

Abstract
Background
Microbial sensing by Toll-like receptors (TLR) and its negative regulation have important role in the pathogenesis of inflammation-related cancer. In this study, we investigated the role of negative regulation of TLR signaling and gut microbiota in the development of colitis-associated cancer in mouse model.
Methods
Colitis-associated cancer was induced by azoxymethane and dextran sodium sulfate in wild-type and in Interleukin-1 receptor associated kinase-M (IRAK-M) deficient mice with or without antibiotic (ATB) treatment. Local cytokine production was analyzed by multiplex cytokine assay or ELISA, and regulatory T cells were analyzed by flow cytometry. Changes in microbiota composition during tumorigenesis were analyzed by pyrosequencing, and β-glucuronidase activity was measured in intestinal content by fluorescence assay.
Results
ATB treatment of wild-type mice reduced the incidence and severity of tumors. As compared with non-treated mice, ATB-treated mice had significantly lower numbers of regulatory T cells in colon, altered gut microbiota composition, and decreased β-glucuronidase activity. However, the β-glucuronidase activity was not as low as in germ-free mice. IRAK-M deficient mice not only developed invasive tumors, but ATB-induced decrease in β-glucuronidase activity did not rescue them from severe carcinogenesis phenotype. Furthermore, IRAK-M deficient mice had significantly increased levels of pro-inflammatory cytokines in the tumor tissue.
Conclusions
We conclude that gut microbiota promotes tumorigenesis by increasing the exposure of gut epithelium to carcinogens and that IRAK-M negative regulation is essential for colon cancer resistance even in conditions of altered microbiota. Therefore, gut microbiota and its metabolic activity could be potential targets for colitis-associated cancer therapy.
Keywords: Cancer in IBD, Mucosal immunity, Colorectal cancer, Microbiota, Germ-free
Introduction
Chronic gut inflammation, as seen in patients with inflammatory bowel diseases (IBD), is a strong risk factor for colon cancer. Therefore, the increase in number of IBD patients observed in last decades, will ultimately lead to increase in number of patients with colitis-associated cancer (CAC) (1, 2). Although the pathogenesis of IBD and the development of CAC are still not completely understood, it is generally accepted that an aberrant immune reaction to intestinal commensal microbiota and subsequent chronic inflammatory responses in the gut play major roles (3).
Microbial stimulation in the gut is important for maintaining physiological functions, including intestinal epithelium growth, mucosal permeability and production of antimicrobial agents, as well as regulation and development of the immune system (3, 4). Various resident bacteria have protective role in the process of inflammation and cancer development but on the other hand there is a great amount of potentially harmful species that can be derived from normal microbiota under specific conditions of imbalance in gut milieu (4–6). An example of a potentially harmful microbe is Helicobacter pylori, which often resides stomach mucosa without clinical consequences. However, this Helicobacter pylori is also confirmed triggering agent of the chronic gastric inflammation and cancer and its treatment with antibiotic leads to cancer prevention(7). Use of broad-spectrum antibiotics, like ciprofloxacin and metronidazole, also brought positive results in the therapy of certain forms of Crohn’s disease and pouchitis (8), decreasing the risk of colon cancer development.
Metabolic activity of microbiota is an important detail of the gut ecosystem. Microbes are equipped with a broad-spectrum of enzymes and therefore can metabolize various substrates (5). Among final products belong potential pro-carcinogens as well as beneficial substances such as short-chain fatty acids (SCFA). SCFA are a crucial source of energy for colonic epithelium and their lack has been implicated in pathogenesis of colorectal carcinoma (9). Protective role is also ascribed to probiotics defined as live bacteria beneficial to health, that have stabilizing effect on gut microbiota during administration with potential to reduce pro-inflammatory response (10, 11). Manipulation of the microbiota therefore brings wide possibilities, although not fully elucidated, of influencing intestinal homeostasis and immune system reactivity.
Detection of conserved microbe-associated molecular patterns is provided by different cellular pattern-recognition receptors, such as family of the Toll-like receptors (TLR) (12). TLR signalization is important in maintaining gut epithelium homeostasis but the stimulation of TLR may also induce cancer development or promote tumor growth (13). Myeloid differentiation factor 88 (MyD88), which is responsible for signal transduction from all TLRs except for TLR3, has been shown to interfere with the pathogenesis of colon inflammation and cancer by triggering pro-inflammatory response via transcription factor NF-κB (14, 15). Interleukin-1 receptor associated kinase-M (IRAK-M) is a molecule crucial in regulation of gut immune response through negative feedback. IRAK-M binds and blocks MyD88/IRAK-4 protein complex thus negatively regulating pro-inflammatory signal transduction by IRAK-1/TRAF6 in various immune cells and gut epithelium (16, 17). The expression of Irak-m gene is closely associated with the presence of intestinal microbiota and TLR signaling (18). Its deficiency enhances the production of pro-inflammatory cytokines in macrophages and intensifies experimentally-induced dextran sodium sulfate (DSS) colitis (19). Recent studies showed that single immunoglobulin IL-1 receptor-related molecule (SIGIRR), another negative regulator of TLR signaling, is involved in inflammation and cancer development (20), which suggests the important role of these molecules in tumorigenesis.
In our previous studies, we found that germ-free condition protected rats from colonic inflammation as well as from cancer (21). Since recognition of microbiota by TLRs plays important role in tumorigenesis, we hypothesized that both dysbiosis and negative regulation of TLR signaling via IRAK-M interferes with colon cancer development. By using inflammation-related mouse model of colon cancer induced by azoxymethane and DSS, we were able to follow the “inflammation-dysplasia-carcinoma” sequence, typical for CAC, under different microbial conditions. Here, we show the impact of gut microbiota composition on colon cancer development and immune system reactivity, and analyze the role of negative regulator IRAK-M in this process.
