

Abstract
Background and Aims
Interleukin-10 (IL-10) signaling genes are attractive inflammatory bowel disease (IBD) candidate genes as IL-10 restricts intestinal inflammation, IL-10 polymorphisms have been associated with IBD in genome-wide association studies, and mutations in IL-10 and IL-10 receptor (IL-10R) genes have been reported in immunodeficient children with severe infantile-onset IBD. Our objective was to determine if IL-10R polymorphisms were associated with early-onset IBD (EO-IBD) and very early-onset IBD (VEO-IBD).
Methods
Candidate-gene analysis of IL10RA and IL10RB was performed after initial sequencing of an infantile onset-IBD patient identified a novel homozygous mutation. The discovery cohort included 188 EO-IBD subjects and 188 healthy subjects. Polymorphisms associated with IBD in the discovery cohort were genotyped in an independent validation cohort of 422 EO-IBD subjects and 480 healthy subjects.
Results
We identified a homozygous, splice-site point mutation in IL10RA in an infantile-onset IBD patient causing a premature stop codon (P206X) and IL-10 insensitivity. IL10RA and IL10RB sequencing in the discovery cohort identified five IL10RA polymorphisms associated with ulcerative colitis (UC) and two IL10RB polymorphisms associated with Crohn’s disease (CD). Of these polymorphisms, two IL10RA SNPs, rs2228054 and rs2228055 were associated with very early-onset UC in the discovery cohort and replicated in an independent validation cohort (OR 3.08, combined p=2×10−4; and OR 2.93, p=6×10−4, respectively).
Conclusions
We identified IL10RA polymorphisms that confer risk for developing VEO-UC. Additionally, we identified the first splice site mutation in IL10RA resulting in infantile-onset IBD. This study expands the phenotype of IL10RA polymorphisms to include both severe arthritis and VEO-UC.
Keywords: IL-10, IL-10 Receptor, inflammatory bowel disease, immunodeficiency
Introduction
Interleukin-10 (IL-10) is an anti-inflammatory cytokine secreted by a variety of cell types and is critical for maintaining immune homeostasis in the gastrointestinal (GI) tract.1 These immunosuppressive effects include restricting T cell proliferation, down-regulating co-stimulatory protein expression on antigen-presenting cells, and limiting pro-inflammatory cytokine production. IL-10 activates downstream signaling by binding to the IL-10 receptor (IL-10R), comprised of two α subunits (encoded by IL10RA) and two β subunits (encoded by IL10RB). This activates JAK1 and TYK2, leading to phosphorylation and nuclear translocation of STAT3 and gene transcription. Intact IL-10 signaling is required for restricting inappropriate Th17 cell expansion – an effector cell type that has been associated with Crohn’s disease (CD).2,3 Consistent with the anti-inflammatory role of IL-10, mice deficient in either IL-10 or IL-10R develop enterocolitis.3-5
Recent studies have shown that loss-of-function mutations in IL10RA and IL10RB6,7 and IL108 in immunodeficient patients are associated with severe, infantile-onset IBD. This has stimulated renewed interest in studying IL-10 pathway genes in the pathogenesis of inflammatory bowel disease (IBD). Genome-wide association studies (GWAS) in IBD populations (including exclusively pediatric-onset disease) have associated single nucleotide polymorphisms (SNPs) in IL10 and STAT3 loci with ulcerative colitis (UC) and CD.9-12
Given the severe phenotype of infantile-onset IBD observed with IL-10R mutations as well as that of an infant described in this paper who was found to have a homozygous, splice site mutation in IL-10RA,6,7 we hypothesized that IL10RA and IL10RB polymorphisms contribute to the risk of IBD developing in the very young. To date, SNPs within IL10RA and IL10RB loci have not been identified by GWAS of IBD. However, the contribution of IL-10R polymorphisms to IBD susceptibility in very young patients may be underestimated in GWAS as this age group represents a very select population that is poorly represented even in GWAS focusing on pediatric IBD.13 Rather than studying only the overall population of patients that constitute “pediatric IBD”, we subdivided patients so that we could concentrate our analyses on extremely young patients (that may be most likely to have the strongest genetic component to IBD risk). Although the Paris classification for IBD stratifies pediatric IBD into those diagnosed younger than 10 years of age and those diagnosed between 10 and 18 years of age,14 we focused on patients diagnosed prior to their 6th birthday - a cohort that comprises ~15% of pediatric IBD and represents a greater extreme of the disease - since the average age at diagnosis of “pediatric IBD patients” is ~10 years old.15 Therefore, we undertook a candidate gene study by deep sequencing of IL10RA and IL10RB in children with early onset IBD (EO-IBD, defined as age of onset <18yo) and subsequently focused on the very early-onset IBD (VEO-IBD, defined as age of onset <6yo).
