ̂(A) and HR, that is, exp(Ψ̂) (B). Here, Ψ̂(s) = 0.79 − 2.02(s + 1)−2 derived from the fitted linear predictor, 0.79r – 0.69s – 2.02{r(s + 1)}−2 + 1.72w for the model (4).
Figure 3.

A to D, the predicted survival curves when receiving thalidomide (solid curves) and no thalidomide (dashed curves) for 4 patients with different values of the predictive score (S) and the prognostic score (W); (s, w) = (0.1, 0.1), (0.1, 0.9), (0.5, 0.1), and (0.5, 0.9). s = 0.1 (0.5) represents a high (small) responsiveness to thalidomide, whereas w = 0.1 (0.9) represents a good (poor) prognosis.
Materials and Methods
We consider a randomized trial to compare an experimental (E) and control treatment (C). We suppose that pretreatment genomic data for signature development is available for a total of n patients. To be specific, we suppose that pretreatment expression levels for a total of G genes are measured using microarrays for each patient, although other types of genomic data, such as single-nucleotide polymorphism genotyping, copy number profiling, and proteomic profiling data can be used similarly.
There are 4 components in our framework:
Development of a continuous signature score,
Estimation of the treatment effects as a function of the developed score,
Test of the strong null hypothesis that the treatment has no effect on any patients,
Prediction of patient-level survival curves for future patients.
The statistical models and procedures used in all the components must be prespecified. Figure 4 outlines our methodology.
Figure 4.

Outline of the proposed methodology.
The last component is a step for developing a basic diagnostic tool to aid selecting appropriate treatments for individual future patients, which is different from the purpose of assessing treatment efficacy for a patient population.
Development of genomic signature score
Because the genomic signature is to predict treatment effects for individual patients, we consider a framework of prediction analysis and apply complete K-fold cross-validation as in the cross-validated ASD because it is more efficient than the split-sample approach (6; see Appendix A). In the K-fold cross-validation, the trial population is split into K roughly equal-sized parts. Patient allocation into the K parts must be prospectively defined. For the kth part, a genomic signature is developed using the data from the other K – 1 parts as a training set, and it is applied to the kth part as a test set (k = 1, …, K). In each fold of cross-validation, the development of the genomic signature is typically composed of selection of predictive gene features from scratch and building of a signature scoring using the selected gene features for the training set (see Appendix B). Without loss of generality, we suppose that the signature score is developed such that, for a patient with a low value of the developed score, the survival probability when receiving E is predicted to be higher than that when receiving C; as such, this patient is predicted to be responsive to E. A large variety of algorithms for developing such scores could be envisaged. The compound covariates predictor is one of the simple, but effective algorithms for high-dimensional genomic data (see Appendix B). After developing a signature scoring function using the training set, we apply it to compute predicted scores for all patients in the test set (denoted by Ui in Appendix B). The value of the predicted score for each patient in the test set is normalized as a quantile (or percentile) value based on the score distribution in the training set.
In each fold of the cross-validation, it is critical that all aspects of the signature development, including selection of gene features, are reconducted (10, 11). When selection of gene features and/or prediction models to develop the signature are optimized on the basis of cross-validated predictive accuracy, the optimization process should be included in the K-fold cross-validation with application of a nested inner loop of K-fold cross-validation (12, 13).
After completion of the K-fold cross-validation, we have the predicted (quantile) scores for all n patients. We denote the score for patient i as Si ∈ (0, 1) (i = 1, …, n). Note that, because Si is a quantile measure, it is essentially continuously valued. Again, for patients with lower values of Si, the survival probabilities when receiving E are predicted to be higher than those when receiving C.
