

Abstract
Intracerebral hemorrhage (ICH), the most common form of hemorrhagic stroke, exhibits the highest acute mortality and the worst long-term prognosis of all stroke subtypes. Unfortunately, treatment options for ICH are lacking due in part to a lack of feasible therapeutic targets. Inflammatory activation is associated with neurological deficits in pre-clinical ICH models and with patient deterioration after clinical ICH. In the present study, we tested the hypothesis that R-7050, a novel cell permeable triazoloquinoxaline inhibitor of the tumor necrosis factor receptor (TNFR) complex, attenuates neurovascular injury after ICH in mice. Up to 2h post-injury administration of R-7050 significantly reduced blood-brain barrier opening and attenuated edema development at 24h post-ICH. Neurological outcomes were also improved over the first three days after injury. In contrast, R-7050 did not reduce hematoma volume, suggesting the beneficial effects of TNFR inhibition were downstream of clot formation/resolution. These data suggest a potential clinical utility for TNFR antagonists as an adjunct therapy to reduce neurological injury and improve patient outcomes after ICH.
Keywords: hemorrhage, stroke, edema, blood-brain barrier, inflammation
INTRODUCTION
Spontaneous intracerebral hemorrhage (ICH), the most prevalent form of hemorrhagic stroke, induces one-year mortality rates >60% and induces the worst long-term neurological outcomes of all stroke subtypes [29]. ICH is caused by the rupture of small vessels damaged by chronic hypertension or amyloid angiopathy, inducing the formation of an intracranial space-occupying hematoma. Neurosurgical clot evacuation improves outcomes in some ICH patients, although many patients are not amenable to surgical intervention due to an inaccessible location or concurrent intraventricular hemorrhage [27]. As such, conservative management remains a clinical mainstay. The lack of efficacious therapeutic options reinforces the notion that ICH is the least treatable form of stroke and stresses the need for improved medical approaches.
Hemolysis promotes spontaneous hematoma resolution, although this process simultaneously induces the release of pro-inflammatory mediators adjacent to the hematoma. Inflammatory activation is associated with increased neurovascular damage, neurological deterioration, and a poor functional recovery after ICH [15, 22, 28, 37], although the precise mechanisms remain undefined. In particular, elevated plasma concentrations of the pro-inflammatory mediator, tumor necrosis factor-α (TNF-α), clinically correlated with acute hematoma enlargement, edema development, and poor patient outcome following ICH [3, 8, 9, 17, 35]. Similarly, increased TNF-α expression was observed in several different species and in multiple experimental models of ICH. Importantly, our laboratory and others reported a functional association between peri-hematomal TNF-α expression and the development brain edema and neurological injury after ICH [16, 19, 26, 34]. As a whole, these findings support the notion that TNF-α represents a rationale therapeutic target after ICH.
Although emerging pre-clinical and clinical evidence suggests a detrimental role, small molecule TNF-α pathway inhibitors remain largely unexplored in the context of a brain hemorrhage. TNF-α induces biological activity via stimulation of the tumor necrosis factor receptor (TNFR) [1, 23]. TNFR interacts with downstream adaptor proteins, including TRADD, TRAF and RIP1, providing specificity of the response toward a pro-inflammatory and/or a cell death response. R-7050 is a novel cell-permeable triazoloquinoxaline compound that selectively inhibited TNF-α induced cellular signaling using differential screening of a 300,000 compound library [14]. Unlike biologic TNF inhibitors (e.g. infliximab, etanercept, adalimumab) that directly bind TNF-α and function as decoy receptors, R-7050 does not affect binding of TNF-α to TNFR. In contrast, R-7050 selectively inhibits the association of TNFR with intracellular adaptor molecules (e.g. TRADD, RIP), limits receptor internalization, and prevents subsequent cellular responses after TNF-α binding [14]. In the present study, we tested the hypothesis that R-7050 reduces neurovascular injury after ICH.
