

Abstract
DNA polymerase η (Polη) functions in the error-free bypass of UV-induced DNA lesions, and a defect in Polη in humans causes the cancer-prone syndrome, the variant form of xeroderma pigmentosum. Both yeast and human Polη replicate through a cis-syn thymine-thymine dimer (TT dimer) by inserting two As opposite the two Ts of the dimer. Polη, however, is a low-fidelity enzyme, and it misinserts nucleotides with a frequency of ≈ 10−2 to 10−3 opposite the two Ts of the TT dimer as well as opposite the undamaged template bases. This low fidelity of nucleotide insertion seems to conflict with the role of Polη in the error-free bypass of UV lesions. To resolve this issue, we have examined the ability of human and yeast Polη to extend from paired and mispaired primer termini opposite a TT dimer by using steady-state kinetic assays. We find that Polη extends from mispaired primer termini on damaged and undamaged DNAs with a frequency of ≈ 10−2 to 10−3 relative to paired primer termini. Thus, after the incorporation of an incorrect nucleotide, Polη would dissociate from the DNA rather than extend from the mispair. The resulting primer-terminal mispair then could be subject to proofreading by a 3′→5′ exonuclease. Replication through a TT dimer by Polη then would be more accurate than that predicted from the fidelity of nucleotide incorporation alone.
The presence of a DNA lesion in the template strand blocks the normal replication machinery. Such lesions can be bypassed by the action of specialized translesion synthesis DNA polymerases (Pols) (1), or by a “copy choice” type of DNA synthesis in which the newly synthesized daughter strand of the undamaged complementary sequence is used as a template to bypass the lesions (2). Alternatively, recombinational mechanisms may be used (3).
The RAD6-Dependent Pathways of Damage Bypass
As indicated from genetic studies in the yeast Saccharomyces cerevisiae, the RAD6 and RAD18 genes are indispensable for error-free as well as mutagenic bypass processes in eukaryotes (4, 5). Rad6, a ubiquitin-conjugating enzyme, forms a tight complex with Rad18, a DNA-binding protein (6, 7). Although the role of the Rad6–Rad18 complex in damage bypass is not known, one possibility is that it modulates the turnover of the replicative Pol stalled at the lesion site and thereby promotes the entry of translesion synthesis Pols to the lesion site. The Rad6–Rad18-dependent bypass of UV lesions involves at least three separate branches, wherein the RAD5 and RAD30 genes function in alternate error-free bypass pathways and REV3 functions in mutagenic bypass (8–10). Rad5, a DNA-dependent ATPase (11), is a member of the Swi–Snf family of proteins (8), but the manner of its action in damage bypass is unknown. RAD30, a member of the umuC/dinB family (9, 12), encodes a DNA Pol, Polη (13), which has the unique ability to replicate through a diversity of DNA lesions.
The Rev3 protein, together with Rev7, constitutes DNA Polζ (14). Polζ also functions in translesion synthesis, but its role in lesion bypass is quite specific. The indispensability of the REV3 and REV7 genes for mutagenesis induced by UV light and other DNA-damaging agents (15, 16) had prompted the generally held notion that Polζ would be a very low-fidelity Pol capable of bypassing DNA lesions. Steady-state kinetic studies, however, have shown that Polζ has a fairly high fidelity, as it misincorporates nucleotides opposite undamaged template bases with a frequency of ≈ 10−4 to 10−5 (17). The fidelity of nucleotide incorporation of Polζ is about the same as that of DNA Polα, required for lagging strand DNA synthesis. Polζ is very inefficient at inserting nucleotides opposite the 3′T of the thymine-thymine dimer (TT dimer) or the (6–4) TT photoproduct, and it is also very poor at inserting nucleotides opposite abasic sites; consequently, Polζ bypasses these lesions very inefficiently (17). Polζ, however, is a very efficient extender of base mispairs, and its ability to extend from base mispairs (f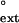 ≈ 10−1 to 10−2) is about 1,000-fold better than its ability to insert a mispaired base (finc ≈ 10−4 to 10−5) (17). Importantly, Polζ is also very adept at extending from nucleotides placed opposite DNA lesions. For example, Polζ extends from a G placed opposite the 3′T of a TT dimer or a (6–4) TT photoproduct almost as efficiently as it extends from an A placed opposite an undamaged T (17). These and other observations have led to the formulation of the principle that mutagenic bypass in eukaryotes involves the action of two different DNA Pols in which a DNA Pol, as for example, human RAD30B-encoded Polι (1), inserts a nucleotide opposite the DNA lesion, whereas Polζ subsequently extends from the inserted nucleotide (17). The indispensability of Polζ for mutagenic bypass in S. cerevisiae derives from the fact that this is the sole enzyme responsible for the extension of “wrong” nucleotides placed opposite DNA lesions.
≈ 10−1 to 10−2) is about 1,000-fold better than its ability to insert a mispaired base (finc ≈ 10−4 to 10−5) (17). Importantly, Polζ is also very adept at extending from nucleotides placed opposite DNA lesions. For example, Polζ extends from a G placed opposite the 3′T of a TT dimer or a (6–4) TT photoproduct almost as efficiently as it extends from an A placed opposite an undamaged T (17). These and other observations have led to the formulation of the principle that mutagenic bypass in eukaryotes involves the action of two different DNA Pols in which a DNA Pol, as for example, human RAD30B-encoded Polι (1), inserts a nucleotide opposite the DNA lesion, whereas Polζ subsequently extends from the inserted nucleotide (17). The indispensability of Polζ for mutagenic bypass in S. cerevisiae derives from the fact that this is the sole enzyme responsible for the extension of “wrong” nucleotides placed opposite DNA lesions.
