

Abstract
We have previously reported that the green tea polyphenol epigallocatechin-3-gallate (EGCG) and the epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) erlotinib had synergistic growth inhibitory effects in cell culture and a nude mouse xenograft model of squamous cell carcinoma of the head and neck (SCCHN). However, the mechanism of their anti-tumor synergism is not fully understood. In the current study, we investigate the mechanism of their synergistic growth inhibitory effects. The treatment of SCCHN cell lines with erlotinib time-dependently increased the expression of cell cycle regulatory proteins p21 and p27 and apoptosis regulatory protein Bim. EGCG alone had very little or no effect on the expression of these proteins among the cell lines. However, simultaneous treatment with EGCG and erlotinib strongly inhibited erlotinib-induced expression of p21 and p27 without affecting the expression of Bim. Moreover, erlotinib increased the expression of p53 protein, the ablation of which by shRNA strongly inhibited EGCG- and erlotinib-mediated growth inhibition and the expression of 21, p27 and Bim. In addition, combined treatment with erlotinib and EGCG inhibited the protein level of p65 subunit of NF-κB and its transcriptional target Bcl-2, but failed to do so in cells with ablated p53. Taken together our results, for the first time, suggest that erlotinib treatment activates p53, which plays a critical role in synergistic growth inhibition by erlotinib and EGCG via inhibiting NK-κB signaling pathway. Characterizing the underlying mechanisms of EGCG and erlotinib synergism will provide an important rationale for chemoprevention or treatment trials using this combination.
Keywords: Chemoprevention, EGFR-TKI, molecular targets, p53
Introduction
Squamous cell carcinoma of the head and neck (SCCHN), a serious healthcare problem in the United States and worldwide, is one of the deadliest of all cancers with more than 45,000 new cases diagnosed each year in the United States and 15,000 deaths annually (1). SCCHN is the sixth most common cancer worldwide and accounts for 3% of all cancers in the United States (1). Despite advances in conventional therapies, including surgery, radiation and chemotherapy, the overall survival rate for SCCHN has not been significantly improved in the past several decades (2). These cancers generally begin as small and often unnoticed lesions inside the mouth. More than a third of untreated precancerous oral lesions undergo malignant transformation into squamous cell cancer. Moreover, a large fraction of these precancerous lesions recur despite complete surgical removal, and the specter of second primary tumor (SPT) development, which occurs in as many as 3–7% of cases, continues to haunt patients who have been successfully treated for their primary tumor (3). It is therefore highly desirable to develop effective preventive approaches using specific natural or synthetic chemical compounds to reduce the incidence of SCCHN.
The tumor suppressor protein p53, which was originally identified as a transcription factor, plays a pivotal role in controlling the cell cycle, apoptosis, genomic integrity, and DNA repair in response to various forms of genotoxic stress. The regulation of p53 is complex and occurs mainly at the post-translational level via multiple phosphorylation and acetylation events that contribute to its stabilization and activation (4, 5). It is widely believed that differently modified forms of p53 differentially regulate patterns of gene expression, which then drive distinct biological responses. p53 can be activated in response to DNA damage (by ATM and Chk2), aberrant growth signals (by p14ARF), and by chemotherapeutic or chemopreventive drugs (6–10). After activation, p53 can bind to regulatory DNA sequences of the target genes and activate their transcription. p53 target genes can be functionally grouped into four categories: cell cycle inhibition (p21, reprimo, cyclin G1, GADD45, and 14-3-3), apoptosis (PERP, NOXA, PUMA, p53AIP1, ASPP1/2, Fas, Bax, and PIDD), genetic stability (p21, DDB2, MSH2, and XPC) and inhibition of angiogenesis (TSP1, Maspin, BAI1, and GD-AIF) (6–11). In addition to its transactivation function, p53 can also act as transrepressor. Accumulating evidence suggests that transcriptional repression of several genes by p53 is critically important for carrying out its functions (12–14). Due to these roles, p53 is regarded as “molecular guardian” of the genome.
Overexpression of epidermal growth factor receptor (EGFR) and its ligands transforming growth factor-α (TGF-α) and epidermal growth factor (EGF) are very common in SCCHN and occur in 80–90% cases, correlating with poor disease-free and overall survival and an increased risk of disease recurrence and metastasis (15–19). Activation of EGFR leads to phosphorylation and activation of phosphatidylinositol 3-kinase (PI3K)/Akt signaling, which in turn activates nuclear factor-κB (NF-κB), leading to the transcription of genes regulating growth, apoptosis, angiogenesis, and invasion and those responsible for chemoresistance and radiation-resistance (20). Transcriptional targets of NF-κB include Bcl-2 and Bcl-xL (21, 22), cyclooxygenase 2 (23), and survivin (24).