MATERIALS AND METHODS
2.1 ICH model
Animal studies were reviewed and approved by the Committee on Animal Use for Research and Education at Georgia Health Sciences University, in compliance with NIH guidelines. A mouse collagenase model of ICH was utilized for all studies, as detailed by our laboratory [19, 32]. Briefly, male CD-1 mice (8–10 weeks old; Charles River, Wilmington, MA, USA) were placed into a stereotactic frame and a 0.5 mm diameter burr hole was drilled over the parietal cortex, 2.2 mm lateral to the bregma. A 26-gauge Hamilton syringe, loaded with 0.04U of bacterial type IV collagenase in 0.5 μL saline, was lowered 3 mm deep from the skull surface directly into the left striatum. The syringe was depressed at a rate of 450 nL/min and left in place for several minutes after the procedure to prevent solution reflux and excess diffusion. Sham animals underwent the same surgical procedure, except that saline was stereotactically injected rather than collagenase. After the syringe was removed, bone wax was used to close the burr hole, the incision was surgically stapled, and mice were kept warm until recovery of the righting reflex. R-7050 (EMD Biosciences, 6–18 mg/kg) was administered via intraperitoneal route at the time of injury or up to 2h post-ICH.
2.2 Hematoma volume
Hematoma volume was quantified using by QuantiChrom Hemoglobin Assay Kit (Bioassay Systems, Hayward, CA), per manufacturer’s recommendations and as routine to our laboratory [19]. The amount of hemoglobin in each hemisphere was calculated using the following: [(optical density of sample/optical density of calibrator)*100.
2.3 Blood-brain barrier (BBB) integrity
BBB integrity was quantified following administration of Evans blue (20 mg/mL in PBS, i.v.) 2h prior to sacrifice, as detailed by our laboratory and others [19, 25]. Absorbance in brain tissue was determined at 620 nm using a Synergy HT plate reader. The amount of Evans blue within brain tissue was calculated using a standard curve and expressed as μg Evans blue/g brain tissue.
2.4 Assessment of cerebral edema
Brain water content, an established measure of cerebral edema, was quantified in a 2 mm coronal tissue sections of the ipsilateral or corresponding contralateral striatum, as routine to our laboratory [18–20]. Tissue was immediately weighed (wet weight), then dehydrated at 65°C. Samples were reweighed 48h later to obtain a dry weight. The percentage of water content in each sample was calculated as follows: % Brain water content = [((wet weight − dry weight)/wet weight)*100].
2.5 Neurological outcomes
Neurological injury was determined using a modified 24-point scale, per our laboratory and others [5, 19, 30]. This scale was comprised of six behavioral tests, each of which was graded from 0 (performs with no impairment) to 4 (severe impairment). A composite score was calculated as the sum of the grades on all six tests. All data was scored independently by two investigators blinded to experimental treatment groups.
2.6 Statistical analysis
One-way analysis of variance (ANOVA) followed by Student Newman-Keuls or Two-way ANOVA followed by Tukey’s post-hoc test were used for multiple group comparisons. Data are expressed as mean +/− SEM. A p value of <0.05 was considered to be significant.
RESULTS
R-7050 attenuates neurovascular injury after ICH
Blood-brain barrier opening contributes to the development of vasogenic edema, an important cause of neurological deterioration after ICH. Evans blue extravasation, a sensitive estimate of blood-brain barrier integrity, increased from 12.2 ± 1.5 μg Evans blue/g brain tissue in sham-operated mice to 47.2 ± 5.8 μg Evans blue/g brain tissue at 24h post-ICH (p<0.01 vs. sham) (Figure 1). R-7050 (6 mg/kg) reduced Evans blue extravasation to 28.7 ± 5.9 μg and 30.3 ± 1.9 μg Evans blue/g brain tissue when administered at 0.5h or 2h post-ICH, respectively (p<0.05 and p<0.01 vs ICH, respectively; not significantly different from sham).
Figure 1. R-7050 maintains blood-brain barrier integrity after ICH.

Mice were administered 6 mg/kg R-7050 at 0.5h or 2h after collagenase-induced ICH. Evans blue extravasation, a sensitive measure of BBB disruption was assessed 24 hours later. Data are expressed as mean ± SEM and were analyzed by one-way ANOVA followed by Student Newman Keul’s post-hoc test (**p<0.01, n=8–9 per group).
