

Abstract
Mammalian cysteine dioxygenase (CDO) is a mononuclear non-heme iron protein that catalyzes the conversion of cysteine (Cys) to cysteine sulfinic acid (CSA) by an unclarified mechanism. One structural study revealed a Cys-persulfenate (or Cys-persulfenic acid) formed in the active site, but quantum mechanical calculations have been used to support arguments that it is not an energetically feasible reaction intermediate. Here, we report a series of high-resolution structures of CDO soaked with Cys at pH values from 4 to 9. Cys binding is minimal at pH≤5 and persulfenate formation is consistently seen at pH values between 5.5 and 7. Also, a structure determined using laboratory-based X-ray diffraction shows that the persulfenate, with an apparent average O-O separation distance of ~1.8 Å is not an artifact of synchrotron radiation. At pH≥8, the active site iron shifts from 4- to 5-coordinate, and Cys soaks reveal a complex with Cys, but no dioxygen, bound. This ‘Cys-only’ complex differs in detail from a previously published ‘Cys-only’ complex which we reevaluate and conclude is not reliable. The high-resolution structures presented here do not resolve the CDO mechanism, but do imply that an iron-bound persulfenate (or persulfenic acid) is energetically accessible in the CDO active site, and that CDO active site chemistry in the crystals is influenced by protonation/deprotonation events with effective pKa values near ~5.5 and ~7.5 that influence Cys binding and oxygen binding/reactivity, respectively. Furthermore, this work provides reliable ligand-bound models for guiding future mechanistic considerations.
Introduction
Mammalian cysteine dioxygenase (CDO) is a non-heme iron protein that in its ferrous form (Fe(II)-CDO) catalyzes the conversion of cysteine (Cys) to cysteine sulfinic acid by incorporating both oxygen atoms of molecular oxygen to form the product (recently reviewed by Stipanuk et al1). The product from this first committed step in cysteine catabolism is then further catabolized to either taurine or pyruvate and sulfate. CDO expression is up-regulated in response to high cysteine levels,2; 3; 4 which if not abated can be toxic.5; 6 In general, tight regulation of CDO plays a key role in intracellular sulfur homeostasis.3 Links between loss-of-function mutations in CDO and rheumatoid arthritis and some neurodegenerative disorders have been hypothesized based on observations of high cysteine and low sulfate levels.5; 7; 8 In the CDO−/− mouse, taurine levels, but not sulfate levels, are low and metabolism of cysteine through alternative desulfhydration pathways is markedly elevated, giving rise to evidence of H2S toxicity.6 CDO−/− mice exhibit a growth deficit, high postnatal mortality, joint hyperlaxity, connective tissue abnormalities including enlarged alveolar air spaces and abnormal organization of elastic fibers in the vasculature and parenchyma of lungs, and fatty acid oxidation defects.6 Recently, CDO has been implicated as a novel tumor suppressor as decreased expression of CDO due to promoter methylation was seen in multiple tumor tissues, and the forced expression of CDO in cancer cells slowed their growth.9
Initial structural studies of recombinant rat CDO (92% identical to human CDO) showed it to have a cupin fold, with one structure revealing an unusual tetrahedral iron coordination involving three His residues and a water as ligands10 and the other structure apparently containing nickel in place of the iron.11 Both structures also revealed an unexpected thioether linkage between the Cys93 and Tyr157 side chains,10; 11 which was later confirmed.4; 12 This crosslink, not present in CDO from prokaryotes,1; 13 is formed during enzyme turnover and is reported to occur in cells4 and to increase catalytic efficiency 10-fold 4 or even more,14 although its impact on activity and physiological relevance has been recently questioned.15 In the unliganded enzyme, the Tyr157 hydroxyl forms a hydrogen bond with the iron-bound water and is part of a Ser153-His155-Tyr157 catalytic triad.10 A Tyr157Phe mutation leads to about a 50-fold loss in activity, consistent with the importance of this residue.4; 16 Two structures of substrate-bound CDO have been reported: the first was a complex of human CDO with cysteine16 and the second a complex of rat CDO with a putative cysteine persulfenate or persulfenic acid molecule (Fig. 1a) formed in the active site during an aerobic soak with cysteine.17 In the persulfenate complex, Tyr157 was centrally located in close contact with both persulfenate oxygens, where it could serve as a catalytic acid/base (Fig. 1a).
Figure 1.
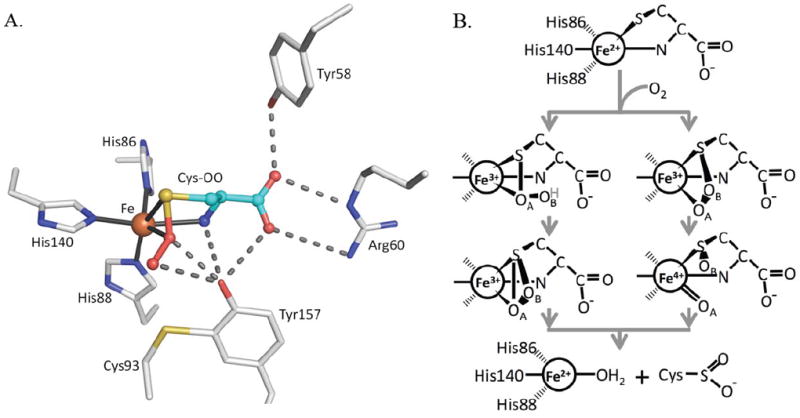
CDO active site geometry and two proposed mechanisms. (A) The CDO active site residues (white carbons) with bound Cys-persulfenate (teal carbons) (PDB entry 3ELN) showing potential H-bond interactions (dashes) and the coordination bonds (black line) to Fe (orange). (B) The key distinction between the two types of mechanisms being considered for CDO is shown. After dioxygen binds to Fe(II)-Cys CDO (top image), one route (left side) involves persulfenate/persulfenic acid formation, where the iron-proximal oxygen (OA) attacks the Cys-Sγ atom.17 The H-atom shown bound to OB (grey text) may or may not be present in this proposed intermediate. The other route (right side), similar to monooxygenase chemistry, proceeds by attack of the Fe-distal oxygen (OB) on the Cys-Sγ atom, followed by creation of a reactive Fe(IV)-oxo intermediate as shown.20; 22
Taking the structural information together with the results of spectroscopic studies18; 19; 20 and chemical considerations,21; 22; 23 there are now two main types of reaction mechanisms under consideration for CDO, both of which involve direct coordination of first Cys then molecular oxygen18 to the active site iron. One mechanism (Fig. 1b, left scheme), similar to that thought to occur for the uncatalyzed reaction and for Ni(II)-catalyzed thiol oxidations,17; 24; 25; 26; 27 includes persulfenate formation which converts to product possibly via a thiadioxirane intermediate; in this mechanism the initial Cys oxidation occurs via sulfur attacking the Fe-proximal oxygen (OA). The other mechanism (Fig. 1b, right scheme), more like that of monooxygenases, has the initial oxidation event involving transfer of the Fe-distal oxygen (OB) to generate Cys-sulfenate and an Fe(IV)-oxo intermediate. This latter mechanism has been supported by recent quantum mechanical calculations,22; 28 but not by a spectroscopic study implying that a Fe(III)-superoxo rather than a Fe(IV)-oxo intermediate facilitates substrate oxidation.19 The properties of small molecule models of the CDO active site are consistent with both mechanisms.29; 30
The challenge of defining the CDO mechanism has been exacerbated by the varied behaviors of recombinant CDO studied by different groups, as well as a lack of consensus in how to best assay CDO activity. The enzyme has been expressed in Escherichia coli using at least five different expression constructs, and the purified proteins range from having 10%11; 31 to 60%19 iron bound that is from <1%15 to 99%18 in the ferrous form, and having amounts of the Cys93-Tyr157 crosslink ranging from minimal15 to dominant.4 The activity assay we use, which includes 0.3 mM Fe2+ and a copper chelator and directly monitors the Cys-sulfinate product, gives consistent results for both natural rat liver CDO2 and our recombinant enzyme,32 and an optimal pH range from ~5.5 to ~6.5. The only other published pH profile of recombinant enzyme did not use added iron, showed much lower activity, and an optimal pH range of ~7 to ~9.33 Although other groups have shown good enzyme activity at pH 6.1 without added iron,18; 34 most current assays are carried out at pH 7.5 without added iron.11; 15; 18; 20; 33 A recent study showed that for Fe(III)-CDO, the inclusion of ascorbate as a reductant in assays can be used to recover enzymatic activity without adding ferrous iron.15 Also, even though Fe(III)-CDO is not enzymatically active,20; 35 spectroscopic titrations have shown Fe(III)-CDO can bind Cys between pH=5.5 and 9.5.19 This implies that for CDO with iron in the ferrous form, this whole pH range (i.e. 5.5 to 9.5) could plausibly support enzyme activity.