Materials and Methods
Animals and experimental schedule
We used two-month-old C57BL/6 male mice reared either in conventional (Institute of Physiology AS CR, Prague, Czech Republic) or in germ-free (GF) conditions (Institute of Microbiology AS CR, Novy Hradek, Czech Republic). The GF mice were reared in controlled sterile conditions, as described previously (22). IRAK-M deficient mice (obtained from the laboratory of Koichi S. Kobayashi) were backcrossed to C57BL/6 background for eleven generations and were held in specific pathogen-free facility in Novy Hradek. All mice received the same diet (ST-1, Velaz, Czech Republic) and tap water ad libitum, and were used according to the procedures approved by the Institute of Microbiology animal care and use committee (No. 094/2008 and 053/2010).
We initiated tumorigenesis using modified protocol published previously by Clapper et al. (23). Briefly, the mice were given single subcutaneous injection of azoxymethane (AOM, 10 mg/kg; Sigma-Aldrich, St. Louis, MO). Starting one week after the AOM injection, mice received 3% dextran sodium sulfate (DSS, MW 36–50 kDa; MP Biomedicals, Illkirch, France) in their drinking water continuously for up to 4 days. DSS was subsequently replaced by tap water for the rest of the experiment. To induce the intestinal microbiota alteration in conventionally-reared mice, we treated a group of animals with antibiotics (ATB): metronidazole (500 mg/L; B. Braun, Melsungen AG, Germany) and ciprofloxacin (100 mg/L; Zentiva, a.s., Hlohovec, Slovak Republic) in their drinking water for the whole experimental period (50 days). Four independent experiments with five mice per group and three independent experiments with at least five mice per group were done in wild-type and IRAK-M deficient mice, respectively.
We recorded changes in body weight, stool consistency and gross bleeding every week, every day during DSS administration, and sacrificed the mice 5 weeks after AOM injection.
Histopathology evaluation
At the end of the experiments, the colon length was measured and fecal samples were tested for occult blood using Okult-Viditest Rapid (Vidia s.r.o., Jesenice u Prahy, Czech Republic). The colon was cut open longitudinally and macroscopically inspected for the presence of pathological lesions. Proximal and distal colon and rectum were fixed in 4% buffered formalin, dehydrated and embedded in paraffin. Histopathological examinations were performed in 4μm sections after hematoxylin/eosin staining. The degree of intestinal alteration was examined by two experienced pathologists (P.R., K.K.) using conventional criteria to determine normal mucosa; low or high-grade dysplasia; non-invasive or invasive carcinoma.
Cultivation and cytokine measurement
We took a part of the colon from every mouse, i.e. from tumor in tumor-bearing mice and from similar locality in the other mice, washed it in cold phosphate-buffered saline (PBS) and weighed. These tissues were then cultivated for 48h at 37°C in RPMI-1640 media (Sigma-Aldrich) containing 10% fetal bovine serum (BioClot GmbH, Aidenbach, Germany) and 1% Antibiotic-Antimycotic solution (Sigma-Aldrich). The supernatants were collected and frozen at −20°C until analysis.
Cytokine profiles were determined using a multiplex cytokine analyzer – Luminex. The Fluorokine MAP Mouse Base Kit was used in accordance with manufacturer’s instructions in combination with recommended bead sets for selected cytokines: IL-1β/IL-1F2, IL-6, IL-10, IL-12 p70, IL-17, TNF-α and IFN-γ (all R&D Systems, Minneapolis, MN) and analyzed using Luminex 200 instrument (Luminex Corporation, Austin, TX). The concentrations of analytes were determined by monitoring the spectral properties of the beads and the amount of phycoerythrin fluorescence.
Levels of TGF-β were measured by commercially available ELISA kit (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions.
Haptoglobin determination
The level of haptoglobin in mouse sera was assessed by Human Haptoglobin ELISA Quantitation Kit (GenWay Biotech., Inc., San Diego, CA). Antibodies used in this kit have high cross-reactivity with mouse haptoglobin that allowed us to use the kit according to the manufacturer’s instructions with minor modifications as described previously (24).
Real-time polymerase chain reaction (PCR)
Samples of colon were placed in RNAlater stabilization reagent (QIAGEN GmbH, Hilden, Germany) and stored in −80°C. Total RNA was extracted by using the RNeasy Mini isolation kit (QIAGEN GmbH) following the manufacturer’s instructions. RNA integrity was determined by gel electrophoresis in 1% agarose gel stained with SYBR-green (Life Technologies) and the concentration of the RNA was assessed by NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). First strand cDNA was synthesized from 0.5 μg of RNA using SuperScript II reverse transcriptase (Life Technologies). Real-time PCR was performed using iQ SYBR-green Supermix (Bio-Rad Laboratories, Hercules, CA) on iQ5 cycler (Bio-Rad). The samples were analyzed in doublets and the expression was normalized to ribosomal protein S12 using iQ5 software (Bio-Rad). All primers were purchased from Generi Biotech (Hradec Kralove, Czech Republic); sequences of the primers were as follows: ribosomal protein S12 forward: 5′-CCTCGATGACATCCTTGGCCTGAG-3′, ribosomal protein S12 reverse: 5′-GGAAGGCATAGCTGCTGGAGGTGT-3′; cyclooxygenase (COX)-2 forward: 5′-AGTGGGGTGATGAGCAACTA-3′, COX-2 reverse: 5′-GGCAATGCGGTTCTGATACT-3′; IL-18 forward: 5′-ACGTGTTCCAGGACACAACA-3′, IL-18 reverse: 5′-ACAAACCCTCCCCACCTAAC-3′.