Estimation of the treatment effects function
Let us say we estimate the treatment effects as a function of Si, Ψ(s). As a basic measure of treatment effects, we consider a logarithm of the HR between the 2 treatment arms under the proportional hazards assumption that the HR is constant over time. Specifically, for a patient with the score value S = s,
| (1) |
represents the treatment effect for that patient. We assume the multivariate Cox proportional hazards model,
| (2) |
where ri is the treatment assignment indicator such that ri = 1 if patient i is assigned to treatment E and ri = 0 otherwise. Here, the functions f2 and f3 capture the main effects of S and the interactions between S and r, respectively. These effects can be nonlinear, but should be monotonic in S, because the score S has been developed such that its lower value represents greater responsiveness to E, on the basis of commonly used linear models in selection of gene features and development of the signature score S (see Appendix B). One simple specification to have monotonic effects for S is to use the fractional polynomials (FP; ref. 14; see Appendix C). The model (2) is fitted by maximum (partial) likelihood. Under the model (2), the treatment effects function Ψ will be expressed as,
| (3) |
Under the specification of monotonic effects for the interaction f3, the estimated function, Ψ̂, will also be monotone. In particular, if the developed signature score S is truly effective in predicting the effect of E, a nondecreasing shape of Ψ̂ is expected for increasing S, such that lower values of s are linked to larger negative values of Ψ̂(s), that is, greater responsiveness to E. If the shape of Ψ̂(s) is decreasing for increasing s, contrary to the intended increasing shape of Ψ̂(s), it indicates that no statistical evidence is obtained from the use of the genomic signature in assessing treatment efficacy. For this case, we remove the terms including the genomic signature from the model and refit the reduced model with only the main effect of ri. Under the reduced model, we have Ψ(si) = β1.
Test of treatment effects
We can conduct a test of treatment efficacy for a patient population based on the estimated treatment effects function Ψ̂. Because our models incorporate varying treatment effects on individual patients, it is natural to consider the strong null hypothesis, H0, that the treatment has no effect on any patients. Under H0, the null distribution of Ψ̂ and P values can be obtained by a permutation method that randomly permutes treatment labels. For permutation data-sets, the entire cross-validation procedure, including the signature development and the estimation of Ψ, is repeated to obtain a null distribution of Ψ̂. We propose to use an average absolute effect size over the entire patient population as the test statistic (see Appendix D). It corresponds to a 2-sided alternative hypothesis to detect treatment effects in both directions where the treatment E (C) is superior to C (E). Another approach is, such as in the second stage of the ASDs (5, 6; see Appendix A), to test treatment effects for a subset of patients with Ψ̂(s) <0, who are predicted to be responsive to E. We can consider an average treatment effect over the responsive patients as the test statistic for a 1-sided test to detect treatment effects in the direction where the treatment E is superior to C (see Appendix D).
Prediction of patient-level survival curves
Although the estimation of the treatment effects function, Ψ(s), provides direct information about the sizes of treatment effects for individual patients in terms of the (logarithm of) HR, information about the survival curves when individual patients receive either one of the two treatments would be more relevant. The patient-level survival curves can be predicted on the basis of the estimates of the baseline hazard function and regression coefficients by fitting the multivariate Cox proportional hazards model (2). However, this model, which can work well for estimating the treatment effects function Ψ, may not be so accurate in predicting the patient-level survival curves because it does not incorporate information about risk or prognostic factors. We therefore, consider the extension of the model (2),
| (4) |
where the function f4 represents an effect of a prognostic index wi. Note that, under the model (4), we have the same form of the treatment effects function (3) with that under the model (2), but the effects β1 and f3 are now interpreted with the use of the prognostic index w (or after adjustment for the prognostic term f4).
The prognostic index, w, can be established on the basis of clinical prognostic factors. However, recent prognostic studies have shown improvement of the accuracy of prognostic prediction by incorporating genomic signatures (15–17). Because large-scale phase III trials with pretreatment genomic data also provide a precious chance for developing reliable prognostic signatures, in addition to reliable predictive signatures, codevelopment of them would be warranted. Our methodology can be easily extended in this direction. Within the K-fold cross-validation, we develop a prognostic signature score, independently of developing the predictive signature score, S, through correlating genomic data with survival outcomes without reference to treatment assignment. An algorithm similar to that for developing the predictive signature is given in Appendix B. At each step of the K-fold cross-validation, we can fit a multivariate Cox model with the developed genomic signature score and clinical prognostic factors as covariates to obtain a composite prognostic signature. A quantile score can also be developed for the prognostic signature. Then, the developed (quantile) prognostic score is used for w in the model (4).
On the basis of the estimates under the model (4), for patient i with (si, wi), we predict a patient-level survival curve or survival rate at a given time point for each treatment. We then compare the predicted survival curve when receiving E and that when receiving C to assess the benefit of receiving E for that patient.