In exploring how to gain additional information about the pH dependence of CDO properties, we found that the CDO crystal form we have studied10; 17 is stable from pH=4 to pH=9 and provides a well-defined system – fully-loaded with iron and fully cross-linked – that can provide independent and direct information about the active site changes associated with variations in pH. Given this, plus the value of accurate structural information for informing mechanistic studies, here we follow up our initial brief report10 of the putative persulfenate complex to describe it more fully, explore the pH dependence of its formation, and prove that it is not a synchrotron-induced artifact. At the higher pH values studied, a complex having only cysteine bound in the active site is observed. Because this complex has Cys bound differently than did the complex reported for human CDO (PDB code 2IC1),16 we reevaluated the human CDO structure and found that structure does not actually represent a reliable Cys-bound complex.
Results and Discussion
Selecting the high resolution limits
This study encompasses nine structures in the main pH series of Cys soaks and four structures designed to provide specific additional insight. For these structures, the data quality and refinement statistics are given in Tables 1 and 2, respectively. Recently, Karplus & Diederichs36 introduced a data quality indicator termed CC1/2 and showed by way of a paired refinement strategy that improved models are obtained by extending the high resolution limit beyond those indicated by the current conventional standards of I/σ ~2 and Rmeas ~60%. Their main test case was one of the CDO data sets collected for this work (the Cys soak at pH=6.2), and they showed that improved models were obtained using data out to CC1/2 between 0.1 and 0.2 even though the merging R-factors become very high. In order to create the best models for the structures in this study, we used such a CC1/2-based cutoff throughout. As one additional test case, paired-refinements were done here for the pH=7.0 structure. As the resolution was extended, Rfree stayed the same, but the gap between Rwork and Rfree decreased, indicating that the model was less over-fit when refinements included the weak high-resolution data (Table 3). Electron density maps also improved at the extended resolutions (e.g. Fig. 2).
Table 1.
Data collection and Refinement Statistics for CDO pH seriesa
| pH=4.0 Cys | pH=4.5 Cys | pH=5.0 Cys | pH=5.5 Cys | pH=6.2 Cys | pH=6.8 Cys | pH=7.0 Cys | |
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| Resolution (Å) | 34-1.55 (1.63-1.55) | 30-1.45 (1.53-1.45) | 34-1.40 (1.48-1.40) | 42-1.45 (1.53-1.45) | 29-1.40 (1.48-1.40) | 42-1.40 (1.48-1.40) | 42-1.25 (1.32-1.25) |
| Unique Obs. | 28050 (4401) | 33437 (4190) | 34382 (3149) | 33402 (2956) | 41131 (5551) | 39197 (5029) | 53507 (6668) |
| Multiplicity | 26.6 (24.8) | 14.4 (6.5) | 19.0 (8.6) | 15.9 (6.4) | 12.6 (4.7) | 21.6 (6.7) | 18.6 (7.0) |
| Completeness | 91.0 (100) | 88.9 (78.2) | 82.3 (53.4) | 88.3 (55.3) | 99.1 (94.3) | 93.0 (83.4) | 92.1 (80.0) |
| <I/σ> | 12.9 (0.5) | 10.3 (0.9) | 12.0 (0.9) | 18.3 (0.5) | 17.5 (0.6) | 13.8 (0.6) | 19.2 (0.6) |
| Rmeasb (%) | 17.4 (812) | 12.9 (303) | 13.2 (406) | 8.0 (428) | 7.9 (294) | 11.5 (277) | 9.9 (365) |
| Rpim (%) | 3.4 (162) | 3.3 (117) | 3.0 (137) | 1.9(162) | 2.1 (129) | 2.1 (105) | 1.9 (135) |
| CC1/2c (%) | 1.00 (0.12) | 1.00 (0.13) | 1.00 (0.13) | 1.00(0.13) | 1.00 (0.15) | 1.00 (0.23) | 1.00 (0.15) |
| Res <I/σ>~2 (Å)d | 1.80 | 1.60 | 1.50 | 1.60 | 1.55 | 1.60 | 1.40 |
| Refinement | |||||||
| Rcryst / Rfree (%) | 20.3/24.1 | 15.2/20.6 | 14.3/19.4 | 13.7/19.8 | 14.5/18.5 | 15.3/20.1 | 14.5/17.6 |
| No. Obs | 27242 | 32582 | 33980 | 32809 | 40816 | 38127 | 52891 |
| No. residues | 186 | 186 | 186 | 186 | 186 | 186 | 186 |
| No. waters | 132 | 165 | 216 | 210 | 240 | 214 | 283 |
| No. atoms | 1723 | 1814 | 1837 | 1761 | 1808 | 1789 | 1874 |
| rmsd angles (°) | 1.44 | 1.23 | 1.26 | 1.25 | 1.23 | 1.26 | 1.28 |
| rmsd lengths (Å) | 0.013 | 0.011 | 0.011 | 0.013 | 0.012 | 0.012 | 0.011 |
| φ,ψ-favored (%)e | 98.5 | 99.5 | 98.9 | 99.5 | 99.5 | 97.9 | 98.9 |
| φ,ψ-outliers (%)e | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| <B> protein (Å2) | 27 | 23 | 22 | 29 | 21 | 25 | 22 |
| <B> ligand (Å2) | - | - | - | 33 | 18 | 21 | 18 |
| PDB code | 4IEO | 4IEP | 4IEQ | 4IER | 4IES | 4IET | 4IEU |
All refinements used space group P43212 with a=b=57.60 Å and c= 122.40 Å. Numbers in parentheses refer to the highest resolution bin.
Rmeas is the multiplicity-weighted merging R-factor55
CC1/2 is the correlation between two datasets each based on half of the data as defined in Karplus & Diederichs.35
The resolution at which <I/σ> ~ 2 is given to allow comparison with previous standards for selecting high resolution limits.
Ramachandran statistics as defined by Molprobity51
Table 2.
Data collection and Refinement Statistics for CDO additional datasetsa
| pH=6.2 Lab-RT | pH=7.0 Cys Lab-LT | pH=8.0 Ala 40 min | pH=8.0 Cys + DT 15 min | |
|---|---|---|---|---|
| Data collection | ||||
| Resolution (Å) | 29-2.15 (2.27-2.15) | 33-1.63 (1.72-1.63) | 31-1.40 (1.48-1.40) | 29-2.00 (2.11-2.00) |
| Unique Obs. | 11697 (1594) | 26578 (3553) | 37124 (4475) | 13313 (1708) |
| Multiplicity | 2.5 (2.5) | 20.9 (13.9) | 11.4 (4.5) | 7.6 (8.0) |
| Completeness | 96.1 (92.0) | 98.6 (92.9) | 87.3 (74.1) | 90.0 (82.4) |
| <I/σ> | 3.6 (0.5) | 8.2 (0.7) | 9.6 (0.5) | 11.2 (2.7) |
| Rmeasb (%) | 27.1 (252) | 23.9 (662) | 12.0 (388) | 11.6 (67.2) |
| Rpim (%) | 16.1 (149) | 5.1 (175) | 3.4 (167) | 4.2 (23.1) |
| CC1/2c (%) | 0.98 (0.13) | 1.00 (0.11) | 1.00 (0.14) | 1.00 (0.85) |
| Res <I/σ>~2 (Å)d | 2.80 | 1.85 | 1.60 | - |
| Refinement | ||||
| Rcryst / Rfree (%) | 17.5/25.6 | 21.3/25.6 | 15.3/20.8 | 17.6/23.3 |
| No. Obs. | 11021 | 25288 | 32519 | 12831 |
| No. residues | 186 | 186 | 186 | 186 |
| No. waters | 53 | 247 | 229 | 127 |
| No. atoms | 1558 | 1788 | 1778 | 1644 |
| rmsd angles (°) | 1.59 | 1.50 | 1.28 | 1.37 |
| rmsd lengths (Å) | 0.017 | 0.015 | 0.014 | 0.014 |
| φ,ψ-favored (%)e | 98.91 | 97.3 | 99.0 | 99.5 |
| φ,ψ-outliers (%)e | 0 | 0 | 0 | 0 |
| <B> protein (Å2) | 41 | 30 | 24 | 33 |
| <B> ligand (Å2) | - | 27 | - | 32 |
| PDB code | 4IEX | 4IEY | 4IEZ | 4JTO |
All refinements used space group P43212, with cell constants a=b=57.60 Å and c= 122.40 Å. Numbers in parentheses refer to the highest resolution bin.
Rmeas is the multiplicity-weighted merging R-factor55
CC1/2 is the correlation between two datasets each based on half of the data as defined in Karplus & Diederichs.35
The resolution at which <I/σ> ~ 2 is given to allow comparison with previous standards for selecting high resolution limits.
Ramachandran statistics as defined by Molprobity51
Table 3.