Flow cytometric analysis
Spleens, mesenteric lymph nodes (MLN), Peyer’s patches (PP) and colonic tissue were collected and processed into single cell suspension. Briefly, cells from MLN, PP and colon were centrifuged and resuspended in 300 μL of cold FACS buffer (PBS containing 0.1% NaN3, 0.5% fetal bovine serum and 0.5M EDTA; pH 7.2 – 7.4). Splenocytes were treated by 5 mL of ACK lysis buffer (0.15M NH4Cl, 10mM KHCO3, 0.5M EDTA in distilled water; pH 7.2 – 7.4), resuspended in 5 mL of cold FACS buffer and kept on ice until staining. Cells were blocked with 10% normal mouse sera and then stained for surface molecules with fluorescent-labeled monoclonal antibodies cocktail containing anti-mouse CD3 – fluorescein isothiocyanate (BD Bioscience, Heidelberg, Germany; clone 145-2C11), CD4 – Qdot 605 (Life Technologies; clone RM4-5), CD8 – PerCP-Cy5.5 (BD Bioscience; clone 53-6.7) and CD25 – allophycocyanin (eBioscience; clone PC61.5). Subsequent intracellular staining for mouse Foxp3 was performed with phycoerythrin-labeled anti-mouse/rat Foxp3 staining set according to the manufacturer’s instructions (eBioscience; clone FJK-16s). Cells were measured using LSRII (BD Bioscience) and data were evaluated by FlowJo software (Tree Star Inc., Ashland, OR).
Microbiota analysis
Stool samples were collected from all mice on days 1, 23 (just before DSS administration), 30 (after DSS treatment) and 50 (the last day of experiment), and total DNA was isolated using ZR Fecal DNA Kit (Zymo Research Corp., Orange, CA) according to the manufacturer’s instruction. The isolated DNA was stored at −20°C for further analyses.
For the detailed determination of microbiota composition, we used high-throughput pyrosequencing described in detail elsewhere (25). Briefly, previously isolated DNA was gel-purified and PCR with broad-range bacterial primers for 16S rDNA including tags for pyrosequencing was performed. PCR product was purified using magnetic beads (AMPure beads, Beckman Coulter Genomics, Danvers, MA) and concentration was measured on Qubit fluorometer (Life Technologies). Equimolar amounts of PCR product from each sample were then used for unidirectional 454 FLX amplicon pyrosequencing using LIB-L emPCR kits following the manufacturer’s protocols (Roche Diagnostics, Basel, Switzerland). Sequence reads were processed using RDP’s pyrosequencing pipeline (http://rdp.cme.msu.edu/) and Greengenes workbench compatible with ARB (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi) following all standard procedures as sequence quality trimming, chimera check, phylotype identification, phylogenetic analysis and diversity analysis.
We performed quantitative PCR (qPCR) with specific primers to determine the numbers of the total bacteria (Eubacteria), Parabacteroides distasonis (P. distasonis) and Faecalibacterium prausnitzii (F. prausnitzii) in the stool samples. The conditions for PCR reactions are listed in Table 1. The qPCR 2x SYBR Master mix (Top-Bio, Prague, Czech Republic) was used along with Stratagene mx3005P (Agilent Technologies, Santa Clara, CA) equipment. Three-log diluted DNA isolated from known number of cells was used as standard for absolute quantification.
Table 1.
Quantitative PCR conditions.
| Target taxon | Primers | Sequence (5′ to 3′) | Annealing temperature (°C) | Standard strain | Reference |
|---|---|---|---|---|---|
| All Eubacteria | Uni331F Uni797R |
TCCTACGGGAGGCAGCAGT GGACTACCAGGGTATCTATCCTGTT |
58 | Clostridium leptum ATCC 29065 | (43) |
| F. prausnitzii | Fprau223F Fprau420R |
GATGGCCTCGCGTCCGATTAG CCGAAGACCTTCTTCCTCC |
58 | F. prausnitzii A2165 | (43) |
| P. distasonis | Bd180F Bd463R |
AAT ACC GCA TGA AGC AGG GAC ACG TCC CGC ACT TTA |
62 | P. distasonis | (44) |
F. prausnitzii, Faecabacterium prausnitzii; P. distasonis, Parabacteroides distasonis.
β-glucuronidase determination
To analyze the activity of β-glucuronidase enzyme in the intestine, the stool samples were collected on the day of AOM injection from wild-type and IRAK-M deficient mice with and without ATB treatment, and from GF mice. Enzyme was extracted from lyophilized and weighed stool pellets into one mL of acetate buffer (50 mM; pH 7) and incubated for two hours at 4°C. 50 μL of extract was added into 100 μL of acetate buffer (50 mM; pH 5) with 50 μL of 2.5 mM MUG substrate (4-methylumbelliferyl-β-D-glucuronide; Glycosynth, Warrington, England) and incubated at 37°C. Product fluorescence was measured at the beginning and after two hours on microplate reader (Tecan GmbH, Grödig, Austria) using 388 nm as excitation and 480 nm as emission wavelength.
Statistical analysis
One-way analysis of variance (ANOVA) with Tukey’s multiple comparison test was used to compare multiple experimental groups. Two-way ANOVA with Bonferoni post-test was used in determination of significant weight changes. Differences between two groups were evaluated using an unpaired two-tailed Student’s t test, and the incidence of invasive carcinoma between AOM/DSS and ATB/AOM/DSS treated IRAK-M deficient mice were compared by Fisher’s exact test. The data are presented either as the mean ± standard deviation, or as percentage of mice with invasive tumors, and differences were considered statistically significant at P ≤ 0.05. GraphPad Prism statistical software (version 5.0, GraphPad Software, Inc., La Jolla, CA) was used for analyses.
Results
Colon cancer incidence is significantly decreased in GF and ATB-treated mice
To follow the effect of microbiota on tumorigenesis, we induced the CAC by AOM/DSS in C57BL/6 mice reared either under conventional, or germ-free conditions, or in conventionally-reared ATB-treated mice. After 5 weeks from AOM injection, the histological examination revealed that either graded dysplasia or carcinoma were present in over 93% of conventionally-reared mice and were not accompanied by any sign of tumor dissemination (Fig. 1A). In all cases, the tumors were situated in the descendent portion of colon and/or in rectum, while no lesions were found in proximal colon. This relatively early tumor development can be ascribed to experimental conditions including sensitive mouse strain, its gut microbiota, diet, or high DSS concentration we used. Therefore, we used shorter experimental period to see shifts in the incidence in early tumor development and to prevent unnecessary loss of mice due to the mortality. We found significantly shorter colon and heavier spleen in AOM/DSS-treated mice as compared with healthy mice (Fig. 1B and 1C). Interestingly, antibiotic treatment of these mice mitigated the DSS-induced weight loss and significantly decreased both the incidence of tumors (16%) and severity of tumor lesions (Fig. 1D, 1A and 1E). Moreover, we did not observe the significant increase in weight of spleen and shortening of colon in these ATB-treated mice (ATB/AOM/DSS-treated) (Fig. 1B and 1C).
