

Abstract
Cells of the Escherichia coli dnaE(Ts) dnaE74 and dnaE486 mutants die after 4 h of incubation at 40°C in Luria-Bertani medium. Cell death is preceded by elongation, is inhibited by chloramphenicol, tetracycline, or rifampin, and is dependent on cell density. Cells survive at 40°C when they are incubated at a high population density or at a low density in conditioned medium, but they die when the medium is supplemented with glucose and amino acids. Deletion of recA or sulA has no effect. We isolated suppressors which survived for long periods at 40°C but did not form colonies. The suppressors protected against hydroxyurea-induced killing. Sequence and complementation analysis indicated that suppression was due to mutation in the cydA gene. The DNA content of dnaE mutants increased about eightfold in 4 h at 40°C, as did the DNA content of the suppressed strains. The amount of plasmid pBR322 in a dnaE74 strain increased about fourfold, as measured on gels, and the electrophoretic pattern appeared to be normal even though the viability of the parent cells decreased 2 logs. Transformation activity also increased. 4′,6′-Diamidino-2-phenylindole staining demonstrated that there were nucleoids distributed throughout the dnaE filaments formed at 40°C, indicating that there was segregation of the newly formed DNA. We concluded that the DNA synthesized was physiologically competent, particularly since the number of viable cells of the suppressed strain increased during the first few hours of incubation. These observations support the view that E. coli senses the rate of DNA synthesis and inhibits septation when the rate of DNA synthesis falls below a critical level relative to the level of RNA and protein synthesis.
Cell division and DNA synthesis are coupled in all organisms, but the mechanism of the coupling is not clear, notwithstanding the sophisticated description of an operon with multiple promoters determining the production of the FtsZ and other proteins involved in cell division (12, 15). In Escherichia coli, the completion of a round of DNA synthesis is usually followed by activation of the cell septation machinery, but synthesis does not trigger septation (4). DNA damage or other blocks to DNA synthesis often lead to inhibition of cell division. For example, induction of the SOS repair pathway activates the sulA gene product, inhibiting cell division (5). Some signal recognizes that the DNA has been damaged (or that synthesis has been inhibited), and as a result division is suspended. Deprivation of thymine necessarily inhibits DNA synthesis and also leads to filamentation (1). Filamentation often accompanies cell death, and there have been numerous studies attempting to elucidate the mechanism of death both as a result of thymine starvation and as a result of other pathways to filamentation.
As part of an investigation of the roles of the different DNA polymerases in mutation (35), we studied the behavior of strains carrying a temperature-sensitive (Ts) mutation in the alpha subunit of the replicative DNA polymerase III. We found that when these strains were incubated at nonpermissive temperatures, they exhibited many of the phenomena previously described as unbalanced growth in connection with thymineless death (10). However, in contrast to the findings with strains undergoing thymineless death, we observed no damage to the DNA. In this paper, we argue that decreasing the rate of DNA synthesis is sufficient to inhibit cell division without the intervention of damaged DNA. We isolated a series of suppressor-modifier mutations which prevented cell death and filamentation. These mutations were predominantly located in the cydA gene controlling the high-efficiency cytochrome bd electron transport protein (3). Loss of this cytochrome system probably reduces the energy supply available for RNA and protein synthesis, thereby restoring the balance in the synthesis of different cellular components. Alternatively, the isolation of numerous independent suppressors in the same gene suggests that there is an as-yet-unknown role for cydA.
MATERIALS AND METHODS
Strains.
The bacterial strains used in this study are shown in Table 1. Plasmid pBR322 was purchased from Fermentas. Plasmid pTK-1, including the cydA gene (22), was kindly provided by R. Gennis. P1 transduction was carried out as described by Miller (26).
TABLE 1.
Strains used
| Identifying genotype of strain | Characteristics | Reference(s) and/or source |
|---|---|---|
| BS40 (dnaE+) | metB metD Δ(proAB lac) rpsL (Smr) | 36 |
| dnaE74 (BS40dnaE74) | metB metD Δ(proAB lac) dnaE74 rpsL (Smr) | 35 |
| dnaE486 Tn10 | dnaE486 zae502::Tn10 | Roger Woodgate |
| dnaE486 | thr-1 leuB6(Am) fhuA21 dnaE486(Ts) lacY1 glnV44 λ−rfbD1 thyA6 rpsL67 thi-1 deoC1 met-89 | E. coli Genetic Stock Center (39) |
| BS40 dnaE486 Tetr | metB metD Δ(proAB lac) rpsL (Smr) dnaE486::Tn10 | From dnaE486 Tn10 strain by transduction |
| BS40 dnaE486 | metB Δ(proAB lac) rpsL (Smr) dnaE486 | From dnaE486 strain by transduction |
| CGSC 7027 ΔrecA (Camr) | λ−recA938::Tn9-200 recD1014 (Nuc−) hsdR2 mdoB202::Tn10 | E. coli Genetic Stock Center (40) |
| BS40 dnaE74 ΔrecA | metB Δ(proAB lac) dnaE74(Ts) rpsL (Smr) ΔrecA (Camr) | By transduction from ΔrecA strain |
| RW82 Δumu | uvrA6 thi-1 thr-1 araD-14 leuB6 lacY1 ilv323(Ts) Δ(gpt-proA)62 mtl-1 xyl-5 rpsL31 tsx-33 supE44 galK2(oc) hisG4(oc) rfbD1 kdgK51 sulA211 Δ(umuDC)595::cat | Roger Woodgate (41) |
| CGSC 7699 ΔsulA | Δ(gpt-lac)5 tsx-35 λ−ompF626 zcb-222::Tn10 pyrD34 or pyrD36 sulA3 e14-rac-0 rfbD1 mg1-51 recA441(Ts) relA1 rpsL31 (Strr) kdgK51 xylA5 mtl-1 spoT1 thy-1 creC510 | E. coli Genetic Stock Center (27) |
| dnaE74 ΔsulA | metB (ΔproAB lac) rpsL (Smr) dnaE74 ΔsulA Tn10 | |
| CGSC7703 ΔrecQ | λ− IN(rrnD-rrnE)1 rph-1 recQ1803::Tn3 | E. coli Genetic Stock Center (28) |
| dnaE74 ΔrecQ | metB Δ(proAB lac) rpsL (Smr) dnaE486 recQ1803::Tn10 | From CGSC 7703 by transduction |
| CGSC 7351 | nadA57::Tn10 λ−rph-1 | E. coli Genetic Stock Center (34) |
| dnaE74 cydA | metB Δ(proAB lac) rpsL (Smr) dnaE74 cydA | This study |
| dnaE74 cydA Tetr | metB Δ(proAB lac) rpsL (Smr) dnaE74 cydA nadA57::Tn10 | By transduction with P1 phage grown on CGSC 7351 |
| dnaE+cydA Tetr | metB metD Δ(proAB lac) rpsL (Smr) cydA nadA57::Tn10 | From BS40 by transduction with P1 phage grown on dnaE74 cydA Tetr cells |
| dnaE74 cydA Tetr (stock) | metB Δ(proAB lac) rpsL (Smr) dnaE74 cydA nadA57::Tn10 | By transduction with P1 phage grown on dnaE+cydA Tetr cells |
| dnaE486 cydA Tetr | metB Δ(proAB lac) rpsL (Smr) dna486 cydA nadA57::Tn10 | From BS40 dnaE486 strain by transduction with P1 grown on dnaE74 cydA Tetr cells |
| dnaE74/pBR322 | metB Δ(proAB lac) dnaE74(Ts) rpsL/pBR322 | By CaCl2 transformation |
| dnaE74/pTK1 | metB Δ(proAB lac) dnaE74(Ts) rpsL/pTK-1 | By CaCl2 transformation with plasmid from R. Gennis (16, 22) |
| BS40/pTK1 | metB Δ(proAB lac) rpsL (Smr)/pTK-1 | By CaCl2 transformation with plasmid from R. Gennis (16, 22) |
Media and general methods.