Comparing standard and CC1/2-based resolution cutoffs for pH=7.0 + Cys dataa.
| Rmeas-based | I/σ-based | CC1/2-based | |
|---|---|---|---|
| Data collection | |||
| Resolution (Å)b | 1.58-1.50 | 1.48-1.40 | 1.32-1.25 |
| CC1/2 | 0.92 | 0.71 | 0.15 |
| Average I/σ | 3.9 | 1.9 | 0.6 |
| Rmeasc | 61% | 119% | 365% |
| R/Rfree @1.5 Å (%)d | 14.5/17.6 | 14.7/17.6 | 15.3/17.6 (14.5/17.6)e |
| Waters built | 237 | 253 | 251 (283) |
The pH 7.0 Cys-bound models were refined using the same automatic phenix.refine strategy which was used as the first step in refinement (see methods) against data truncated at 1.50 Å (based on Rmeas ~60%), 1.40 Å (based on I/σ ~2), or 1.25 Å (based on CC1/2 criteria)35.
The resolution range for the data used in refinement was 42 Å to the high resolution limit
the multiplicity-weighted merging R-factor55
Rcryst and Rfree of the indicated model calculated at 1.50 Å resolution
Numbers in parentheses are for the final refined model
Figure 2.

Electron density map improvement using a more generous high resolution cutoff. (A) Shown is 2Fo-Fc active site electron density for the pH=6.8+Cys soak at 1.60 Å resolution (contoured at 1.0 ρrms) along with the refined model (atom coloring as in figure 1). At this resolution, the <I/σ> is 2.1, Rmeas is 154%, and CC1/2 is 0.78. (B) Same as (A) but using data out to 1.40 Å resolution with the map contoured at 1.2 ρrms (see statistics in Table 1). Many details of the map show subtle improvement, but especially notable is the improvement for the distal persulfenate oxygen, which becomes resolved from the rest of the persulfenate.
Conformational changes associated with Cys-persulfenate binding
Simmons et al. (2008)17 was a brief report describing the observation of the Cys-persulfenate complex of CDO but did not include broader descriptions of conformational changes associated with ligand binding; we describe these more fully here. In the difference maps for persulfenate bound versus unliganded CDO (Fig. 3a), the largest peak is due to the Fe atom shifting about 0.5 Å toward His86. The second strongest peak is for the sulfur atom of the bound Cys-persulfenate, and the whole Cys-persulfenate has strong positive features except for the OA (proximal) oxygen. The OA oxygen sits in negative density because its presence is more than offset by the loss of the tightly held iron-bound water that is displaced from nearly the same position upon substrate binding.
Figure 3.
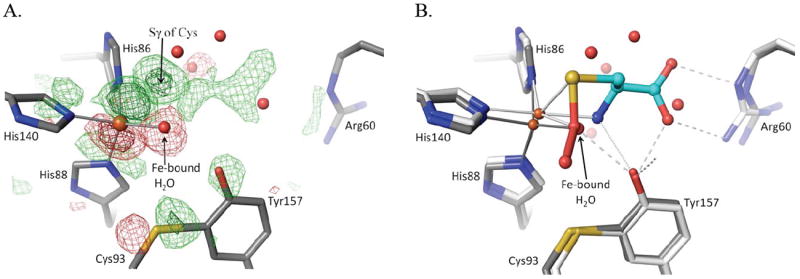
Active site changes upon Cys binding. (A) Fo-Fo difference map between the pH6.2-Cys soak and unliganded CDO (PDB entry 2B5H; Simmons 2006) using phases calculated from the unliganded structure. The unliganded model is shown (grey carbons) and the map contour levels are ±4.0 ρrms (light green/red) and ±10.0 ρrms (dark green/red). (B) Overlay, in the same view as panel A, of the models of unliganded (grey carbon atoms) and Cys-persulfenate bound (white/cyan carbon atoms) CDO. All waters shown are from the unliganded structure and are displaced by the bound ligand. The visible shifts of the iron and other active site residues and the placement of the persulfenate are consistent with the difference density shown in panel A.
The Cys93/Tyr157 pair, Arg60 and Tyr58 all shift slightly (~0.3 Å) upon substrate binding and become more ordered in positions where they hydrogen bond with the persulfenate OA oxygen and α–amino group (Tyr157) or with its α-carboxylate (Arg60, Tyr58 and Tyr157) (Fig. 3b). The Ser and His of the Ser-His-Tyr catalytic triad do not move as much as the Tyr, so the His-Tyr hydrogen bond elongates slightly from 2.7 Å to 2.9 Å. Additional difference map peaks distributed throughout the protein are associated with small global shifts in three lobes of the protein driven by the movement of the iron atom and the ligating His side chains. If the lobe including the iron ligands (residues 82-91, 100-124, 132-143, 161-179) is taken as a reference, the atoms at the edges of the other two lobes are shifted by ~1.0 Å (not shown).
pH rapidly equilibrates through the crystal
To probe how the formation or binding mode of the persulfenate ligand is influenced by the protonation state of active site residues, we solved the structures of CDO soaked with 100 mM Cys at pH values from 4.0 to 9.0. Evidence that the pH of the soaking buffer did equilibrate through the crystal was fortuitously provided by structural changes of residues 66-71 (Fig. 4) and residue 79 which are both far from the active site. Residues 66-69 adopt a Type I’ turn in all soaks at pH≤6.2, with the Asn67 side chain pointing outward to be near the His20 imidazole (Fig. 4a). In contrast, for the pH=8.0 and pH=9.0 soaks, the backbone conformation shifts so that the Asn67-sidechain points inward and accepts an H-bond from the backbone amide of Lys69 (Fig. 4b). The intermediate pH structures have a mix of the conformations, with ~60% of the high pH conformation seen at pH=7 (Fig. 4c) and ~25% at pH=6.8. These differences are plausibly related to a deprotonation of His20.
Figure 4.
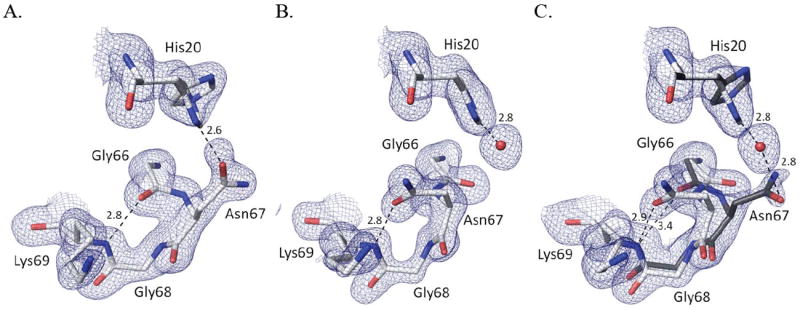
pH-dependent changes in the Asn67 loop. (A) 2Fo-Fc electron density and refined model from the pH5.5-Cys soak for residues 66 – 69 and His20; contoured at 1.2 ρrms, with potential H-bond interactions (dashed lines; distance in Å) shown. (B) Equivalent image based on the pH9-Cys soak. (C) Equivalent image based on the pH7-Cys soak contoured at 0.8 ρrms. The estimated occupancies of His20 alternate conformations are 100%/0% at pH=9.0, 80%/20% at pH=7.0, and a 50%/50% at pH=5.5. Assuming a protonated His20 adopts a 50/50 mix of conformations, the occupancies of the two turn conformations imply that His20 would be ~0, ~60 and ~100% deprotonated at pH=5.5, 7.0 and 9.0, respectively. Also at pH≤4.5 (not shown) His20 adopts a single conformation, a change we attribute to a protonation of the Asp64 carboxylate, which H-bonds to His20-Nε2 in all structures.
For Glu79, at pH≤5.5 the side chain oxygens are close (~3.1 Å) to the backbone carbonyls of residues 147 and 148. However, in all soaks above pH=6.2, the side chain shifts to displace a water and form an H-bond with its own backbone amide. (In the pH=6.2 structure, only the high-pH conformation of Glu79 was modeled, even though ~10% of the low pH conformation appears to still be present). The movement away from the electronegative carbonyl groups is consistent with the trigger being a deprotonation of the Glu79 to form the carboxylate. This Glu79 shift occurred even for a ~30 s soak at pH=4.5 (data not shown), indicating that pH equilibration through the crystal is quite rapid.
pH dependence of the persulfenate complex formation
pH range 5.5-7.0
In the active site, the persulfenate is formed at pH values ranging from 5.5-7.0 with minimal variations from the structure described by Simmons et al. (Fig. 5d-g).10 Compared to that structure, all of the structures presented here have a slightly higher occupancy of the bound persulfenate and the Cys93-Tyr157 crosslink, which we attribute to a minor difference between protein preparations. A notable feature is that in the pH=6.2, 6.8 and 7.0 structures (Fig. 5e-g), the electron density for the persulfenate group is better resolved than was seen previously,17 and the observed persulfenate OA–OB distance is a rather long 1.7 – 1.9 Å (see further discussion below). One pH dependent change seen is that the Fe-S ligation distance complex varies among the persulfenate complexes: from 2.57 Å at pH=5.5, to 2.49 Å at pH=6.2, and ~2.38 Å at pH=6.8 and 7.0. These are all slightly longer than typical Fe-S bonds in high resolution protein structures (2.33 for S-Fe2+ and 2.29 for S-Fe3+).37 Since the resolutions of these structures are similar, we think the changes are not an artifact due to limited resolution of some data sets, and may represent some heterogeneity due to partial protonation of an active site group as pH decreases.
