

Abstract
Apolipoprotein E (APOE) is the major risk factor for sporadic Alzheimer's disease. Among other functions, APOE is proposed to sequester neurotoxic amyloid-β (Aβ) peptides in the brain, delivering them to cellular catabolism via neuronal APOE receptors. Still, the receptors involved in this process remain controversial. Here, we identified the pro-neurotrophin receptor sortilin as major endocytic pathway for clearance of APOE/Aβ complexes in neurons. Sortilin binds APOE with high affinity. Lack of receptor expression in mice results in accumulation of APOE and of Aβ in the brain and in aggravated plaque burden. Also, primary neurons lacking sortilin exhibit significantly impaired uptake of APOE/Aβ complexes despite proper expression of other APOE receptors. Despite higher than normal brain APOE levels, sortilin-deficient animals display anomalies in brain lipid metabolism (e.g., accumulation of sulfatides) seen in APOE-deficient mice, indicating functional deficiency in cellular APOE uptake pathways. Together, our findings identified sortilin as an essential neuronal pathway for APOE-containing lipoproteins in vivo and suggest an intriguing link between Aβ catabolism and pro-neurotrophin signaling converging on this receptor.
Introduction
Sortilin is a member of the family of vacuolar protein sorting 10 protein (VPS10P) domain receptors, a group of type-1 membrane proteins with important roles in regulation of neuronal viability and function (Nykjaer and Willnow, 2012). Sortilin is expressed in neurons of the CNS and PNS but also in extra-neuronal tissues, including liver and fat (Petersen et al., 1997; Morris et al., 1998; Sarret et al., 2003; Kjolby et al., 2010). Most notably, sortilin is recognized for its activity as a receptor for pro-nerve growth factor (proNGF) and pro-brain-derived neurotrophic factor required for induction of neuronal cell death by these proapoptotic molecules (Nykjaer et al., 2004; Teng et al., 2005). Sortilin-dependent induction of cell death has been documented during development and aging, as well as in acute insults to the nervous system (Jansen et al., 2007). Surprisingly, recent studies uncovered a distinct function for this receptor in hepatocytes in which it interacts with apolipoprotein (APO) B100 to control assembly and hepatic release of lipoproteins into the circulation (Kjolby et al., 2010; Musunuru et al., 2010). This observation suggested a mechanistic link between lipoprotein metabolism and regulation of neuronal viability that converges on this receptor pathway.
With respect to neuronal viability, the relevance of sortilin as a death receptor in acute neuronal injury (e.g., spinal cord injury) is well appreciated (Jansen et al., 2007). However, its contribution to chronic neurodegenerative processes as in Alzheimer's disease (AD) remains less clear. Such a role had been contemplated based on the fact that proNGF concentrations are increased in the brain of AD patients (Fahnestock et al., 2001). In support of this hypothesis, sortilin levels in brain specimens from patients with AD or with mild cognitive impairment were positively correlated with the severity of neuropathology, suggesting increased proNGF signaling through sortilin as an underlying cause of neurodegeneration (Mufson et al., 2010). As an alternative hypothesis, sortilin was reported to regulate trafficking of BACE1 (β-site APP-cleaving enzyme), and the enzyme responsible for initiating amyloidogenic cleavage of the amyloid precursor protein (APP), the main etiologic agent in AD (Finan et al., 2011). The latter model is in line with functions of related VPS10P domain receptors SORLA (sorting protein-related receptor) and SORCS1 (sortilin-related receptor CNS expressed) that modulate amyloidogenic processing through control of intracellular sorting of APP (Andersen et al., 2005; Schmidt et al., 2007; Lane et al., 2010).
Although intriguing, studies on the role of sortilin in AD have so far solely relied on in vitro studies. Now, we applied mouse models with targeted Sort1 gene disruption to query the relevance of this receptor pathway for AD-related processes in vivo. Mice lacking sortilin exhibit increased brain levels of amyloid-β (Aβ) peptides and show aggravated plaque burden, substantiating the significance of this receptor for amyloidogenic processes. However, contrary to previous hypotheses, our data revealed that sortilin does not affect APP processing in cells or in vivo. Rather, it acts as an apolipoprotein E (APOE) receptor in neurons, constituting a major endocytic pathway for catabolism of Aβ bound to this APO in the brain.