Brain water content, a measure of brain edema, increased from 75.6 ± 0.3% in sham-operated mice to 81.5 ± 0.5% at 24h post-ICH (p<0.05 vs. sham). 6, 12, or 18 mg/kg R-7050 reduced brain water content to 78.5 ± 0.3%, 78.3 ± 0.3%, or 79.3 ± 0.5%, respectively (all treatments p<0.05 vs. ICH; treatments not significantly different from each other) (Figure 2B). Notably, mice treated with 18 mg/kg exhibited a reduction in general activity/locomotion; thus, follow up studies did not utilized this dose. As was observed with Evans blue extravasation, R-7050 (6 mg/kg) significantly reduced brain water content after ICH. Administration of R-7050 at 0.5h or 2h post-ICH attenuated brain water content to levels observed in sham-operated mice (p<0.05 vs ICH, not significantly different from sham) (Figure 2B).
Figure 2. R-7050 reduces edema development after ICH.

(A) Mice were administered R-7050 (6, 12, 18 mg/kg) just prior to collagenase-induced ICH. Brain water content, a measure of cerebral edema, was assessed in the ipsilateral hemisphere at 24h post-ICH. (B) Mice were administered 6 mg/kg R-7050 at 0.5h or 2h after collagenase-induced ICH. Brain edema was assessed 24h later. Comparisons within each hemisphere between different treatments groups were done using a one-way ANOVA followed by Student Newman Keul’s post-hoc test (# p<0.05 vs sham, *p<0.05, **p<0.01). No significant differences were observed between groups in the contralateral hemispheres. Data are expressed as mean ± SEM from 8–9 mice/group
R-7050 does not reduce hematoma volume after ICH
Hematoma volume is directly correlated with functional outcomes; thus, the effect of R-7050 on hematoma volume was ascertained. In contrast to the reduction in BBB opening and edema formation, R-7050 (6, 12 mg/kg) did not significantly reduce hematoma volume over the first 72h, as assessed by quantification of hemoglobin content within the ipsilateral hemisphere (Figure 3). Specifically, hemoglobin content was increased within the injured hemisphere from 30.1 ± 2.0 mg/dL in sham-operated mice to 117.9 ± 16.7 mg/dL following ICH (p<0.05 vs. sham). Similarly, neither 6 mg/kg nor 12 mg/kg R-7050 affected hemoglobin content, as compared to placebo-treated ICH mice (101.7 ± 17.0 mg/dL and 111.1 ± 17.3 mg/dL, respectively).
Figure 3. R-7050 does not affect hematoma volume after ICH.
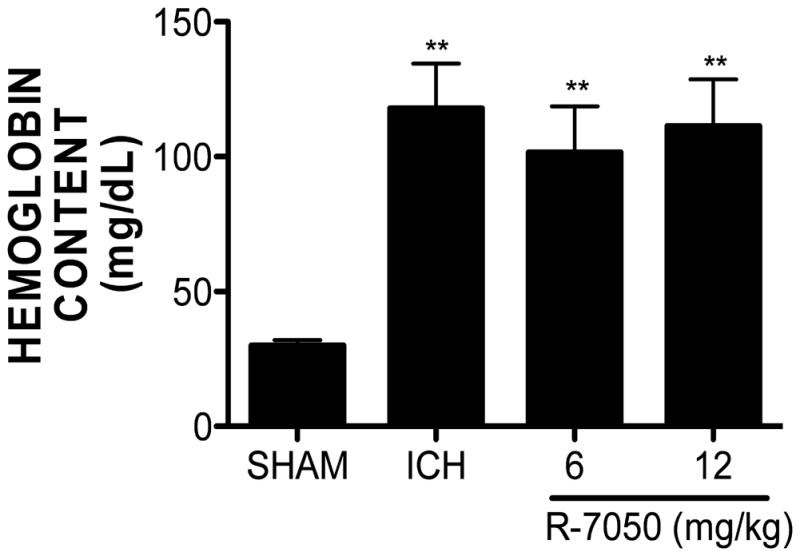
R-7050 (6, 12 mg/kg) administration at the time of ICH failed to reduce hematoma size at 72h post-ICH. Hematoma volume was quantified by determining the hemoglobin content of each hemisphere at 72 hours post-ICH. Data are expressed as mean ± SEM (*p<0.05,***p<0.001 vs. Sham; n=8 per group).