Methods
Identification of Novel IL-10RA Mutation in Patient with Infantile Colitis
We identified an IL-10RA mutation in a Caucasian female presenting with infantile colitis who was not recognized a priori to be the product of a consanguineous marriage, but whose parents came from the same geographically-isolated region of Canada. Her first year of life was complicated by recurrent fevers (initially at six days of life). At three months old, the patient developed cellulitis and scalp eczema and generalized erythroderma (Figure 1A). Bloody diarrhea and a perianal skin tag developed at five months of age (Figure 1B) that progressed to perianal and recto-vaginal fistulae. Further complications included urinary tract infections, scalp pustules, and severe, effusive, large joint polyarthritis (Figure 1C). Joint aspirations demonstrated high neutrophil counts (>100,000/mm3) despite sterile cultures. The patient did not manifest thyroid involvement. Endoscopic evaluation revealed patchy colonic ulcerations (Figure 1D) and biopsies showed chronic colitis (Figure 1E) with the absence of small bowel inflammation. Gastrointestinal symptoms were refractory to antibiotics, intravenous immunoglobulins, corticosteroids, anakinra, sulfasalazine, and azathioprine, and the patient underwent a loop ileostomy at 23 months old.
Figure 1. Clinical Features of Patient with Severe, Infantile-Onset IBD.
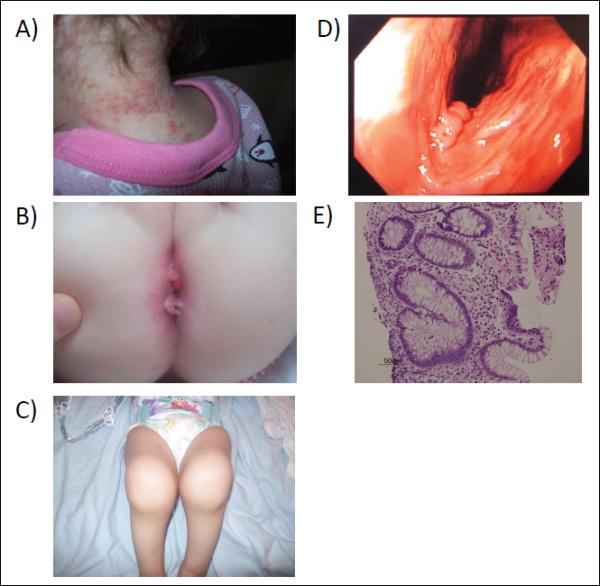
The patient had skin folliculitis (A), perianal disease with fistulae (B), joint effusions (C), and colonic erosions (D). Histology showed colitis with gland branching and mixed lamina propria cell infiltrate with fibropurulent exudate (arrow) (E).
An extensive immunological evaluation included normal immunoglobulin levels and lymphocyte subsets, normal NADPH oxidase function, a normal thymic biopsy, normal karyotyping and negative FISH for 22q11. Screening for JAK3, IPEX, RMRP, IL2RA, IRAK4, and TRAPS mutations and mutations associated with chronic granulomatous disease were negative.
Genomic DNA was purified from whole blood using Puregene Blood Kit (Qiagen, USA). IL10RA and IL10RB were amplified using intronic primers flanking each exon and sequenced with the ABI3730 DNA analyzer (Applied Biosystems, Melbourne, Australia). IL10RA and IL10RB variants are numbered according to GeneBank accession numbersNM_001588 and NM_00628, respectively. Amino acid numbering refers to position in the immature protein (including signal peptide).
RNA was isolated from whole blood by PAXgene Blood RNA kit (Qiagen, USA) according to the manufacturer’s instructions. cDNA was synthesized using SuperScript III Reverse Transcriptase (Life Technologies, Carlsbad, California, USA). Primers for full-length IL10RA, full-length IL10RB, and for Exon 4 to Exon 7 of IL10RA were designed and synthesized at The Centre for Applied Genomics, Toronto. PCR was performed according to standard protocol, and the purified PCR product was cloned into pJET cloning vector (Fermentas, USA) and sequenced by ABI 3730 DNA analyzer (Applied Biosystems, Melbourne, Australia).
Functional IL-10 Testing in IL-10R-Deficient Patient
PBMCs from the patient and a healthy control were isolated using Polymorphoprep (Nycomed Pharma AS, Norway). The PBMCs were stimulated with IL-10 (40ng/mL) or IL-6 (20ng/mL) for 10 and 30 minutes. Cellular extracts were prepared following standard protocol. The protein concentration was measured by protein assay kit (Bio-Rad, Hercules, Canada), and 50μg protein was separated by 8% SDS-PAGE gel and transferred to membrane. The membrane was incubated with antibodies to STAT3 and phosphorylated (Tyr705) STAT3 (p-STAT3) (Cell Signaling, USA) followed by HRP-conjugated secondary antibodies. The membrane was read using a chemiluminescence system (GE Healthcare, UK). β-actin was used as a loading control.