Figure 5.
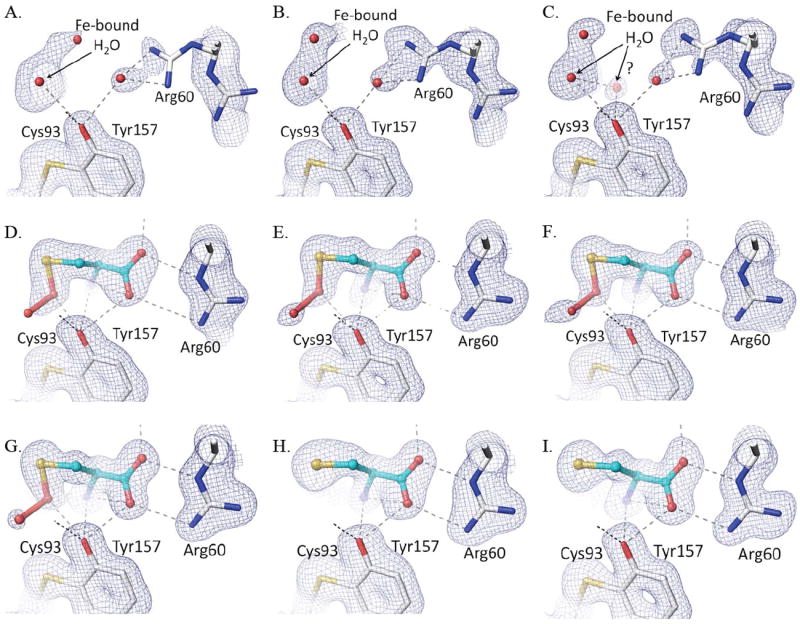
Active site density of Cys soaks as a function of pH. Each panel shows, for the relevant structure, the refined model (white carbon atoms for protein and cyan carbons for the ligand, and potential H-bond interactions as dashed lines) and the 2Fo-Fc density contoured at 1.2 ρrms. Shown are Cys soaks at (A) pH=4.0, (B) pH=4.5, (C) pH=5.0, (D) pH=5.5, (E) pH=6.2, (F) pH=6.8, (G) pH=7.0, (H) pH=8.0, and (I) pH=9.0. Although His155-Nε2 is not shown, the initial trajectory of its H-bond to the Tyr157 hydroxyl is also shown.
pH range 4.0-5.0
At pH=4.0, 4.5, and 5.0 (Fig. 5a-c), the Cys soaks showed little to no ligand binding and, as was observed at pH=6.2,10 a four-coordinate Fe with a solvent ligation distance of ~2.1 Å matching that expected for an Fe(II)-H2O interaction.37 At these low pHs, Arg60 partially adopts a second conformation not compatible with Cys binding. At pH 4.0 and 4.5, difference map features indicate that about 20% of the molecules have lost iron with the iron-ligating His86 side chain moved away (Supplemental Fig. 1). Because crystals soaked at pH=4.0 for an hour no longer diffracted, that soak was done for only 20 min (Table 1). At pH=5.0, some weak density (1.5 ρrms) is present at the position where the Cys α-amino group coordinates the Fe (Fig. 5c). We modeled this as a low-occupancy water, but it may reflect some binding of substrate. The lack of Cys binding in the crystals at pH≤5 is consistent with the lack of activity of CDO below pH=52; 31; 32 as well as with the decreased binding of Cys to Fe(III)-CDO at acidic pHs.19 In the latter study, the decreased binding was attributed to protonation of the Cys α-amino and/or thiol group, and we agree that this is a reasonable assignment.
pH range 8.0-9.0
At the higher pH values of 8.0 and 9.0, Cys binds strongly, but no persulfenate is formed and there is also no binding of dioxygen (Fig. 5h,i). Compared with the persulfenate complex at pH=6.2, the Fe-atom and Cys in these Cys-only complexes are shifted ~0.15 Å toward Arg60 with the Arg-carboxylate H-bonds shortening by about 0.2 Å (Fig. 6). The bound Cys has additional slight shifts as the N- and S- coordination geometries adjust due to the lack of dioxygen. The Fe-S ligation distances at both pHs are 2.35 Å. Through a subtle domain motion, Tyr157 moves to accommodate the shift in the Cys (Fig. 6). Interestingly, the new iron position is ~0.6 Å from that of the unliganded enzyme, meaning that the largest iron atom movement occurs upon Cys binding, with a smaller additional movement occurring upon dioxygen binding and persulfenate formation.
Figure 6.
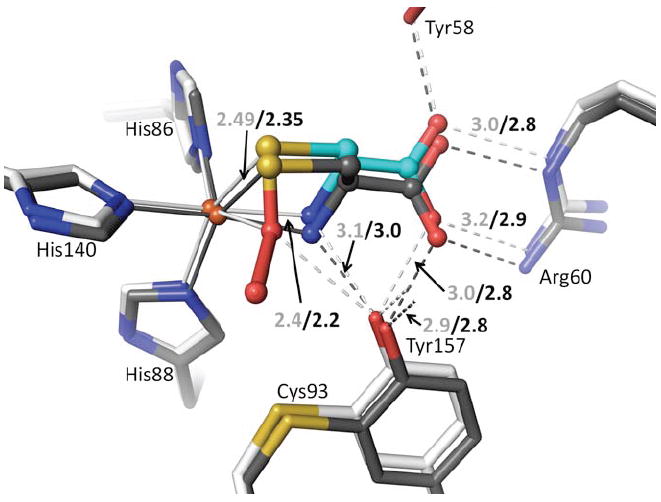
Active site differences between Cys-bound and Cys-persulfenate bound CDO. The persulfenate complex at pH=6.2 (cyan and white carbons) is overlaid on the Cys-bound complex pH=9.0 (dark carbons). Coordination and H-bond distances are given for the persulfenate complex (light text) and for the Cys-bound complex (black text). In the Cys-bound complex the Cys N- and Sγ-ligands are moved slightly to partly occupy the space left by the missing sixth ligand. This movement causes a slight shift in the whole Cys orientation and of the Cys-Tyr and Arg that interact with it.
The natural conclusion would be that deprotonation of an active site residue, such as Tyr157, is responsible for the lack of oxygen binding and persulfenate formation at pH=8 and 9. However, because Fe(III)-CDO binds Cys but is not active,19; 20 we also considered whether the lack of persulfenate formation could be due to hydroxide facilitated oxidation of the active site iron – as can occur for free iron in aerobic solutions38. To test this we collected data from crystals soaked with Cys at pH=8.0 in the presence of the reductant dithionite, and it also yielded a Cys-only complex (Fig. 7b). A control Cys+dithionite soak at a lower pH yielded a bound Cys-persulfenate (data not shown), showing that even though dithionite is an oxygen scavenger, it does not sufficiently decrease oxygen levels to hinder persulfenate formation during these long soaks. We also monitored UV-visible absorbance spectra on the pH=8 Cys-complex crystals, and it did not exhibit the 640 nm absorbance band associated with the Fe(III)-CDO-Cys complex19; 20 and looked no different than that of the pH=6.2 Cys-persulfenate complex (Supplementary Figure 2).
Figure 7.
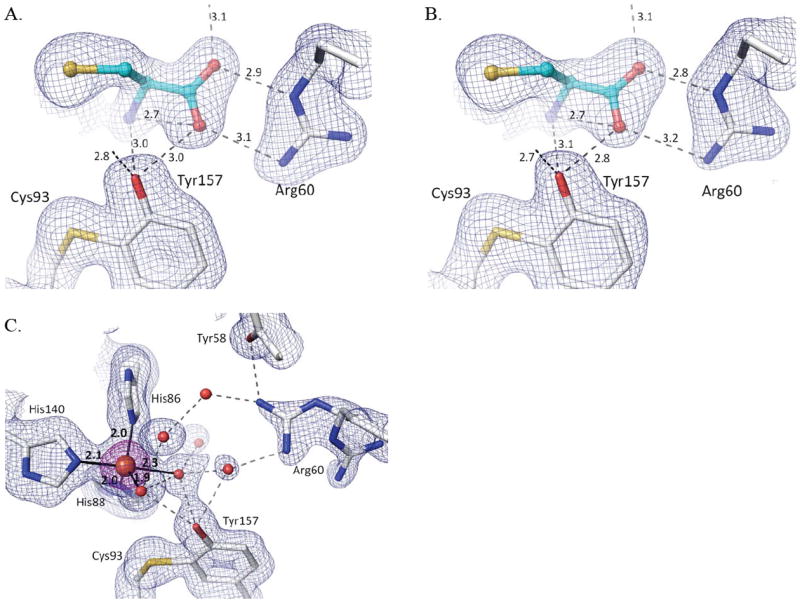
Cys-only complex and 5-coordinate iron at pH=8.0 with and without dithionite. (A) Structure and 2Fo-Fc density for the pH=8.0 Cys soak contoured at 1.0 ρrms (same as figure 5h but with H-bond distances given). (B) Same as A, but for the pH=8.0 + Cys + dithionite soak. (C) 2Fo-Fc density contoured at 1.4 ρrms (blue) and 5.0 ρrms (purple) for ligand-free CDO at pH=8.0. Iron ligating bonds (solid black lines) are shown with distances.