Role of DNA Polη in the Error-Free Bypass of UV Lesions
Genetic studies in S. cerevisiae have indicated a role for RAD30-encoded Polη in the error-free bypass of UV lesions. Although the rad30Δ mutation confers a moderate degree of UV sensitivity, a synergistic increase in UV sensitivity occurs in the rad5Δ rad30Δ double mutant, and the frequency of UV-induced mutations is much higher in the double mutant than in the rad5Δ or rad30Δ single mutants (9, 10). UV light induces the formation of cyclobutane TT dimers, and Polη efficiently replicates through the TT dimer by inserting two As opposite the two Ts of the dimer (13). UV, however, also induces the formation of lesions at 5′-TC-3′ and 5′-CC-3′ dipyrimidine sites, and the 3′C in both these sequence contexts is highly mutagenic. In both yeast and humans, UV-induced mutations occur predominantly by a 3′ C → T transition that results from the insertion of an A opposite the 3′C during DNA replication (18). In vitro bypass studies with a TC or CC cis-syn cyclobutane dimer, however, are difficult to perform because the C in the dimer is quite unstable, and in vitro, it rapidly deaminates to U. To determine whether Polη also mediates the error-free bypass of UV-induced TC and CC photoproducts, we have used two ura3 mutations, ura3-210 and ura3-364, which revert to wild-type URA3 by the incorporation of an A residue opposite the 3′C of the TC or CC UV-induced lesion, respectively (18). The incidence of UV-induced mutations at both of these sites is about 5-fold higher in the rad30Δ strain than in wild type, indicating a role for Polη in the error-free bypass of UV lesions formed at TC and CC sites (18).
Cells from the variant form of xeroderma pigmentosum (XP-V) are much slower than normal cells in the replication of UV-damaged DNA (19–21), and they are hypermutable with UV light (22, 23). As a consequence, XP-V individuals suffer from a high incidence of skin cancers. XP-V cells harbor nonsense or frameshift mutations in Polη, which produce a severely truncated protein and concomitantly result in the loss of DNA Pol activity (24, 25). Thus, by contributing to the accurate bypass of UV lesions, Polη plays an indispensable role in the prevention of sunlight-induced cancers.
Fidelity and Processivity of DNA Polη
The proficient ability of yeast and human Polη to bypass a TT dimer and other DNA lesions that distort the DNA helix suggested that Polη is refractory to geometric distortions imposed on DNA by these lesions (26–28). The unusual tolerance of Polη to geometric distortions may confer a low nucleotide insertion fidelity on the enzyme, and in fact, both yeast and human Polη are low-fidelity enzymes, misincorporating nucleotides on undamaged DNA with a frequency of ≈ 10−2 to 10−3 (26, 27). In a subsequent study, human Polη was found to make many errors in an in vitro DNA synthesis reaction (29). Also, Polη synthesizes DNA with a low processivity, and we have estimated that about 25% of Polη molecules dissociate from DNA after each nucleotide incorporation event (26). Remarkably, both yeast and human Polη insert As opposite the two Ts of the TT dimer with the same efficiency and accuracy as opposite undamaged template bases (27, 28).
Inefficient Extension of Mismatched Base Pairs by DNA Polη
The accuracy of synthesis by DNA Pols depends not only on the frequency of incorporation of incorrect nucleotides into DNA, but also on the frequency of extension of mismatched primer termini. In the absence of efficient extension, the mismatched nucleotide will be subject to removal by a proofreading exonuclease; thus, extension of mismatched primers is critical for mutation fixation. Previously, we examined the ability of Polη to extend from base mispairs on undamaged DNAs. These steady-state kinetics studies have shown that both yeast and human Polη extend from mismatched base pairs with frequencies ranging from 10−2 to 10−3 relative to matched base pairs (30). To better understand how Polη, a low-fidelity enzyme, can function in the error-free replication of UV-damaged DNA, here we examine the ability of Polη to extend from paired and mispaired termini on cis-syn TT dimer containing DNA substrates.
Materials and Methods
DNA Substrates.
The following synthetic oligodeoxynucleotides were used in this study. The four 45-nt oligomers, used as primers for the paired and mispaired primer-template substrates at the position of the 3′T of the TT dimer, had the following sequences: 5′-GTTTT CCCAG TCACG ACGAT GCTCC GGTAC TCCAG TGTAG GCATN, where N is G, A, T, or C. The four 46-nt oligomers, used as primers for the paired and mispaired primer-template substrates at the position of the 5′T of the TT dimer, had the following sequences: 5′-GTTTT CCCAG TCACG ACGAT GCTCC GGTAC TCCAG TGTAG GCATA N, where N is G, A, T, or C. The 75-nt template had the following sequence: 5′-AGCAA GTCAC CAATG TCTAA GAGTT CGTAT TATGC CTACA CTGGA GTACC GGAGC ATCGT CGTGA CTGGG AAAAC, wherein the underlined TT was either normal or contained a cis-syn TT dimer. The 75-mer template was derived from the 10-nt oligomer 5′-CGTATTATGC-3′, which was ligated to flanking 25-nt and 40-nt oligomers, respectively. The cis-syn TT dimer was incorporated into the 10-nt oligomer by treatment with 360 nm UV light and purified by HPLC. These oligodeoxynucleotides were PAGE-purified, and their concentrations were determined by measuring A260. The primer strands were 5′ 32P-end-labeled by using polynucleotide kinase (Roche Molecular Biochemicals) and [32P-γ]ATP (≈6,000 Ci/mmol) (Amersham Pharmacia) and were subsequently purified by using a BioGel P30 spin column. The 32P-end-labeled primer (0.05 μM) was annealed to the template (0.08 μM) in 50 mM Tris⋅Cl, pH 7.5, and 100 mM NaCl by incubating at 90°C for 2 min and slowly cooling to room temperature over several hours. The identity of the cis-syn TT dimer was confirmed by treatment with T4 UV endonuclease V, and the purity of the damaged substrate was verified by the inability of yeast Polδ to bypass the lesion.