Finally, we regard the entire n patients in the clinical trial as a training set and apply the entire procedure. That is, we develop predictive and prognostic scoring functions and compute scores for all n patients. When feature selection and/or prediction models are optimized in the prior K-fold cross-validation, we invoke a new session of K-fold cross-validation for n patients to determine optimal values of the tuning parameters using the same optimization procedure, and then compute scores at these values for n patients. The empirical distributions of those scores serve as reference distributions. For any new patient, the scoring functions are used to compute predictive and prognostic scores, which are then normalized using the reference distributions. For the new patient, we then assess the expected treatment effect by plugging these values into the estimated treatment effects function, Ψ̂, obtained in the cross-validated prediction analysis. From (4) one can also compute the predicted survival curves for the new patient under each treatment based on the cross-validated prediction analysis.
Results
A large-scale randomized phase III trial was conducted to assess whether the addition of thalidomide, which has activity against advanced and refractory multiple myeloma, improves survival in the up-front management of patients with multiple myeloma undergoing melphalan-based tandem transplantation (8, 9). Despite significantly higher complete response rate and superior event-free survival among patients randomized to thalidomide, compared with the control patients with no thalidomide, overall survival (OS) was similar between treatment groups at the time of first publication, with a median follow-up of 42 months (8), although, with longer follow-up of 72 months, a tendency of long-term effect of thalidomide on OS was observed (9). As another unique feature of this phase III trial, pretreatment RNA from highly purified plasma cells was applied to Affymetrix U133Plus2.0 microarrays for 351 patients, out of 668 randomized patients. Because the efficacy of thalidomide on OS has been relatively uncertain, we conducted a predictive analysis for OS using the data with a median follow-up of 72 months for 351 patients with microarray gene-expression data. Figure 1 shows an OS curve for 351 patients by treatment arm [175 with thalidomide and 176 with no thalidomide (control)].
For each of 54,675 gene features on the microarray, we standardized gene-expression levels after normalization to have mean 0 and SD 1 across all 351 patients. We first developed the predictive signature score, S, and the prognostic signature score, W, using a 5-fold cross-validation. For the training set, we screened predictive genes using the significance level of 0.001 for a score test for no interaction between the gene feature and treatment and developed a compound covariates predictor (see Appendix B). Similarly, but independently, we screened prognostic genes using the significance level of 0.001 for a score test for no association of the gene feature and OS developed a compound covariates predictor (see Appendix B). We then obtained the predicted (quantile) signature scores S and W for the test set. After the completion of the 5-fold cross-validation, we had obtained the predicted values of these scores for all 351 patients.
We fit the multivariate Cox proportional hazards model with both S and W in (4) for the entire patient cohort. We specified linear terms for the main effects of S and W, such that f2(si) = β2si and f4(wi) = β4wi, but the FPs with one term (FP1) for the interaction of R and S, f3(si) (see Appendix C). The results were similar for the FPs with 2 terms (FP2). The estimated treatment effects function, Ψ̂(s), is provided in Fig. 2. Ψ̂(s) <0 represents thalidomide’s effects that prolong OS. The estimates Ψ̂(s) for 0 ≤ s ≤ 1 represent the underlying smooth function about varying treatment effects for the whole range of the score S (i.e., the entire patient population). For approximately the half of patients with lower values of S, thalidomide is expected to prolong OS by varying degrees. On the other hand, for the rest of the patients with larger values of S, it could have small adverse effects on OS.
We conducted a test of treatment efficacy for the subset of the patient population predicted to benefit from thalidomide based on Ψ̂(s). Because the interest in this randomized trial was to assess improvement in survival by adding thalidomide for patients with high-dose therapies, compared with the control arm with no thalidomide, it is reasonable to test treatment efficacy for a subset of patients who are considered to be responsive to thalidomide using the 1-sided test statistic [see (A4) in Appendix D]. The observed value of the test statistic was −0.47 in log HR (0.62 in HR). The P value obtained from 2,000 permutations was 0.019, which is significant if our test is used for a significance level 2% at the second stage of cross-validated ASD (6; see Appendix A). For reference, the permutation-based P value for the observed 2-sided test statistic, 0.35 [see (A3) in Appendix D], was 0.038.