The media and general microbiological techniques used were described previously by Miller (26). All hydroxyurea solutions were prepared from solid material immediately before use. Quantitative DNA analysis was performed by the diphenylamine method of Burton (9). Gel electrophoresis was carried out in 0.8% agarose gels at 90 V in a Tris-borate buffer. Quantitative analysis of gels was performed with a Macintosh computer by using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/).
Sequencing.
DNA sequencing was done by the University of Chicago Cancer Research Center Sequencing Facility by using Applied Biosystems capillary electrophoresis. PCR products were purified with a Qiagen Qiaquick Spin purification kit. Primers used for identification of the cydA mutations and for verification of the dnaE74 and dnaE486 strains are shown in Table 2. Oligonucleotides were obtained from Integrated DNA Technologies.
TABLE 2.
Primers used
| Gene or region | PCR primer | Sequencing primer | Sequence |
|---|---|---|---|
| dnaE486 | 486F | PCR primer | 5′CGAAATCGTGTATGGTATTGGC |
| 486R | PCR primer | 5′ATACTGGTTGATAGGGTGTCC | |
| dnaE74 | Forward 486(37) | PCR primer | 5′ATGGCGGCGCTGGATATTGTTTTC |
| Reverse 486 | PCR primer | 5′CACCGCTTTCGCCGCCATTGTA | |
| dnaE74 seq | 5′TTGCTGCGGCGAATAGTT | ||
| cydA | 10739F | PCR primer | 5′TTCCTTTTTGTGCCACTGACG |
| 11819R | PCR primer | 5′TGGAGTCTTTGGTTGCCTGTTG | |
| 11104F | 5′ACTGTGGATTCTGGTTGCG |
PCR.
Thermocycling was carried out by using a Eppendorf Master Cycler with a heated lid. Each mixture contained 1 U of high-fidelity Platinum Taq DNA polymerase (Invitrogen), 2.5 μl of 10× buffer, 0.5 μl of a mixture containing each deoxynucleoside triphosphate at a concentration of 10 mM (Invitrogen), 1 μl of 50 mM MgSO4, 5 ng of each primer (Table 2), 25 ng of genomic DNA template, and enough double-distilled H2O to bring the final volume to 25 μl. The thermal cycling program included an initial denaturation step consisting of 95°C for 45 s, followed by 35 cycles of 45 s at 95°C, 40 s at 52°C, and 150 s at 68°C. Following thermal cycling the samples were kept at 4°C. PCR primers were used for sequencing of the cydA product along with an internal primer, as shown in Table 2.
Mutagenesis and mapping.
N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG)-induced mutagenesis was done as described by Miller (26). A freshly prepared MNNG stock solution (50 μl of a 1-mg/ml solution) was added to 1 ml of a washed overnight culture of dnaE74 cells in citrate buffer. After approximately 30 min the cells were harvested, resuspended in Luria-Bertani (LB) medium, and incubated for 2 h at 27°C. They were then diluted 1:20 in LB medium and incubated overnight at 27°C. Cells were plated and incubated at 40°C overnight and then switched to 27°C. Colonies which appeared were then tested for growth at 40 and 27°C. We selected strains which could not grow at 40°C but formed colonies at 27°C. The suppressor in strain PS3 was mapped by using the Gross collection of mapping strains (34) obtained from the E. coli Genetic Stock Center. Screening of suppressed colonies was made more difficult by the population effect described below (see Results). Since high population densities of the dnaE74 strain were not killed by incubation at 40°C, we used the following procedure for tests of segregants from conjugation and transduction experiments. Exconjugants were selected on LB medium containing tetracycline. The strains were then transferred with toothpicks onto plates containing LB medium supplemented with tetracycline, and after at least 24 h of incubation at 27°C, each of the colonies was swiped with a toothpick and the toothpick was used to seed 1 ml of LB medium. A 5-μl drop of the medium was then placed on two LB medium plates. One plate was incubated at 27°C as a control. The second plate was incubated at 40°C for 24 h and then at 27°C for a second 24 h. Drops which did not show growth after 24 h at 40°C but did show confluent or almost confluent growth after the switch to 27°C were classified as carrying the suppressor. The original dnaE74 strain gave only a few, individually distinguishable colonies. We confirmed the classification by quantitative tests of survival in critical cases and found that the results agreed with the diagnosis obtained by screening. We found that it was necessary to use either LB medium plates or LB medium plates containing streptomycin for the screening analysis since growth, even growth of the tetracycline-resistant strains, appeared to be slowed on LB medium plates containing tetracycline.
Nucleoid staining.
The method of Jaffe et al. (20) and Donachie and Begg (14) was used to develop a protocol for nucleoid staining. Following 4 h of incubation at 40°C in LB broth at a 1:1,000 dilution, cells were treated with chloramphenicol (20 μg/ml for 10 min), harvested, and washed in phosphate-buffered saline (pH 7.4). Resuspended cultures (10 μl) were spread and dried on the surfaces of glass slides and fixed with ice-cold 100% methanol. The slides were then immersed into tap water 10 times, dried, and treated with poly-l-lysine (10 μl of a 5-μg/ml solution). Once the slides were dry, 10 μl of 4′,6′-diamidino-2-phenylindole (DAPI) (10 μg/ml in saline) was added, followed by 1 drop of Vectashield mounting medium (Vector Laboratories, Inc.), and then a coverslip was added immediately. The preparations were examined and photographed by using fluorescence with a Zeiss Axioskop 0100W/2 with a Photometrix camera attachment. Photographs were captured by using the IPLAB Spectrum software and were edited by using Adobe Photoshop 7.