Materials and Methods
Materials.
Recombinant human APOE4 (Calbiochem, MBL), APOE3 (Alpha Diagnostics, Sigma), and HiLyte Fluor 488-labeled β-amyloid (1–40) (Anaspec) were obtained commercially. The pro-domain of murine NGF (amino acids E19 to R121) was expressed in Escherichia coli as a glutathione S-transferase (GST) fusion protein and purified on glutathione–agarose beads. APOE-deficient high-density lipoprotein (HDL) was isolated from plasma of healthy donors as described previously (Rellin et al., 2008). APOE-conditioned media were produced by transient transfection of HEK293 cells with expression constructs encoding human APOE2, APOE3, or APOE4 (Chen et al., 2010) and collected in DMEM without fetal calf serum for 48 h. Proteoliposomes containing APOE2, APOE3, and APOE4 were prepared according to published protocols (Heeren et al., 1999).
Mouse and cell models.
PDAPP (mice expressing human APP V717F variant under control of the human platelet derived growth factor β-chain gene promoter) (Games et al., 1995), Sorl1−/− (Andersen et al., 2005), Apoe−/− (Plump et al., 1992), 5xFAD (Oakley et al., 2006), and Sort1−/− (Jansen et al., 2007) lines of mice have been described previously. Mice carrying a loxP-modified Lrp1 allele (Rohlmann et al., 1996) were bred to animals transgenic for Cre recombinase under control of the neuron-specific enolase promoter (Nse–creCK1) (Kwon et al., 2006). Experimental procedures involving animals were performed following approval by local authorities. Primary hippocampal neurons were prepared from newborn mice (P1–P2) and cultured for 7–10 d in Neurobasal-A medium (Invitrogen) supplemented with B27 (Invitrogen) and GlutaMAX (Invitrogen). Primary cultures of cortical astrocytes were prepared from cortices of P0 mice by digestion in trypsin/EDTA solution. Tissues were triturated in DMEM with 10% FCS and DNase and seeded in poly-l-lysine-coated T75 flask in DMEM with 10% FCS and primocin.
Metabolic labeling of primary astrocyte cultures.
Astrocyte cultures in six-well 36-mm plate format were metabolically labeled using [35S]Met/[35S]Cys in DMEM without Met/Cys supplemented with 2% dialyzed FCS and Brefeldin A (10 μg/ml) for 4 h at 37°C. Cells were washed twice in DMEM before addition of DMEM plus 2% FCS and incubation at 37°C. At indicated time points, conditioned media and cell lysate were harvested and subject to immunoprecipitation with 1 μg of anti-APOE IgG (Calbiochem) coupled to γ-bind beads. Precipitated proteins were visualized by SDS-PAGE and autoradiography.
BIAcore.
Surface plasmon resonance analysis was performed on a BIAcore 2000 instrument (GE Healthcare) equipped with CM5 sensor chips coupled with the recombinant extracellular domain of sortilin (0.046 pmol/mm2) as published previously (Nykjaer et al., 2004). Proteoliposomes, in the absence or presence of 20 μm neurotensin (NT), were diluted in 10 mm HEPES, 150 mm NaCl, 1.5 mm CaCl2, and 1 mm EGTA, pH 7.4 (running buffer) and injected at 5 pmol/min for 5 min, at 4°C. Kinetic parameters were determined using the BIAevaluation 3.0 software. The number of APOE isoforms bound per moles of immobilized sortilin was estimated by dividing the ratio RUligand/massligand with RUsortilin/masssortilin (in which RU is response units).
Quantitative RT-PCR.