Figure 1. Commensal microbiota is required for the progression of colitis-associated cancer in AOM/DSS-induced colorectal carcinoma model.

(A) Total percentage of cases of high-grade dysplasia and tumor incidence was higher in antibiotic (ATB)-non-treated group. (B, C) The colon length and spleen weight were measured at the end of the experiment. Significant shortening of colon length and increase in spleen weight was found in ATB-non treated mice. ** P<0.01, *** P<0.001. (D) ATB-treated mice showed significantly smaller decrease in body weight after DSS treatment and during tumorigenesis when compared to ATB-non-treated mice. The values are relative to the weight before DSS treatment. * P<0.05, ** P<0.01, *** P<0.001 for ATB/AOM/DSS compared with non-treated control, † P<0.05, †† P<0.01, ††† P<0.001 for AOM/DSS compared with non-treated control, ‡ P<0.05, ‡‡ P<0.01, ‡‡‡ P<0.001 for ATB/AOM/DSS compared with AOM/DSS. (E) Histology screening showed decrease of lesions severity in ATB-treated mice as compared with ATB-non-treated mice. Data shown are combined results from three separate experiments with at least five mice per group.
To investigate the role of microbiota in carcinogenesis further, we used the same protocol to induce the colon cancer in GF mice. After 5 weeks, the incidence of tumors in GF mice (10%) was even lower than in ATB-treated conventional animals, confirming the significance of microbiota in the pathogenesis of inflammation associated colorectal carcinoma. Moreover, if present, the tumors had polypoid exophytic character, which corresponded to low-grade dysplasia (data not shown), they were less numerous and smaller in size as compared with those in conventionally-reared mice.
Effect of ATB on microbiota during tumorigenesis
Since dysbiosis is characteristic for IBD and colon cancer, uncovering changes in microbiota composition seems to be crucial for understanding of immune mechanisms involved in pathogenesis of these diseases (4). To address this issue, we compared the fecal microbiota composition between ATB-treated (ATB/AOM/DSS-treated) and control (AOM/DSS-treated) wild-type mice by high-throughput pyrosequencing at different time-points during tumorigenesis (Fig. 2A). ATB treatment caused an increase in occurrence of Bacteriodes, Parabacteroides, Prevotella, Blautia, Desulfovibrio and Subdoligranulum genera and decrease in occurrence of Allobaculum, Alistipes, Ruminoccocus and Johnsonella genera as compared with non-treated mice. In accordance with the previous report, we observed decreasing levels of Bacteroidetes and increasing levels of Firmicutes at the end of the experiment during tumor progression (26). In order to quantify these changes, we performed quantitative analysis with qPCR. Using all Eubacteria and species specific primers, we found that ATB treatment did not decrease the total count of bacteria (Fig. 2B), but resulted in changes of particular bacteria (Fig. 2C). We analyzed P. distasonis and F. prausnitzii as prevalent representatives of Bacteroidetes and Firmicutes phyla, respectively, because of their evident biological activity associated with intestinal inflammation (24, 27). P. distasonis showed increase in abundance during the first 30 days of ATB treatment whereas F. prausnitzii was markedly reduced. On the other hand, ATB-non-treated mice showed opposite trend as P. distasonis moderately decreased whereas F. prausnitzii abundance continuously grew during tumorigenesis.
Figure 2. Microbiota changes play an important role in the induction of colitis-associated cancer.

We collected and analyzed feces at the beginning, before and after DSS treatment and at the end of the experiment to describe the bacterial profile changes during tumor development. (A) Pyrosequencing demonstrates changes among the main fecal phyla of Bacteroidetes, Firmicutes and Proteobacteria during tumorigenesis determined on the level of genera. The heatmap is colored according to the relative abundance of the bacterial genera (green for low, red for high). (B, C) Continuous changes of fecal microbiota were assessed by quantitative PCR using specific primers. The data are normalized by the day 1. The total count of bacteria and the relative numbers of Parabacteroides distasonis and Faecalibacterium prausnitzii showed variability during tumorigenesis. (D) Activity of β-glucuronidase was measured in the colon content collected in the day of AOM injection from germ-free (GF), antibiotic (ATB)-treated and ATB-non-treated mice. The enzyme activity is significantly decreased by ATB treatment as well as in GF mice. The data are presented as mean ± standard deviation with * P<0.05, *** P<0.001.
Microbiota metabolism increases tumor incidence
The genotoxic effect of AOM can be increased by microbial β-glucuronidase, which releases active compound – methylazoxy-methanol from AOM metabolite (28). Thus, we analyzed the specific activity of this enzyme in germ-free mice and in ATB-treated and non-treated wild-type and IRAK-M deficient mice at the time of carcinogen introduction. For this purpose, we collected samples of feces just before AOM injection and compared the metabolic activity by measuring specific fluorescent product. We found decrease of β-glucuronidase activity in GF as well as in ATB-changed conditions (Fig. 2D). Almost 3-times lower activity, when compared with non-treated mice, was linked with lower tumor incidence in the group of ATB-treated wild-type mice and confirmed that ATB treatment decreased the amount of β-glucuronidase-equipped bacteria in the gut. Although the β-glucuronidase activity was more than 2-times lower in ATB-treated IRAK-M deficient mice, this change was insufficient to reduce the tumor development in these mice. Therefore, the IRAK-M seems to be the effector molecule promoting tumor resistance after ATB treatment in this model, i.e. under the conditions of reduced carcinogen load.