We conclude that even if any iron oxidation occurred, it is minimal and it is not the cause of the lack of persulfenate. Given that Cys soaks yielded a different complex at pH=8 and 9 compared to pH=7, we also investigated if the coordination geometry of the unliganded active site changed at these pHs. The unliganded structure at pH=8.0 revealed a 5-coordinate iron with the fifth ligand modeled as a water (d ~ 2.3 Å) at a position equivalent to that occupied by the Cys α-amino group in the complex (Fig 7c). Although we cannot be confident what deprotonation event is causing the change in coordination, we note that Tyr157 is hydrogen-bonded to the water/hydroxide at the fourth coordination site and this ligand has a shorter coordination distance of ~1.95 Å (compared with 2.1 Å for the pH=6.2 unliganded enzyme), consistent with it being a hydroxide (Fig. 7c).37;39 These features are also seen in an equivalent soak at pH=9 (data not shown).
Laboratory X-ray source control data sets
It is known that protein structures solved at cryogenic temperatures using synchrotron radiation can display artifactual changes in structure due to both radiation damage40; 41 and freezing.42; 43 To assess if the features seen in the unliganded or liganded CDO active site were significantly impacted by these factors, we used data from a rotating anode X-ray source to analyze a frozen low-temperature (LT) Cys-soaked crystal and also room temperature (RT) unliganded and Cys-soaked crystals. The LT control structure shows unequivocally that the persulfenate is not an artifact of synchrotron radiation. Indeed, the laboratory-based LT view of the Cys persulfenate complex is virtually identical to those based on synchrotron data (Fig. 8a), including the observed OA–OB distance of ~1.8 Å.
Figure 8.
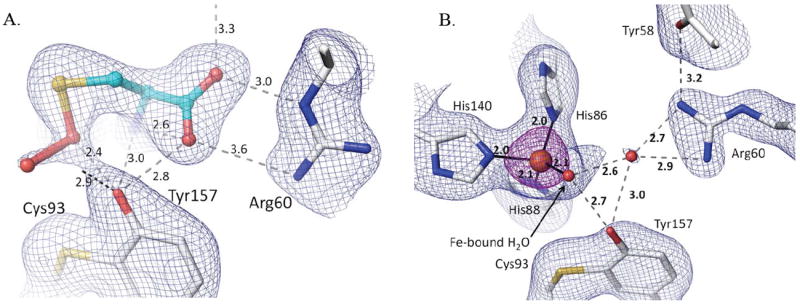
The persulfenate is not an artifact of freezing or synchrotron radiation. Each panel shows 2Fo-Fc density along with the corresponding refined model with protein carbons (white) and ligand carbons (cyan) and H-bonds (dashed lines with distances). (A) Lab-LT-Cys structure with density for bound Cys-persulfenate contoured at 0.7 ρrms. (B) Lab-RT structure of unliganded CDO with density contoured at 1.4 ρrms (blue) and 5.0 ρrms (purple). The view in panel B matches that of figure 6c and d.
We describe this ~1.8 Å OA-OB separation as a distance rather than a bond length, because the electron density with the apparent ~1.8 Å distance could represent a mixture of structures having a range of OA-OB distances and geometries and levels of disorder. Structures that could be present are, for instance, Cys plus unreacted dioxygen, a Cys-persulfenate, a Cys-persulfenic acid, a Cys-sulfenate (Cys-SOH) plus a very weakly bonded oxygen, and even a thiadioxirane form. It is, however, of worth noting that for this unusual compound, the bond may actually be rather long, and indeed in the quantum mechanical calculations modeling a persulfenate intermediate,22 the optimized OA-OB bond was a nearly non-bonded 2.3 Å distance (and the Fe-S bond also lengthened to 2.42 Å). Kumar et al. interpreted these results to imply that a persulfenate species would not be stable in the active site and that “it seems likely the experimentally found ‘persulfenate’ structure actually refers to a protonated species.”22 As we already have noted, crystallography at this resolution cannot distinguish a protonated versus a deprotonated species, and it was an oversight of our original report17 to not make it clear that the bound intermediate may be a persulfenate or a persulfenic acid. Indeed, a protonated neutral form could be favored by the OB environment, which is a weakly-polar pocket loosely bounded by Leu95-Cδ1, C93-Sγ, His140-Cε1, Y157-OH and His155-Cε1.
Regarding possible temperature-dependent changes, the unliganded RT structure at 2.15 Å resolution (Table 1) has several side chains with increased disorder as expected,42; 43 but the Fe coordination is essentially unchanged (Fig. 8b). The most notable conformational change involves the active site residue Arg60 (and the nearby Met179) which mostly shifts to an alternate position (pointing toward the iron) that was weakly occupied in the low pH soaks (Fig 5 a-c) and fully occupied in the structure with Cys164 modified by disulfide formation with Cys (see Fig. 9c,d).10;12 The RT Cys soak was not informative, because due to the long data collection time (days), it yielded a structure that had a perturbed active site because of disulfide formation at Cys164 (data not shown).
Figure 9.
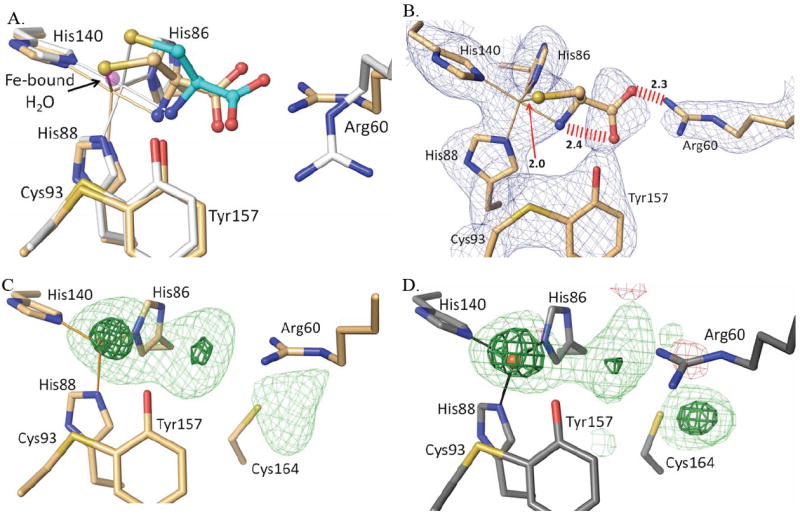
Reevaluation of the published human CDO-Cys complex. (A) An overlay of active site structure of PDB entry 2IC1 (orange carbon atoms and orange bonds to the iron atom) on the pH=9 CDO-Cys complex reported here (white and cyan carbon atoms and white bonds to the iron atom), with the iron bound water from unliganded CDO (PDB entry 2GH2) shown for reference (semi-transparent purple atom) and with the iron positions indicated by the apex of the coordination bonds. (B) A 2Fo-Fc omit map of the active site of 2IC1 (calculated after omitting the ligand from the model) contoured at 1.5 ρrms. Also indicated are three unreasonably close approaches in the 2IC1 structure (red hash marks or arrow with distances). Note that at this contour level density for the Cys in 2IC1 is not continuous even though the iron ligating His residues and other active site residues have strong continuous density. (C) Difference map for PDB entry 2IC1 (Ye et al. 2007) with the ligand atoms removed from the model; the contour levels are 4.0 ρrms (lightgreen) and 10.0 ρrms (darkgreen). (D) Same as C, but calculated for PDB entry 2GH2 (unliganded disulfide modified rat CDO)10 after removing active site waters 505, 651, 656, 828, 830 and truncating the data to 2.7 Å resolution to match that of PDB entry 2IC1.
Re-examination of structure reported for human CDO in complex with cysteine
The consistency of the binding mode of Cys across a wide pH range with and without persulfenate formation leads us to hypothesize that these structures are giving us a reliable view of the physiologically relevant binding mode of Cys to CDO. Surprisingly, the binding mode seen here differs in key details from that reported in the 2.7 Å resolution analysis of Cys binding at pH=6.5 to human CDO (PDB entry 2IC1).16 Three striking differences (Fig. 9a) are the Arg60 side chain position, the cysteine Sγ-atom placement which in PDB entry 2IC1 matches the position of the iron bound water seen in unliganded structures, and the iron atom position which in the 2IC1 model also matches that seen in unliganded CDO structures. Interestingly, the 2IC1 model also has steric problems with bad contacts between the Cys carboxylate oxygens and both Arg60 (2.27 Å) and the Cys α-amino group (2.45 Å), as well as an implausibly short Fe-S ligation distance of just 2.02 Å (Fig. 9b). Upon further inspection, we noticed that the positions of Arg60 and Met79 (which also differs from our Cys-only structure) match those that were previously shown in a high resolution analysis10 to be associated with an artifactual disulfide formation at Cys164. Indeed, Ye et al.16 comment on (and show in their figure 3a) a substantial modification of Cys164 that they left uninterpreted.