RESULTS
The dnaE(Ts) dnaE74 and dnaE486 mutants are the result of single nucleotide substitutions in the alpha subunit of the E. coli replicative polymerase (37). It was reported some years ago that when the dnaE486 strain was incubated at 42°C, it lost the ability to produce colonies after a lag period of 1 h and that death occurred without a lag when temperature stress was combined with thymine starvation (7). Death occurs at 40°C for the dnaE486 and dnaE74 strains, both of which are single-nucleotide-substitution mutants (37), after 2 h or more. Growth at 40°C, as measured by turbidity, was practically identical for both the dnaE74 mutant and the isogenic wild-type parent (Fig. 1, top panel). The number of cells began to increase after about 90 min for the wild type but not for the mutant. When the increase in mass (turbidity) without an increase in cell number was plotted as turbidity divided by viable cell number as a function of time (Fig. 1, bottom panel), the data illustrated that there was activation of some signal in the wild type which was missing in the dnaE(Ts) mutant incubated at a nonpermissive temperature.
FIG. 1.

(Top panel) Growth of wild-type strain BS40 and the dnaE74(Ts) mutant at 40°C. Cultures were incubated overnight at 27°C in LB medium with streptomycin to obtain a density of ∼5 × 109 cells/ml (BS40) or 6.4 × 109 cells/ml (dnaE74 mutant) and then diluted 1:100 in 200 ml of prewarmed LB medium with streptomycin. Turbidity and viable cell counts were determined at intervals, as shown. (Bottom panel) Turbidity divided by viable cell count as a function of time. Squares, BS40; triangles, the dnaE74 mutant; open symbols, turbidity; solid symbols, viable cell count. OD@600nm, optical density at 600 nm.
Cell death requires growth.
The death of the dnaE(Ts) mutants incubated at 40°C required growth. We found that the antibiotics tetracycline and rifampin were protective at 40°C, whereas hydroxyurea, an inhibitor of DNA synthesis, potentiated cell death (Table 3). Measurement of cell death was complicated by an effect of cell density. Overnight cultures that were harvested, washed, and resuspended in LB medium at a 1:10 dilution did not die, and the cell number may actually have increased during incubation at 40°C. Death was limited at a dilution of 1:100 and was extensive only at a higher dilution (Table 4). We hypothesized that this population effect was due to the limited amount of growth possible at the 1:10 dilution rather than to some more complex quorum-sensing mechanism (13).
TABLE 3.
Effects of antibiotics on survival
| Addition (concn) |
N/N0 fora:
|
|
|---|---|---|
| dnaE+ strain | dnaE74 strain | |
| None | 256b | 0.0069b |
| Chloramphenicol (20 μg/ml) | 0.85 | 1.06 |
| Rifampin (50 μg/ml) | 0.49 | 0.92 |
| Tetracycline (15 μg/ml) | 1.13 | 0.89 |
| Hydroxyurea (40 mM)c | 1.6 × 10−4 | 9.1 × 10−6 |
N, viable count at 5 h; N0, viable count at zero time. Overnight cultures in LB medium (dnaE+ strain, ∼6 × 109 cells/ml; dnaE strain, ∼7 × 109 cells/ml) were diluted 1:1,000 in 10 ml of LB medium, and the cultures were supplemented with antibiotics and incubated with shaking for 5 h at 40°C. At the end of the experiment the cultures were diluted and plated on LB medium plates.
Average of three determinations.
Data also shown in Table 9.
TABLE 4.
Effect of cell density on viability of dnaE(Ts) strains incubated at 40°C
| dnaE allele | Length of incubation at 40°C (h) |
N/N0 at a culture dilution ofa:
|
|||
|---|---|---|---|---|---|
| 10−1 | 10−2 | 10−3 | 10−4 | ||
| dnaE+ | 5 | 4.9 | 22.7 | 96.6 | 386 |
| dnaE74 | 5 | 5.2 | 0.048 | 0.00078 | 0.00061 |
| dnaE486 | 4 | 1.08 | 0.0058 | 0.001 | |
N, viable count after incubation at 40°C; N0, viable count for nonincubated culture. Cultures were grown overnight at 27°C in LB medium. The cultures were then harvested, washed, resuspended in fresh LB medium, and diluted. Cultures were incubated with shaking at 40°C. The viable counts for nonincubated cultures were 8.8 × 109 cells/ml for the dnaE+ strain, 6.9 × 109 cells/ml for the dnaE74 strain, and 4.0 × 109 cells/ml for the dnaE486 strain.
In order to test this hypothesis, an overnight culture of the dnaE74 strain in LB medium was harvested, washed, diluted 1:10 in LB medium, and incubated for 4 h at 40°C. The cells were collected by centrifugation, and the supernatant was filtered. An overnight culture of either the dnaE74 or dnaE486 strain was diluted 1:1,000 in this conditioned medium and incubated 4 h at 40°C. The cells survived this treatment when they were compared to controls diluted in fresh LB medium. However, supplementation of the conditioned medium with glucose and Casamino Acids was sufficient to convert the medium into a medium lethal for cells diluted 1:1,000 (Table 5). Since killing occurred in conditioned medium supplemented with glucose and Casamino Acids, the results of this experiment support the view that the population effect was due to the limited amount of growth possible at a 1:10 dilution.
TABLE 5.
Effect of conditioned medium
| Medium |
N/N0 fora:
|
||
|---|---|---|---|
| dnaE+ strain | dnaE74 strain | dnaE486 strain | |
| Filtered conditioned medium | 3.3 | 1.42 | 1.61 |
| Filtered conditioned medium plus glucose (0.2%) and Casamino Acids (10 μg/ml) | 15.2 | 0.044 | 0.063 |
| Fresh LB medium | 94 | 0.0026 | 0.0021 |
N, viable count at 4 h; N0 viable count at the start of incubation. Conditioned medium was prepared by diluting an overnight culture of the dnaE74 strain 1:10 in 100 ml of LB medium and incubating the preparation for 4 h at 40°C. The bacteria were removed by centrifugation, and the conditioned medium was filter sterilized. Overnight cultures (dnaE74 strain, 6.9 × 109 cells/ml; dnaE486 strain, 3.8 × 109 cells/ml; dnaE+ strain, 6.6 × 109 cells/ml) were diluted 1:1,000 in the test media and incubated for 4 h at 40°C.
Several gene products have been implicated in the programmed death of E. coli after damage to the DNA (24). These products include the product of recA involved in induction of the SOS repair system and the product of sulA involved in the inhibition of FtsZ, the division ring protein. We tested the effects of deletion of recA, sulA, and recQ (involved in thymineless death [29]) when they were combined singly with the dnaE74 mutation and found that there was no measurable effect on the survival time at 40°C (data not shown).
Isolation and analysis of suppressor mutations.