IRAK-M deficient mice developed invasive tumors and ATB treatment was insufficient to protect them from cancer
Since negative regulator of TLR-signaling, IRAK-M, and gut microbiota regulate each other and since IRAK-M molecule mitigates intestinal inflammation in chronic colitis model, as we recently published (18, 19), we further analyzed the importance of IRAK-M in ATB-associated protection against colon cancer development. We used AOM/DSS to induce colon cancer and performed the ATB treatment in conventionally-reared IRAK-M deficient mice. An increased infiltration of mononuclear cells was observed in lamina propria of control non-treated IRAK-M deficient mice as compared with wild-type controls. IRAK-M deficient mice were also more sensitive to AOM/DSS treatment and the incidence of tumors reached 100% after 5 weeks (Fig. 3A). Moreover, the favorable effect of ATB treatment on tumor development was not presented in IRAK-M deficient mice where there was no difference in the tumor incidence, colon length, spleen weight and body weight between ATB-treated and ATB-non-treated group (Fig. 3). Detailed histological analysis showed widespread flat tumor lesions with high infiltration of inflammatory cells and invasion of crypt bases into submucosa, which had never been seen in wild-type mice (Fig. 4). The invasive carcinoma are significantly more common in the AOM/DSS as compared to ATB/AOM/DSS-treated IRAK-M deficient mice (53% vs. 10%, P=0.005, Fisher’s exact test) (Fig. 4D). We also followed the mice for additional 6 weeks observing that both the 100 % incidence of tumors and histological finding are similar as 6 weeks earlier (data not shown).
Figure 3. IRAK-M regulation was important for ATB-induced colon cancer resistance.
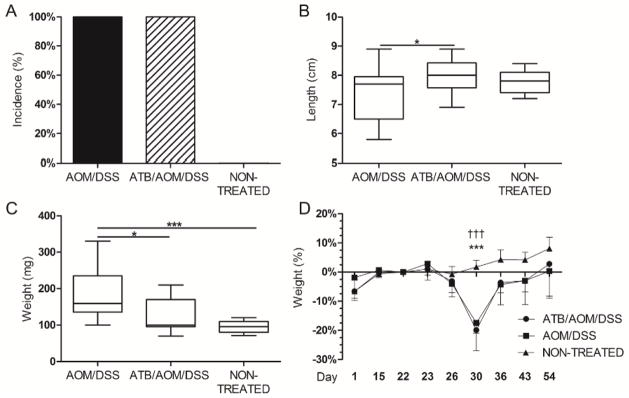
We used the same protocol as in wild-type mice to induce colitis-associated cancer in IRAK-M deficient mice. (A) IRAK-M deficient mice were more sensitive to the AOM/DSS treatment, which is documented by the tumor incidence. (B, C) Significant shortening of the colon and increase in the weight of spleen were found in both AOM/DSS and antibiotic (ATB)/AOM/DSS-treated mice when compared with non-treated mice. * P<0.05, *** P<0.001. (D) Weight changes during tumorigenesis. The values are relative to the weight before DSS treatment. *** P<0.001 ATB/AOM/DSS compared with non-treated control, ††† P<0.001 AOM/DSS compared with non-treated control. Data shown are compiled from two independent experiments with at least six mice per group.
Figure 4. IRAK-M deficient mice developed invasive type of tumor lesions in AOM/DSS model.

(A) Macroscopic views of distal colon and rectum of IRAK-M deficient mice show healthy intestine, polypoid lesion and flat lesion. (B) Hematoxylin/eosin-stained representative images of intestines in low magnification show the differences among normal mucosa, polypoid and flat tumor lesions. (C) Detail of the sections shows normal mucosa of non-treated mice, and high-grade carcinoma and invasive character of tumor lesions in AOM/DSS-treated IRAK-M deficient mice. Magnification 10x. (D) Histological examination of colon tissue showed that only 10% of ATB-treated IRAK-M deficient mice have massive widespread tumor lesions with invasion to submucosa, as compared to 53% of ATB-non-treated mice (P=0.005, Fisher’s exact test).
IRAK-M deficient mice showed enhanced pro-inflammatory response
As the chronic inflammation plays a crucial role in tumorigenesis (1), we analyzed the inflammatory response on both local (production of cytokines in the gut) and systemic level (serum acute phase protein – haptoglobin). We found a significant increase in the pro-inflammatory cytokines – IL-1β, IL-6, TGF-β and TNF-α in supernatants from tumor tissues of AOM/DSS-treated IRAK-M deficient mice as compared with wild-type mice (Fig. 5A). The increased inflammatory reaction in IRAK-M deficient mice became more visible in ATB-treated groups where the inflammation and tumorigenesis were reduced in wild-type mice. On the other hand, ATB-treated wild-type mice had significantly higher production of IL-10 and IL-12 than IRAK-M deficient mice. TGF-β is an important factor for regulatory T cells response and function under chronic inflammatory conditions, and for tumor invasion (29). In our experiments we observed low levels of TGF-β at the sites of cancer and inflammation in wild-type mice, while we found much higher production in tumor-bearing IRAK-M deficient mice (Fig. 5B). Moreover, we found unchanged expression of IL-18 in colon tissue of both wild-type and IRAK-M deficient mice, which correlates with the lower levels of IFN-γ found in supernatants (Fig. 5C). These findings suggest increased suppression of anti-tumor immunity in IRAK-M deficient mice. The expression of COX-2 in colon tissue was significantly increased in tumor-bearing wild-type as well as IRAK-M deficient mice confirming local pro-inflammatory activation (Fig. 5D).
Figure 5. IRAK-M deficient mice showed increased pro-inflammatory response to AOM/DSS treatment at the end of the experiment.