Flow Cytometry
Thawed PBMC were stained in 2% FBS with CD4-PerCP (eBioscience, USA) and CD25-PE (Miltenyi Biotech, USA) followed by intracellular staining with FoxP3-FITC (eBioscience, USA). Flow cytometry was performed using a FACSCalibur (BD Biosciences, USA). Co-expression of CD25 and intracellular FoxP3 was assessed in cells within CD4+ lymphocyte gate.
IL-10R Sequencing in Discovery Cohort
The discovery cohort included DNA samples from 188 EO-IBD patients from the Hospital for Sick Children (Toronto, Canada) (49 patients) including some subjects as part of the NEOPICS consortium (Supplemental Table 1) and Children’s Hospital of Wisconsin (Milwaukee, Wisconsin) (139 patients). Control DNA samples were obtained from The Centre for Applied Genomics (Ontario Population Genomics Platform, plates used: 1-4); 95% of this cohort are Caucasians of European ancestry. Complete description can be found at http://www.tcag.ca/facilities/cyto_population_control_DNA.html.
The coding sequences (and intervening sequences and 3′-untranslated regions) of IL10RA and IL10RB were amplified. PCR products were sequenced by Sanger method (Beckman-Coulter Genomics, Danvers, MA). SNPs were detected using Polyphred software and all chromatograms with reliability score <99 were manually reviewed.
Allelic frequencies were determined for SNPs identified in discovery cohort. Testing for Hardy-Weinberg equilibrium was done by χ2 analysis. Odds ratios (OR) were calculated by comparing the ratio of individuals with two copies of the major allele to those with one or two copies of a variant allele (DD vs. Dd/dd model) between EO-IBD and control groups. OR were determined for UC and CD and specific age groups: children diagnosed before their 18th birthday (<18yo) and children diagnosed before their 6th birthday (<6yo). p values were calculated (but not adjusted for multiple testing in discovery cohort). SNPs with p value <0.05 were considered to be associated with IBD risk in the discovery cohort. Linkage between SNPs was assessed by χ2 analysis using a 3×3 table of genotypes. Linkage disequilibrium (LD) plots were created with Golden Helix SVS 7.0 (Golden Helix, Montana) using expectation-maximization (EM) algorithm to calculate r2 and D’ values.
Genotyping of IL10RA and IL10RB of Replication Cohort
DNA samples were collected from 422 pediatric IBD patients at the Hospital for Sick Children and genotyped using Taqman probes for SNPs associated with IBD from the discovery cohort. Control DNA samples (distinct from discovery cohort controls) were obtained from The Centre for Applied Genomics. Allelic frequencies were calculated and Hardy-Weinberg equilibrium was assessed by χ2 analysis. OR were calculated (DD vs. Dd/dd model) and analyzed by χ2 analysis. p values were adjusted for multiple testing by Bonferroni method (based on the number of SNPs associated with IBD in discovery cohort), and an adjusted p value <0.05 was considered statistically significant. As quality control, allele frequencies in discovery and validation cohort were compared by χ2 analysis. LD analysis and haplotype testing was performed as above.
All probands had a confirmed diagnosis of IBD fulfilling standard diagnostic criteria. Phenotypic characterization was based on Montreal classification.16 Age categories reflect age at diagnosis: early-onset IBD (EO-IBD) defined as diagnosis before 18th birthday; very early-onset IBD (VEO-IBD) defined diagnosis before 6th birthday; infantile-onset IBD defined as diagnosis before 1st birthday.14 Institutional review boards at Hospital of Sick Children, Children’s Hospital of Wisconsin, Mount Sinai Hospital (Toronto, Canada), and Massachusetts General Hospital approved these studies.
Results
Novel, Aberrant IL10RA Splice Site Mutation Identified in Patient with Infantile Colitis
We identified a patient with severe, infantile-onset IBD with significant arthritis and folliculitis (Figure 1). Sequencing of IL10RA and IL10RB in this patient identified a homozygous mutation in the intervening sequence of intron 5 at the second base of the conserved GT splice donor site (g.IVS5+2T>C) (Figure 2A). This novel mutation was confirmed by sequencing in an independent laboratory (data not shown) and has not been identified in the 1000 Genomes Project.
Figure 2. Identification of IL10RA g.IVS5+2T>C Mutation in Genomic DNA.
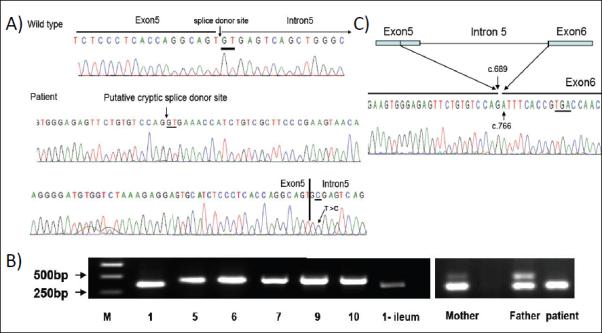
(A) Sequencing of Exon 5 of IL10RA revealed a homozygous g.IVS5+2T>C mutation (Bottom), compared to wild-type (Top). The splice donor site, putative cryptic splice donor site, and T>C mutation are underlined. (B) RT-PCR of IL10RA (Exon 4-7) from RNA isolated from blood and ileum. Lane 1 and Lane 1-ileum is from the patient. Lanes 2-6 are controls. Right: RT-PCR of Exon 4-7 of IL10RA from RNA from the patient and her parents. (C) Sequence of abnormal PCR product shows 76bp deletion (c.690_765del) leading to a premature stop codon (underlined).