On the basis of the estimates obtained from fitting the model (4), we predicted patient-level survival rates for patients with S = 0.1, 0.5, or 0.9 and W = 0.1, 0.5, or 0.9. Again, lower values of S represent larger effects of prolonging OS by receiving thalidomide. Lower values of W represent higher survival rates (better prognosis). Figure 3 shows the predicted survival curves when receiving either of the 2 treatments (thalidomide and no thalidomide) for 4 combinations with (S, W) = (0.1, 0.1), (0.1, 0.9), (0.5, 0.1), and (0.5, 0.9). Table 1 summarizes the predicted 5-year survival rates with thalidomide and no thalidomide and their difference for all the combinations. Generally, even for the same level of the predictive score, S, the effect size of thalidomide was larger (larger absolute difference in the predicted survival rate under each treatment) for patients with poor prognosis (larger W). For example, for patients with S = 0.1, the difference in the predicted survival rate was 17.9% for W = 0.1, whereas it was 27.8% for W = 0.9 (see also Fig. 3A and B).
Table 1.
Predicted 5-year survival rates for each treatment
| S | W | Survival rate with thalidomide | Survival rate with no thalidomide | Difference |
|---|---|---|---|---|
| 0.1 | 0.1 | 85.3% | 67.4% | 17.9% |
| 0.5 | 79.5% | 56.6% | 22.9% | |
| 0.9 | 71.9% | 44.1% | 27.8% | |
| 0.5 | 0.1 | 75.8% | 73.8% | 2.1% |
| 0.5 | 67.1% | 64.5% | 2.6% | |
| 0.9 | 56.4% | 53.2% | 3.1% | |
| 0.9 | 0.1 | 73.7% | 79.1% | −5.4% |
| 0.5 | 64.5% | 71.4% | −6.9% | |
| 0.9 | 53.2% | 61.5% | −8.3% |
Finally, we applied the procedures for selecting genes and developing signature-scoring functions to the entire 351 patients. Eighty-one and 662 genes with no overlap were selected using the significance level of 0.001 for developing the predictive and prognostic signature scoring functions, respectively. The empirical distributions of those scores will serve as reference distributions for predicting patient-level survival curves under each treatment for any new patient on the basis of the fitted model of (4) obtained in the cross-validated prediction analysis.
For treatment selection, it would be reasonable to withhold thalidomide for approximately half of the patients with Ψ̂(s) > 0 because no improvement in survival is expected by receiving thalidomide. For the rest of patients, the decision of whether to use thalidomide would take into consideration the estimated sizes of thalidomide’s effects for individual patients as well as other factors, such as safety issues, including severe peripheral neuropathy and deep-vein thrombosis (8).
Discussion
Although development of diagnostic tests to predict patients’ responsiveness to anticancer therapies on clinical endpoints is particularly important, it is generally difficult to develop such tests in early clinical trials. If a signature to identify responsive patients cannot be developed in early clinical trials, the use of genomic data from large-scale randomized clinical trials can be a useful approach for developing reliable diagnostic tests. The ASDs (5, 6) have provided a new framework that allows this, while strictly controlling the false positive rate in assessing treatment efficacy for the entire patients or a subset of responsive patients. In this article, we have considered another framework designed to estimate treatment effects quantitatively over the entire patient population, rather than qualitatively classifying patients as in or not in a responsive subset.
To achieve this framework, we have proposed a methodology to develop and validate continuous genomic signatures within a single randomized clinical trial. These signatures can be used for prospectively assessing treatment efficacy for the entire patient population or a subset of responsive patients and also for predicting patient-level survival curves for treating individual patients. We have provided an implementation of this approach with the use of relatively simple but effective statistical procedures for developing genomic signatures. While there are usually multiple signature development methods (both in feature selection and prediction algorithm) with comparative predictive accuracy (18), further research on the application of more complex alternatives is warranted.
Our methodology may be used on a stand-alone basis for the analysis or as the second stage of the ASDs (5, 6). In the former circumstance, the test of the strong null hypothesis for the overall population at a 2-sided 0.05 significance level controls the study-wise type I error. The hypothesis of no heterogeneity in treatment effect can be tested using a test statistic, such as those for testing the interaction term in the multivariate Cox model in (2) or (4) and the variance of the cross-validated treatment effects (var{Ψ̂(si)}), and evaluating its distribution under permutations of the genomic covariate vectors. We plan to study this test in more detail subsequently.