(A, B) Cytokine levels in colonic tissue culture supernatants from wild-type (WT) and IRAK-M deficient mice with or without antibiotic (ATB) treatment were measured by Luminex and ELISA. (C, D) The changes in the expression of IL-18 and COX-2 in the colon tissue of WT and IRAK-M deficient mice were analyzed using real-time PCR. (E) Haptoglobin levels were determined by ELISA method in the sera of wild-type and IRAK-M deficient mice ± ATB treatment. Data are presented as mean ± standard deviation with *P<0.05, **P<0.01, ***P<0.001. Data shown are compiled from two independent experiments with at least six mice per group.
To determine the systemic response on tumorigenesis we measured serum haptoglobin levels and we confirmed increased inflammatory activity in tumor-bearing mice in both wild-type and IRAK-M deficient mice (Fig. 5E). Moreover, IRAK-M deficient mice showed significantly higher levels of haptoglobin when compared with wild-type mice. Thus, IRAK-M deficiency seems to maintain an increased pro-inflammatory reactivity affecting the whole organism, and a decreased anti-tumor immunity, regardless of ATB treatment.
Regulatory T cells accumulated in tumor tissue and local lymph nodes
Tumor-bearing mice are known to have different organ distribution and numbers of regulatory T (Treg) cells as compared with healthy controls (30). Therefore, we measured the numbers of Treg cells (CD4+Foxp3+) in spleen, MLN, PP, and directly in the tumor tissue. We did not observe any significant differences in Treg cell populations in spleens and PPs among the experimental groups (data not shown). In contrast to significant increase of Treg cells populations that we found in MLN and colon tissue of tumor-bearing wild-type as well as IRAK-M deficient mice (Fig. 6A). Generation of Foxp3+ Tregs in the periphery is associated with TGF-β-induced expression of Foxp3 transcription factor in CD4+ cells (31). Interestingly, we found significant increase in this initial population of CD4+CD25−Foxp3+ cells in MLN of IRAK-M deficient mice (Fig. 6B). These findings are consistent with other measured factors inducing immune suppression in tumor microenvironment (e.g. high TGF-β, low IL-12).
Figure 6. Tumor-bearing mice showed higher levels of Treg cells.
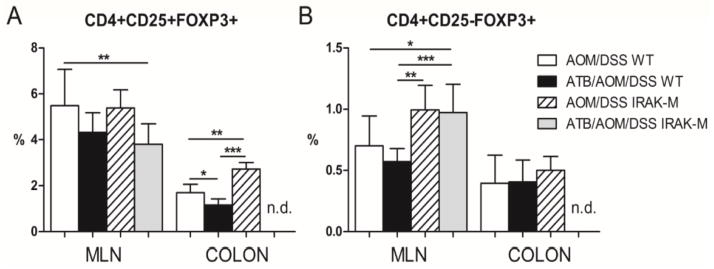
Percentage of CD4+Foxp3+ regulatory T (Treg) cells was measured by flow cytometry in mesenteric lymph nodes (MLN), and colon tissue of treated wild-type (WT) mice and IRAK-M deficient mice. (A) Significantly increased population of CD4+CD25+Foxp3+ Treg cells was found in colon tissue of treated IRAK-M deficient mice. (B) Local induction of immunosuppressive milieu is documented by increase in CD4+CD25−Foxp3+ cells in the MLN of treated IRAK-M deficient mice. Differences in Treg cells are presented as bars ± standard deviation and *P<0.05, **P<0.01, ***P<0.001. n.d.: not done. Data shown are compiled from two independent experiments with at least six mice per group.
Discussion
Microbiota-derived signals are crucial for the development and setting of mucosal immune system and the gut is an important primary site of host – microbe interaction. Disruption of balance between intestinal mucosa and commensal bacteria can result in inflammatory disorders (4). Chronic inflammation, as IBD has long been recognized as a risk factor for cancer development at various sites. For example, individuals suffering from ulcerative colitis have a 2 – 8 fold increased risk of developing colorectal cancer through inflammation-related mechanisms, which increases with the extent and duration of the disease (2). Both these processes, intestinal homeostasis and colitis-associated carcinogenesis, are closely associated with the presence of stimulatory signals derived from intestinal bacteria. In our study, we showed that defect in regulation of signaling cascades involved in bacteria recognition resulted in increased pro-inflammatory response and extensive colon tumorigenesis. This is in agreement with the fact that epithelial cell damage triggers the TLR/MyD88 dependent pathway to enhance regeneration of the epithelium and in the situation of IRAK-M deficiency, which means lack of negative regulation, can cause uncontrolled proliferation resulting in dysplasia and cancer (32). The suggestion that defect in IRAK-M regulation could play a role in inflammatory disease pathogenesis was later supported by Balaci et al., who found polymorphisms in genes encoding IRAK-M in early-onset persistent asthma patients (33). Nevertheless, the function of IRAK-M may not be the same in lung as it is in colon. Xie and coworkers in their study didn’t find any tumor growth in IRAK-M deficient mice after simple inoculation of tumor cells (34) and another recently published study about IRAK-M function in lung macrophages confirmed these results (35). Therefore, further research is needed to reveal the exact role of IRAK-M in different types of tissues. We have previously shown that the expression of IRAK-M in colon is low in GF condition and increases after colonization (18). One can hypothesize that changes in the type and level of antigenic stimulation after antibiotic treatment will increase the tumor development either because of lower negative regulation or by inhibition of pro-inflammatory signals. Surprisingly, we found that ATB treatment had protective effect on tumor development in wild-type mice. Subsequent investigation confirmed the important role of IRAK-M in tumorigenesis, because we showed that ATB treatment was not sufficient to decrease the tumor incidence in IRAK-M deficient mice. We also observed increased production of pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α), and COX-2 expression in the colons, and haptoglobin level in the sera of IRAK-M deficient mice, which suggests their increased sensitivity to inflammatory stimuli. These results suggest that not only microbiota composition but also fine tuning of MyD88-dependent signaling plays important role in the development of colon cancer in this model.