Erlotinib (OSI-774 or Tarceva), an EGFR tyrosine kinase inhibitor (TKI), has strong antitumor and promising chemopreventive efficacies in a variety of cancer types including SCCHN. It is currently being tested in the clinic either as single agent or in combination with other drugs (25–26). Heterogeneity of the tumor cell population, redundant cellular growth, and constitutive activation of survival pathways severely limit the efficacy of erlotinib monotherapy. Epigallocatechin-3-gallate (EGCG), the most abundant and most active polyphenol present in green tea, has been extensively studied for its chemopreventive and antitumor effects in chemical-induced rodent carcinogenesis models and in cell culture (8, 12, 27–30). We have previously reported that the combination of erlotinib and EGCG is a promising strategy for chemoprevention of SCCHN (30). The combination of the two compounds synergistically inhibited growth of SCCHN by inducing cell-cycle arrest and apoptosis (30). However, the mechanism of this synergistic growth inhibition was incompletely characterized. In the present manuscript, for the first time, we have demonstrated that erlotinib induces the expression of cell cycle regulatory proteins p21 and p27, and the induction of apoptosis regulatory protein Bim, in a p53-dependent manner. EGCG enhances the proapoptotic potential of erlotinib by inhibiting the expression of p21 and p27 without affecting the expression of Bim. Moreover, we have identified crosstalk between p53 and NF-κB signaling pathways induced by these compounds, which suggests that p53 inhibits expression of the p65 subunit of NF-κB and its gene product Bcl-2.
Materials and Methods
Cell lines
Tu177 and Tu212 cell lines established from a laryngeal and hypopharyngeal tumor, respectively, were provided by Dr. Gary L. Clayman (University of Texas M.D. Anderson Cancer Center, Houston, TX). Tu686 and 686LN are paired cell lines from a primary tongue cancer and its lymph node metastasis, respectively. These were gifts from Dr. Peter G. Sacks (New York University College of Dentistry). 1986LN and 886LN cell lines, also provided by Dr. Peter G. Sacks, were derived from lymph node metastasis of squamous cell carcinomas of the tongue and larynx, respectively. SQCCY1 and SCC38 cell lines derived from oral cavity and tonsil fossa, respectively, were obtained from Dr. Shi-Yong Sun (Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA). M4e cell was derived from 686LN via an in vivo selection in nude mice (31). All cell lines were maintained in DMEM/F12 (1:1) medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (streptomycin and penicillin G) in a 37°C, 5% CO2 humidified incubator.
Reagents
EGCG (Sigma Chemical, St. Louis, MO) and erlotinib (Genentech, South San Francisco, CA) were dissolved in autoclaved water and dimethyl sulphoxide (DMSO), respectively, as stock solutions for in vitro studies. The reagents were further diluted in DMEM/F12 medium immediately before use. The final concentration of DMSO was less than 0.1%.
Cell growth inhibition assay
To test the effects of single agent EGCG and erlotinib on cell growth of SCCHN, sulforhodamine B (SRB) cytotoxicity assays were adapted from Skehan et al. (32). Cells maintained in medium with 5% FBS were seeded in 96-well plates at a density of 4,000 cells/well overnight prior to drug treatment. Afterwards, drugs were added as single agents in various concentrations (30 μM for EGCG and 2 μM for erlotinib), followed by incubation at 37°C and 5% CO2 for 96 hr. Cells were fixed for 1 hr with 10% cold trichloroacetic acid. Plates were washed 5 times in water, air-dried and then stained with 0.4% SRB for 10 min. After washing 4 times in 1% acetic acid and air-drying, bound SRB was dissolved in 10 mM unbuffered Tris base (pH 10.5). Plates were read in a microplate reader by measuring absorbance at 492 nm. The percent survival was then calculated based upon the absorbance values relative to the untreated samples.
Annexin V-PE staining for apoptosis
Tu686 cells were treated with EGCG (30 μM), erlotinib (2 μM) or their combination (EGCG 30 μM + erlotinib 2 μM) for 4 days, then trypsinized and washed in cold 1× PBS. The cells were then resuspended in 1× Annexin binding buffer (BD PharMingen), and then stained with Annexin V-phycoerythrin (Annexin V-PE; BD PharMingen) and 7-AAD (BD PharMingen) for 15 min at room temperature. The stained samples were measured using a fluorescence-activated cell sorting (FACS) caliber bench-top flow cytometer (Becton Dickinson, Franklin Lakes, NJ). FlowJo software (Tree Star, Ashland, OR) was used for apoptosis analysis.
Western blot analysis
Whole cell lysates were extracted from drug-treated cells using lysis buffer. 25 μg of protein was separated on 8–12% SDS-PAGE, transferred onto a PVDF membrane (Millipore, Bedford, MA) and immunoblotted with specific antibodies. Mouse anti-β-actin antibody (Trevigen, Gaithersburg, MD) was used as a sample loading control. Immunostained protein bands were detected with an enhanced chemiluminescence kit (Amersham, Buckinghamshire, UK).
Transfection of packaging cells for viral production and infection of cells with virus
Packaging cells 293T were plated in 10 cm plates at a cell density of 5×106 a day prior to transfection in DMEM containing 10% heat inactivated FBS but no antibiotics. shp53 and shGFP constructs in lentivirus vector were generous gifts from Dr. Didier Trono, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland. Transfection of packaging cells and infection of mammalian cells were carried out using standard protocols described in http://tronolab.com/index.php. In brief, 293T cells were transfected with ~6 μg of plasmids (1.6 μg pCMV-dR8.74, 1 μg pMD2G and ~3 μg of lentiviral vector) using lipid transfection (lipofectamin/Plus reagent, Invitrogen Corp) as per the protocol supplied by the manufacturer. The virus-containing media was used for infecting Tu686 cells.