Proteins and Other Reagents.
Yeast and human Polη were expressed and purified as described (13, 27). Solutions of each dNTP (100 mM) were purchased from Roche Molecular Biochemicals, and their concentrations were confirmed by measuring A260.
Steady-State Kinetics Assays.
To measure the relative efficiency of mispair extension, the steady-state kinetics of incorporation of the correct nucleotide after either the paired or the mispaired primer-template termini were examined. Yeast or human Polη (1 nM) was incubated with either the paired or mispaired primer-template substrate (10 nM) and with various concentrations of the next correct nucleotide at 25°C in 25 mM Tris⋅Cl (pH 7.5), 5 mM MgCl2, 5 mM DTT, 100 μg/ml BSA, and 10% glycerol. After 5 min, reactions were quenched with 10 vol of formamide loading buffer (80% deionized formamide/10 mM EDTA, pH 8.0), boiled for 2 min, and chilled on ice. Products were separated on a 10% polyacrylamide sequencing gel containing 6 M urea, and gel band intensities were quantified by using the PhosphorImager (Molecular Dynamics). The rate of incorporation was plotted as a function of nucleotide concentration, and the Vmax and Km steady-state parameters were obtained by fitting these data to the Michaelis–Menten equation using nonlinear regression (SIGMA PLOT 4.0). The intrinsic efficiency of mismatch extension, f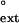 , is a constant that represents the efficiency of extending a mispaired primer-terminus in competition with an equal concentration of paired primer terminus, and it is calculated by using the following equation: f
, is a constant that represents the efficiency of extending a mispaired primer-terminus in competition with an equal concentration of paired primer terminus, and it is calculated by using the following equation: f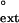 = (Vmax/Km)mispaired/(Vmax/Km)paired (31–33).
= (Vmax/Km)mispaired/(Vmax/Km)paired (31–33).
This analysis assumes that the binding affinities for the paired and mispaired primer termini are similar, which has been shown to be true for other DNA Pols (32, 34, 35).
Results and Discussion
We used steady-state kinetics to evaluate the ability of human and yeast Polη to extend from matched or mismatched primer termini opposite damaged and undamaged TT residues. The extension of the primer terminus opposite from the 3′T of a nondamaged or damaged TT sequence by human Polη was examined by measuring the incorporation of the correct nucleotide, A, opposite the 5′ T of the TT sequence after an A⋅T primer-terminal base pair or at a G⋅T, T⋅T, or C⋅T primer-terminal mispair. The concentration of dATP was varied from 0 to 0.5 μM for the paired primer terminus and from 0 to 200 μM for the mispaired primer termini. The rate of nucleotide incorporation was plotted as a function of nucleotide concentration to obtain the Vmax and Km steady-state parameters (Table 1). The relative efficiency of mispair extension, f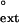 , is a constant and represents the efficiency of extending a mispaired primer terminus in competition with an equal concentration of paired primer terminus in the limit of zero concentration of next nucleotide (31–33). For each mispaired primer terminus, we determined the f
, is a constant and represents the efficiency of extending a mispaired primer terminus in competition with an equal concentration of paired primer terminus in the limit of zero concentration of next nucleotide (31–33). For each mispaired primer terminus, we determined the f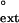 value from the ratio of the efficiency (Vmax/Km) of mispaired primer extension to the efficiency (Vmax/Km) of paired primer extension. As shown in Table 1, human Polη extends about equally efficiently from mispaired primer termini opposite the 3′ T on damaged and undamaged DNAs. Thus, for the undamaged DNA, the f
value from the ratio of the efficiency (Vmax/Km) of mispaired primer extension to the efficiency (Vmax/Km) of paired primer extension. As shown in Table 1, human Polη extends about equally efficiently from mispaired primer termini opposite the 3′ T on damaged and undamaged DNAs. Thus, for the undamaged DNA, the f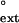 values for the G⋅T, T⋅T, and C⋅T mispaired primer termini opposite the 3′ T were 1.1 × 10−2, 1.7 × 10−3, and less than 4 × 10−4, respectively, and for the damaged DNA, the respective f
values for the G⋅T, T⋅T, and C⋅T mispaired primer termini opposite the 3′ T were 1.1 × 10−2, 1.7 × 10−3, and less than 4 × 10−4, respectively, and for the damaged DNA, the respective f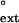 values were 4.4 × 10−2, 5.5 × 10−3, and 7.2 × 10−4 (Table 1).
values were 4.4 × 10−2, 5.5 × 10−3, and 7.2 × 10−4 (Table 1).
Table 1.