In order to analyze the cell death phenomenon, we decided to look for genetic suppressors. We adapted a two-pronged selection process. First, we looked for strains which survived for long periods at 40°C. However, in order to avoid isolating revertants of the dnaE mutation, we also required that strains not form colonies at 40°C. Cultures were treated with MNNG as described by Miller (26) and were incubated overnight in LB medium, and then dilutions were plated and incubated at 40°C for 24 h before they were switched to incubation at 27°C. We repeated this experiment several times to minimize selection of sisters. We isolated 51 strains, some of which had clearly different phenotypes (Fig. 2) but all of which survived for long periods at 40°C relative to the survival time of the dnaE74 strain.
FIG. 2.

Survival of the dnaE74(Ts) strain and its suppressor strains at 40°C. Strains were incubated overnight in LB medium containing streptomycin at 40°C. The cultures were then washed, resuspended in LB medium, plated on prewarmed LB medium plates, and incubated at 40°C for different times. The plates were then incubated for 24 h at 27°C, and the organisms were counted. No colonies were visible at the time of the switch from 40 to 27°C. N, viable count after incubation; No, viable count at zero time.
We mapped one of these strains (PS3) by standard genetic techniques, first using the Gross collection of Hfr strains carrying a Tn10 insertion and then using a collection of Tn10 insertions at particular positions (30, 34). Our screening methodology is described in Materials and Methods. Successful screening required attention to the population effect (Table 4) and precluded direct replica plating. We found a cotransduction frequency of about 90% with Tn10 inserted into the nadA gene (30). Utilizing this high cotransduction frequency, we were able to transfer the suppressor gene to a wild-type (BS40) background to create a strain carrying the suppressor in a dnaE+ background. We then transferred the suppressor back to the dnaE74 strain to create a suppressed strain properly designated the dnaE cydA strain. The suppressor was not allele specific since transfer to the dnaE486 strain resulted in strains with properties like those of the suppressed dnaE74 strain (data not shown). Suppressed and nonsuppressed strains had similar growth rates at 27°C, but in contrast to the growth rate of the suppressed strain, the growth rate of the nonsuppressed strain increased markedly at 40°C (data not shown).
Sequence analysis of the suppressed strain revealed a G→A mutation at position 257 in the cydA gene (Table 6). In contrast to some other mutations observed in our original suppressor isolate, this mutation cotransduced with the biological suppressor effect. We screened our 51 suppressor isolates for linkage to the nadA gene. Forty-nine of the 51 isolates showed over 60% cotransduction. Twenty-seven isolates with high cotransduction frequencies were sequenced in both directions through much of the cydA gene (Table 6). We observed 13 different changes in the gene, including three repeats. The different mutations observed indicate that the different isolates were, for the most part, the result of independent events. All but two of the observed mutations in the cydA product appeared to be located in the portion of the protein found in the periplasmic space (16, 17, 22), and considering the stop codons observed, it is likely that the mutations inactivated the enzyme. The wild-type cydA+ gene was able to complement the suppressed strain to restore filamentous killing. Plasmid pTK-1 included the functional cydA+ gene (22) and was derived from pBR322 (16). Transformation of the dnaE74 cydA strain with pTK-1, but not transformation with pBR322, resulted in a strain which was killed by incubation at 40°C with the production of filaments (Fig. 3 and Table 7). The pTK-1 plasmid introduced into a dnaE+ strain had no effect on septation during incubation at 40°C. We concluded that most of the suppressors which we isolated were due to inactivation of the cydA gene. We are continuing our attempts to identify the remaining isolates.
TABLE 6.
cydA sequence changes observed in suppressor isolatesa
| Suppressor isolate | Cotransduction frequency | Base change | Base position | Amino acid position | Amino acid change |
|---|---|---|---|---|---|
| PS1 | 0 | ||||
| PS3 | 0.89 | C→T | 770934 | 85 | Ser→Phe |
| 1A | None | ||||
| 1B | 0.96 | G→A | 771129 | 150 | Trp→stop |
| 1C | 0.83 | None | |||
| 1D | 0.79 | C→T | 770934 | 85 | Ser→Phe |
| 1E | 0.75 | None | |||
| 1F | G→A | 771295 | 205 | Trp→stop | |
| 1G | 0.91 | None | |||
| 1H | 0.85 | None | |||
| 1I | 0.79 | C→T | 771003 | 108 | Ser→Phe |
| 1K | 0.83 | ||||
| 1L | G→A | 771392 | 238 | Gly→Ser | |
| 1M | 0.88 | None | |||
| 1Nb | 0.79 | C→T | 771402 | 241 | Ser→Phe |
| 1Nb | C→T | 771477 | 266 | Pro→Leu | |
| 1O | 0.79 | None | |||
| 1P | 0.96 | None | |||
| 1Q | 0.95 | None | |||
| 1R | 0.78 | G→T | 770888 | 70 | Gly→Cys |
| 1S | 0.87 | C→T | 771668 | 330 | Gln→stop |
| 1Z | 0.85 | G→A | 771460 | 261 | Trp→stop |
| 2A | 0.84 | G→A | 771460 | 261 | Trp→stop |
| 2B | 0 | None | |||
| 2E | 0.85 | None | |||
| 2F | 0.94 | G→A | 771395 | 240 | Asp→Asn |
| 2J | 0.79 | G→A | 771392 | 239 | Gly→Ser |
| 2K | 0.8 | None | |||
| 2N | 0.93 | G→A | 770922 | 82 | Trp→stop |
| 2Q | 0.65 | G→A | 771788 | 371 | Ala→Thr |
Sequences were determined for the cydA gene from E. coli nucleotides 770738 to 772249 by using PCR products and primers described in Table 2 and sequencing in both directions. The cydA gene runs from nucleotide 770678 to nucleotide 772249. The cotransduction frequency is the fraction of replacements of cydA by cydA+ in a cross with phage P1 grown on a cydA+ nadA57::Tn10 strain. Frequencies were determined from 96 isolates screened as described in Materials and Methods. The nucleotide positions are the positions in the sequence published by Blattner et al. (6). The amino acid positions are the positions in the protein rather than the positions calculated from the gene sequence since the first ATG does not appear in the protein.
Two base changes were found in the cydA region of this isolate.
FIG. 3.