This mutation was located in the 5′ splice donor site - a critical position for RNA splicing. Mutation of the invariant dinucleotide GT at the 5′ splice donor site impairs normal splicing and allows activation of cryptic splice site(s).17 We identified a cryptic splice donor site (CAGgtgaaa) in Exon 5 of IL10RA using Human Splicing Finder (Figure 2A).18 To confirm aberrant IL10RA splicing, RNA was isolated from whole blood and IL10RA transcripts were analyzed by RT-PCR. The expected 433bp PCR fragment was detected in 5 healthy donors. However, only a smaller fragment was observed in the patient (Figure 2B) and sequencing of this fragment revealed a 76bp deletion (c.690_765del) from the 3′ end of Exon 5 (Figure 2C). The resulting reading frame-shift mutation led to incorporation of 3 aberrant amino acids of Exon 6 (p.204ISP206) and a premature stop codon at P206X (Figure 2C). The truncated protein contained only the extracellular domain of IL10RA (Figure 3A). The patient’s asymptomatic parents were g.IVS5+2T>C carriers displaying wild-type and altered splicing variants (Figure 2B).
Figure 3. Altered IL-10 Signaling in Patient with IL-10RA Mutation.

(A) Truncated IL10RA protein product found in the patient compared to full-length IL10RA in parents/controls. Extracellular (ECD), intracellular (ICD), signaling peptide (SP), and transmembrane (TM) domains are shown. (B) Western blot analysis of IL-10-induced STAT3 phosphorylation in PBMCs from patient and control. (C) Flow cytometric analysis of peripheral blood FoxP3+ Tregs of patient and mother. Cells depicted are CD4+ in small lymphocyte gate and are plotted CD25 vs. intracellular FoxP3.
IL10RA Splice Mutation Prevents IL-10-Dependent STAT3 Phosphorylation
In order to determine the functional effects of P206X IL10R1, we assessed in vitro STAT3 phosphorylation, a required step in the IL-10 signaling cascade for downstream immunosuppressive effects.19 PBMC were stimulated with IL-10, and STAT3 phosphorylation was measured by Western blot analysis. IL-10-induced STAT3 phosphorylation was defective in the patient. IL-6-induced STAT3 phosphorylation was intact (Figure 3B). Thus, P206X IL-10R1 leads to defective IL-10 downstream signaling.
FoxP3+ Regulatory T cells are Present in the Absence of IL-10 Signaling
Regulatory T cells (Tregs) that express the transcription factor FoxP3 possess suppressive ability, and defects in the FOXP3 gene (in mice and humans) cause systemic autoimmunity.20,21 As some but not all studies have suggested that IL-10 signaling is critical for the stability of FoxP3 expression,3,22 we sought to determine whether FoxP3+ Tregs were present in the absence of IL-10 signaling. FoxP3+ Tregs were identified in the peripheral blood of the affected patient (Figure 3C). This provides evidence in humans that FoxP3+ Tregs can develop in the absence of intact IL-10 signaling.
IL10RA and IL10RB Polymorphisms are Associated with IBD
In order to determine whether IL10RA and IL10RB mutations are associated more generally with IBD and extend beyond the rare patients with infantile-onset IBD that have homozygous, loss-of-function mutations, we performed deep sequencing of coding and flanking intronic regions of IL10RA and IL10RB in a cohort of 188 EO-IBD patients and 188 controls (Supplementary Table 2). The EO-IBD group included 121 children with CD, 2 with IBD-unclassified, and 65 with UC. The ages of children ranged from 6.5 months to 11 years old, with 68 VEO-IBD subjects (including two infants).
In this discovery cohort, 59 SNPs (28 novel) were found in IL10RA and 39 SNPs (15 novel) in IL10RB (Supplemental Table 3). There were 14 (6 novel) non-synonymous SNPs (12 in IL10RA and 2 in IL10RB). Each novel, non-synonymous SNP was identified only in the heterozygous state, and none of the resulting amino acid changes were predicted to be deleterious by SIFT23 or Polyphen.24 Additionally, we found 5 SNPs in IL10RA were associated with EO-UC and 2 SNPs in IL-10RB were associated with EO-CD (Table 1 and Supplemental Table 3). Three IL10RA SNPs (rs10892202, rs4252249, and rs4252270) were protective for EO-UC while the remaining two IL10RA SNPs (rs2228054 and rs2228055) were associated with increased risk for EO-UC and VEO-IBD. In subgroup analysis of these five IL10RA SNPs, four of the SNPs were found to be associated with very early-onset UC (VEO-UC) while there was a positive trend for VEO-UC in rs2228055 (Table 2). Of the IL10RB SNPs that were associated with EO-CD, rs1058867 was associated with increased CD risk and rs8178561 was protective for EO-CD. The minor allelic frequencies of the SNPs described above did not differ between the IBD cohorts from Wisconsin and Toronto (data not shown).