Kinetics of mispair extension by human Pol η on undamaged and TT dimer-containing DNA substrates
| Primer | Template | Vmax, nM/min | Km, μM | Vmax/Km |
f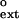
|
|---|---|---|---|---|---|
| G | 3′ T (nondamaged) | 1.4 ± 0.09 | 5.1 ± 0.8 | 0.27 | 1.1 × 10−2 |
| A | 3′ T (nondamaged) | 1.2 ± 0.04 | 0.048 ± 0.006 | 25 | 1.0 |
| T | 3′ T (nondamaged) | 0.27 ± 0.01 | 6.4 ± 1.3 | 0.042 | 1.7 × 10−3 |
| C | 3′ T (nondamaged) | ND | >100 | <0.01 | <4 × 10−4 |
| G | 3′ T (TT dimer) | 0.80 ± 0.02 | 1.0 ± 0.1 | 0.80 | 4.4 × 10−2 |
| A | 3′ T (TT dimer) | 0.79 ± 0.05 | 0.045 ± 0.010 | 18 | 1.0 |
| T | 3′ T (TT dimer) | 0.44 ± 0.01 | 4.2 ± 0.5 | 0.10 | 5.5 × 10−3 |
| C | 3′ T (TT dimer) | 0.22 ± 0.007 | 17 ± 2 | 0.013 | 7.2 × 10−4 |
| G | 5′ T (nondamaged) | 1.4 ± 0.03 | 3.3 ± 0.2 | 0.42 | 8.9 × 10−3 |
| A | 5′ T (nondamaged) | 1.6 ± 0.03 | 0.034 ± 0.002 | 47 | 1.0 |
| T | 5′ T (nondamaged) | 1.0 ± 0.1 | 12 ± 3 | 0.083 | 1.8 × 10−3 |
| C | 5′ T (nondamaged) | 1.2 ± 0.06 | 53 ± 7 | 0.023 | 4.9 × 10−4 |
| G | 5′ T (TT dimer) | 1.0 ± 0.04 | 3.7 ± 0.4 | 0.27 | 9.0 × 10−3 |
| A | 5′ T (TT dimer) | 1.3 ± 0.2 | 0.043 ± 0.015 | 30 | 1.0 |
| T | 5′ T (TT dimer) | 1.1 ± 0.1 | 9.5 ± 2.3 | 0.12 | 4.0 × 10−3 |
| C | 5′ T (TT dimer) | 1.3 ± 0.03 | 13 ± 1 | 0.1 | 3.3 × 10−3 |
Mispair extension was examined in the presence of the next correct nucleotide: dATP for mispairs at the 3′ T and dTTP for mispairs at the 5′ T. ND, not determined.
The efficiency of extension of the primer terminus opposite from the 5′ T of the undamaged or damaged TT sequence by human Polη was determined by examining the incorporation of the correct nucleotide, T, opposite a template A after an A⋅T primer-terminal base pair or at a G⋅T, T⋅T, or C⋅T primer-terminal mispair. Here also, the f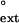 values, which range from 9.0 × 10−3 for the G⋅T mispair opposite the 5′ T of the dimer to 5.0 × 10−4 for C⋅T mispair opposite the 5′ T in the undamaged TT sequence, are approximately the same for the damaged and nondamaged DNA substrates (Table 1).
values, which range from 9.0 × 10−3 for the G⋅T mispair opposite the 5′ T of the dimer to 5.0 × 10−4 for C⋅T mispair opposite the 5′ T in the undamaged TT sequence, are approximately the same for the damaged and nondamaged DNA substrates (Table 1).
Yeast Polη resembles the human enzyme in its ability to extend from mispaired primer termini on damaged and undamaged TT sequences. As shown in Table 2, the f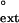 values for yeast Polη range from 1.4 × 10−1 for the G⋅T mispair opposite the 3′ T of the TT dimer to <6 × 10−4 for C⋅T mispair opposite the 5′ T of the undamaged DNA, and overall the f
values for yeast Polη range from 1.4 × 10−1 for the G⋅T mispair opposite the 3′ T of the TT dimer to <6 × 10−4 for C⋅T mispair opposite the 5′ T of the undamaged DNA, and overall the f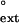 values are quite similar for the damaged and undamaged DNA substrates.
values are quite similar for the damaged and undamaged DNA substrates.
Table 2.
Kinetics of mispair extension by yeast Pol η on undamaged and TT dimer-containing DNA substrates
| Primer | Template | Vmax, nM/min | Km, μM | Vmax/Km |
f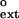
|
|---|---|---|---|---|---|
| G | 3′ T (nondamaged) | 0.66 ± 0.03 | 13 ± 2 | 0.05 | 2.9 × 10−2 |
| A | 3′ T (nondamaged) | 0.50 ± 0.02 | 0.29 ± 0.03 | 1.7 | 1.0 |
| T | 3′ T (nondamaged) | 0.17 ± 0.01 | 76 ± 21 | 0.002 | 1.2 × 10−3 |
| C | 3′ T (nondamaged) | ND | >200 | <0.001 | <6 × 10−4 |
| G | 3′ T (TT dimer) | 0.42 ± 0.03 | 5.3 ± 1.9 | 0.079 | 1.4 × 10−1 |
| A | 3′ T (TT dimer) | 0.45 ± 0.02 | 0.82 ± 0.08 | 0.55 | 1.0 |
| T | 3′ T (TT dimer) | 0.25 ± 0.009 | 130 ± 20 | 0.0019 | 3.5 × 10−3 |
| C | 3′ T (TT dimer) | 0.15 ± 0.013 | 120 ± 40 | 0.0013 | 2.4 × 10−3 |
| G | 5′ T (nondamaged) | 0.43 ± 0.006 | 8.8 ± 3.3 | 0.048 | 2.7 × 10−2 |
| A | 5′ T (nondamaged) | 0.51 ± 0.03 | 0.28 ± 0.04 | 1.8 | 1.0 |
| T | 5′ T (nondamaged) | 0.53 ± 0.05 | 76 ± 21 | 0.0070 | 3.9 × 10−3 |
| C | 5′ T (nondamaged) | ND | >200 | <0.001 | <6 × 10−4 |
| G | 5′ T (TT dimer) | 0.55 ± 0.05 | 49 ± 12 | 0.011 | 7.9 × 10−3 |
| A | 5′ T (TT dimer) | 0.49 ± 0.06 | 0.35 ± 0.12 | 1.4 | 1.0 |
| T | 5′ T (TT dimer) | 0.078 ± 0.02 | 140 ± 50 | 0.00056 | 4.0 × 10−4 |
| C | 5′ T (TT dimer) | 0.37 ± 0.04 | 65 ± 17 | 0.0057 | 4.1 × 10−3 |
Mispair extension was examined in the presence of the next correct nucleotide: dATP for mispairs at the 3′ T and dTTP for mispairs at the 5′ T. ND, not determined.