Given these observations, we explored if the active site electron density from the 2IC1 data might be better explained by a modification of Cys164 rather than the binding of Cys as a substrate. An inspection of the electron density maps for entry 2IC1 shows that while the density for the modeled Arg60 and Met179 positions is strong, the density for the modeled Cys is quite weak for the Cα, Cβ, and N-atoms (Fig. 9b). Also notable, is that the strong density into which the Cys Sγ-atom is modeled should not be taken as evidence for Cys-binding, because it sits at the same position that is occupied by the coordinating water of unliganded CDO. Furthermore, the maximal density of the iron peak indicates that the iron should be slightly shifted toward the Sγ-atom, which would make that already too short coordination distance even shorter. These observations support the conclusion that the electron density the Sγ-atom has been built into is actually due to the coordinating water present in unliganded CDO rather than a sulfur atom.
At the same time, the electron density is remarkably similar to that of the Cys164 modified enzyme of Simmons et al.10 when those data are truncated to 2.7 Å (Fig 9c versus d). This similarity shows that all of the active site features of the 2IC1 structure can be explained by rearranged side chains and water molecules associated with Cys164 modification. Based on these observations, we conclude that 2IC1 is not a Cys complex.
Implications for understanding the mechanism of CDO
We undertook these studies to better understand the mechanism of CDO and related dioxygenases. The most significant results are the capturing of a Cys-only complex at pH 8 and above, and demonstrating that the CDO:persulfenate complex, until now seen only in a single crystal structure, is a robust result that is not an artifact of synchrotron radiation, and is consistently formed in the active site of CDO between pH 5.5 and 7.0, corresponding roughly with the pH range over which CDO shows activity in our standard assay done in the presence of 0.3 mM Fe2+.32 As representatives of the complexes have high occupancies and are determined at high (1.25-1.4 Å) resolution, the conformations of the Cys-only and Cys-persulfenate ligands and their interactions with the active site residues have coordinate uncertainties of ~0.05-0.10 Å for the C, N, and O atoms and even better for the S and Fe atoms (see methods).
From the pH series, we conclude that Cys binding requires the deprotonated form of a group with an apparent pKa near 5.5, and that the coordination of the active site iron and the oxygen binding and reactivity of the Cys complex are both influenced by the protonation state of a group with an apparent pKa near 7.5. Unfortunately, crystal structures even at a resolution of ~1.4 Å do not show hydrogen positions, so without obvious changes in interactions (which we do not have here), we are not able to infer what groups are becoming protonated/deprotonated. As mentioned above, we postulate that the group with an apparent pKa near 5.5 is the Fe-bound Cys α-amino and/or thiol group, and that the group becoming deprotonated near pH=7.5 is the coordinating water in the unliganded complex and is Tyr157-OH in the Cys-bound complex. As previously described,17 Tyr157 is well placed to interact with either of the persulfenate oxygens (OA and OB in Figure 1b) and serve as a catalytic acid/base during catalysis, and a pKa value near 7 or 8 is reasonable for a Tyr side chain acting as an acid/base catalyst (e.g.44; 45).
Unfortunately, correlating our structurally-defined effective pKa values with those seen to influence the activity of CDO is difficult, because as noted in the introduction there is not agreement on the pH dependence of activity. Based on the pH-profile showing activity limited by apparent pKa values of 5.5 and 6.5,2; 4; 32 we would infer that the deprotonation occurring in the crystal near pH=7.5 is blocking catalysis. However, based on the pH-profile with activity occurring between pH=7 and 9,33 we would infer that the deprotonation event was generating the active form of the enzyme. Clearly, coming to agreement on how to best prepare and assay CDO and obtaining an accurate pH profile of its kcat and KM values are important next steps in helping define the mechanism of CDO.
Our discovery that the reported complex of human CDO with bound Cys16 is not correct is also significant because that model served as the basis of quantum mechanical studies of the CDO mechanism.21; 22; 23; 28 Geometry optimization done in setting up the recent quantum mechanics study did correct the chemically unreasonable 2.0 Å S-Fe bond length to be ~2.3 Å (see figures 5 of Kumar et al.),22 but other features of the ‘optimized geometry’ active site structure in the simulations of the persulfenate-based mechanism are different from the interactions consistently seen in the crystal structures presented here. Prominent examples are the interaction of Arg60 with the Cys α-carboxylate and the relative positions of the persulfenate oxygens and Tyr157 (see figure 11 of Kumar et al.).22 Additional evidence for the critical nature of Tyr157 is that the residue is conserved in all CDO sequences13 and is sitting in much the same position even in a bacterial CDO structure having <20% sequence identity with rat CDO (PDB entry 2GM6). Since catalytic proficiency can depend exquisitely on the precise placement of active site residues, it will be of interest for some calculations to be guided more closely by the positions of the protein side chains seen here and in other reliable CDO structures.
As we see it, despite ambiguity about the protonation state of the persulfenate/persulfenic complex and uncertainty as to its relevance to the catalytic mechanism, the ability of the CDO active site to make and stabilize it proves that its formation is energetically accessible in the CDO active site. That an intrinsically less-stable molecule like the persulfenate rather than substrate or product would be stably trapped in the active site of the crystalline enzyme can be explained in that the on-enzyme equilibrium between substrate, various on and off-pathway intermediates, and product can be highly perturbed from that which would occur in solution. Also important to note is that these equilibria would also be temperature-dependent, so that the dominant form present in the active site may be influenced by the freezing process.
In considering the relevance to catalysis of this complex, one piece of evidence in favor of it being an intermediate would be to show that the crystalline enzyme is active. Unfortunately, our attempts thus far to show that crystalline CDO can convert Cys to the Cys-sulfinate product have not been successful (RB Cooley, unpublished). This could be due to a variety of factors, including something as simple as the high ionic strength and/or high PEG concentrations of the mother liquor causing technical challenges with the analyses (which they do), or something as subtle as the crystal lattice hindering certain dynamic modes of the enzyme that are required for complete turnover. Given the principle that active sites are optimized for stabilizing the transition state of a reaction, we think that this complex is closely related to an intermediate of the true catalytic mechanism even if it is not directly on-pathway. As was noted in a recent review,30 the proposal that CDO operates by monooxygenase like chemistry with the involvement of an Fe(IV)=O intermediate (figure 1b, right path) is associated with a conceptual difficulty: why would such a powerful oxidizing agent as Fe(IV)=O be generated for such a relatively easy oxidation reaction? The mechanism of CDO has been surprisingly challenging to elucidate, but the accurate structures described here together with the insight into protonation/deprotonation events influencing CDO chemistry provide a solid structural foundation that will support more accurate computational work and guide the design and interpretation of further experimental studies using CDO and small molecule CDO mimics.
Materials and Methods
Expression, purification and crystallization
Expression, purification and crystallization of recombinant rat CDO was done as described previously,17; 32; 46 but with seeds from crushed CDO crystals added to facilitate nucleation. The enzyme was stored at 8 mg/mL in 10 mM Tris pH=7.4 buffer and crystallized at room temperature in hanging drops formed from 0.5 μL seed stock in reservoir buffer,1 μL protein stock and 1 μL of a 0.1M tri-sodium citrate pH=5.6, 24% PEG 4000, and 0.15 M ammonium acetate reservoir (final pH=6.2).17; 32 The crystals were isomorphous with those previously described,17; 46 having spacegroup P43212 and one chain in the asymmetric unit.
Crystal soaks and data collection
Soaks were done at room temperature using the reservoir condition as an artificial mother liquor (AML). For cryo-crystallographic data collection, crystals were flash frozen by plunging into liquid nitrogen. For room temperature data collection, crystals were soaked as described and then scooped using a MicroRT™ capillary system (Mitegen).
For an initial pH series, crystals were incubated for 1 h in solutions with 100 mM cysteine added to AML that had its pH adjusted to 4.5, 5.5, 6.5, 7.5, 8.5 or 9.5. After data were collected, we discovered that the added Cys had altered the pH; by recreating the solutions, we found that the final pH values were 5.5, 6.2, 6.8, 7.0, 7.0 and 7.0. Additional 1 h Cys soaks were carried out in solutions made with final pH values (after cysteine addition) of 4.0, 4.5, 5.0, 7.0, 8.0, or 9.0. Crystals did not diffract after some 1 h soaks at pH=4.0 and 8.0 so shorter soaks of 15 or 20 min were done. Also, at pH=4.5 a series of short control soaks ranging from 30 s to 12 min were performed in addition to the 1 h soak. Ligand-free structures at a few pH values were obtained from crystals soaked 40 – 60 min in AML at the targeted pH. For the ligand-free pH=8.0 structure, 100 mM Ala was added to AML to create buffering more equivalent to the Cys soak. For soaks at pH=8.0 with dithionite, crystals were incubated in a Cys- (20 min) or Ala- (60 min) containing degassed AML with 20 mM dithionite and 200 μM methyl viologen (as a color indicator of the redox status of the solution) and the final pH adjusted to pH=8 with degassed NaOH. Although crystals were soaked and handled in an aerobic environment, solutions remained deep blue for the duration of the soak.