Results
EGCG inhibits erlotinib-induced expression of cell cycle regulatory proteins
In our earlier study, we showed that erlotinib and EGCG had synergistic growth inhibitory properties. Moreover, our study suggested that treatment of SCCHN with erlotinib time-dependently induced G1 arrest with little apoptosis (30). The presence of EGCG had time-dependent effects on erlotinib response. Initially, EGCG slightly increased erlotinib-induced G1 arrest and later increased apoptosis (30). In order to study the mechanism of cell cycle arrest, we examined the expression of cell-cycle regulatory proteins p21 and p27 in a panel of SCCHN cell lines. As expected, erlotinib time-dependently increased the expression of both p21 and p27 in all except two of the cell lines tested, 886LN and M4e. Both 886LN and M4e lack detectable p53 expression. Treatment with EGCG alone also increased p21 expression to some extent in most cell lines, but less than that induced by erlotinib (Fig. 1 and Fig. 4E). This effect of EGCG on p21 and p27 expression is corroborated by the potentiation of erlotinib-induced cell cycle arrest by EGCG at earlier time points. However, combined treatment with erlotinib and EGCG inhibited the expression of p21 and p27 as compared with erlotinib treatment alone (Fig. 1 and Fig. 4E). These results suggest that EGCG inhibited erlotinib-induced expression of p21 and p27.
Figure 1.
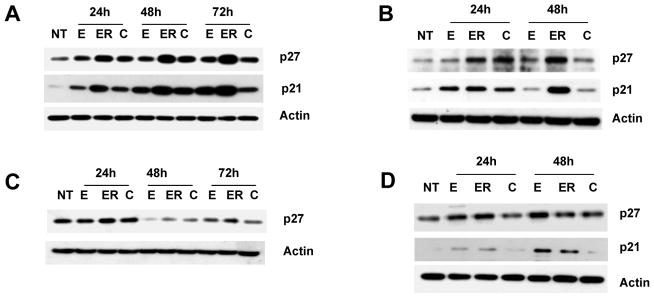
Inhibition of erlotinib-induced cell cycle regulatory protein p21 and p27 by EGCG. (A) Tu686 (B) 1986LN (C) 886LN and (D) SQCCY1 cells were treated with 30 μM EGCG (E), 0.5 μM erlotinib (ER) or a combination of 30 μM EGCG and 0.5 μM erlotinib (C) for the indicated times. Total cell lysates were immunoblotted with anti-p27 (Santa Cruz) and anti-p21 (Santa Cruz). NT is untreated cells. Reproducibility of the data were confirmed with at least three independent experiments for each cell lines.
Figure 4.

p53 is required for the growth inhibitory effects of erlotinib, EGCG and their combination. (A) Tu686 and M4e cells were treated with 2 μM erlotinib, 30 μM EGCG and a combination of 2 μM of erlotinib and 30 μM EGCG for 120 h, stained with annexin V-PE and the apoptotic population was determined by flow cytometry. (B) The expression of p53 was knocked down in Tu686 cells by a lentivirus-based shRNA specific for p53. shGFP was used as control. Expression of p53 was determined in total cell lysates. (C) Tu686 cells transduced with shGFP and shp53 were treated with 2 μM erlotinib, 30 μM EGCG and a combination of 2 μM of erlotinib and 30 μM EGCG for 72 h. Expression of p27, p21 and Bim in the total cell lysates was measured by Western blotting. (D) Upper panel: shGFP- and shp53-transduced cells were treated with 2 μM erlotinib, 30 μM EGCG and a combination of 2 μM of erlotinib and 30 μM EGCG for 96 h. Cell growth was measured by SRB assay. Experiments were done in triplicate for each agent and reproducibility was confirmed by multiple independent experiments. Lower panel: cells were treated as in upper panel and the plates were stained with methylene blue. (E) 686LN and M4e cells were treated with 0.5 μM erlotinib, 30 μM EGCG and a combination of 0.5 μM of erlotinib and 30 μM EGCG for 72 h and expression of p21 and p27 were determined by Western blotting.
Erlotinib and EGCG induce the expression of proapoptotic protein Bim
We have reported previously that combined treatment with erlotinib and EGCG synergistically induced apoptosis of SCCHN (30). To understand the mechanism of apoptosis, we examined the expression of Bim after treatment with erlotinib and EGCG, either as a single agent or in combination. Bim is a proapoptotic protein previously reported to be induced by EGFR-TKI to mediate apoptosis (33, 34). As shown in Figure 2, treatment with erlotinib time-dependently increased the expression of Bim in a number of SCCHN cell lines. EGCG as a single agent had very minimal effect on the expression of Bim (Figure 2). Unlike the expression of p21 and p27, combined treatment with erlotinib and EGCG did not inhibit erlotinib-induced expression of Bim. In some cell lines, such as 886LN, combined treatment with EGCG and erlotinib increased the expression of Bim as compared with either agent.
Figure 2.