Both yeast and human Polη incorporate wrong nucleotides opposite the two Ts of a TT dimer or those of an undamaged TT sequence with a frequency of ≈ 10−2 to 10−3 (27, 28). In Fig. 1A, we compare the finc values for the insertion of nucleotides opposite the two Ts of the TT dimer or the undamaged TT sequence with the f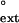 values for subsequently extending from the same mispair by human Polη. Points lying above the dashed line indicate mispairs that have a higher efficiency of extension than insertion, while those below the dashed line indicate mispairs with a lower efficiency of extension than insertion. For human Polη, the efficiency of inserting a wrong nucleotide and of extending from that mispair are approximately the same; and this similarity of the finc and f
values for subsequently extending from the same mispair by human Polη. Points lying above the dashed line indicate mispairs that have a higher efficiency of extension than insertion, while those below the dashed line indicate mispairs with a lower efficiency of extension than insertion. For human Polη, the efficiency of inserting a wrong nucleotide and of extending from that mispair are approximately the same; and this similarity of the finc and f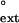 values holds for both undamaged and TT dimer-containing DNAs. Fig. 1B compares the finc and f
values holds for both undamaged and TT dimer-containing DNAs. Fig. 1B compares the finc and f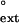 values for yeast Polη. Again, the finc and f
values for yeast Polη. Again, the finc and f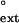 values are about the same for a given mispair for both the damaged and undamaged DNA substrates.
values are about the same for a given mispair for both the damaged and undamaged DNA substrates.
Figure 1.

Comparison of the finc and f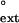 values for each mispair (primer base⋅template base) opposite each T of a TT dimer or the undamaged TT sequence. (A) Human Polη. (B) Yeast Polη. The dashed line corresponds to finc = f
values for each mispair (primer base⋅template base) opposite each T of a TT dimer or the undamaged TT sequence. (A) Human Polη. (B) Yeast Polη. The dashed line corresponds to finc = f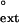 . The finc values for human and yeast Polη are from refs. 27 and 26, respectively. ○, TT dimer; ●, undamaged TT sequence.
. The finc values for human and yeast Polη are from refs. 27 and 26, respectively. ○, TT dimer; ●, undamaged TT sequence.
In summary, both yeast and human Polη misinsert nucleotides opposite a TT dimer with a frequency of ≈ 10−2 to 10−3, and they extend from the mismatched nucleotides opposite a TT dimer with about the same frequency. Because of its low processivity and the less efficient extension of mismatched primer termini than of matched primer termini from opposite a TT dimer, Polη would have a much higher probability of dissociating from the primer terminus after the incorporation of an incorrect nucleotide than a correct nucleotide. Any mispairs would then be subject to the proofreading exonuclease activity of Polδ or other proofreading exonucleases. Recent studies with human cell extracts have suggested that misincorporations introduced by Polη are subject to such exonucleolytic removal (36). Thus, in vivo, replication through a TT dimer by Polη would be more accurate than 10−2 to 10−3, its fidelity for nucleotide incorporation. Also, the Rad6–Rad18 complex, essential for damage bypass, may limit synthesis by Polη to lesion sites by promoting the ubiquitin-dependent dissociation of Polη once the lesion has been bypassed. The bypass of cyclobutane pyrimidine dimers by Polη thus could be quite accurate.
The ability of human Polη to incorporate nucleotides opposite the TT dimer and to extend from these nucleotides was previously examined by others (37). Our observations differ from that study in several important aspects. First, it was reported that human Polη incorporates all four nucleotides opposite nondamaged template T residues about equally well, and that it also incorporates all four nucleotides opposite the 3′ T of the TT dimer with nearly the same frequency (37). These conclusions were based on qualitative studies performed by using a single, saturating concentration of each nucleotide. Consequently, in that study (37), one could only have observed changes in kcat between the correct and incorrect nucleotides, but not the changes in the Km. Because the specificity for the incorporation of the correct nucleotide over the incorrect nucleotide depends both on the kcat and Km changes, the incorporation fidelity must be determined by kinetic studies performed over a broad range of nucleotide concentrations. Our steady-state kinetics studies with both yeast and human Polη have indicated that incorporation of the correct nucleotide A opposite both the 3′ T and 5′ T of the TT dimer and the nondamaged TT sequence is favored 100–1,000-fold over the incorrect nucleotide (27, 28). The previous qualitative studies performed at a single-saturating concentration of nucleotide (37) also concluded that misextension on the TT dimer-containing DNA substrate is less efficient than misextension on the nondamaged DNA substrate. The steady-state kinetics studies reported here indicate that there is no significant difference in the ability of Polη to extend from mispairs on nondamaged or TT dimer-containing DNA substrates. Overall, we find that with respect to both the efficiency of incorrect nucleotide incorporation and the efficiency of mispaired primer extension, human and yeast Polη behave essentially the same on nondamaged and TT dimer-containing DNA substrates.
Error-Free Bypass of 8-Oxoguanine by Polη.
7,8-Dihydro 8-oxoguanine (8-oxoG) is one of the adducts formed by the attack of oxygen-free radicals on bases in DNA. Eukaryotic replicative DNA Pols replicate through an 8-oxoG lesion by inserting an A opposite the lesion: consequently, 8-oxoG is highly mutagenic and causes G·C to T·A transversions. Genetic studies in yeast have implicated a role for Polη in minimizing the incidence of these mutations. In S. cerevisiae, deletion of OGG1, which encodes a DNA glycosylase involved in the removal of 8-oxoG when paired with C, causes an increase in the incidence of G·C to T·A transversions, and a synergistic increase in the rate of these mutations occurs in the absence of Polη in the yeast ogg1Δ mutant (38).