Real time RT-PCR was performed on cDNA reversely transcribed from total RNA using a 7900HT Real-Time PCR system (Applied Biosystems) (Rohe et al., 2009). Transcripts were determined using commercial Assay-on-Demand primer/probe sets (Applied Biosystems). Each template was run in triplicates, and relative expression levels were calculated by normalization to the internal control β2 microglobulin.
Determination of APP processing and plaque burden.
Intensities of immunoreactive bands of Western blot analyses were quantified by optical densitometry using NIH ImageJ software (http://rsb.info.nih.gov/ij/). Aβ40/42 [soluble (s) fraction] as well as sAPPα and sAPPβ levels in brain extracts or cell media were analyzed using commercial ELISA kits (Mesoscale Discovery) and BACE1 activity by BACE1 fluorescence resonance energy transfer (FRET) assay kit (Invitrogen). For determination of plaque burden, two to six hippocampal paraffin sections (5 μm) from 5xFAD; Sort1+/+ (2 and 10 months) and 5xFAD; Sort1−/− (2 and 10 months) mice were stained with 1% Thioflavin S (Sigma). Quantification of density of thioflavin S staining was performed using NIH ImageJ software in single optical images. Plaque density was related to the hippocampal area and averaged for each animal studied.
Uptake studies in established and primary cell cultures.
For determination of APOE uptake, cells were incubated with supernatant from mock transfectants or HEK293 cells transfected with human APOE expression constructs. After 30 min incubation at 37°C, the ligand-containing medium was removed, and cells were washed in PBS containing 500 U/ml heparin and subjected to standard confocal immunofluorescence microscopy. For detection of surface binding, Triton X-100 was omitted from the incubation buffers. To determine cellular uptake of Aβ, human recombinant APOE4 was associated with HDL in a ratio of 1:1 with respect to HDL protein content (Rellin et al., 2008). HiLyte–Aβ was added to the lipidated APOE mixture and incubated at 37°C for 1 h to allow complex formation. As a negative control, free HiLyte–Aβ was mixed with APOE-free HDL. Internalization of HiLyte–Aβ was evaluated by confocal immunofluorescence microscopy of cells treated with 10 μg/ml (lipidated APOE) or 1 μm HiLyte–Aβ40 for 1 h at 37°C. For quantification of uptake (see Fig. 10B), unbound Aβ was removed by dialysis in PBS for 4 h. Aβ concentration in the dialyzed fraction was measured by ELISA.
Figure 10.

Sortilin mediates cellular uptake of Aβ40 bound by human APOE. A, Replicate layers of parental CHO cells or CHO cells overexpressing murine sortilin (CHO–mSort) were incubated with HiLyte–Aβ40 complexed with APOE-free HDL or with HiLyte–Aβ40 complexed with APOE4- or APOE3-containing HDL. Uptake of APOE-complexed HiLyte–Aβ40 is seen in cells expressing sortilin but not in parental cells. Scale bar, 4 μm. DAPI staining is shown in blue. The inset depicts detection of HiLyte–Aβ40 using antiserum 4G8 (red). B, Levels of Aβ40 were determined by ELISA in CHO or CHO–mSort cells incubated with APOE4-complexed Aβ40 or the same concentration of free Aβ40. Statistical differences were determined by Bonferroni's post hoc test (total degrees of freedom = 15; F = 27.47). C, Levels of Aβ40 were determined by ELISA in CHO–mSort cells incubated with lipidated APOE4-complexed Aβ40. When indicated, the medium also included 10 μm NT or GST–pro. Statistical differences were determined by Bonferroni's post hoc test (degrees of freedom = 8; F =67.05) (**p < 0.01; ***p < 0.001).
Lipid analyses.