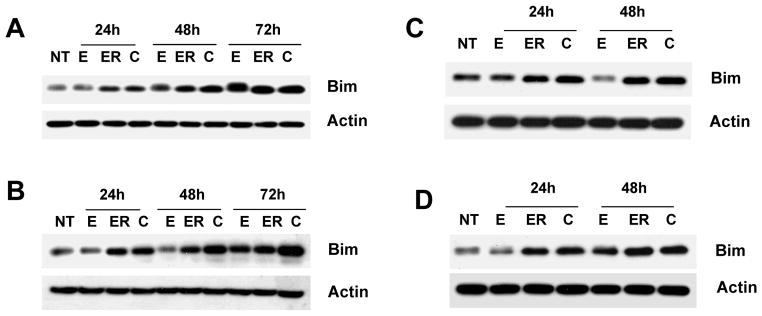
Expression of Bim following erlotinib and EGCG treatment. (A) Tu686 (B) 886LN (C) Tu212 and (D) SCC38 cells were treated as in Figure 1. Total cell lysates were immunoblotted with anti-Bim (abcam). Reproducibility of the data were confirmed with at least three independent experiments for each cell lines.
p53 is required for the growth inhibition by erlotinib and EGCG
Since p21 and p27 are well established transcriptional targets of p53 and the Bim promoter has a binding site for p53, we next examined the expression of p53 in SCCHN. As shown in Figure 3A, treatment with erlotinib time-dependently increased the protein level of p53 in SCCHN cell lines. EGCG alone had variable effects on the expression of p53, depending on the cell type. The transcriptional activity of p53 is regulated by complex posttranslational modifications by phosphorylation and acetylation (4, 5). Next, we tested the phosphorylation of p53 at Ser15 after treatment with erlotinib and EGCG, a site frequently phosphorylated by stress signaling and that regulates cell cycle arrest and apoptosis. Treatment with both erlotinib and EGCG increased the phosphorylation of p53 at Ser15 in Tu686 cells, which was more potent after combined treatment (Figure 3B). To further explore whether p53 is important for the growth inhibitory and apoptotic effect induced by erlotinib, EGCG and their combination, we measured apoptosis of Tu686 and M4e cells by annexin V-PE staining. As shown in Figure 4A, EGCG alone induced almost no apoptosis (7.98% as compared to 4.84% in the untreated control) of Tu686 cells when compared with untreated control. However, erlotinib alone induced moderate apoptosis (30.38%), which was further increased after combination with EGCG (78.66%). In contrast, p53-negative M4e cells were resistant to apoptosis induced by EGCG, erlotinib or their combination. These results suggest that erlotinib and EGCG have synergistic/additive apoptotic effect in cells with wild-type p53, while p53-null cells are resistant.
Figure 3.

Expression of p53 following erlotinib and EGCG treatment. (A) SCCHN cells were treated as in Figure 1 and total cell lysates were immunoblotted with anti-p53 (Santa Cruz). (B) Tu686 cells were treated with 1 μM erlotinib, or 30 μM EGCG or a combination of 1 μM erlotinib and 30 μM EGCG. Total cell lysates were immunoblotted with anti-phospho p53 (Ser 15). Reproducibility of the data were confirmed with three independent experiments.
To further confirm the role of p53 in EGCG- and erlotinib-induced apoptosis, we downregulated the expression of p53 in Tu686 cells using a lentivirus-based shRNA construct, and a pool of cells with ablated p53 was established by GFP selection (Figure 4B). Cells transduced with shGFP were used as control. These cells were treated with erlotinib, EGCG or their combination and total cell lysates were used to study the expression of p21, p27 and Bim by Western blotting. As shown in Figure 4C, treatment with erlotinib induced the expression of p27, p21 and Bim in cells transduced with shGFP. However, in cells with ablated p53, there were very little or no increase in the levels of p27, p21, and Bim, suggesting that p53 is transcriptionally active in this cell line and is required for the erlotinib-induced expression of p21, p27, and Bim. The combination of EGCG with erlotinib inhibited erlotinib-induced p27 and p21 but not Bim expression in shGFP-transduced cells. This result is consistent with the previous results described in Figure 1 and 2. Furthermore, the basal level of p27 was increased after p53 knockdown. This is consistent with the observations in 686LN and M4e cells (Figure 4E). Both Tu686 transduced with shp53 and M4e cells have high proliferation rates and since they are not affected by the treatments, become confluent at the time of harvesting. Confluent cells were arrested in G1 phase and this might be responsible for the increased p27 level.
We next examined the growth of these cells after treatment with erlotinib, EGCG, and their combination by SRB assay and methylene blue staining. As shown in Figure 4D, treatment with erlotinib strongly inhibited the growth of shGFP-transduced cells, but not of shp53-transduced cells. EGCG also inhibited the growth of control cells to some extent, but not of p53 knockdown cells. The combination of erlotinib and EGCG more potently inhibited the growth of shGFP cells, but only slightly of shp53 cells. These results suggest that p53 is required for the growth inhibitory effects of erlotinib, EGCG, and their combination. In order to further confirm the role of p53 in erlotinib- and EGCG-mediated expression of p21, p27 and Bim, we employed another cell line, M4e, which originated from 686LN and lost p53 during natural selection in nude mice (31). These cells were treated with the compounds and total cell lysates were used to study the expression of p27, p21, and Bim. As shown in Figure 4E, there was no increase in the levels of p21, p27, or Bim after treatment with both compounds alone or in combination in M4e cells. These cells were also resistant to erlotinib- and EGCG-mediated growth inhibition (Fig. 4A). These results again suggest that p53 is required for erlotinib- and EGCG-mediated growth inhibition and for the expression of p21, p27, and Bim.