It is known that administration of ATB influences the diversity of intestinal microbiota (36). Using high-throughput pyrosequencing, we showed that the microbiota composition changes in both groups of ATB-treated and ATB-non-treated mice during tumor development. We showed that the lower tumor incidence is associated with these changes in the microbiota like an increase in proportions of Bacteroidetes and decrease in Firmicutes and Proteobacteria. The capability of certain microbes to metabolize carcinogens in the intestine can increase the exposure of the epithelium by cancer causing substances (5, 37). Bile acids can serve as an example of endogenous substances influencing tumor development. Mainly secondary bile acids, products of bacterial metabolism, are suspect to promote epithelial cells mutations and its transition to cancer (38, 39). High fat diet leads in increased secretion of bile and enhances its prolonged contact with the intestinal mucosa, which is, thus, exposed to oxidative stress, DNA damage, and activation of NF-κB pathway. Similar mechanisms are known for pro-carcinogen AOM, but may not be limited to this particular compound. The AOM is metabolized in liver to methylazoxy-methanol, which is conjugated with glucuronic acid and eliminated from organism in urine or bile. Epithelial and bacterial enzyme β-glucuronidase can dissociate the conjugate and release free methylazoxy-methanol, which is the main DNA methylating compound (28). We found that there is significant decrease in β-glucuronidase enzymatic activity after ATB treatment, which correlates with the decrease in tumor incidence, size and severity. Analysis of microbiota composition confirmed that our combination of ATB decreased the bacteria having β-glucuronidase activity – e.g. Clostridiaceae. It is important to mention, that β-glucuronidase could be produced also by gut epithelium (40). But since the β-glucuronidase activity is extremely low in GF mice, the majority of this activity seems to be generated by gut microbiota and not by epithelium. Changing the microbiota composition with ATB could therefore decrease the local production of β-glucuronidase, thus decreasing the exposure of gut epithelium to carcinogens.
The impaired regulation of TLR downstream pathway promotes the activation of mucosal and systemic immune response, which results in chronic pro-inflammatory stimulation. This effect, together with potent immune suppression, then leads to massive tumor progression (41). Here, we showed that IRAK-M deficient mice have lower levels of IFN-γ and decreased expression of IL-18 in their colons, which suggests inhibition of anti-tumor immunity. Host’s immune response is also tuned by regulatory T cells, which maintain the immune tolerance to harmless antigens in gut lumen; this is substantially influenced by microbiota presence and composition. As germ-free mice, which are known to have decreased counts of lymphocytes including regulatory T cells (42), have also reduced tumor incidence. Thus, immune suppression becomes a disadvantage in such disorders like cancer, when active suppression of immune response enables cancer cells to grow and spread (30). In our study, we found higher levels of Foxp3+ regulatory T cells in tumors and local lymph nodes of tumor-bearing mice as compared with healthy controls. We also observed higher counts of CD4+CD25−Foxp3+ cells in tumor-bearing mice suggesting induction of local suppression immune response. Interestingly, as compared with wild-type controls, IRAK-M deficient mice showed increased immune suppression. Our data are consistent with the observation made by Berglund et al. who found upregulated expression of Foxp3 transcription factor, a marker of regulatory T cells, in IRAK-M deficient mice during acute DSS-induced colitis (19). This suggests that impaired regulation of TLR downstream pathway renders IRAK-M deficient mice more susceptible to tumorigenesis due to the active immune suppression, which is enhanced by inflammation.
We conclude that metabolic activity of certain commensal microbes substantially influences the process of CAC. We showed that antibiotic treatment changes the microbiota composition, and that this change is responsible for the beneficial effect on tumorigenesis. IRAK-M deficient mice developed increased local and systemic pro-inflammatory response to microbial stimulation via TLR. Although the antibiotic treatment reduced the population of bacteria with beta-glucuronidase in the intestine, IRAK-M deficiency, enhancing inflammatory response of the host, led to the aggressive tumor development. We showed that IRAK-M is one of the important effector molecules promoting tumor resistance under the conditions of reduced carcinogen load. Further investigation is needed to reveal how host’s genetic background shapes the intestinal microbiota and its role in maintaining the balance in local immune response, and in the induction of inflammation or CAC. Therefore, understanding of this host-microbiota crosstalk could bring new strategies in IBD-related cancer therapy and prevention.
Acknowledgments
The study was supported by grants from the Czech Science Foundation (P304/11/1252, P303/08/0367, and P303/12/0535), and the Academy of Sciences of the Czech Republic (No. M200201207), NIH (R01DK074738 and R03TW006833), and Institutional Research Concept (RVO: 61388971).
The authors thank Dagmar Srutkova, Zaneta Ruzickova, Jan Svoboda, Jiri Dvorak and Tomas Vetrovsky for excellent technical assistance.