Synchrotron data sets were collected at beamlines 5.0.1, 5.0.2, and 8.2.1 at the Advanced Light Source (Lawrence Berkeley National Laboratory) and beamline X26C at the National Synchrotron Light Source (Brookhaven National Laboratory). In-house diffraction data were collected on a Rigaku RU-H3R rotating Cu-anode operating at 50 kV and 100 mA and an R-Axis IV image plate detector. Data were processed and scaled using iMosflm47 and SCALA.48 For all refinements, the same 10% of the data as used previously17 were flagged for use in Rfree. Also, a random 10% of reflections beyond the previous 1.42 Å resolution limit were flagged for Rfree calculations. Data statistics are in Tables 1 and 2.
Crystallographic refinement
The unit cell constants for individual crystals were mostly within the ranges a=b=57.6-58.2 Å and c=121.7-122.9 Å, and these refinements all used a=b=57.60 Å and c=122.40 Å. Refinements were done using PHENIX49 and for each dataset were initiated using an automated protocol that when applied to a refinement of the previously published 1.42 Å resolution CDO:Cys complex (PDB code 3eln) yielded a better R/Rfree.36 Refinements began with the 1.5 Å resolution unliganded CDO model (PDB code 2b5h) with, when appropriate, the coordinates for the Cys-persulfenate or cysteine added. The occupancy of the persulfenate was fixed at 0.7 in all persulfenate-containing models. The Cys-only models at pH=8 and 9 set the Cys occupancy at 1.0. Geometric restraints for the ligand as well as the Cys-Tyr crosslink were created by Elbow50 and manual editing to match restraints used previously.17 For the OA-OB bond, the restraint of 1.47 Å was initially used,17 but was changed to 1.8 Å with a relatively lose weight (estimated standard deviation=0.10 Å) after observing a distinct density peak for the O2 atom in some of the structures (eg. see Figs. 2 and 5).
The first stage of refinement specified isotropic B-factors, riding hydrogens, automatic water adding, and rotamer fitting. This result was then further refined using anisotropic B-factors. The anisotropic B-factors were only accepted if their use resulted in an Rfree improvement of at least 1% compared with isotropic plus TLS refinements with 1 TLS group defining the entire chain. After these automated stages, the refinements were completed using manual rebuilding in Coot51 and PHENIX-refine minimization. Some water sites were removed and others were added using standard criteria (>1 ρrms intensity in the 2Fo –Fc map, >2.4 Å distance from nearest contact), and all difference map peaks above 5*ρrms were checked. Molprobity52 was used to find problems with model geometry in these final rounds. Refinement statistics are in Tables 1 and 2.
Estimates of coordinate uncertainties for individual atoms were calculated using the Cruickshank method53 implemented in Refmac.54; 55 As examples of the coordinate uncertainties in the active site, for the pH=5.5+Cys / pH=9.0 Cys-only structures the residual uncertainties (to one significant figure or the nearest 0.01 Å and in order of the two structures) are as follows: Fe 0.01 / 0.005 Å; Tyr157-OH 0.04 / 0.03 Å; Arg60-NH2 0.16 / 0.09 Å; Cys-Sγ 0.04 / 0.02 A; and for the persulfenate-OA and OB atoms in the pH=5.5 structure, 0.07 and 0.10 Å.
Supplementary Material
-
►
Controversy exists about the mechanism of Fe(II)-dependent cysteine dioxygenase
-
►
A Cys-persulfenate is formed in the active site of crystalline CDO from pH=5.5 to 7
-
►
An iron-bound persulfenate is not an artifact of synchrotron radiation.
-
►
A complex of unreacted Cys is formed in the active site of CDO from pH 8 to 9
-
►
The titration behavior and ligand-bound models can guide mechanistic considerations
Acknowledgments
We thank Dr. Dale Tronrud for useful scientific discussions and help with methods. We also thank Dr. Babak Andi and Dr. Allan Orville for collecting coordinated spectroscopic and diffraction data at the National Synchrotron Light Source (NSLS). This project was supported in part by Grant DK-056649 to MHS from the National Institute of Diabetes and Digestive and Kidney Diseases. It was also aided by the Collaborative Crystallography Project of the Berkeley Center for Structural Biology supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. Synchrotron data were collected at the Advanced Light Source and the NSLS, respectively supported by contracts DE-AC02-98CH10886 and DE-AC02-05CH11231 from the Office of Basic Energy Sciences of the U.S. Department of Energy
Footnotes
Accession Numbers. Coordinates and structure factors for the CDO models have been deposited in the Protein Data Bank with accession numbers as follows: pH=4.0 + Cys 20 min (PDB code 4IEO); pH=4.5 + Cys 1 h (PDB code 4IEP); pH=5.0 + Cys 1 h (PDB code 4IEQ); pH=5.5 + Cys 1 h (PDB code 4IER); pH=6.2 + Cys 1 h (PDB code 4IES); pH=6.8 + Cys 1 h (PDB code 4IET); pH=7.0 + Cys 1 h (PDB code 4IEU); pH=8.0 + Cys 1 h (PDB code 4IEV); pH=9.0 + Cys 1 h (PDB code 4IEW); pH=6.2 + Lab-RT (PDB code 4IEX); pH=7.0 + Cys Lab-LT (PDB code 4IEY); pH=8.0 + Ala (PDB code 4IEZ); pH=8.0 + Ala + DT (PDB code 4JTN); pH=8.0 + Cys + DT (PDB code 4JTO).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stipanuk MH, Simmons CR, Karplus PA, Dominy JE., Jr Thiol dioxygenases: unique families of cupin proteins. Amino Acids. 2011;41:91–102. doi: 10.1007/s00726-010-0518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagley PJ, Stipanuk MH. The activities of rat hepatic cysteine dioxygenase and cysteinesulfinate decarboxylase are regulated in a reciprocal manner in response to dietary casein level. J Nutr. 1994;124:2410–21. doi: 10.1093/jn/124.12.410. [DOI] [PubMed] [Google Scholar]
- 3.Dominy JE, Jr, Hwang J, Stipanuk MH. Overexpression of cysteine dioxygenase reduces intracellular cysteine and glutathione pools in HepG2/C3A cells. Am J Physiol Endocrinol Metab. 2007;293:E62–9. doi: 10.1152/ajpendo.00053.2007. [DOI] [PubMed] [Google Scholar]
- 4.Dominy JE, Jr, Hwang J, Guo S, Hirschberger LL, Zhang S, Stipanuk MH. Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity. J Biol Chem. 2008;283:12188–201. doi: 10.1074/jbc.M800044200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stipanuk MH. Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu Rev Nutr. 2004;24:539–77. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 6.Ueki I, Roman HB, Valli A, Fieselmann K, Lam J, Peters R, Hirschberger LL, Stipanuk MH. Knockout of the murine cysteine dioxygenase gene results in severe impairment in ability to synthesize taurine and an increased catabolism of cysteine to hydrogen sulfide. Am J Physiol Endocrinol Metab. 2011;301:E668–84. doi: 10.1152/ajpendo.00151.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, Sturman SG. Plasma cysteine and sulphate levels in patients with motor neurone, Parkinson’s and Alzheimer’s disease. Neurosci Lett. 1990;110:216–20. doi: 10.1016/0304-3940(90)90814-p. [DOI] [PubMed] [Google Scholar]
- 8.Bradley H, Gough A, Sokhi RS, Hassell A, Waring R, Emery P. Sulfate metabolism is abnormal in patients with rheumatoid arthritis. Confirmation by in vivo biochemical findings. J Rheumatol. 1994;21:1192–6. [PubMed] [Google Scholar]
- 9.Brait M, Ling S, Nagpal JK, Chang X, Park HL, Lee J, Okamura J, Yamashita K, Sidransky D, Kim MS. Cysteine dioxygenase 1 is a tumor suppressor gene silenced by promoter methylation in multiple human cancers. PLoS One. 2012;7:e44951. doi: 10.1371/journal.pone.0044951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmons CR, Liu Q, Huang Q, Hao Q, Begley TP, Karplus PA, Stipanuk MH. Crystal structure of mammalian cysteine dioxygenase. A novel mononuclear iron center for cysteine thiol oxidation. J Biol Chem. 2006;281:18723–33. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]
- 11.McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN., Jr Structure and mechanism of mouse cysteine dioxygenase. Proc Natl Acad Sci U S A. 2006;103:3084–9. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleffmann T, Jongkees SA, Fairweather G, Wilbanks SM, Jameson GN. Mass-spectrometric characterization of two posttranslational modifications of cysteine dioxygenase. J Biol Inorg Chem. 2009;14:913–21. doi: 10.1007/s00775-009-0504-x. [DOI] [PubMed] [Google Scholar]