Yeast Polη replicates through 8-oxoG efficiently and accurately by inserting a C across from the lesion and by efficiently extending from this base pair. Steady-state kinetic studies have shown that yeast Polη inserts a C opposite 8-oxoG as efficiently as opposite the undamaged G, and it extends from the C·8-oxoG or C·G base pair equally efficiently (38). Compared with the insertion of C, A is incorporated opposite 8-oxoG 20-fold less well, and compared with extension from C opposite 8-oxoG, extension from A is about 6-fold less efficient (38). Thus, yeast Polη discriminates against the incorporation of an A opposite 8-oxoG both at the insertion and extension steps. Human Polη, however, is less accurate in bypassing 8-oxoG, as in addition to the insertion of C, it inserts some A opposite this lesion (38, 39). The A nucleotide inserted opposite 8-oxoG by human Polη could be subject to removal by a MutY-related DNA glycosylase or by the mismatch repair system, affording Polη another opportunity to insert the correct nucleotide C.
Bypass of 6O-Methylguanine (m6G) by DNA Polη.
Treatment of cells with alkylating agents such as N-methyl-N′-nitro-N-nitrosoguanidine forms m6G in DNA. m6G is a highly mutagenic lesion, and in both yeast and humans, it specifically induces G⋅C to A⋅T transition mutations. Whereas m6G presents a strong block to replicative DNA Pols, both yeast and human Polη replicate through the lesion quite efficiently by inserting a C or a T opposite the lesion (40). Steady-state kinetic analyses have indicated that compared with the insertion of a C opposite a template G, yeast Polη inserts a C or T opposite m6G about 20-fold and 15-fold less efficiently, respectively, and compared with extension from a C⋅G base pair, extension from the C⋅m6G base pair is only about 2-fold less efficient and extension from the T⋅m6G base pair is about 5-fold less efficient (40).
Chloroethylating agents, in combination with methylating agents such as procarbazine and temozolomide, are used for the treatment of malignant tumors such as lymphomas, brain tumors, and gastrointestinal carcinomas. The clinical effectiveness of these agents derives at least in part from their ability to form m6G in DNA. The involvement of Polη in m6G bypass raises the possibility that inactivation of this enzyme may be useful for increasing the effectiveness of alkylating agents in cancer treatment.
Conclusions and Perspectives.
Polη is unique among eukaryotic DNA Pols in its proficient ability to replicate through a variety of lesions that distort the geometry of the DNA helix. Polη bypasses a cis-syn TT dimer by inserting two As opposite the dimer. No other eukaryotic polymerase is able to efficiently bypass a TT dimer, presumably because of the geometric distortion conferred on DNA by this lesion. Although Polζ has been reported to bypass a cis-syn TT dimer, it does so very inefficiently (14, 17), and that is because Polζ is highly inefficient at inserting nucleotides opposite the 3′ T of the TT dimer (17). In one study, Polι was reported to bypass a cis-syn TT dimer (41); however, our studies have indicated that Polι does not even insert a nucleotide opposite the 3′ T of the TT dimer, and thus it is unable to bypass this lesion (17). The highly inefficient bypass of the TT dimer reported for Polι (41) could have been due to a contamination of the damaged DNA preparation with undamaged DNA.
Our genetic studies in yeast have indicated a role for Polη in the error-free bypass of cis-syn cyclobutane dimers formed at 5′-TC-3′ and 5′-CC-3′ sequences. Further, Polη inserts a G opposite the 3′ T of a (6–4) TT photoproduct with a modest efficiency, and Polζ efficiently extends from the resulting base pair (42). Very possibly, Polη also inserts a G opposite the 3′ C of TC or CC (6–4) photoproducts and thus promotes the error-free bypass of this lesion, which is formed quite frequently at the TC site. By contrast to cis-syn cyclobutane pyrimidine dimers, which have only a modest effect on DNA structure and in which the two pyrimidine residues in the dimer can still form base pairs with the correct nucleotides, a (6–4) photoproduct induces a large structural distortion in DNA, and the 3′ T in the (6–4) photoproduct is held perpendicular to the 5′ T (43). The ability of Polη to insert a nucleotide opposite the 3′ T of a (6–4) TT photoproduct implies that this enzyme is refractory even to the severe distortion of this lesion. Polη also differs from other eukaryotic Pols in its proficient ability to bypass an 8-oxoG lesion, and by contrast to the replicative DNA polymerases, which bypass this lesion poorly and which insert an A opposite from the lesion, Polη (particularly the yeast protein) bypasses an 8-oxoG by predominantly inserting a C. Although the 8-oxoG⋅A base pair has the correct geometry, 8-oxoG in this conformation mimics a T and forms the same two hydrogen bonds with A as in the T⋅A base pair. On the other hand, in the 8-oxoG⋅C base pair, the template strand is highly distorted in the vicinity of the lesion, but the 8-oxoG⋅C base pair involves the same three hydrogen bonds as in the G⋅C base pair (44–47). Yeast Polη is somewhat more efficient at inserting a C than a T opposite the m6G lesion, even though the m6G⋅T base pair retains the Watson–Crick geometry more closely than the m6G⋅C base pair. The m6G⋅C base pair, however, is more hydrogen-bonded than the m6G⋅T base pair (48, 49). The ability of Polη to bypass these distorting DNA lesions by preferentially inserting the correct nucleotide may reflect an active site that is quite insensitive to geometric distortions in DNA, but which depends more on the formation of normal Watson–Crick hydrogen bonds between the bases.
Most DNA Pols, including eukaryotic replicative Pols, are very sensitive to geometric distortions in DNA, and consequently, they are unable to replicate through DNA lesions. Moreover, the structures of several DNA Pols have indicated that although the dNTP first binds the primer-template bound Pol in a nontemplate-dependent fashion, only in the presence of correct Watson–Crick geometry between the incoming dNTP and the template base does a catalytically essential conformational change in the Pol occur (50). By contrast to most DNA Pols, which selectively incorporate the correct nucleotide by an induced fit conformational change, Polη may not require as stringent a fit for catalysis.