p53-dependent inhibition of NF-κB by erlotinib and EGCG
We next examined the protein levels of the p65 subunit of NF-κB and its transcriptional target Bcl-2 in Tu686 cells after treatment with erlotinib, EGCG, and their combinations. As shown in Figure 5A, treatment with erlotinib or the combination of erlotinib and EGCG inhibited the protein level of both p65 and Bcl-2. To investigate any possible crosstalk between p53 and NF-κB pathways, we next examined the expression of p65 and Bcl-2 in Tu686 cells with ablated p53. Treatment with the compounds efficiently inhibited p65 and Bcl-2 in cells with shGFP, but not in cells with shp53, suggesting that the inhibition of NF-κB pathway by erlotinib and EGCG is mediated via p53 (Figure 5B and C). To further confirm that p53 is required for NF-κB inhibition, we examined the expression of p65 and Bcl-2 in M4e cells. Both erlotinib and EGCG or their combination failed to inhibit p65 and Bcl-2 in M4e cells (Figure 5D). These results again confirm that p53 is required for the inhibition of NF-κB pathway by erlotinib, EGCG or the combination of erlotinib and EGCG.
Figure 5.
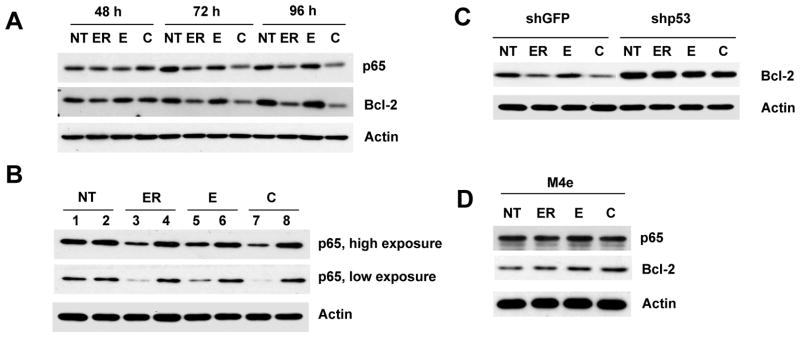
p53-dependent inhibition of NF-κB. (A) Tu686 cells were treated with 2 μM erlotinib, 30 μM EGCG and a combination of 2 μM of erlotinib and 30 μM EGCG for the indicated times. Expression of p65 and Bcl-2 was measured by immunoblotting. (B) shGFP- and shp53-transduced Tu686 cells were treated with 2 μM erlotinib, 30 μM EGCG and a combination of 2 μM of erlotinib and 30 μM EGCG for 72 h. Total cell lysates were immunoblotted with anti-p65. Lanes 1, 3, 5, 7 are shGFP and lanes 2, 4, 6, 8 are shp53. (C) Cells were treated as in Figure 5B and total cell lysates were used for the expression of Bcl-2. (D) M4e cells were treated with 0.5 μM erlotinib, 30 μM EGCG and a combination of 0.5 μM of erlotinib and 30 μM EGCG for 48 h and total cell lysates were immunoblotted with anti-p65 and anti-Bcl-2. Reproducibility of all results were confirmed by three independent experiments.
Discussion
EGFR is overexpressed in 80–90% of SCCHN and is one of the best established and most widely accepted biomarker for premalignant lesions of the head and neck and SCCHN (15–19). Thus, inhibition of EGFR by using antibodies or specific EGFR-TKI such as erlotinib is a promising approach for prevention and therapy of SCCHN. Unfortunately, only a very small percentage of patients (~5%) respond to EGFR-TKI therapy even if they have high EGFR expression (35, 36). In order to make erlotinib therapy clinically more relevant and to select the optimal patients who could benefit from this approach, it is therefore important to understand the mechanism of erlotinib-mediated growth inhibition and to identify agents that could additively/synergistically increase the efficacy of erlotinib. We have previously reported that EGCG had a synergistic growth inhibitory effect with erlotinib (30). In the current study, we have demonstrated several novel aspects of erlotinib-mediated cell signaling and the mechanism of synergistic growth inhibition by erlotinib and EGCG. Here, we have shown for the first time that erlotinib activates p53 and that erlotinib- and EGCG-induced cell-cycle arrest and apoptosis are mediated via p53. Knockdown of p53 protected cells from erlotinib- and EGCG-mediated growth inhibition (Fig. 4). Previously, we tested five SCCHN cell lines, four of which showed synergistic response to the combination of erlotinib and EGCG at all doses, while the 886LN cell line, which has no detectable p53 expression (30), showed synergism only at higher doses of the combined agents. Moreover, the M4e cell line, which lost p53 during natural selection in mice (31), was resistant to the erlotinib and EGCG combination, whereas the parental cell line 686LN was sensitive. Together, these results suggest that p53 has some role in erlotinib- and EGCG-induced anti-tumor effect.