Our methodology can be used as the second stage of the ASDs when the overall treatment effect is not significant. The 1-sided test [see (A4) in Appendix D] is similar to the test of the average treatment effect for a subset of responsive patients proposed in the ASDs (5, 6). These 1-sided tests would be relevant typically when developing a new molecularly targeted agent, or more generally, when assessing improvement in survival by prescribing the experimental treatment, as in the multiple myeloma example. Our methodology also provides a 2-sided test for the entire patient population [see (A3) in Appendix D]. This could be relevant when comparing 2 competitive treatments (or treatment regimens) to establish standard therapy. In such settings, the overall treatment effect for the entire population can be small (and thus not detected) due to combining reversed effects from distinct subsets of responsive patients to either 1 of the 2 treatments. The 2-sided test would be particularly effective for such situations. This would indicate a direct use of this test as the primary analysis without conducting the classical test for the overall treatment effect. There are many methodologic issues, including sample size determination that can provide interesting directions for further research.
For licensing studies sponsored by pharmaceutical companies, although the proposed method can detect treatment effects for a subset of responsive patients, uncertainty in identifying the responsive subset based on Ψ̂(s) (due to possible model misspecification and random variation) may limit confidence in labeling of the new treatment for the identified patient subset (i.e., Ψ̂(s) <0). The proposed method, however, can provide useful information for designing a second confirmatory trial, possibly with a targeted or enrichment design with small sample sizes, especially when there is no overall treatment effect for the entire population.
Our methodology can also be useful even if the overall average treatment effect is significant. To avoid overtreatment of the population, it is useful to identify patient subsets that receive large, small, or no benefits from the treatment and to predict patient-level survival curves to aid selecting treatment for individual patients. Provided that some conditions are met (19), our methodology could also be applied to genomic data from past randomized trials to analyze the heterogeneity in treatment effects over the study population and to develop diagnostic tools for established treatments.
Confidence intervals for the treatment effects function and patient-level survival curves are particularly important both in assessing the heterogeneity of treatment effects across patients and in developing diagnostic tools for individual patients. Construction of these intervals via resampling methods is another subject for future research.
Finally, for using our framework, it is essential to plan for collection of genomic data in designing randomized clinical trials. The rapid development of high-throughput technologies, which has reduced the cost of microarrays and exome sequencing, and infrastructure development for genomic analysis in the context of clinical studies have allowed collection of large-scale genomic data in randomized trials, such as the illustrative example of the multiple myeloma trial. Through providing the new framework for developing and validating continuous genomic signatures in randomized trials, we hope this article could contribute to accelerating modern clinical studies toward predictive medicine.
Translational Relevance.
This article proposes a new framework of design and analysis of phase III randomized clinical trials with embedded DNA microarrays toward personalized or predictive medicine. The proposed framework allows randomized clinical trials to assess treatment efficacy for a patient population in a manner that takes into consideration the heterogeneity in patients’ responsiveness to treatment on survival outcomes and also predicts patient-level survival curves and treatment effects for individual patients. This new framework for developing and validating continuous genomic signatures in randomized trials could help to accelerate modern clinical studies toward predictive medicine. This work also provides an illustration of the new framework, indicating one of the first successes in exploring signatures that predict treatment efficacy on overall survival with the use of microarray gene-expression data in phase III randomized trials. This strategy would encourage clinical investigators to plan for collection of genomic data in designing future randomized clinical trials.
Acknowledgments
The authors thank the reviewers for their contributions in improving an earlier draft of the article.
Appendix A: Adaptive Signature Designs
These designs first test the overall treatment effects for the entire study population using a significant level α1 (e.g., 0.03), out of the overall type I error rate 0.05. If it is not significant, these designs proceed to the second stage. A genomic classifier is developed for a portion of samples (training set) and the treatment efficacy for the patient subset classified as “responsive” to the new treatment is tested at significance level 0.05 – α1 (e.g., 0.02) for the rest samples (test set). The second stage can be based on split-sample (5) or cross-validation (6). The latter, called the cross-validated ASD, is expected to be more efficient because it maximizes the portion of samples contributing to the signature development (6).
Appendix B: Algorithms for Developing Genomic Signatures
Selection of predictive gene features
A test of interaction between treatment and gene features is conducted for each gene feature separately, and a set of the most significant gene features is selected for the training set. Specifically, for a particular gene feature, a standard multivariate Cox proportional hazards model is commonly assumed:
| (A1) |
where ri the treatment assignment indicator such that ri = 1 if patient i is assigned to treatment E and ri = 0 otherwise, xi the expression level of the gene feature, and the product rixi representing an interaction term of treatment and gene feature (i = 1, …, n). The parameters ψ’s are the regression parameters and h0(t) is the baseline hazard function. We calculate a test statistic for testing a null interaction effect, ψ3 = 0, such as a score or Wald type of statistics. A standardized test statistic, Z, approximately follows the standard normal distribution under the null interaction effect. Without loss of generality, we suppose that a negative (positive) Z represents such a gene that overexpression is linked to a reduction (elevation) of risk of developing the event by receiving E. The significance level of this test for gene selection can be regarded as a tuning parameter that is determined to maximize cross-validated prediction partial likelihood. A simpler approach is using an arbitrary, fixed significance level, such as 0.001 (20).