Because the frequency of UV-induced mutations rises in the absence of Polη in both yeast and humans, Polη must bypass these DNA lesions predominantly in an error-free manner. Although Polη misincorporates nucleotides opposite either T of the TT dimer with a frequency of ≈ 10−2 to 10−3, it extends from mispaired bases much less readily than from the correctly paired bases. Compared with the extension from an A, Polη extends from a G, a C, or a T nucleotide placed opposite either of the Ts of the TT dimer with a frequency of ≈ 10−2 to 10−3. The less efficient extension of mispairs would promote the dissociation of Polη from DNA, which in turn would favor the excision of mismatched nucleotides by a proofreading exonuclease. UV lesions then would be bypassed in a much more accurate fashion than that predicted from the fidelity of nucleotide incorporation alone. It remains to be seen whether the Polδ 3′→5′ exonuclease, in fact, functions in the removal of mismatched nucleotides incorporated opposite UV lesions and other distorting DNA lesions, or whether there are specialized nucleases more suited for such a task. Genetic studies in yeast also have implicated a role for Polη in the error-free bypass of an 8-oxoG lesion, and compared with the insertion of C, Polη inserts the G and T nucleotides opposite this lesion with a frequency of ≈ 10−2, and an A is inserted with a frequency of 5 × 10−2. Also, Polη is much more efficient at extending from the correct 8-oxoG⋅C base pair than from the 8-oxoG⋅A base pair. Thus, for both the TT dimer and the 8-oxoG lesion, where genetic studies have convincingly shown a role for Polη in the error-free bypass, Polη extends from the correct base pair better than from the incorrect base pair.
The bypass of DNA lesions by Polη is not always error-free. Although Polη bypasses an m6G lesion correctly by inserting a C, it also inserts a T residue quite frequently, and consistent with these biochemical observations, genetic studies in yeast support a role of Polη in the mutagenic bypass of this DNA lesion. Polη, thus, is a translesion synthesis DNA Pol that promotes the error-free bypass of some lesions and the mutagenic bypass of others. However, Polη appears to be specifically adapted for bypassing the more frequently formed lesions, such as those induced by UV light and those resulting from oxidative DNA damage, efficiently and accurately.
Acknowledgments
This work was supported by National Institutes of Health Grants GM19261 and CA80882.
Abbreviations
- Pol
polymerase
- TT dimer
thymine-thymine dimer
- 8-oxoG
7,8-dihydro 8-oxoguanine
- m6G
6O-methylguanine
Footnotes
This paper results from the National Academy of Sciences colloquium, “Links Between Recombination and Replication: Vital Roles of Recombination,” held November 10–12, 2000, in Irvine, CA.
References
- 1.Johnson R E, Washington M T, Prakash S, Prakash L. Proc Natl Acad Sci USA. 1999;96:12224–12226. doi: 10.1073/pnas.96.22.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Higgins N P, Kato K, Strauss B. J Mol Biol. 1976;101:417–425. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- 3.Rupp W D, Wilde C E I, Reno D L, Howard-Flanders P. J Mol Biol. 1971;61:25–44. doi: 10.1016/0022-2836(71)90204-x. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence C W. Adv Genet. 1982;21:173–254. doi: 10.1016/s0065-2660(08)60299-0. [DOI] [PubMed] [Google Scholar]
- 5.Prakash S, Sung P, Prakash L. Annu Rev Genet. 1993;27:33–70. doi: 10.1146/annurev.ge.27.120193.000341. [DOI] [PubMed] [Google Scholar]
- 6.Bailly V, Lamb J, Sung P, Prakash S, Prakash L. Genes Dev. 1994;8:811–820. doi: 10.1101/gad.8.7.811. [DOI] [PubMed] [Google Scholar]
- 7.Bailly V, Lauder S, Prakash S, Prakash L. J Biol Chem. 1997;272:23360–23365. doi: 10.1074/jbc.272.37.23360. [DOI] [PubMed] [Google Scholar]
- 8.Johnson R E, Henderson S T, Petes T D, Prakash S, Bankmann M, Prakash L. Mol Cell Biol. 1992;12:3807–3818. doi: 10.1128/mcb.12.9.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald J P, Levine A S, Woodgate R. Genetics. 1997;147:1557–1568. doi: 10.1093/genetics/147.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson R E, Prakash S, Prakash L. J Biol Chem. 1999;274:15975–15977. doi: 10.1074/jbc.274.23.15975. [DOI] [PubMed] [Google Scholar]
- 11.Johnson R E, Prakash S, Prakash L. J Biol Chem. 1994;269:28259–28262. [PubMed] [Google Scholar]
- 12.Roush A A, Suarez M, Friedberg E C, Radman M, Siede W. Mol Gen Genet. 1998;257:686–692. doi: 10.1007/s004380050698. [DOI] [PubMed] [Google Scholar]
- 13.Johnson R E, Prakash S, Prakash L. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- 14.Nelson J R, Lawrence C W, Hinkle D C. Science. 1996;272:1646–1649. doi: 10.1126/science.272.5268.1646. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence C W, Hinkle D C. Cancer Surv. 1996;28:21–31. [PubMed] [Google Scholar]