R-7050 improves neurological outcomes after ICH
A protective effect of R-7050 was observed across the first three days post-ICH, as compared to placebo treated mice, with a complete reduction in neurological deficits observed by 72h (p<0.05 vs ICH, not significantly different from sham) (Figure 4). Similarly, an intermediate protective effect was observed with both 6 mg/kg and 12 mg/kg R-7050 at 24h and 48h post-ICH (p<0.01 vs ICH, p<0.05 vs. sham). Placebo treatment had no significant effect on neurobehavioral outcomes, as compared to ICH with no treatment.
Figure 4. R-7050 improves neurological outcome after ICH.

Mice were administered R-7050 (6, 12 mg/kg, i.p.) 0.5h after collagenase-induced ICH. Neurological outcomes were assessed at 24h, 48h, or 72h following sham or ICH using a 24 point neurological scale score. Data are the mean ± SEM (n=9–10/group) and were analyzed using repeated measures ANOVA followed by Bonferroni’s post hoc test (*p<0.05, **p<0.01).
DISCUSSION
ICH induces the highest mortality of all stroke subtypes and <20% ICH survivors recover functional independence after six months [13, 29]. Hematoma volume directly correlates with neurological deterioration and patient mortality and neurosurgical clot evacuation produces more favorable outcomes in subsets of ICH patients [4, 29]; however, many patients remain poor surgical candidates and the efficacy of surgical intervention for spontaneous supratentorial ICH remains controversial [2, 27]. These data emphasize the devastating nature of ICH and indicate the dire need for efficacious medical treatment options to improve long-term patient prognoses.
Erythrocyte extravasation induces the formation of a space-occupying hematoma within the brain parenchyma that may result in microvascular compression, herniation, and neurovascular dysfunction [4, 36]. Spontaneous hematoma resolution alleviates the mass effect of the clot, although the simultaneous release of toxic hemoglobin metabolites enhances immune activation and exacerbates neurological injury. Along these lines, acute expression of TNF-α was associated with increased neurovascular injury after ICH and elevated cerebrospinal fluid and plasma TNF-α concentrations correlated with patient mortality after spontaneous ICH [3, 9, 16]. Consistent with a possible detrimental role in acute brain injury, TNF-α expression temporally preceded glial cell death following exposure to hemin, a major hemoglobin oxidation product within intracranial hematomas [21]. Similarly, heme exposure increased programmed necrosis through autocrine production of TNF-α [12]. Furthermore, unconjugated bilirubin, a heme catabolite that contributed to inflammatory activation and increased brain edema after ICH in mice [24], was associated with increased microglial TNF-α production and enhanced TNFR-dependent gliotic responses [10, 11, 31]. These findings support the assertion that TNF-α signaling exerts an acute detrimental role after ICH.
In the present study, a single administration of R-7050 reduced neurovascular injury after ICH. In contrast to biologic approaches that directly bind and neutralize TNF-α activity, R-7050 selectively inhibits the association of TNFR with intracellular adaptor molecules, such as TRADD and RIP1, limiting receptor internalization and preventing subsequent cellular responses after TNF-α binding [14]. Consistent with the assertion that TNFR signaling promotes acute brain injury after ICH, we found that necrostatin-1, a selective RIP1 inhibitor [7], limited peri-hematoma cell death, attenuated neuroinflammatory activation, reduced neurovascular injury, and improved neurobehavioral outcomes after ICH (King and Dhandapani, unpublished results). RIP1 functionally mediates TNF-α-induced activation of the pro-inflammatory transcription factor, NFκB [6]; thus, our pre-clinical findings are in line with clinical reports suggesting a direct correlation between inflammatory activation at the time of admission and early neurological deterioration in ICH patients [22]. Furthermore, our laboratory and others reported that NFκB activation reduces blood-brain barrier integrity, increases edema development, and exacerbates neurobehavioral deficits after experimental ICH [15, 19, 37]. Taken together, these results suggest TNF-α may initiate a detrimental signaling cascade after ICH.