Compound covariates predictor for developing genomic signatures
To cope with the interaction effects of a number of predictive gene features, we consider a composite score. Specifically, for patient i,
| (A2) |
where Ω is the set of selected genes in the step 1 and zg is a standardized test statistic of the interaction test for gene g obtained from the training set. This is the compound covariates predictor (21–23). If the patient i belongs to the test set, the value of score Ui is regarded as a predicted value. Smaller values of Ui correspond to greater chance of benefiting from E relative to C.
Development of prognostic signatures
Screening of prognostic genes is typically based on a score (log-rank) test or Wald test derived from a univariate Cox regression that correlates a single gene with survival outcome (24), without regard to treatment assignment. The test is predicated on the definition of prognostic genes that have similar effects on survival, irrespective of which treatment is received. There are a large variety of algorithms for developing prognostic signatures (24). Again, the compound covariates predictor is one of simple, but effective algorithms for high-dimensional genomic data, in which the standardized test statistic from a univariate Cox regression is used for zg in (A2) for a set of selected prognostic genes Ω.
Appendix C: Fractional Polynomials
One term FPs functions (FP1), f3(s) = β3sa (a ∈ {−2, −1, −0.5, 0. 0.5, 1, 2, 3} with a = 0 identical to log(s), the natural logarithm of (s) are always monotonic. The 2 term FP (FP2) functions, f3(s) = β31sa1 + β32sa2, with different powers a1 ≠ a2 is monotonic when sign(β31β32) sign(a2) = sign(a1) (14). Because of an acute change in the FP functions when s is close to 0 for a ≤ 0, we shall add a constant 1 to S. For example, f3(s) = β3(s + 1)a for FP1.
Appendix D: Test Statistics and P values
As a test statistic for a 2-sided alternative hypothesis to detect treatment effects in both directions where the treatment E (C) is superior to C (E), we propose to use the following test statistic,
| (A3) |
which represents a summation of absolute effect sizes, or equivalently, an average absolute effect size over the entire patient population. Another approach is, such as in the second stage of the ASDs (5, 6; see Appendix A), to test treatment effects for a subset of patients with Ψ̂(s) <0, who are predicted to be responsive to E. Let I be a collection of s that satisfies Ψ̂(s) <0, which represents a group of responsive patients to E, and L be the size or length of I, which represents the size of the group of responsive patients. Then, as the counterpart of T, we can consider a 1-sided test statistic, 1
| (A4) |
which represents an average treatment effect over the responsive patients.
In calculating P values of the 2-sided statistic T using the permutation method, we count the number of permutations with the values of T are equal or larger than the observed value of T. For the 1-sided statistic TR, we count the number of permutations with the values of TR are equal or less than the observed value of TR.