Second, we showed that p53 mediated erlotinib- and EGCG-induced expression of p21, p27, and Bim. p21 and p27 are well established effectors that induce G1 and G2/M arrest in response to various genotoxic stress. A growing body of evidence suggests that erlotinib induces the expression of p21 and p27 (37–39). Moreover, several recent studies demonstrated that erlotinib-induced apoptosis is mediated by Bim in non small cell lung cancers (NSCLC) (33, 34). However, the mechanism of induction of p21/p27 and Bim by erlotinib is unknown. Ablation of p53 by shRNA inhibited the expression of p21, p27 and Bim. In two more cell lines (886LN and M4e) which lack detectable expression of p53, erlotinib failed to induce p21 and p27, and Bim. Again, these cells are resistant to erlotinib or a combination of erlotinib and EGCG. These results suggest that p53 certainly plays a role in erlotinib and EGCG response in SCCHN cell lines. However, erlotinib also induced the expression of p21, p27, and Bim in SCCHN cell lines with mutated p53 (Tu177 and Tu212, data not shown). In addition to p53, these proteins can be induced by other p53 family members such as p73 (40) and by the members of FOXO (41), and some members of STAT family of transcription factors (42). Further studies are warranted to study the mechanism of expression of these proteins in SCCHN with mutated p53.
Third, we explored the mechanism of anti-tumor synergy exhibited by erlotinib and EGCG. The overall response of cells to a particular stress is the balance between two sets of proteins, such as the cell cycle regulatory proteins like p21 and p27, and proapoptotic proteins like Bim, and is dependent on the magnitude of stress (dose). At lower dose, the growth arrest signal generally predominates over the apoptotic signal. But at higher dose, the apoptotic signal outplays the growth arrest signal. It is possible to shift the phenotype from growth arrest to apoptosis by inhibiting the targets responsible for arrest, such as p21. Several previous studies support this hypothesis. For example, expression of p53 in colorectal cancer cells induces both p21 and Puma, but cells undergo growth arrest through p21. Disruption of p21 via gene targeting induces apoptosis of these cells in a Puma-dependent manner (43). Inactivation of p21 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl-2 ratio in response to chemotherapeutic drugs (44). Some studies also suggest that absence of p21 favors apoptosis (45). Consistent with these studies, our results suggest that erlotinib induced the expression of cell-cycle regulatory proteins p21, p27, and apoptosis regulatory protein Bim. However, cells underwent G1 arrest with minimal apoptosis in response to erlotinib. Simultaneous use of erlotinib and EGCG time-dependently inhibited erlotinib-induced expression of p21 and p27 without affecting the expression of Bim and the phenotype shifted from growth arrest to apoptosis. How EGCG inhibits p21 and p27 remains to be elucidated. However, it is also possible that erlotinib protects Bim from EGCG-mediated inhibition by stabilizing it via posttranslational modification. Particularly, studies conducted by Gong et al. suggest that erlotinib not only induces Bim, but also stabilizes it by posttranslational modification (35).
Finally, we explored a novel erlotinib-dependent crosstalk between p53 and NF-κB signaling. We found that treatment with erlotinib and EGCG inhibited the protein level of p65 subunit of NF-κB and its target Bcl-2 in Tu686 cells with p53. However, these compounds failed to inhibit p65 and Bcl-2 after ablation of p53 by shRNA. Moreover, the compounds also failed to inhibit NF- B (p65) and Bcl-2 in M4e cells. Both M4e and Tu686 cells with knockdown p53 were resistant to the compounds. These results suggest that p53 is required for the inhibition of NF-κB signaling by erlotinib and EGCG. It was reported previously that constitutive activation of NF-κB signaling prevented the transcriptional activation of p53, and the inactivation of NF-κB activates p53 to induce apoptosis (46). Other studies suggest that activation of p53 is required for inhibition of NF-κB signaling (47, 48). NF-κB plays a central role in the regulation of apoptotic pathways. Activation of NF-κB gene products in cancer cells is one of the major causes of chemoresistance. Inhibition of NF-κB gene products particularly Bcl-2, Bcl-xL and survivin favors apoptosis. Several studies suggest that inhibition of AKT is important for the inhibition of NF-κB (49, 50). AKT activates IκB kinase (IKK), which phosphorylates IκB leading to the activation of NF-κB. Our study suggests that inhibition of NF-κB by erlotinib and EGCG is not mediated by AKT since the compounds also inhibit AKT in cells with ablated p53 (data not shown). The decrease in the level of NF-κB (p65) by erlotinib and EGCG suggest that the inhibition might be due to proteosomal degradation or at the transcriptional level.
In conclusion, we have shown that erlotinib induces cell-cycle and apoptosis via p53-dependent induction of p21, p27, and Bim, and via p53-dependent inhibition of NF-κB and its anti-apoptotic target Bcl-2 in some SCCHN cell lines. The potentiation of erlotinib effects by EGCG is mediated via inhibition of the cell-cycle regulatory signaling pathway. Drug-associated toxicities arising from high-dose single agent intervention continuously pose challenges for the development of effective chemoprevention strategies. It was established by multiple studies in preclinical and clinical settings that combinations of two or more compounds are effective in lower doses with minimal toxicities (51, 52). By describing the mechanism of synergy between these compounds as well as uncovering how they induce p53-dependent apoptosis, we have made the case for the rational combination of erlotinib with green tea polyphenols for further preclinical and clinical development. Indeed, a phase I clinical trial using low dose erlotinib (100 mg TID) plus EGCG has been planned in an attempt to prevent carcinogenesis in patients with premalignant lesions of the head and neck at Emory Winship Cancer Institute and another phase I/II trial in advanced non-small cell lung cancer is ongoing at Louisiana State University (NCT00707252).