Although most accurately mimicking the spontaneous intracerebral bleeding and evolving hematoma expansion observed in patients, the intrastriatal injection of bacterial collagenase used in this model may induce an exaggerated inflammatory response, as compared to the blood injection model of ICH. Thus, the beneficial effects of R-7050 may be overestimated. Nonetheless, TNF-α was elevated in ICH patients [9] and increased peri-hematoma TNF-α expression contributed to edema development after ICH in rodents [16]. These data suggest the beneficial effects of R-7050 observed herein are unlikely to be model specific and rather, may have more widespread therapeutic implications. Despite a clear damaging role in acute brain injury, TNF-α may also prime cerebral endothelial cells for erythropoietin-induced angiogenesis and enhance functional recovery after a brain hemorrhage [33]. This potential caveat indicates both beneficial and detrimental effects of TNF after brain injury and suggests the need for additional research prior to clinical translation of these data. Future work by our laboratory will further delineate the specific roles of TNF-TNFR signaling after ICH.
In conclusion, we identified a novel role for the triazoloquinoxaline TNF-α inhibitor, R-7050, in the attenuation of neurovascular injury and neurobehavioral improvement in a pre-clinical model of ICH. These data suggest therapeutic targeting of TNF-α represents a feasible means to limit neurological injury after a brain hemorrhage; however, we cannot exclude the possibility that R-7050 may exert neuroprotective effects via off-target effects independent of TNFR. Future work by our laboratory will continue to explore the translational potential of this compound, including further studies of drug specificity, as well as the establishment of a full therapeutic window and dosing paradigm in multiple pre-clinical models of ICH, including both collagenase and blood injection models of ICH
HIGHLIGHTS.
R-7050, a novel TNFR antagonist, reduces neurovascular injury after ICH
Pharmacological inhibition of TNFR improves outcomes after ICH
TNFR may represent a viable therapeutic target after ICH
Acknowledgments
This work was supported in part by grants from the National Institute of Health (NS065172, NS075774) and from the American Heart Association (BGIA2300135) to KMD. The funding agencies played no role in study design, data collection and interpretation, or publication.
ABBREVIATIONS
- BBB
Blood-brain barrier
- ICH
Intracerebral hemorrhage
- RIP1
Receptor interacting protein 1
- TNF-α
Tumor necrosis factor-α
- TNFR
Tumor necrosis factor-α receptor
- TRADD
Tumor necrosis factor receptor type 1 associated DEATH domain protein
- TRAF
Tumor necrosis factor receptor associated factor
Footnotes
The authors declare no competing interests or conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 2.Broderick J, Connolly S, Feldmann E, Hanley D, Kase C, Krieger D, Mayberg M, Morgenstern L, Ogilvy CS, Vespa P, Zuccarello M. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke; a journal of cerebral circulation. 2007;38:2001–2023. doi: 10.1161/STROKEAHA.107.183689. [DOI] [PubMed] [Google Scholar]
- 3.Castillo J, Davalos A, Alvarez-Sabin J, Pumar JM, Leira R, Silva Y, Montaner J, Kase CS. Molecular signatures of brain injury after intracerebral hemorrhage. Neurology. 2002;58:624–629. doi: 10.1212/wnl.58.4.624. [DOI] [PubMed] [Google Scholar]
- 4.Christoforidis GA, Slivka A, Mohammad Y, Karakasis C, Avutu B, Yang M. Size matters: hemorrhage volume as an objective measure to define significant intracranial hemorrhage associated with thrombolysis. Stroke; a journal of cerebral circulation. 2007;38:1799–1804. doi: 10.1161/STROKEAHA.106.472282. [DOI] [PubMed] [Google Scholar]
- 5.Clark W, Gunion-Rinker L, Lessov N, Hazel K. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke; a journal of cerebral circulation. 1998;29:2136–2140. doi: 10.1161/01.str.29.10.2136. [DOI] [PubMed] [Google Scholar]