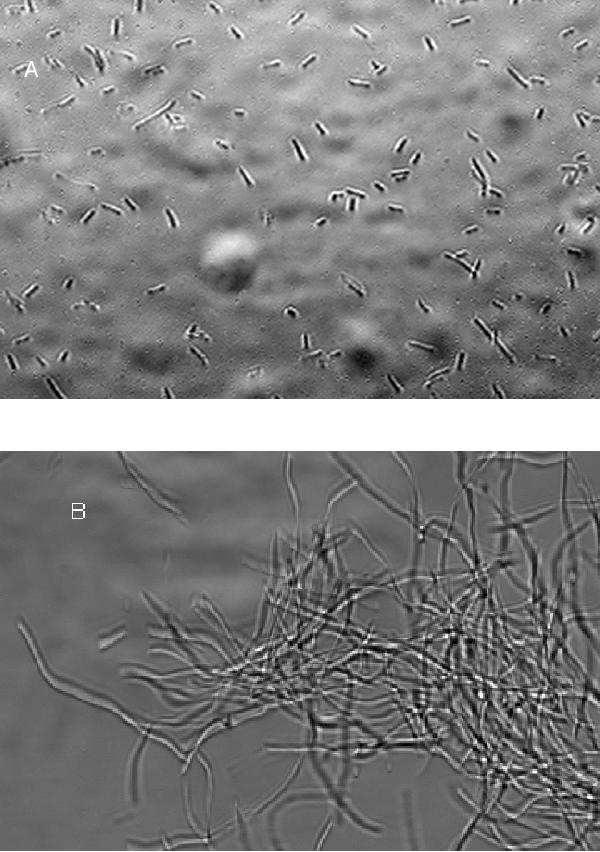
Effect of cydA on suppressor action. A plasmid was introduced into the dnaE74 cydA strain by CaCl2 transformation and selection on LB medium containing ampicillin. Transformants were picked, cultured overnight in LB medium containing ampicillin, and then incubated for 4 h with shaking at 40°C. (A) Plasmid pBR322; (B) plasmid pTK-1. Magnification, ×400.
TABLE 7.
Wild-type cydA reverses the suppressor effect
| Genotype of strain |
N/N0 with the following plasmida:
|
||
|---|---|---|---|
| None | pBR322 | pTK-1 | |
| dnaE74 cydA | 8.5 × 10−1 | 7.5 × 10−1 | 2.5 × 10−3 |
| dnaE74 | 4.8 × 10−6 | 2.5 × 10−5 | 1.4 × 10−5 |
N, viable count after incubation; N0, viable count at zero time. Freshly transformed strains were incubated overnight at 27°C in LB medium or LB medium containing ampicillin for cells with a plasmid. Cultures were then diluted and plated. The plates were incubated either at 27°C or at 40°C for 24 h and then switched to 27°C. Strains were transformed with plasmids by the CaCl2 method (26) and selected on LB medium plates containing ampicillin incubated at 27°C, and colonies were picked and used immediately after transformation.
DNA synthesis in temperature-sensitive mutants.
dnaE mutants were first characterized as quick-stop mutants since thymine incorporation was shown to quickly plateau after a shift to a nonpermissive temperature, 42.5°C (39). However, direct determination of DNA synthesis in cells incubated in LB medium at a dilution of 1:1,000 indicated that on average there was an eightfold increase in the total DNA during 4 h of incubation at 40°C, notwithstanding a loss of viability of over 2 logs. The increase (at a 1:10 dilution) was approximately linear for the 4-h period (Table 8). Cells of the original dnaE74 cydA suppressed strain exhibited a more-than-eightfold increase in total DNA in 4 h. The number of viable dnaE74 cydA cells (in which the cydA mutation was transferred to the dnaE74 strain by transduction) increased about fivefold during the same period of incubation at 40°C. These increases were compared with the approximately 50-fold increase in the amount of hot-acid-soluble material absorbing at 260 nm after extraction of dnaE74 cells and with the 40-fold increase in the total number of viable cells in a wild-type culture incubated at 40°C for 4 h (Fig. 1, top panel). The increase in DNA content was therefore about one-fifth to one-tenth the increase expected for a normal culture.
TABLE 8.
Synthesis of DNA by dnaE(Ts) strains incubated at 40°Ca
| Genotype of strain | Dilution | Vol (ml) | Total amt of DNA (μg) in culture after incubation at 40°C for:
|
N/N0b | |||||
|---|---|---|---|---|---|---|---|---|---|
| 0 h | 1.5 h | 2.5 h | 3 h | 4 h | 4.5 h | ||||
| dnaE74 | 1:10 | 50 | 279 | 1,250 | |||||
| 1:10 | 10 | 80.7 | 221 | 324 | 417 | 3.6 | |||
| 1:1,000 | 25 | 1.06 | 7.4 | 0.0063 | |||||
| 1:1,000 | 100 | 4.25 | 32.6 | 0.0044 | |||||
| dnaE486 | 1:10 | 10 | 463 | 834 | 1.08 | ||||
| 1:100 | 50 | 46 | 247 | 0.0058 | |||||
| 1:1,000 | 100 | 4.6 | 38.9 | 0.001 | |||||
| dnaE74 cydA | 1:10 | 10 | 25.8 | 211 | 5 | ||||
| 1:100 | 50 | 12.9 | 143 | 5.7 | |||||
| 1:1,000 | 100 | 2.6 | 25.0 | 5.7 | |||||
| 1:100 | 50 | 9.8 | 31.6 | 45 | 86.6 | 5.7c | |||
Cultures were grown overnight to a concentration of ≈5 × 109 cells/ml for the dnaE74 strain or 2 × 109 to 3 × 109 cells/ml for the dnaE74 cydA strain, collected, resuspended in LB medium, diluted, and incubated in different volumes of prewarmed medium for different times. Cultures were harvested, washed with 1× SSC, and extracted first with cold perchloric acid and then with hot perchloric acid. The perchloric acid extracts were analyzed by the diphenylamine method of Burton (9).
N, viable count after incubation for 4 h, unless indicated otherwise; N0, viable count at zero time.
The viable count after incubation was determined after incubation for 4.5 h.
In order to determine whether dnaE(Ts) mutants can synthesize biologically normal DNA, we investigated the propagation of plasmid pBR322 in cells incubated at a nonpermissive temperature. This plasmid contains both ampicillin resistance and tetracycline resistance genes which can be selected and scored and has a readily recognizable electrophoretic profile. Replication is complex. The first 200 to 400 nucleotides are synthesized by polymerase I, and the polymerase III replication complex (25) synthesizes the remainder of the 4,361 bp. We transformed the dnaE74 strain with pBR322, selecting for ampicillin resistance. To minimize formation of concatemers by recombination in this recA+ strain, we used the resulting cultures immediately. Incubation at 40°C of a 1:1,000 dilution of an overnight culture of the dnaE74 strain containing pBR322 resulted in an approximately 4.5-fold increase in the amount of supercoiled plasmid DNA without any change in the electrophoretic pattern (Fig. 4). We repeated this experiment five times and observed on average a 390% increase in the supercoiled band as measured by using the NIH Image program. The transforming activity for ampicillin resistance increased on average 3.8-fold in these experiments. During this period of incubation the viability of the host cells decreased by over 2 orders of magnitude. In order to see whether there was any widespread damage in nonselected regions of the genome, we tested 153 transformed colonies for tetracycline resistance. No tetracycline-sensitive colonies were observed.
FIG. 4.