Acknowledgments
This work was supported by grants from the NIH (P50 CA128613, U01 CA101244, and R01 CA112643). DMS, GC and FRK are Distinguished Cancer Scholars of the Georgia Cancer Coalition (GCC). We also wish to give our thanks and appreciation to Dr. Didier Trono for providing the lentivirus based gene silencing system and Dr. Anthea Hammond for her critical and editorial review of this article.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008;359:1143–54. doi: 10.1056/NEJMra0707975. [DOI] [PubMed] [Google Scholar]
- 3.Chuang SC, Scelo G, Tonita JM, et al. Risk of second primary cancer among patients with head and neck cancers: A pooled analysis of 13 cancer registries. Int J Cancer. 2008;123:2390–6. doi: 10.1002/ijc.23798. [DOI] [PubMed] [Google Scholar]
- 4.Olsson A, Manzl C, Strasser A, Villunger A. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 2007;14:1561–75. doi: 10.1038/sj.cdd.4402196. [DOI] [PubMed] [Google Scholar]
- 5.Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–21. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000;10:94–9. doi: 10.1016/s0959-437x(99)00038-6. [DOI] [PubMed] [Google Scholar]
- 7.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485–95. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 8.Hastak K, Agarwal MK, Mukhtar H, Agarwal ML. Ablation of either p21 or Bax prevent p53-dependent apoptosis induced by green tea polyphenol epigallocatechin-3-gallate. FASEB J. 2005;19:789–91. doi: 10.1096/fj.04-2226fje. [DOI] [PubMed] [Google Scholar]
- 9.Shin DM, Lee JS, Lippman SM, et al. p53 expressions: predicting recurrence and second primary tumors in head and neck squamous cell carcinoma. J Natl Cancer Inst. 1996;88:519–29. doi: 10.1093/jnci/88.8.519. [DOI] [PubMed] [Google Scholar]
- 10.Lippman SM, Shin DM, Lee JJ, et al. p53 and retinoid chemoprevention of oral carcinogenesis. Cancer Res. 1995;55:16–9. [PubMed] [Google Scholar]
- 11.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 12.Agarwal MK, Amin AR, Agarwal ML. DNA replication licensing factor minichromosome maintenance deficient 5 rescues p53-mediated growth arrest. Cancer Res. 2007;67:116–21. doi: 10.1158/0008-5472.CAN-06-2835. [DOI] [PubMed] [Google Scholar]
- 13.Hammond EM, Mandell DJ, Salim A, et al. Genome-wide analysis of p53 under hypoxic conditions. Mol Cell Biol. 2006;26:3492–504. doi: 10.1128/MCB.26.9.3492-3504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho JS, Ma W, Mao DY, Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol. 2005;2005:7423–31. doi: 10.1128/MCB.25.17.7423-7431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666–72. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- 16.Kim S, Grandis JR, Rinaldo A, Takes RP, Ferlito A. Emerging perspectives in epidermal growth factor receptor targeting in head and neck cancer. Head Neck. 2008;30:667–74. doi: 10.1002/hed.20859. [DOI] [PubMed] [Google Scholar]
- 17.Grandis JR, Melhem MF, Gooding WE, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90:824–32. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 18.Cohen EE. Role of epidermal growth factor receptor pathway-targeted therapy in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2006;24:2659–65. doi: 10.1200/JCO.2005.05.4577. [DOI] [PubMed] [Google Scholar]
- 19.Shin DM, Ro JY, Hong WK, Hittelman WN. Dysregulation of epidermal growth factor receptor expression in premalignant lesions during head and neck tumorigenesis. Cancer Res. 1994;54:3153–9. [PubMed] [Google Scholar]
- 20.Bharti AC, Aggarwal BB. Nuclear factor-kappa B and cancer: its role in prevention and therapy. Biochem Pharmacol. 2002;64:883–8. doi: 10.1016/s0006-2952(02)01154-1. [DOI] [PubMed] [Google Scholar]
- 21.Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20:7342–51. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 22.Sohur US, Dixit MN, Chen CL, Byrom MW, Kerr LA. Rel/NF-kappaB represses bcl-2 transcription in pro-B lymphocytes. Gene Expr. 1999;8:219–29. [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–33. doi: 10.1196/annals.1352.026. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Kandasamy K, Srivastava RK. Differential roles of RelA (p65) and c-Rel subunits of nuclear factor kappa B in tumor necrosis factor-related apoptosis-inducing ligand signaling. Cancer Res. 2003;63:1059–66. [PubMed] [Google Scholar]
- 25.Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 26.Jackman DM, Kindler HL, Yeap BY, et al. Erlotinib plus bevacizumab in previously treated patients with malignant pleural mesothelioma. Cancer. 2008;113:808–14. doi: 10.1002/cncr.23617. [DOI] [PubMed] [Google Scholar]
- 27.Lambert JD, Lee MJ, Lu H, et al. Epigallocatechin-3-gallate is absorbed but extensively glucuronidated following oral administration to mice. J Nutr. 2003;133:4172–7. doi: 10.1093/jn/133.12.4172. [DOI] [PubMed] [Google Scholar]
- 28.Conney AH. Enzyme induction and dietary chemicals as approaches to cancer chemoprevention: the seventh DeWitt S. Goodman Lecture Cancer Res. 2003;63:7005–31. [PubMed] [Google Scholar]