- 6.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature chemical biology. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dziedzic T, Bartus S, Klimkowicz A, Motyl M, Slowik A, Szczudlik A. Intracerebral hemorrhage triggers interleukin-6 and interleukin-10 release in blood. Stroke. 2002;33:2334–2335. doi: 10.1161/01.str.0000027211.73567.fa. [DOI] [PubMed] [Google Scholar]
- 9.Fang HY, Ko WJ, Lin CY. Inducible heat shock protein 70, interleukin-18, and tumor necrosis factor alpha correlate with outcomes in spontaneous intracerebral hemorrhage. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2007;14:435–441. doi: 10.1016/j.jocn.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 10.Fernandes A, Barateiro A, Falcao AS, Silva SL, Vaz AR, Brito MA, Silva RF, Brites D. Astrocyte reactivity to unconjugated bilirubin requires TNF-alpha and IL-1beta receptor signaling pathways. Glia. 2011;59:14–25. doi: 10.1002/glia.21072. [DOI] [PubMed] [Google Scholar]
- 11.Fernandes A, Falcao AS, Silva RF, Gordo AC, Gama MJ, Brito MA, Brites D. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. Journal of neurochemistry. 2006;96:1667–1679. doi: 10.1111/j.1471-4159.2006.03680.x. [DOI] [PubMed] [Google Scholar]
- 12.Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, Chan FK, Bozza MT. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood. 2012;119:2368–2375. doi: 10.1182/blood-2011-08-375303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebel JM, Jr, Jauch EC, Brott TG, Khoury J, Sauerbeck L, Salisbury S, Spilker J, Tomsick TA, Duldner J, Broderick JP. Relative edema volume is a predictor of outcome in patients with hyperacute spontaneous intracerebral hemorrhage. Stroke; a journal of cerebral circulation. 2002;33:2636–2641. doi: 10.1161/01.str.0000035283.34109.ea. [DOI] [PubMed] [Google Scholar]
- 14.Gururaja TL, Yung S, Ding R, Huang J, Zhou X, McLaughlin J, Daniel-Issakani S, Singh R, Cooper RD, Payan DG, Masuda ES, Kinoshita T. A class of small molecules that inhibit TNFalpha-induced survival and death pathways via prevention of interactions between TNFalphaRI, TRADD, and RIP1. Chemistry & biology. 2007;14:1105–1118. doi: 10.1016/j.chembiol.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Hickenbottom SL, Grotta JC, Strong R, Denner LA, Aronowski J. Nuclear factor-kappaB and cell death after experimental intracerebral hemorrhage in rats. Stroke; a journal of cerebral circulation. 1999;30:2472–2477. doi: 10.1161/01.str.30.11.2472. discussion 2477–2478. [DOI] [PubMed] [Google Scholar]
- 16.Hua Y, Wu J, Keep RF, Nakamura T, Hoff JT, Xi G. Tumor necrosis factor-alpha increases in the brain after intracerebral hemorrhage and thrombin stimulation. Neurosurgery. 2006;58:542–550. doi: 10.1227/01.NEU.0000197333.55473.AD. discussion 542–550. [DOI] [PubMed] [Google Scholar]
- 17.Kim JS, Yoon SS, Kim YH, Ryu JS. Serial measurement of interleukin-6, transforming growth factor-beta, and S-100 protein in patients with acute stroke. Stroke. 1996;27:1553–1557. doi: 10.1161/01.str.27.9.1553. [DOI] [PubMed] [Google Scholar]
- 18.Kimbler DE, Shields J, Yanasak N, Vender JR, Dhandapani KM. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PloS one. 2012;7:e41229. doi: 10.1371/journal.pone.0041229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King MD, McCracken DJ, Wade FM, Meiler SE, Alleyne CH, Jr, Dhandapani KM. Attenuation of hematoma size and neurological injury with curcumin following intracerebral hemorrhage in mice. Journal of neurosurgery. 2011;115:116–123. doi: 10.3171/2011.2.JNS10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laird MD, Sukumari-Ramesh S, Swift AE, Meiler SE, Vender JR, Dhandapani KM. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? Journal of neurochemistry. 2010;113:637–648. doi: 10.1111/j.1471-4159.2010.06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laird MD, Wakade C, Alleyne CH, Jr, Dhandapani KM. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free radical biology & medicine. 2008;45:1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Leira R, Davalos A, Silva Y, Gil-Peralta A, Tejada J, Garcia M, Castillo J. Early neurologic deterioration in intracerebral hemorrhage: predictors and associated factors. Neurology. 2004;63:461–467. doi: 10.1212/01.wnl.0000133204.81153.ac. [DOI] [PubMed] [Google Scholar]