- 13.Dominy JE, Jr, Simmons CR, Karplus PA, Gehring AM, Stipanuk MH. Identification and characterization of bacterial cysteine dioxygenases: a new route of cysteine degradation for eubacteria. J Bacteriol. 2006;188:5561–9. doi: 10.1128/JB.00291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siakkou E, Rutledge MT, Wilbanks SM, Jameson GN. Capturing crosslink formation with enzymatic activity in cysteine dioxygenase. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbapap.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 15.Imsand EM, Njeri CW, Ellis HR. Addition of an external electron donor to in vitro assays of cysteine dioxygenase precludes the need for exogenous iron. Arch Biochem Biophys. 2012;521:10–17. doi: 10.1016/j.abb.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Ye S, Wu X, Wei L, Tang D, Sun P, Bartlam M, Rao Z. An insight into the mechanism of human cysteine dioxygenase. Key roles of the thioether-bonded tyrosine-cysteine cofactor. J Biol Chem. 2007;282:3391–402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 17.Simmons CR, Krishnamoorthy K, Granett SL, Schuller DJ, Dominy JE, Jr, Begley TP, Stipanuk MH, Karplus PA. A putative Fe2+-bound persulfenate intermediate in cysteine dioxygenase. Biochemistry. 2008;47:11390–2. doi: 10.1021/bi801546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierce BS, Gardner JD, Bailey LJ, Brunold TC, Fox BG. Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: electronic structure of the active site and mechanistic implications. Biochemistry. 2007;46:8569–78. doi: 10.1021/bi700662d. [DOI] [PubMed] [Google Scholar]
- 19.Crawford JA, Li W, Pierce BS. Single turnover of substrate-bound ferric cysteine dioxygenase with superoxide anion: enzymatic reactivation, product formation, and a transient intermediate. Biochemistry. 2011;50:10241–53. doi: 10.1021/bi2011724. [DOI] [PubMed] [Google Scholar]
- 20.Gardner JD, Pierce BS, Fox BG, Brunold TC. Spectroscopic and computational characterization of substrate-bound mouse cysteine dioxygenase: nature of the ferrous and ferric cysteine adducts and mechanistic implications. Biochemistry. 2010;49:6033–41. doi: 10.1021/bi100189h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aluri S, de Visser SP. The mechanism of cysteine oxygenation by cysteine dioxygenase enzymes. J Am Chem Soc. 2007;129:14846–7. doi: 10.1021/ja0758178. [DOI] [PubMed] [Google Scholar]
- 22.Kumar D, Thiel W, de Visser SP. Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. J Am Chem Soc. 2011;133:3869–82. doi: 10.1021/ja107514f. [DOI] [PubMed] [Google Scholar]
- 23.de Visser SP, Straganz GD. Why do cysteine dioxygenase enzymes contain a 3-His ligand motif rather than a 2His/1Asp motif like most nonheme dioxygenases? J Phys Chem A. 2009;113:1835–46. doi: 10.1021/jp809700f. [DOI] [PubMed] [Google Scholar]
- 24.Mirza SA, Pressler MA, Kumar M, Day RO, Maroney MJ. Oxidation of Nickel Thiolate Ligands by Dioxygen. Inorganic Chemistry. 1993;32:977–987. [Google Scholar]
- 25.Clennan EL. Persulfoxide: Key intermediate in reactions of singlet oxygen with sulfides. Accounts of Chemical Research. 2001;34:875–884. doi: 10.1021/ar0100879. [DOI] [PubMed] [Google Scholar]
- 26.Grapperhaus CA, Darensbourg MY. Oxygen capture by sulfur in nickel thiolates. Accounts of Chemical Research. 1998;31:451–459. [Google Scholar]
- 27.Farmer PJ, Solouki T, Soma T, Russell DH, Darensbourg MY. Divergent Pathways for the Addition of Dioxygen to Sulfur in Nickel Cis-Dithiolates - an Isotopomeric Analysis. Inorganic Chemistry. 1993;32:4171–4173. [Google Scholar]
- 28.Kumar D, Sastry GN, Goldberg DP, de Visser SP. Mechanism of S-oxygenation by a cysteine dioxygenase model complex. J Phys Chem A. 2012;116:582–91. doi: 10.1021/jp208230g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McQuilken AC, Jiang Y, Siegler MA, Goldberg DP. Addition of dioxygen to an N4S(thiolate) iron(II) cysteine dioxygenase model gives a structurally characterized sulfinato-iron(II) complex. J Am Chem Soc. 2012;134:8758–61. doi: 10.1021/ja302112y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McQuilken AC, Goldberg DP. Sulfur oxygenation in biomimetic non-heme iron-thiolate complexes. Dalton Trans. 2012;41:10883–99. doi: 10.1039/c2dt30806a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chai SC, Jerkins AA, Banik JJ, Shalev I, Pinkham JL, Uden PC, Maroney MJ. Heterologous expression, purification, and characterization of recombinant rat cysteine dioxygenase. J Biol Chem. 2005;280:9865–9. doi: 10.1074/jbc.M413733200. [DOI] [PubMed] [Google Scholar]
- 32.Simmons CR, Hirschberger LL, Machi MS, Stipanuk MH. Expression, purification, and kinetic characterization of recombinant rat cysteine dioxygenase, a non-heme metalloenzyme necessary for regulation of cellular cysteine levels. Protein Expr Purif. 2006;47:74–81. doi: 10.1016/j.pep.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 33.Chai SC, Bruyere JR, Maroney MJ. Probes of the catalytic site of cysteine dioxygenase. J Biol Chem. 2006;281:15774–9. doi: 10.1074/jbc.M601269200. [DOI] [PubMed] [Google Scholar]
- 34.Siakkou E, Wilbanks SM, Jameson GN. Simplified cysteine dioxygenase activity assay allows simultaneous quantitation of both substrate and product. Anal Biochem. 2010;405:127–31. doi: 10.1016/j.ab.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 35.Tchesnokov EP, Wilbanks SM, Jameson GN. A strongly bound high-spin iron(II) coordinates cysteine and homocysteine in cysteine dioxygenase. Biochemistry. 2012;51:257–64. doi: 10.1021/bi201597w. [DOI] [PubMed] [Google Scholar]
- 36.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–3. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W. Data mining of metal ion environments present in protein structures. J Inorg Biochem. 2008;102:1765–76. doi: 10.1016/j.jinorgbio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan B, Lahav O. The effect of pH on the kinetics of spontaneous Fe(II) oxidation by O2 in aqueous solution--basic principles and a simple heuristic description. Chemosphere. 2007;68:2080–4. doi: 10.1016/j.chemosphere.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Green MT. Application of Badger’s rule to heme and non-heme iron-oxygen bonds: an examination of ferryl protonation states. J Am Chem Soc. 2006;128:1902–6. doi: 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- 40.Weik M, Berges J, Raves ML, Gros P, McSweeney S, Silman I, Sussman JL, Houee-Levin C, Ravelli RB. Evidence for the formation of disulfide radicals in protein crystals upon X-ray irradiation. J Synchrotron Radiat. 2002;9:342–6. doi: 10.1107/s0909049502014589. [DOI] [PubMed] [Google Scholar]
- 41.Hall A, Sankaran B, Poole LB, Karplus PA. Structural changes common to catalysis in the Tpx peroxiredoxin subfamily. J Mol Biol. 2009;393:867–81. doi: 10.1016/j.jmb.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci U S A. 2011;108:16247–52. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halle B. Biomolecular cryocrystallography: structural changes during flash-cooling. Proc Natl Acad Sci U S A. 2004;101:4793–8. doi: 10.1073/pnas.0308315101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ibarra CA, Chowdhury P, Petrich JW, Atkins WM. The anomalous pKa of Tyr-9 in glutathione S-transferase A1-1 catalyzes product release. J Biol Chem. 2003;278:19257–65. doi: 10.1074/jbc.M301566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlegel BP, Jez JM, Penning TM. Mutagenesis of 3 alpha-hydroxysteroid dehydrogenase reveals a “push-pull” mechanism for proton transfer in aldo-keto reductases. Biochemistry. 1998;37:3538–48. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- 46.Simmons CR, Hao Q, Stipanuk MH. Preparation, crystallization and X-ray diffraction analysis to 1.5 A resolution of rat cysteine dioxygenase, a mononuclear iron enzyme responsible for cysteine thiol oxidation. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:1013–6. doi: 10.1107/S1744309105033737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leslie A. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4+ ESF-EAMCB newsletter on protein crystallography. 1992;26 [Google Scholar]
- 48.Evans PR. Recent advances in phasing. In: Wilson KS, Davies G, Ashton AW, Bailey S, editors. Proc CCP4 Study Weekend. Daresbury Laboratory; Warrington, UK: 1997. pp. 97–102. [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr. 2009;65:1074–80. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol. 1999;285:1711–33. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 53.Cruickshank DWJ. Protein precision re-examined: Luzzati plots do not estimate final errors. Proceedings of the CCP4 Study Weekend. 1996 Jan [Google Scholar]
- 54.Murshudov GN, Dodson EJ. Simplified error estimation a la Cruickshank in macromolecular crystallography. CCP4 Newsletter. 1997 Jan [Google Scholar]
- 55.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.