Table 1. IL-10R Polymorphisms Associated with EO-IBD.
Odds ratios using DD vs. Dd/dd model. Discovery Cohort: EO-IBD (n=188), UC (n=65), CD (n=121), and controls (n=188). Validation Cohort: EO-IBD (n=422), UC (n=122), CD (n=300), and controls (n=480). Threshold for significance in Validation cohort was 0.007 to correct for multiple testing.
| Discovery Cohort | Validation Cohort | Combined Cohorts | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| IL10RA SNP | OR | p value | OR | p value | OR | p value | |
| rs10892202 | EO-IBD | 0.60 | 0.067 | 0.88 | 0.405 | 0.79 | 0.057 |
| EO-UC | 0.30 | 0.011 | 0.82 | 0.437 | 0.62 | 0.034 | |
| EO-CD | 0.79 | 0.469 | 0.90 | 0.562 | 0.87 | 0.358 | |
| rs2228054 | EO-IBD | 1.87 | 0.056 | 0.93 | 0.711 | 1.15 | 0.360 |
| EO-UC | 3.06 | 0.003 | 1.25 | 0.452 | 1.71 | 0.019 | |
| EO-CD | 1.24 | 0.691 | 0.83 | 0.432 | 0.92 | 0.686 | |
| rs2228055 | EO-IBD | 1.60 | 0.158 | 1.03 | 0.885 | 1.17 | 0.295 |
| EO-UC | 2.52 | 0.016 | 1.36 | 0.302 | 1.71 | 0.021 | |
| EO-CD | 1.10 | 0.848 | 0.90 | 0.668 | 0.95 | 0.820 | |
| rs4252249 | EO-IBD | 0.68 | 0.143 | 0.86 | 0.360 | 0.82 | 0.104 |
| EO-UC | 0.30 | 0.012 | 0.78 | 0.329 | 0.60 | 0.024 | |
| EO-CD | 0.94 | 0.774 | 0.92 | 0.640 | 0.92 | 0.605 | |
| rs4252270 | EO-IBD | 0.63 | 0.107 | 0.88 | 0.409 | 0.79 | 0.060 |
| EO-UC | 0.30 | 0.012 | 0.77 | 0.306 | 0.59 | 0.021 | |
| EO-CD | 0.84 | 0.665 | 0.91 | 0.592 | 0.89 | 0.438 | |
|
| |||||||
| IL10RB SNP | OR | p value | OR | p value | OR | p value | |
|
| |||||||
| rs1058867 | EO-IBD | 1.78 | 0.011 | 0.91 | 0.492 | 1.18 | 0.109 |
| EO-UC | 1.35 | 0.373 | 0.96 | 0.863 | 1.06 | 0.740 | |
| EO-CD | 2.12 | 0.004 | 0.89 | 0.460 | 1.12 | 0.369 | |
| rs8178561 | EO-IBD | 0.34 | 0.004 | 0.92 | 0.680 | 0.75 | 0.060 |
| EO-UC | 0.85 | 0.834 | 1.07 | 0.814 | 1.00 | 1.0 | |
| EO-CD | 0.10 | <0.001 | 0.90 | 0.646 | 0.64 | 0.030 | |
Table 2. IL-10R Polymorphisms Associated with VEO-IBD.
Odds ratios using DD vs. Dd/dd model. Discovery Cohort: VEO-IBD (n=65), VEO-UC (n=41), VEO-CD (n=24), and controls (n=188). Validation Cohort: VEO-IBD (n=28), VEO-UC (n=18), VEO-CD (n=10), and controls (n=480). Threshold for significance in Validation cohort was 0.007 to correct for multiple testing.