We modified the method described by Han et al. (2003). Thus, frozen brain tissues were homogenized in a precooled methanol/chloroform/water (5:2:1) mixture at a final ratio of 50 mg of tissue per 1 ml of solvent. Samples were homogenized in a Precellys 24 homogenizer (Bertin Technologies) two times for 20 s at 5.500 Hz. Solid debris were removed by centrifugation for 10 min at 10,000 × g, and 100 μl of supernatant were dried under vacuum overnight. Dried samples were resuspended in 1 ml of methanol containing 1 μg/ml N-palmitoyl-sulfatide (Matreya) as internal standard for the following analysis. For mass spectrometric analysis, 5 μl of sample were injected at 20 μl/min by an Agilent 1200 UPLC system (Agilent Technologies) directly into a TSQ Vantage mass spectrometer (Thermo Fisher Scientific) using a flow injection analysis. Acquisition was performed in negative mode, scanning Q3 quadrupole from 500–1050 mass/charge. The spray voltage was set at 3000 V, sheath gas at 13 arbitrary units, capillary temperature at 260°C, and S-Lens RF amplitude at 71 V. No gas was used in the collision cell. Abundance values for N24:1 sulfatide were calculated as the ratio of their intensities and the intensity of the internal standard. Intensities were calculated by automatic integration by Xcalibur software (Thermo Fisher Scientific) and were validated manually.
Statistical analyses of data.
Results comparing two groups were analyzed by Student's t test (see Figs. 3C, 14) or Mann–Whitney U test (all others). To compare the means of more than two groups, we used one-way (see Fig. 10C) or two-way (see Figs. 10B, 13B) ANOVA. F values and degrees of freedom are stated in the respective figure legends. When applicable, post hoc differences were compared using Bonferroni's test. All statistical analyses were performed using GraphPad Prism 5 software.
Figure 3.
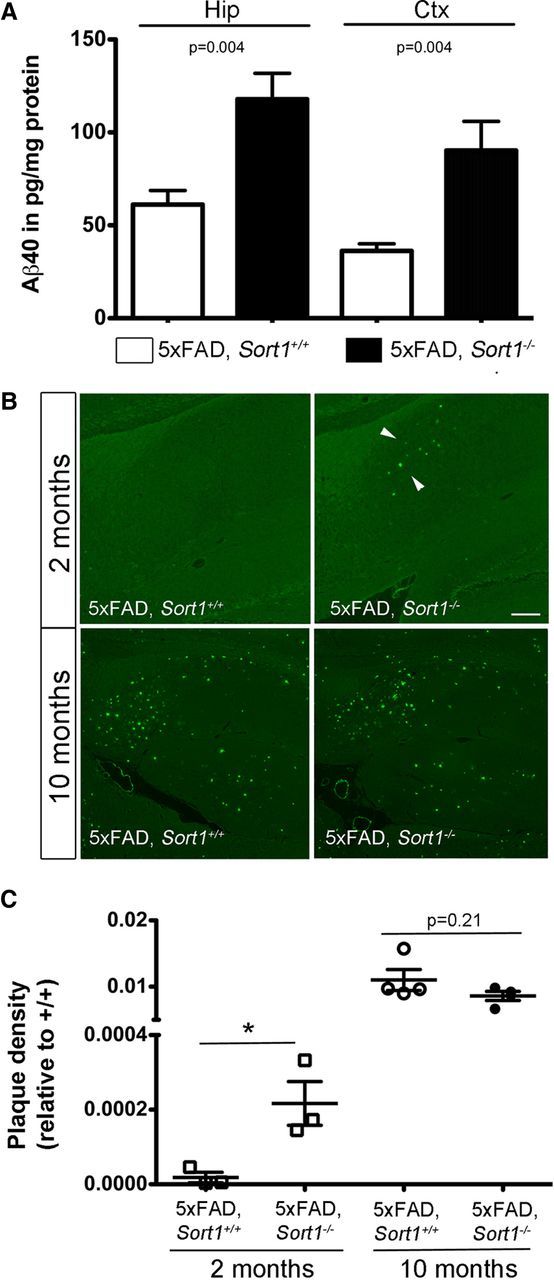
Sortilin deficiency increases plaque burden in the brain. A, Levels of Aβ40 were determined by ELISA in extracts of hippocampus (Hip) and cortex (Ctx) of 5xFAD mice either wild-type (5xFAD; Sort1+/+) or homozygous for the Sort1 null allele (5xFAD; Sort1−/−) at 2 months of age (n = 5–6 per group). B, Thioflavin S staining of sagittal sections through the brain of three to four individual mice at 2 and 10 months of age. Deposition of plaques (arrowheads) in the subiculum is seen in 2-month-old 5xFAD mice lacking sortilin (5xFAD; Sort1−/−) but not in the age- and gender-matched controls (5xFAD; Sort1+/+). At 10 months of age, a massive plaque burden is seen in both genotypes. Scale bar, 150 μm. C, Quantification of plaque burden in individual mice of the indicated age and genotype based on thioflavin S staining as exemplified in B (n = 2–6 sections per animal). *p < 0.05.