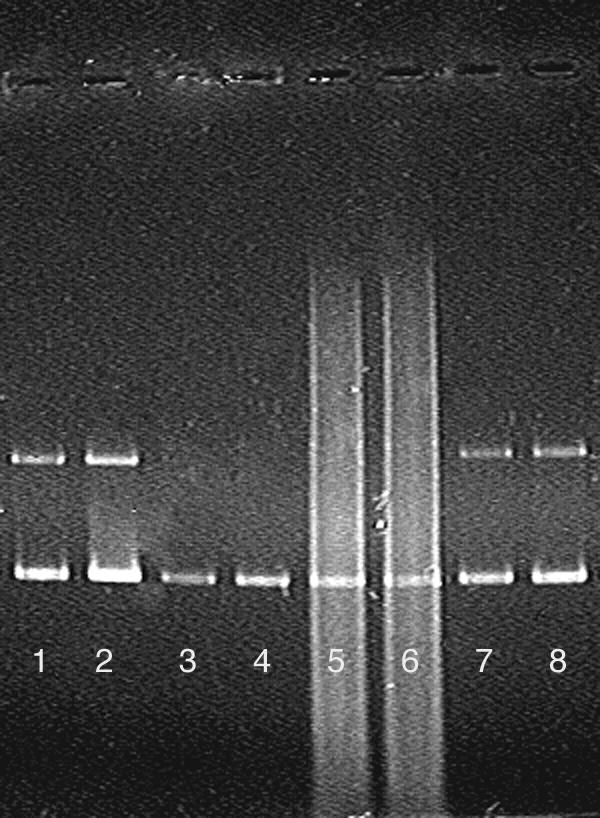
The dnaE74 strain was transformed with pBR322, and a freshly transformed isolate was cultured overnight in LB medium containing ampicillin at 27°C, washed, suspended in 10 ml of 1× SSC (0.15 M NaCl plus 0.015 M sodium citrate), diluted 1:1,000 in 100 ml of prewarmed medium, and incubated 4 h at 40°C with shaking. A sample was removed and used for a viability analysis, and two 3-ml portions were used as zero-time samples. The plasmid was extracted with a Qiagen minprep kit. Dilutions of the product were electrophoresed along with a pBR322 standard (Fermentas), and the gel was stained with Sybr Green (1:10,000) after electrophoresis for 3.75 h at 60 mV. Lane 1, 75 ng of pBR; lane 2, 100 ng of pBR; lane 3, 10 μl of a 1:10 dilution of an extract of a nonincubated culture; lane 4, 15 μl of a 1:10 dilution of an extract of a nonincubated culture; lane 5, 10 μl of an undiluted extract of a culture incubated for 4 h at 40°C; lane 6, 15 μl of an undiluted extract of a culture incubated for 4 h at 40°C; lane 7, 25 ng of pBR; lane 8, 50 ng of pBR322. The ratio of the viable count after 4 h of incubation at 40°C to the viable count at zero time was 7.6 × 10−3.
Although a normal plasmid was apparently synthesized under conditions in which the viability of the host cells decreased, it could be argued that there was a defect in DNA synthesis at the level of chromosomal segregation, as has been observed with other mutations (23). We therefore looked for nucleoid segregation by DAPI staining (14). The methodology used included 10 min of incubation with chloramphenicol prior to staining to condense nucleoids. Examination of filaments from the dnaE74 strain incubated at 40°C for 4 h revealed some filaments with multiple nuclei spaced equally along the filaments (Fig. 5A). Experiments without the chloramphenicol treatment gave similar results, but the nucleoids were less condensed (data not shown). Examination of the suppressed dnaE74 cydA strain revealed elongated cells with one or two nucleoids (Fig. 5B). It appeared that nucleoids were being formed and segregated.
FIG. 5.

DAPI staining of cells incubated for 4 h at 40°C in LB medium. Magnification, ×400. (A) dnaE74 strain; (B) dnaE74 cydA strain.
We mimicked the effect of slowing DNA synthesis while permitting rapid protein and RNA synthesis by incubating strains with hydroxyurea at 40°C. Hydroxyurea was toxic to both the wild type and the dnaE74 mutant at 40°C (Table 1). Examination of the dying BS40 wild-type cultures showed that there was extensive filamentation (data not shown). Introduction of the cydA mutation protected both the dnaE74 mutant and the wild-type strain from hydroxyurea toxicity (Table 9), indicating that there was some commonality between the mechanism of cell death induced by introducing a temperature-sensitive polymerase allele and the mechanism of cell death induced by treatment with a drug which reduces the supply of deoxynucleoside triphosphates. We also noted a population effect similar to that seen with the dnaE(Ts) mutant. When a 1:100 dilution of an overnight culture was used, hydroxyurea was bacteriostatic but not bactericidal for a dnaE+ strain.
TABLE 9.
Effect of hydroxyurea on survivala
| Genotype of strain | N0 (109) |
N/N0 witha:
|
|
|---|---|---|---|
| No hydroxyurea | Hydroxyurea (40 mM) | ||
| dnaE+su+ | 7.9 | 66.4 | 1.6 × 10−4 |
| dnaE+su | 9.6 | 7.0 | 2.6 |
| dnaE+su+ | 10 | 1.0 × 10−3 | 9.1 × 10−5 |
| dnaE su | 4.2 | 2.2 | 1.1 |
N, viable count after incubation for 5 h; N0, viable count at zero time. Overnight cultures grown at 27°C were diluted 1:1,000 in LB medium and incubated for 5 h at 40°C.
DISCUSSION
dnaE(Ts) mutants are able to synthesize normal DNA at a low rate at 40°C. Incubation at this temperature does not inhibit the accumulation of cellular mass or RNA but does result in inhibition of cell division. Death ultimately occurs. A suppressor mutation prevents death of a dnaE(Ts) mutant when it is incubated at 40°C. Of 51 suppressors isolated, 49 had a mutation in cydA or in a closely linked locus. How does cell death occur? What is the significance of the independent reisolation of suppressor mutations within the same gene?
We suggest that the phenomenon which we describe in this paper is essentially the unbalanced growth phenomenon, a concept described in relation to thymineless death 50 years ago (10). Thymine-requiring bacteria that are incubated in the absence of thymine die, and this death is accompanied by extensive protein and RNA synthesis. Inhibition of protein synthesis prevents death. Cohen developed the concept that in some way the synthesis of the major macromolecular constituents of the cell needs to be kept in step. This finding led to an extensive series of papers (1), but there was never a definitive explanation of how cells die as a result of thymine starvation. Two of the hallmarks of thymineless death are the occurrence of DNA damage and the instability of the DNA growing point (31). Much, if not all, of this damage can be ascribed to the incorporation of uracil into the DNA in the absence of thymine (1). This incorporation, followed by the action of uracil glycosylase, leads to abasic sites and breaks in the DNA, which in turn lead to aberrant recombination. A recent explanation attributes cell death to activation of the mazF gene product due to the loss of the mazE inhibitor as a result of inhibition of transcription of these genes (19, 33). Such inhibition has previously been reported for cases in which RNA synthesis and protein synthesis were reduced (2, 32), and this is not the case in thymineless death. A special mechanism, possibly involving altered DNA structures, was proposed to explain MazE transcription inhibition in this case.