| Discovery Cohort | Validation Cohort | Combined Cohorts | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| IL10RA SNP | OR | p value | OR | p value | OR | p value | |
| rs10892202 | VEO-IBD | 0.41 | 0.044 | 1.17 | 0.701 | 0.60 | 0.086 |
| VEO-UC | 0.28 | 0.030 | 0.71 | 0.585 | 0.40 | 0.032 | |
| VEO-CD | 0.62 | 0.230 | 2.35 | 0.179 | 0.98 | 0.951 | |
| rs2228054 | VEO-IBD | 2.69 | 0.016 | 2.58 | 0.024 | 2.49 | 3×10−3 |
| VEO-UC | 3.29 | 0.013 | 3.86 | 0.005 | 3.08 | 2×10−4 | |
| VEO-CD | 1.85 | 0.140 | 1.93 | 0.404 | 1.66 | 0.273 | |
| rs2228055 | VEO-IBD | 2.38 | 0.032 | 2.80 | 0.014 | 2.56 | 2×10−3 |
| VEO-UC | 2.58 | 0.058 | 4.21 | 0.003 | 2.93 | 6×10−4 | |
| VEO-CD | 2.08 | 0.057 | 2.10 | 0.345 | 2.01 | 0.106 | |
| rs4252249 | VEO-IBD | 0.41 | 0.045 | 1.18 | 0.701 | 0.61 | 0.882 |
| VEO-UC | 0.28 | 0.046 | 0.71 | 0.585 | 0.40 | 0.033 | |
| VEO-CD | 0.63 | 0.240 | 2.35 | 0.179 | 0.98 | 0.958 | |
| rs4252270 | VEO-IBD | 0.41 | 0.045 | 1.16 | 0.722 | 0.60 | 0.081 |
| VEO-UC | 0.28 | 0.046 | 0.70 | 0.572 | 0.40 | 0.031 | |
| VEO-CD | 0.62 | 0.240 | 2.32 | 0.186 | 0.97 | 0.938 | |
|
| |||||||
| IL10RB SNP | OR | p value | OR | p value | OR | p value | |
|
| |||||||
| rs1058867 | VEO-IBD | 1.55 | 0.186 | 0.94 | 0.868 | 1.16 | 0.515 |
| VEO-UC | 1.28 | 0.596 | 1.13 | 0.814 | 1.10 | 0.748 | |
| VEO-CD | 2.11 | 0.058 | 0.56 | 0.364 | 1.26 | 0.519 | |
| rs8178561 | VEO-IBD | 0.70 | 0.528 | 1.23 | 0.687 | 0.92 | 0.804 |
| VEO-UC | 1.04 | 1.000 | 1.89 | 0.267 | 1.32 | 0.447 | |
| VEO-CD | 0.23 | 0.212 | 0.74 | 0.260 | 0.37 | 0.160 | |
Linkage analysis of these SNPs showed that rs2228054 and rs2228055 were in LD (p<0.0001) (Haplotype Block 1 AG) as were rs10892202, rs4252249, and rs4252270 (Haplotype Block 2 CAT) (p<0.0001) (LD plot in Supplementary Figure 1). Haplotype Block 1 AG was associated with VEO-IBD (OR 2.35, p=0.020), UC (OR 2.41, p=0.016) and VEO-UC (OR 2.48, p=0.025) but not for EO-IBD or EO-CD (Supplementary Table 4). Haplotype Block 2 had a protective effect for VEO-IBD (OR 0.40, p=0.025), UC (OR 0.29, p=0.007) and VEO-UC (OR 0.28, p=0.025), EO-IBD (OR 0.57, p=0.030) but had no significant effect in EO-CD.
Validation Study Confirms IL10RA SNP Associations with VEO-UC
We determined the frequency of the SNPs (found in the discovery cohort) in an independent cohort of 422 children with EO-IBD and 480 healthy controls (validation cohort) by Taqman genotyping. This EO-IBD cohort was comprised of 122 children with UC and 300 children with CD (including 18 VEO-UC and 10 VEO-CD). χ2 analysis of discovery and validation controls showed that they were similar with respect to the studied SNPs (data not shown).
Although the association with the 7 IL10R SNPs and EO-IBD did not replicate in the validation cohort, combined analysis of the discovery and showed nominal association with all the IL10RA SNPs (Table 1) and EO-UC. Furthermore, the association with VEO-UC with both rs2228054 and rs2228055 was replicated in the validation VEO-UC cohort, (rs2228054 OR 3.86 (95% CI 1.39-10.71, p=0.005, adjusted p=0.044) and rs2228055 OR 4.21 (95% CI 1.51-11.69, p=0.003, adjusted p=0.024) (Table 2)) and remained significant in a combined analysis of the discovery and validation cohorts (OR 3.08, combined p=2×10−4; and OR 2.93, p=6×10−4 , respectively).
Linkage analysis of Haplotype Block 1 AG and Haplotype Block 2 CAT showed that these SNPs were also in LD in the validation cohort (p<0.0001). The Haplotype Block 1 AG was also associated with VEO-IBD (OR 2.43, p=0.023), and VEO-UC (OR 3.40, p=0.006) but not for overall IBD or EO-UC (Supplementary Table 4). Haplotype Block 2 CAT was not associated with VEO-IBD (p=0.89) or VEO-UC (p=0.51).
Discussion
Homozygous, loss-of-function mutations in IL10RA and IL10RB have been reported to cause severe, infantile-onset IBD.6,7 In this report, we identify a novel splice site mutation in IL-10RA resulting in defective IL-10 signaling. In addition to the colitis, severe perianal disease, and recurrent infections that have been previously described in patients with IL-10R mutations, this patient also suffered from severe arthritis.