Footnotes
Disclosure of Potential Conflicts of Interest
J.D. Shaughnessy Jr is employed as a Founder in Signal Genetics, LLC, has honoraria from speakers bureau from Caris Life Sciences, Inc., and has ownership interest (including patents) in Signal Genetics, LLC, Esoterix, Inc., and Novartis, Inc. No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: S. Matsui, R. Simon
Development of methodology: S. Matsui, R. Simon, J.D. Shaughnessy Jr
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): P. Qu, J.D. Shaughnessy Jr, B. Barlogie, J. Crowley
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S. Matsui, R. Simon, J.D. Shaughnessy Jr, J. Crowley
Writing, review, and/or revision of the manuscript: S. Matsui, R. Simon, B. Barlogie, J. Crowley
Study supervision: S. Matsui
References
- 1.Balis FM. Evolution of anticancer drug discovery and the role of cell-based screening. J Natl Cancer Inst. 2002;94:78–9. doi: 10.1093/jnci/94.2.78. [DOI] [PubMed] [Google Scholar]
- 2.Schilsky RL. End points in cancer clinical trials and the drug approval process. Clin Cancer Res. 2002;8:935–8. [PubMed] [Google Scholar]
- 3.Rothenberg ML, Carbone DP, Johnson DH. Improving the evaluation of new cancer treatments: challenges and opportunities. Nat Rev Cancer. 2003;3:303–9. doi: 10.1038/nrc1047. [DOI] [PubMed] [Google Scholar]
- 4.Hoering A, Leblanc M, Crowley JJ. Randomized phase III clinical trial designs for targeted agents. Clin Cancer Res. 2008;14:4358–67. doi: 10.1158/1078-0432.CCR-08-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freidlin B, Simon R. Adaptive signature design: an adaptive clinical trial design for generating and prospectively testing a gene expression signature for sensitive patients. Clin Cancer Res. 2005;11:7872–8. doi: 10.1158/1078-0432.CCR-05-0605. [DOI] [PubMed] [Google Scholar]
- 6.Freidlin B, Jiang W, Simon R. The cross-validated adaptive signature design. Clin Cancer Res. 2010;16:691–8. doi: 10.1158/1078-0432.CCR-09-1357. [DOI] [PubMed] [Google Scholar]
- 7.Janes H, Pepe MS, Bossuyt PM, Barlow WE. Measuring the performance of markers for guiding treatment decisions. Ann Intern Med. 2011;154:253–9. doi: 10.1059/0003-4819-154-4-201102150-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barlogie B, Tricot G, Anaissie E, Shaughnessy J, Rasmussen E, van Rhee F, et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N Engl J Med. 2006;354:1021–30. doi: 10.1056/NEJMoa053583. [DOI] [PubMed] [Google Scholar]
- 9.Barlogie B, Anaissie E, van Rhee F, Shaughnessy JD, Jr, Szymonifka J, Hoering A, et al. Reiterative survival analyses of total therapy 2 for multiple myeloma elucidate follow-up time dependency of prognostic variables and treatment arms. J Clin Oncol. 2010;28:3023–7. doi: 10.1200/JCO.2009.26.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambroise C, McLachlan GJ. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc Natl Acad Sci U S A. 2002;99:6562–6. doi: 10.1073/pnas.102102699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon R, Radmacher MD, Dobbin K, McShane LM. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Natl Cancer Inst. 2003;95:14–8. doi: 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]
- 12.Dudoit S, Fridlyand J. Classification in microarray experiments. In: Speed TP, editor. Statistical analysis of gene expression microarray data. Boca Raton, FL: Chapman & Hall/CRC; 2003. pp. 93–158. [Google Scholar]
- 13.Varma S, Simon R. Bias in error estimation when using cross-validation for model selection. BMC Bioinformatics. 2006;7:91. doi: 10.1186/1471-2105-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Royston P, Altman DG. Regression using fractional polynomials of continuous covariates: parsimonious parametric modelling (with discussion) Appl Stat. 1994;43:429–67. [Google Scholar]
- 15.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 16.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 17.Shaughnessy JD, Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–84. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 18.Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, Nobel AB, et al. Concordance among gene-expression–based predictors for breast cancer. N Engl J Med. 2006;355:560–9. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- 19.Simon RM, Paik S, Hayes DF. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst. 2009;101:1446–52. doi: 10.1093/jnci/djp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon RM, Korn EL, McShane LM, Radmacher MD, Wright GW, Zhao Y. Design and analysis of DNA microarray investigations. New York: Springer; 2004. [Google Scholar]
- 21.Tukey JW. Tightening the clinical trial. Control Clin Trials. 1993;14:266–85. doi: 10.1016/0197-2456(93)90225-3. [DOI] [PubMed] [Google Scholar]
- 22.Radmacher MD, McShane LM, Simon R. A paradigm for class prediction using gene expression profiles. J Comput Biol. 2002;9:505–911. doi: 10.1089/106652702760138592. [DOI] [PubMed] [Google Scholar]
- 23.Matsui S. Predicting survival outcomes using subsets of significant genes in prognostic marker studies with microarrays. BMC Bioinformatics. 2006;7:156. doi: 10.1186/1471-2105-7-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witten DM, Tibshirani R. Survival analysis with high-dimensional covariates. Stat Methods Med Res. 2010;19:29–51. doi: 10.1177/0962280209105024. [DOI] [PMC free article] [PubMed] [Google Scholar]