Incubation of the dnaE(Ts) mutants at 40°C appears to mimic much of the effect of thymine deficiency, but we concluded that this occurs without the introduction of DNA damage. There is at least a fourfold increase in the amount of supercoiled pBR322 and a concomitant increase in transforming activity during incubation at a nonpermissive temperature and during a period in which the viable cell count decreases by at least 2 logs. This observation fits the hypothesis that it is failure of the cell division mechanism, not the DNA synthesis mechanism itself, which leads to cell death. An experiment measuring the stability of nascent and genomic DNA (11) for dnaE(Ts) and dnaB(Ts) mutants at 42.5°C showed that in contrast to the dnaB mutant in which the growing point was unstable, the dnaE mutant’s growing point showed no evidence of degradation (J. Courcelle, personal communication). Second, the total amount of DNA increases 5- to 10-fold in 4 h. The increase is roughly linear, and the amount indicates that the increase is not due solely to completion of already initiated chromosomes. There is an eightfold increase in the DNA content of a suppressed strain, and this increase is associated with a more-than-fivefold increase in cell number. In order for a cell to be counted as viable, its DNA needs to be capable of indefinite replication. Since the suppressor mutation is in the cydA gene, which has no known direct effect on DNA metabolism, we consider this additional evidence that the new DNA is normal. After completion of DNA replication, the newly replicated circles need to separate and be partitioned, after which cell division normally occurs. In the absence of the partition function, cells arrest in a filamentous state with a large nucleoid mass made of intertwined chromosomes in the center of the filament (23). The filaments produced by the dnaE(Ts) mutants growing at a nonpermissive temperature did include what appeared to be nucleoid masses in the process of division at the center (Fig. 5A), but they also included numerous nucleoids distributed throughout the filament as though there were no impediment to normal segregation. The problem appears to be not with DNA segregation but with the actual formation of septa. It should be noted that we have no evidence concerning whether it is actually DNA polymerase III that makes this DNA. It has been reported that given the appropriate suppressors, DNA polymerase I can carry out extensive synthesis (8).
The question of what factor(s) inhibits the formation of septa is therefore critical. A great many studies on division in E. coli point to the central role of the FtsZ protein in producing the ring whose constriction leads to division. A recent review (15) lists 10 proteins involved in this process in E. coli. The question is why lowering the rate of DNA synthesis inhibits the formation of the ring and how this is accomplished. We suppose that the sensing mechanism measures the rate of DNA synthesis and compares it to the rate of RNA and/or protein synthesis. Restricting the amount of growth with antibiotics, by population size, or by removal of cytochrome bd prevents elongation of the filaments to the point where the process becomes irreversible, possibly because of activation of one of the toxins associated with cell death in E. coli (19).
We found multiple recurrences of the same suppressor. Not only were the mutations in 49 of 51 isolates closely linked to the nadA gene, but also in the 27 closely linked isolates sequenced, there were 14 different mutations close to each other in the cydA gene with 2 repeats. We are currently sequencing the remainder of the gene in these isolates, as well as the surrounding region. The repeated isolation of mutants with different mutations in the same gene appears to imply that cytochrome bd has some specific and novel role in cell death under unbalanced growth conditions. However, a simpler explanation is based on the nature of the selection involved in the isolation of the suppressors. Suppressed strains were required to survive but not to form colonies at 40°C. The cydAB genes along with their cydCD regulators have been determined to be non-heat-shock proteins required for growth of E. coli at an elevated temperature (18, 38). Cytochrome bd, the product of the cydAB gene, is a high-efficiency cytochrome active at a relatively low oxygen tension, like that which occurs in cultures having a high density and at an elevated temperature. It may be that in the absence of this cytochrome the production of the nucleoside triphosphates required for production of RNA and DNA at a high temperature becomes limiting. The specificity of the suppressor is due to the requirement that it must be effective at a relatively high temperature but not at a low temperature. However, this explanation does not exclude the possibility that there is an additional and more direct role. E. coli has three different ribonucleotide reductases and selects which enzyme to use depending the availability of oxygen (21). Inhibiting DNA synthesis when an efficient cytochrome oxidase makes oxygen readily available might recruit ribonucleotide reductase into some cell-killing pathway. It may be that forced use of a different ribonucleotide reductase in dnaEcydA mutants inhibits this pathway, an explanation which also accounts for the protective effect of cydA against the lethal effects of hydroxyurea, an inhibitor of DNA synthesis. In any case, we concluded that DNA damage is not required to set in motion a chain of reactions that leads to cell death. Rather, a differential decrease in the rate of DNA synthesis appears to be sufficient.
Acknowledgments
We especially acknowledge the help of Mary Berlyn and the E. coli Genetic Stock Center. Without the strains from their collection, this work would not have been possible. We thank Robert Gennis for providing plasmid pTK-1 and J. Courcelle for providing unpublished data. We especially thank Ralph Weichselbaum, Stephen Kron, and Paul Mueller for many stimulating discussions.
This work was supported in part by grant 5RO1 CA32436-21 from the National Cancer Institute and by a grant to the Center for Molecular Oncology from the Ludwig Institute for Cancer Research.