The contribution of IL10R variants to IBD susceptibility more generally in children was heretofore unclear. Our study is the first to specifically examine by deep sequencing the role of IL-10R SNPs in EO-IBD and VEO-IBD. We have identified two IL10RA SNPs associated with VEO-UC and these results have been validated in an independent replication cohort with OR of 2.9 and 3.1. These two SNPs, rs2228054 and rs2228055, were frequently found in the heterozygous state among IBD patients and inherited as a haplotype. The conferred risk may be due to one or both SNPs. Alternatively, the increased risk may reside in a regulatory region (e.g., promoter) in LD with these SNPs. We propose that this risk haplotype exerts a mild phenotype in the general population resulting in disease only in the presence of another genetic variant(s) or environmental trigger. In contrast to the complete loss-of-function IL-10R mutations (described here and previously6,7) which present with symptoms more typical of Crohn’s disease, these SNPs are associated with UC, which is consistent with the association of SNPs in IL-10 signaling genes and UC.10,12 Although this study points to an important role of the IL-10 pathway in VEO-UC, due to the limited number of VEO-IBD patients, this association with VEO-UC will require further functional investigations and replication.
Although GWAS of adult and pediatric IBD have identified novel pathways important in the pathogenesis of IBD, the SNPs detected by GWAS only account for ~25% of the assumed genetic heritability.11 The risk for IBD associated with SNPs in IL10, TYK2, and STAT3 loci demonstrate the importance of IL-10 in IBD pathogenesis.9,12 However, GWAS have not identified an association between SNPs in the IL10RA or IL10RB loci and IBD risk in the pediatric population.13,25 Similarly, the recently published meta-analysis of GWAS of UC patients failed to show a statistically significant increase in risk for rs2228054.26 This is most likely due to the unique subset of patients examined in our study, and highlights the importance of candidate gene studies examining rare presentations of IBD. GWAS evaluating exclusively-pediatric IBD focus primarily on EO-IBD patients which has been shown to be phenotypically more similar to adult-onset IBD than VEO-IBD.14 Therefore, the contribution of IL-10R SNPs to the susceptibility of IBD in very young children has been underestimated in recent adult and late-onset pediatric GWAS as VEO-IBD represents a very unique population that is poorly represented even in GWAS focusing on pediatric IBD.13
Although defective IL-10 signaling causes a strong inflammatory phenotype in the GI tracts of both humans and mice, it is still unclear which is the most critical cell types(s) that require IL-10 signaling. While Murai et al suggested that an IL-10-dependent signal was required for the maintenance of FoxP3+ Tregs,22 Chaudhry et al demonstrated in mice with a Treg lineage-specific deletion of IL-10R that IL-10-dependent signals were required for the function but not the maintenance of Tregs.3 Peripheral blood FoxP3+ Tregs were present in the patient studied in this report with defective IL-10 signaling demonstrating that this signal is not required for the development of FoxP3+ Tregs.
In summary, we have described a novel splice site mutation in IL10RA that broadens the existing phenotype of IL-10R deficiency to include severe arthritis. The discovery of this patient in a cohort of VEO-IBD patients reinforces that IL-10R deficiency should be considered in all infantile-onset IBD cases, especially those with severe perianal disease and now arthritis, and that this diagnosis should prompt consideration of stem cell transplantation. Additionally, the association of SNPs in IL10RA with VEO-UC implicates a role for IL-10R in the pathogenesis of IBD in a broader population of patients than recently appreciated. Such information may permit stratification of patients for clinical trials that target the IL-10 pathway.
Supplementary Material
Supplementary Figure 1. Linkage Disequilibrium (LD) Plot for IL10RA Polymorphisms in Discovery Cohort. Haplotype Block 1 AG (rs2228054, rs2228055) with D’ value=0.976 and Haplotype Block 2 CAT (rs4252249, rs10892202, rs4252270) with D’ values >0.999 for rs4252249 with rs10892202 and rs4252270 and D’ value=0.984 for rs10892202 with rs4252270). Red signifies r2>0.5, blue signifies r2<0.5.
Acknowledgements
We would like to thank BP and her family and all patients and families who participated. Special thanks to Karoline Fielder, Kathleen Kelleher and Karen Frost for collection of patient samples and to Joanne Stempak for sample handling. Thanks to Dr. Ken Croitoru and members of Dr. Snapper’s laboratory for critical reading of this manuscript. We would like to thank the Massachusetts General Hospital Clinical Research Program for assistance with biostatistical calculations. CJM is supported by an award from the Crohn’s and Colitis Foundation of America (CCFA). AMM is supported by a transition award from the Crohn’s and Colitis Foundation of Canada (CCFC)/ Canadian Association of Gastroenterology (CAG)/ Canadian Institute for Health Research (CIHR), a Canadian Child Health Clinician Scientist Program (Strategic Training Initiatives in Health Research Program – CIHR) award and an Early Researcher Award from the Ontario Ministry of Research and Innovation and a CDHNF/NASPGHAN George Ferry Young Investigator Development Award. MSS is supported in part by the Gale and Graham Wright Research Chair in Digestive Diseases. Partial funding to recruit subjects and collect samples was provided by MSS through NIDDK grant DK. This work was funded in part by funding to Dr. Snapper through the Wolpow Chair in IBD Research and Treatment as well as through funding made possible from the Harvard Digestive Disease Center P30 DK034854.