References
- 1.Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 2.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 4.Tlaskalova-Hogenova H, Stepankova R, Kozakova H, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. 2011;8:110–120. doi: 10.1038/cmi.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arthur JC, Jobin C. The struggle within: microbial influences on colorectal cancer. Inflamm Bowel Dis. 2011;17:396–409. doi: 10.1002/ibd.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tannock GW. The bowel microbiota and inflammatory bowel diseases. Int J Inflam. 2010;2010:954051. doi: 10.4061/2010/954051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsonnet J. Helicobacter pylori and gastric cancer. Gastroenterol Clin North Am. 1993;22:89–104. [PubMed] [Google Scholar]
- 8.Gionchetti P, Rizzello F, Lammers KM, et al. Antibiotics and probiotics in treatment of inflammatory bowel disease. World J Gastroenterol. 2006;12:3306–3313. doi: 10.3748/wjg.v12.i21.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thangaraju M, Cresci GA, Liu K, et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009;69:2826–2832. doi: 10.1158/0008-5472.CAN-08-4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barc MC, Charrin-Sarnel C, Rochet V, et al. Molecular analysis of the digestive microbiota in a gnotobiotic mouse model during antibiotic treatment: Influence of Saccharomyces boulardii. Anaerobe. 2008;14:229–233. doi: 10.1016/j.anaerobe.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Azcarate-Peril MA, Sikes M, Bruno-Barcena JM. The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol. 2011;301:G401–424. doi: 10.1152/ajpgi.00110.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 14.Fukata M, Abreu MT. TLR4 signalling in the intestine in health and disease. Biochem Soc Trans. 2007;35:1473–1478. doi: 10.1042/BST0351473. [DOI] [PubMed] [Google Scholar]
- 15.Salcedo R, Worschech A, Cardone M, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubbard LL, Moore BB. IRAK-M regulation and function in host defense and immune homeostasis. Infect Dis Rep. 2010:2. doi: 10.4081/idr.2010.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi K, Hernandez LD, Galan JE, et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 18.Biswas A, Wilmanski J, Forsman H, et al. Negative regulation of Toll-like receptor signaling plays an essential role in homeostasis of the intestine. Eur J Immunol. 2011;41:182–194. doi: 10.1002/eji.201040479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berglund M, Melgar S, Kobayashi KS, et al. IL-1 receptor-associated kinase M downregulates DSS-induced colitis. Inflamm Bowel Dis. 2010;16:1778–1786. doi: 10.1002/ibd.21287. [DOI] [PubMed] [Google Scholar]
- 20.Xiao H, Gulen MF, Qin J, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26:461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 21.Vannucci L, Stepankova R, Kozakova H, et al. Colorectal carcinogenesis in germ-free and conventionally reared rats: Different intestinal environments affect the systemic immunity. Int J Oncol. 2008;32:609–617. [PubMed] [Google Scholar]
- 22.Stepankova R, Powrie F, Kofronova O, et al. Segmented filamentous bacteria in a defined bacterial cocktail induce intestinal inflammation in SCID mice reconstituted with CD45RBhigh CD4+ T cells. Inflamm Bowel Dis. 2007;13:1202–1211. doi: 10.1002/ibd.20221. [DOI] [PubMed] [Google Scholar]
- 23.Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007;28:1450–1459. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 24.Kverka M, Zakostelska Z, Klimesova K, et al. Oral administration of Parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. Clin Exp Immunol. 2011;163:250–259. doi: 10.1111/j.1365-2249.2010.04286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zakostelska Z, Kverka M, Klimesova K, et al. Lysate of Probiotic Lactobacillus casei DN-114 001 Ameliorates Colitis by Strengthening the Gut Barrier Function and Changing the Gut Microenvironment. PLoS One. 2011;6:e27961. doi: 10.1371/journal.pone.0027961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen XJ, Rawls JF, Randall T, et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1:138–147. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiala E. Investigations into the metabolism and mode of action of the colon carcinogen 1, 2-dimethylhydrazine. Cancer. 1975;36:2407–2412. doi: 10.1002/1097-0142(197512)36:6<2407::aid-cncr2820360620>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 29.Chen W, Perruche S, Li J. CD4+CD25+ T regulatory cells and TGF-beta in mucosal immune system: the good and the bad. Curr Med Chem. 2007;14:2245–2249. doi: 10.2174/092986707781696591. [DOI] [PubMed] [Google Scholar]
- 30.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 31.Murai M, Krause P, Cheroutre H, et al. Regulatory T-cell stability and plasticity in mucosal and systemic immune systems. Mucosal Immunol. 2010;3:443–449. doi: 10.1038/mi.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asquith M, Powrie F. An innately dangerous balancing act: intestinal homeostasis, inflammation, and colitis-associated cancer. J Exp Med. 2010;207:1573–1577. doi: 10.1084/jem.20101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balaci L, Spada MC, Olla N, et al. IRAK-M is involved in the pathogenesis of early-onset persistent asthma. Am J Hum Genet. 2007;80:1103–1114. doi: 10.1086/518259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie Q, Gan L, Wang J, et al. Loss of the innate immunity negative regulator IRAK-M leads to enhanced host immune defense against tumor growth. Mol Immunol. 2007;44:3453–3461. doi: 10.1016/j.molimm.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Standiford TJ, Kuick R, Bhan U, et al. TGF-beta-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene. 2011;30:2475–2484. doi: 10.1038/onc.2010.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dethlefsen L, Huse S, Sogin ML, et al. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamer HM, De Preter V, Windey K, et al. Review article: Functional analysis of colonic bacterial metabolism: Relevant to health? Am J Physiol Gastrointest Liver Physiol. 2011 doi: 10.1152/ajpgi.00048.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venturi M, Hambly RJ, Glinghammar B, et al. Genotoxic activity in human faecal water and the role of bile acids: a study using the alkaline comet assay. Carcinogenesis. 1997;18:2353–2359. doi: 10.1093/carcin/18.12.2353. [DOI] [PubMed] [Google Scholar]
- 39.Bernstein C, Holubec H, Bhattacharyya AK, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011;85:863–871. doi: 10.1007/s00204-011-0648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rod TO, Midtvedt T. Origin of intestinal beta-glucuronidase in germfree, monocontaminated and conventional rats. Acta Pathol Microbiol Scand B. 1977;85:271–276. doi: 10.1111/j.1699-0463.1977.tb01973.x. [DOI] [PubMed] [Google Scholar]
- 41.Vannucci L, Stepankova R, Grobarova V, et al. Colorectal carcinoma: Importance of colonic environment for anti-cancer response and systemic immunity. J Immunotoxicol. 2009;6:217–226. doi: 10.3109/15476910903334343. [DOI] [PubMed] [Google Scholar]
- 42.Hrncir T, Stepankova R, Kozakova H, et al. Gut microbiota and lipopolysaccharide content of the diet influence development of regulatory T cells: studies in germ-free mice. Bmc Immunol. 2008;9 doi: 10.1186/1471-2172-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartosch S, Fite A, Macfarlane GT, et al. Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Appl Environ Microbiol. 2004;70:3575–3581. doi: 10.1128/AEM.70.6.3575-3581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreader CA. Design and evaluation of Bacteroides DNA probes for the specific detection of human fecal pollution. Appl Environ Microbiol. 1995;61:1171–1179. doi: 10.1128/aem.61.4.1171-1179.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]