- 29.Park AJ, Surh YJ. Chemopreventive potential of eppigallocatechin gallate and genistein: evidence from epidemiological and laboratory studies. Toxicol Lett. 2004;150:43–56. doi: 10.1016/j.toxlet.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Zhang H, Tighiouart M, et al. Synergistic inhibition of head and neck tumor growth by green tea (−)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int J Cancer. 2008;123:1005–14. doi: 10.1002/ijc.23585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Su L, Pirani AA, et al. Understanding metastatic SCCHN cells from unique genotypes to phenotypes with the aid of an animal model and DNA microarray analysis. Clin Exp Metastasis. 2006;23:209–22. doi: 10.1007/s10585-006-9031-0. [DOI] [PubMed] [Google Scholar]
- 32.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–12. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 33.Costa DB, Halmos B, Kumar A, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669–79. doi: 10.1371/journal.pmed.0040315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gong Y, Somwar R, Politi K, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rapidis AD, Vermorken JB, Bourhis J. Targeted therapies in head and neck cancer: past, present and future. Rev Recent Clin Trials. 2008;3:156–66. doi: 10.2174/157488708785700285. [DOI] [PubMed] [Google Scholar]
- 36.Saba NF, Khuri FR, Shin DM. Targeting the epidermal growth factor receptor. Trials in head and neck and lung cancer. Oncology (Williston Park) 2006;20:153–61. discussion 162, 166, 169 passim. [PubMed] [Google Scholar]
- 37.Sutter AP, Höpfner M, Huether A, Maaser K, Scherübl H. Targeting the epidermal growth factor receptor by erlotinib (Tarceva) for the treatment of esophageal cancer. Int J Cancer. 2006;118:1814–22. doi: 10.1002/ijc.21512. [DOI] [PubMed] [Google Scholar]
- 38.Thomas F, Rochaix P, Benlyazid A, et al. Pilot study of neoadjuvant treatment with erlotinib in nonmetastatic head and neck squamous cell carcinoma. Clin Cancer Res. 2007;13:7086–92. doi: 10.1158/1078-0432.CCR-07-1370. [DOI] [PubMed] [Google Scholar]
- 39.Ling YH, Li T, Yuan Z, Haigentz M, Jr, Weber TK, Perez-Soler R. Erlotinib, an effective epidermal growth factor receptor tyrosine kinase inhibitor, induces p27KIP1 up-regulation and nuclear translocation in association with cell growth inhibition and G1/S phase arrest in human non-small-cell lung cancer cell lines. Mol Pharmacol. 2007;72:248–58. doi: 10.1124/mol.107.034827. [DOI] [PubMed] [Google Scholar]
- 40.Amin AR, Thakur VS, Paul RK, et al. SHP-2 tyrosine phosphatase inhibits p73-dependent apoptosis and expression of a subset of p53 target genes induced by EGCG. Proc Natl Acad Sci U S A. 2007;104:5419–24. doi: 10.1073/pnas.0700642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–50. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 42.Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–92. doi: 10.2174/1566524023362456. [DOI] [PubMed] [Google Scholar]
- 43.Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100:1931–6. doi: 10.1073/pnas.2627984100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Javelaud D, Besancon F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl-2 ratio. J Biol Chem. 2002;277:37949–54. doi: 10.1074/jbc.M204497200. [DOI] [PubMed] [Google Scholar]
- 45.Mahyar-Roemer M, Roemer K. p21 Waf1/Cip1 can protect human colon carcinoma cells against p53-dependent and p53-independent apoptosis induced by natural chemopreventive and therapeutic agents. Oncogene. 2001;20:3387–98. doi: 10.1038/sj.onc.1204440. [DOI] [PubMed] [Google Scholar]
- 46.Gurova KV, Hill JE, Guo C, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci U S A. 2005;102:17448–53. doi: 10.1073/pnas.0508888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dey A, Wong ET, Bist P, Tergaonkar V, Lane DP. Nutlin-3 inhibits the NFkappaB pathway in a p53-dependent manner: implications in lung cancer therapy. Cell Cycle. 2007;6:2178–85. doi: 10.4161/cc.6.17.4643. [DOI] [PubMed] [Google Scholar]
- 48.Plesnila N, von Baumgarten L, Retiounskaia M, et al. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-kappaB transcriptional activity. Cell Death Differ. 2007;14:1529–41. doi: 10.1038/sj.cdd.4402159. [DOI] [PubMed] [Google Scholar]
- 49.Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 50.Kim D, Chung J. Akt: versatile mediator of cell survival and beyond. J Biochem Mol Biol. 2002;35:106–15. doi: 10.5483/bmbrep.2002.35.1.106. [DOI] [PubMed] [Google Scholar]
- 51.Torrance CJ, Jackson PE, Montgomery E, et al. Combinatorial chemoprevention of intestinal neoplasia. Nat Med. 2000;6:1024–8. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]
- 52.Meyskens FL, Jr, McLaren CE, Pelot D, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila Pa) 2008;1(1):32–8. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]