- 16.Johnson R E, Torres-Ramos C A, Izumi T, Mitra S, Prakash S, Prakash L. Genes Dev. 1998;12:3137–3143. doi: 10.1101/gad.12.19.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson R E, Washington M T, Haracska L, Prakash S, Prakash L. Nature (London) 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 18.Yu S-L, Johnson R E, Prakash S, Prakash L. Mol Cell Biol. 2001;21:185–188. doi: 10.1128/MCB.21.1.185-188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehmann A R, Kirk-Bell S, Arlett C F, Paterson M C, Lohman P H M, de Weerd-Kastelein E A, Bootsma D. Proc Natl Acad Sci USA. 1975;72:219–223. doi: 10.1073/pnas.72.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cleaver J E, Greene A E, Coriell L L, Mulivor R A. Cytogenet Cell Genet. 1981;31:188–192. doi: 10.1159/000131646. [DOI] [PubMed] [Google Scholar]
- 21.Boyer J C, Kaufmann W K, Brylawski B P, Cordeiro-Stone M. Cancer Res. 1990;50:2593–2598. [PubMed] [Google Scholar]
- 22.Wang Y-C, Maher V M, Mitchell D L, McCormick J J. Mol Cell Biol. 1993;13:4276–4283. doi: 10.1128/mcb.13.7.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waters H L, Seetharam S, Seidman M M, Kraemer K H. J Invest Dermatol. 1993;101:744–748. doi: 10.1111/1523-1747.ep12371686. [DOI] [PubMed] [Google Scholar]
- 24.Johnson R E, Kondratick C M, Prakash S, Prakash L. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 25.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. Nature (London) 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 26.Washington M T, Johnson R E, Prakash S, Prakash L. J Biol Chem. 1999;274:36835–36838. doi: 10.1074/jbc.274.52.36835. [DOI] [PubMed] [Google Scholar]
- 27.Johnson R E, Washington M T, Prakash S, Prakash L. J Biol Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- 28.Washington M T, Johnson R E, Prakash S, Prakash L. Proc Natl Acad Sci USA. 2000;97:3094–3099. doi: 10.1073/pnas.050491997. . (First Published March 21, 2000; 10.1073/pnas.050491997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuda T, Bebenek K, Masutani C, Hanaoka F, Kunkel T A. Nature (London) 2000;404:1011–1013. doi: 10.1038/35010014. [DOI] [PubMed] [Google Scholar]
- 30.Washington M T, Johnson R E, Prakash S, Prakash L. J Biol Chem. 2001;276:2263–2266. doi: 10.1074/jbc.M009049200. [DOI] [PubMed] [Google Scholar]
- 31.Goodman M F, Creighton S, Bloom L B, Petruska J. Crit Rev Biochem Mol Biol. 1993;28:83–126. doi: 10.3109/10409239309086792. [DOI] [PubMed] [Google Scholar]
- 32.Creighton S, Goodman M F. J Biol Chem. 1995;270:4759–4774. doi: 10.1074/jbc.270.9.4759. [DOI] [PubMed] [Google Scholar]
- 33.Mendelman L V, Petruska J, Goodman M F. J Biol Chem. 1990;265:2338–2346. [PubMed] [Google Scholar]
- 34.Wong I, Patel S S, Johnson K A. Biochemistry. 1991;30:526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 35.Huang M-M, Arnheim N, Goodman M F. Nucleic Acids Res. 1992;20:4567–4573. doi: 10.1093/nar/20.17.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bebenek K, Matsuda T, Masutani C, Hanaoka F, Kunkel T A. J Biol Chem. 2001;276:2317–2320. doi: 10.1074/jbc.C000690200. [DOI] [PubMed] [Google Scholar]
- 37.Masutani C, Kusumoto R, Iwai S, Hanaoka F. EMBO J. 2000;19:3100–3109. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haracska L, Yu S-L, Johnson R E, Prakash L, Prakash S. Nat Genet. 2000;25:458–461. doi: 10.1038/78169. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Yuan F, Wu X, Rechkoblit O, Taylor J-S, Geachintov M E, Wang Z. Nucleic Acids Res. 2000;28:4717–4724. doi: 10.1093/nar/28.23.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haracska L, Prakash S, Prakash L. Mol Cell Biol. 2000;20:8001–8007. doi: 10.1128/mcb.20.21.8001-8007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tissier A, Frank E G, McDonald J P, Iwai S, Hanaoka F, Woodgate R. EMBO J. 2000;19:5259–5266. doi: 10.1093/emboj/19.19.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson R E, Haracska L, Prakash S, Prakash L. Mol Cell Biol. 2001;21:3558–3563. doi: 10.1128/MCB.21.10.3558-3563.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J-K, Patel D, Choi B-S. Photochem Photobiol. 1995;62:44–50. doi: 10.1111/j.1751-1097.1995.tb05236.x. [DOI] [PubMed] [Google Scholar]
- 44.Kouchakdjian M, Bodepudi V, Shibutani S, Eisenberg M, Johnson F, Grollman A P, Patel D J. Biochemistry. 1991;30:1403–1412. doi: 10.1021/bi00219a034. [DOI] [PubMed] [Google Scholar]
- 45.Lipscomb L A, Peek M E, Morningstar M L, Verghis S M, Miller E M, Rich A, Essignman J M, Williams L D. Proc Natl Acad Sci USA. 1995;92:719–723. doi: 10.1073/pnas.92.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McAuley-Hecht K E, Leonard G A, Gibson N J, Thomson J B, Watson W P, Hunter W N, Brown T. Biochemistry. 1994;33:10266–10270. doi: 10.1021/bi00200a006. [DOI] [PubMed] [Google Scholar]
- 47.Oda Y, Uesugi S, Ikehara M, Nishimura S, Kawase Y, Ishikawa H, Inoue H, Ohtsuka E. Nucleic Acids Res. 1991;19:1407–1412. doi: 10.1093/nar/19.7.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel D J, Shapiro L, Kozlowski S A, Gaffney B L, Jones R A. Biochemistry. 1986;25:1027–1036. doi: 10.1021/bi00353a012. [DOI] [PubMed] [Google Scholar]
- 49.Patel D J, Shapiro L, Kozlowski S A, Gaffney B L, Jones R A. Biochemistry. 1986;26:1036–1042. doi: 10.1021/bi00353a013. [DOI] [PubMed] [Google Scholar]
- 50.Steitz T A. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]