Abbreviations
- CD
Crohn’s disease
- IBD
inflammatory bowel disease
- IL-10
Interleukin-10
- IL10RA
IL-10 Receptor A
- IL10RB
IL-10 Receptor B
- PBMC
peripheral blood mononuclear cells
- PBS
phosphate-buffered saline
- PMN
polymorphonuclear neutrophils
- SNP
single nucleotide polymorphism
- STAT3
signal transducer and activator of transcription 3
- p-STAT3
STAT3 phosphorylated at tyrosine 705
- UC
ulcerative colitis
Footnotes
Competing Interests: CJM has nothing to disclose.
CHG has nothing to disclose.
SK has nothing to disclose.
CK has nothing to disclose.
VMW has nothing to disclose.
TWD has nothing to disclose.
RHB has nothing to disclose.
MM has nothing to disclose.
JCL has nothing to disclose.
EC has nothing to disclose.
SMB has nothing to disclose.
CMR has nothing to disclose.
MSS has nothing to disclose.
AMG has nothing to disclose.
SBS has nothing to disclose.
AMM has nothing to disclose.
Author Contribution: AMM, and SBS conceived and designed all experiments. MSS, AMM, AMG, TDW, SK provided study samples. CJM and TDW analyzed the data. CJM, VMW, CHG performed functional analysis under supervision of AMM, and SBS. CJM, SBS and AMM wrote the manuscript with contributions from all authors.
References
- 1.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 2.Huber S, Gagliani N, Esplugues E, et al. Th17 Cells Express Interleukin-10 Receptor and Are Controlled by Foxp3(-) and Foxp3(+) Regulatory CD4(+) T Cells in an Interleukin-10-Dependent Manner. Immunity. 2011;34(4):554–65. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaudhry A, Samstein RM, Treuting P, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of th17 cell-mediated inflammation. Immunity. 2011;34(4):566–78. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 5.Spencer SD, Di Marco F, Hooley J, et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187(4):571–8. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361(21):2033–45. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Begue B, Verdier J, Rieux-Laucat F, et al. Defective IL10 Signaling Defining a Subgroup of Patients With Inflammatory Bowel Disease. Am J Gastroenterol. 2011 doi: 10.1038/ajg.2011.112. [DOI] [PubMed] [Google Scholar]
- 8.Glocker EO, Frede N, Perro M, et al. Infant colitis--it’s in the genes. Lancet. 2010;376(9748):1272. doi: 10.1016/S0140-6736(10)61008-2. [DOI] [PubMed] [Google Scholar]
- 9.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke A, Balschun T, Karlsen TH, et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40(6):713–5. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 11.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40(8):955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40(11):1319–23. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 13.Imielinski M, Baldassano RN, Griffiths A, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41(12):1335–40. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine A, Griffiths A, Markowitz J, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis. 2011;17(6):1314–21. doi: 10.1002/ibd.21493. [DOI] [PubMed] [Google Scholar]
- 15.Heyman MB, Kirschner BS, Gold BD, et al. Children with early-onset inflammatory bowel disease (IBD): analysis of a pediatric IBD consortium registry. J Pediatr. 2005;146(1):35–40. doi: 10.1016/j.jpeds.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 16.Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19(Suppl A):5–36. doi: 10.1155/2005/269076. [DOI] [PubMed] [Google Scholar]
- 17.Roca X, Sachidanandam R, Krainer AR. Intrinsic differences between authentic and cryptic 5′ splice sites. Nucleic Acids Res. 2003;31(21):6321–33. doi: 10.1093/nar/gkg830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172(1):567–76. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- 20.Godfrey VL, Wilkinson JE, Russell LB. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am J Pathol. 1991;138(6):1379–87. [PMC free article] [PubMed] [Google Scholar]
- 21.Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet. 2002;39(8):537–45. doi: 10.1136/jmg.39.8.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murai M, Turovskaya O, Kim G, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10(11):1178–84. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kugathasan S, Baldassano RN, Bradfield JP, et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat Genet. 2008;40(10):1211–5. doi: 10.1038/ng.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43(3):246–52. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Linkage Disequilibrium (LD) Plot for IL10RA Polymorphisms in Discovery Cohort. Haplotype Block 1 AG (rs2228054, rs2228055) with D’ value=0.976 and Haplotype Block 2 CAT (rs4252249, rs10892202, rs4252270) with D’ values >0.999 for rs4252249 with rs10892202 and rs4252270 and D’ value=0.984 for rs10892202 with rs4252270). Red signifies r2>0.5, blue signifies r2<0.5.