REFERENCES
- 1.Ahmad, S. I., S. H. Kirk, and A. Eisenstark. 1998. Thymine metabolism and thymineless death in prokaryotes and eukaryotes. Annu. Rev. Microbiol. 52:591-625. [DOI] [PubMed] [Google Scholar]
- 2.Aizenman, E., H. Engelberg-Kulka, and G. Glaser. 1996. An Escherichia coli chromosomal “addiction module” regulated by guanosine 3′,5′-bispyrophosphate: a model for programmed bacterial cell death. Proc. Natl. Acad. Sci. 93:6059-6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexeeva, S., K. J. Hellingwerf, and M. J. Teixeira de Mattos. 2002. Quantitative assessment of oxygen availability: perceived aerobiosis and its effect on flux distribution in the respiratory chain of Escherichia coli. J. Bacteriol. 184:1402-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernander, R., and K. Nordstrom. 1990. Chromosome replication does not trigger cell division in E. coli. Cell 60:365-374. [DOI] [PubMed] [Google Scholar]
- 5.Bi, E., and J. Lutkenhaus. 1993. Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. J. Bacteriol. 175:1118-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 7.Bouvier, F., and N. Sicard. 1975. Interference of dna ts mutations of Escherichia coli with thymineless death. J. Bacteriol. 124:1198-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryan, S., H. Chen, Y. Sun, and R. E. Moses. 1988. Alternate pathways of DNA replication in Escherichia coli. Biochim. Biophys. Acta 951:249-254. [DOI] [PubMed] [Google Scholar]
- 9.Burton, K. 1956. A study of the conditions and mechanism of the diphenylamine reaction for the colorimetric estimation of deoxyribonucleic acid. Biochem. J. 62:315-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen, S. S., and H. D. Barner. 1954. Studies on unbalanced growth in Escherichia coli. Proc. Natl. Acad. Sci. USA 40:885-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Courcelle, J., and P. C. Hanawalt. 1999. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol. Gen. Genet. 262:543-551. [DOI] [PubMed] [Google Scholar]
- 12.Dewar, S. J., and R. Dorazi. 2000. Control of division gene expression in Escherichia coli. FEMS Microbiol. Lett. 187:1-7. [DOI] [PubMed] [Google Scholar]
- 13.Donabedian, H. 2003. Quorum sensing and its relevance to infectious diseases. J. Infect. 46:207-214. [DOI] [PubMed] [Google Scholar]
- 14.Donachie, W. D., and K. J. Begg. 1989. Cell length, nucleoid separation, and cell division of rod-shaped and spherical cells of Escherichia coli. J. Bacteriol. 171:4633-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Errington, J., R. A. Daniel, and D. J. Scheffers. 2003. Cytokinesis in bacteria. Microbiol. Mol. Biol. Rev. 67:52-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang, H., R. J. Lin, and R. B. Gennis. 1989. Location of heme axial ligands in the cytochrome d terminal oxidase complex of Escherichia coli determined by site-directed mutagenesis. J. Biol. Chem. 264:8026-8032. [PubMed] [Google Scholar]
- 17.Ghaim, J. B., D. P. Greiner, C. F. Meares, and R. B. Gennis. 1995. Proximity mapping the surface of a membrane protein using an artificial protease: demonstration that the quinone-binding domain of subunit I is near the N-terminal region of subunit II of cytochrome bd. Biochemistry 34:11311-11315. [DOI] [PubMed] [Google Scholar]
- 18.Goldman, B. S., K. K. Gabbert, and R. G. Kranz. 1996. The temperature-sensitive growth and survival phenotypes of Escherichia coli cydDC and cydAB strains are due to deficiencies in cytochrome bd and are corrected by exogenous catalase and reducing agents. J. Bacteriol. 178:6348-6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayes, F. 2003. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301:1496-1499. [DOI] [PubMed] [Google Scholar]
- 20.Jaffe, A., R. D'Ari, and S. Hiraga. 1988. Minicell-forming mutants of Escherichia coli: production of minicells and anucleate rods. J. Bacteriol. 170:3094-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jordan, A., and P. Reichard. 1998. Ribonucleotide reductases. Annu. Rev. Biochem. 67:71-98. [DOI] [PubMed] [Google Scholar]
- 22.Kaysser, T. M., J. B. Ghaim, C. Georgiou, and R. B. Gennis. 1995. Methionine-393 is an axial ligand of the heme b558 component of the cytochrome bd ubiquinol oxidase from Escherichia coli. Biochemistry 34:13491-13501. [DOI] [PubMed] [Google Scholar]
- 23.Levine, C., and K. J. Marians. 1998. Identification of dnaX as a high-copy suppressor of the conditional lethal and partition phenotypes of the parE10 allele. J. Bacteriol. 180:1232-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis, K. 2000. Programmed death in bacteria. Microbiol. Mol. Biol. Rev. 64:503-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marians, K. J. 1992. Prokaryotic DNA replication. Annu. Rev. Biochem. 61:673-719. [DOI] [PubMed] [Google Scholar]
- 26.Miller, J. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 27.Miller, J. H., and K. B. Low. 1984. Specificity of mutagenesis resulting from the induction of the SOS system in the absence of mutagenic treatment. Cell 37:675-682. [DOI] [PubMed] [Google Scholar]
- 28.Nakayama, K., N. Irino, and H. Nakayama. 1985. The recQ gene of Escherichia coli K12: molecular cloning and isolation of insertion mutants. Mol. Gen. Genet. 200:266-271. [DOI] [PubMed] [Google Scholar]
- 29.Nakayama, K., S. Shiota, and H. Nakayama. 1988. Thymineless death in Escherichia coli mutants deficient in the RecF recombination pathway. Can. J. Microbiol. 34:905-907. [DOI] [PubMed] [Google Scholar]
- 30.Nichols, B. P., O. Shafiq, and V. Meiners. 1998. Sequence analysis of Tn10 insertion sites in a collection of Escherichia coli strains used for genetic mapping and strain construction. J. Bacteriol. 180:6408-6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reiter, H., and G. Ramareddy. 1970. Loss of DNA behind the growing point of thymine-starved Bacillus subtilis 168. J. Mol. Biol. 50:533-548. [DOI] [PubMed] [Google Scholar]
- 32.Sat, B., R. Hazan, T. Fisher, H. Khaner, G. Glaser, and H. Engelberg-Kulka. 2001. Programmed cell death in Escherichia coli: some antibiotics can trigger mazEF lethality. J. Bacteriol. 183:2041-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sat, B., M. Reches, and H. Engelberg-Kulka. 2003. The Escherichia coli mazEF suicide module mediates thymineless death. J. Bacteriol. 185:1803-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singer, M., T. A. Baker, G. Schnitzler, S. M. Deischel, M. Goel, W. Dove, K. J. Jaacks, A. D. Grossman, J. W. Erickson, and C. A. Gross. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol. Rev. 53:1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strauss, B. S., R. Roberts, L. Francis, and P. Pouryazdanparast. 2000. Role of the dinB gene product in spontaneous mutation in Escherichia coli with an impaired replicative polymerase. J. Bacteriol. 182:6742-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strauss, B. S., D. Sagher, and S. Acharya. 1997. Role of proofreading and mismatch repair in maintaining the stability of nucleotide repeats in DNA. Nucleic Acids Res. 25:806-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vandewiele, D., A. R. Fernandez de Henestrosa, A. R. Timms, B. A. Bridges, and R. Woodgate. 2002. Sequence analysis and phenotypes of five temperature sensitive mutator alleles of dnaE, encoding modified alpha-catalytic subunits of Escherichia coli DNA polymerase III holoenzyme. Mutat. Res. 499:85-95. [DOI] [PubMed] [Google Scholar]
- 38.Wall, D., J. M. Delaney, O. Fayet, B. Lipinska, T. Yamamoto, and C. Georgopoulos. 1992. arc-dependent thermal regulation and extragenic suppression of the Escherichia coli cytochrome d operon. J. Bacteriol. 174:6554-6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wechsler, J. A., and J. D. Gross. 1971. Escherichia coli mutants temperature-sensitive for DNA synthesis. Mol. Gen. Genet. 113:273-284. [DOI] [PubMed] [Google Scholar]
- 40.Wertman, K. F., A. R. Wyman, and D. Botstein. 1986. Host/vector interactions which affect the viability of recombinant phage lambda clones. Gene 49:253-262. [DOI] [PubMed] [Google Scholar]
- 41.Woodgate, R. 1992. Construction of a umuDC operon substitution mutation in Escherichia coli. Mutat. Res. 281:221-225. [DOI] [PubMed] [Google Scholar]