- 23.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 24.Loftspring MC, Johnson HL, Feng R, Johnson AJ, Clark JF. Unconjugated bilirubin contributes to early inflammation and edema after intracerebral hemorrhage. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:1133–1142. doi: 10.1038/jcbfm.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manaenko A, Chen H, Kammer J, Zhang JH, Tang J. Comparison Evans Blue injection routes: Intravenous versus intraperitoneal, for measurement of blood-brain barrier in a mice hemorrhage model. Journal of neuroscience methods. 2011;195:206–210. doi: 10.1016/j.jneumeth.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayne M, Ni W, Yan HJ, Xue M, Johnston JB, Del Bigio MR, Peeling J, Power C. Antisense oligodeoxynucleotide inhibition of tumor necrosis factor-alpha expression is neuroprotective after intracerebral hemorrhage. Stroke; a journal of cerebral circulation. 2001;32:240–248. doi: 10.1161/01.str.32.1.240. [DOI] [PubMed] [Google Scholar]
- 27.Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, Karimi A, Shaw MD, Barer DH. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. 2005;365:387–397. doi: 10.1016/S0140-6736(05)17826-X. [DOI] [PubMed] [Google Scholar]
- 28.Platt N, da Silva RP, Gordon S. Recognizing death: the phagocytosis of apoptotic cells. Trends Cell Biol. 1998;8:365–372. doi: 10.1016/s0962-8924(98)01329-4. [DOI] [PubMed] [Google Scholar]
- 29.Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. The New England journal of medicine. 2001;344:1450–1460. doi: 10.1056/NEJM200105103441907. [DOI] [PubMed] [Google Scholar]
- 30.Rosenberg GA, Mun-Bryce S, Wesley M, Kornfeld M. Collagenase-induced intracerebral hemorrhage in rats. Stroke. 1990;21:801–807. doi: 10.1161/01.str.21.5.801. [DOI] [PubMed] [Google Scholar]
- 31.Silva SL, Vaz AR, Barateiro A, Falcao AS, Fernandes A, Brito MA, Silva RF, Brites D. Features of bilirubin-induced reactive microglia: from phagocytosis to inflammation. Neurobiology of disease. 2010;40:663–675. doi: 10.1016/j.nbd.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 32.Sukumari-Ramesh S, Alleyne CH, Jr, Dhandapani KM. Astrocyte-specific expression of survivin after intracerebral hemorrhage in mice: a possible role in reactive gliosis? Journal of neurotrauma. 2012;29:2798–2804. doi: 10.1089/neu.2011.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, Chopp M, Teng H, Bolz M, Francisco MA, Aluigi DM, Wang XL, Zhang RL, Chrsitensen S, Sager TN, Szalad A, Zhang ZG. Tumor necrosis factor alpha primes cerebral endothelial cells for erythropoietin-induced angiogenesis. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:640–647. doi: 10.1038/jcbfm.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wasserman JK, Zhu X, Schlichter LC. Evolution of the inflammatory response in the brain following intracerebral hemorrhage and effects of delayed minocycline treatment. Brain research. 2007;1180:140–154. doi: 10.1016/j.brainres.2007.08.058. [DOI] [PubMed] [Google Scholar]
- 35.Woiciechowsky C, Schoning B, Cobanov J, Lanksch WR, Volk HD, Docke WD. Early IL-6 plasma concentrations correlate with severity of brain injury and pneumonia in brain-injured patients. J Trauma. 2002;52:339–345. doi: 10.1097/00005373-200202000-00021. [DOI] [PubMed] [Google Scholar]
- 36.Xi G, Fewel ME, Hua Y, Thompson BG, Jr, Hoff JT, Keep RF. Intracerebral hemorrhage: pathophysiology and therapy. Neurocritical care. 2004;1:5–18. doi: 10.1385/ncc:1:1:5. [DOI] [PubMed] [Google Scholar]
- 37.Zhao X, Zhang Y, Strong R, Zhang J, Grotta JC, Aronowski J. Distinct patterns of intracerebral hemorrhage-induced alterations in NF-kappaB subunit, iNOS, and COX-2 expression. Journal of neurochemistry. 2007;101:652–663. doi: 10.1111/j.1471-4159.2006.04414.x. [DOI] [PubMed] [Google Scholar]