Figure 14.

Sortilin deficiency results in accumulation of N24:1 sulfatides in the brain. Levels of N24:1 sulfatides in the hippocampus of 5-month-old (n = 6–7 per group) and 10-month-old (n = 13–14 per group) mice of the indicated genotypes. **p < 0.01; ***p < 0.001.
Figure 13.

Sortilin is a major APOE uptake pathway relevant for neuronal catabolism of Aβ. A, Expression of APP, LRP1, and SORLA in the primary neuronal cell cultures of the indicated Sort1 genotypes as determined by Western blotting. Detection of tubulin served as loading control. B, Levels of Aβ40 were determined by ELISA in primary neurons from Sort1+/+ and Sort1−/− newborn mice incubated with the indicated two concentrations of lipidated APOE4 complexed with Aβ40. The data are mean ± SEM (n = 6) expressed as percentage of the respective (Sort1+/+) control. Statistically significant differences between genotypes were determined by Bonferroni's post hoc test (total degrees of freedom = 21; F = 31.06) and are indicated (**p < 0.01, ***p < 0.001). C, D, Levels of sAPPα and sAPPβ (C) and Aβ40 and Aβ42 (D) were determined by ELISA in primary hippocampal neurons either wild-type (PDAPP+/−; Sort1+/+; open bars) or homozygous for the Sort1 null allele (PDAPP+/−; Sort1−/−; filled bars). No statistically significant differences were seen for any of the APP processing products comparing genotypes (n = 9–14).
Results
Loss of sortilin increases brain Aβ levels in mice
Mice with targeted disruption of Sort1 (the gene encoding sortilin) have been generated by us previously (Jansen et al., 2007). Here, we crossed Sort1−/− animals with the PDAPP line of mice, an established model of amyloidogenic processing carrying the human APPV717F gene variant (Games et al., 1995). Loss of receptor expression did not alter total brain levels of endogenous murine (Fig. 1A,B) or human APP (Fig. 1C,D) in these mice compared with controls. However, lack of sortilin resulted in a significant increase in Aβ40 levels in brain extracts of PDAPP+/−; Sort1−/− mice compared with PDAPP+/−; Sort1+/+ controls (Fig. 1E). Intriguingly, the increase in Aβ40 was not paralleled by corresponding changes in the levels of sAPPα or sAPPβ fragments in affected animals (Fig. 1E). A rise in Aβ40 levels in PDAPP+/−; Sort1−/− mice was seen both in the hippocampus and in the cortex at 5 months (Fig. 1F) and 9 months (Fig. 1G) of age. Unimpaired proteolytic activity in the sortilin-deficient mouse brain was substantiated by Western blot analysis of C-terminal fragments C83 and C99, the products of α- and β-secretases, respectively (Fig. 2A,B), and by determination of BACE1 activity using FRET assay (Castellano et al., 2011) (Fig. 2C).
Figure 1.

Sortilin deficiency selectively increases Aβ levels in the murine brain. A, B, Levels of murine APP protein (A; mAPP) and App mRNA (B) are unchanged in extracts of sortilin-deficient (Sort1−/−) compared with control (Sort1+/+) mouse brains as shown by Western blotting and quantitative RT-PCR, respectively (n = 4 per genotype for B). C, D, Cortical levels of human APP protein (hAPP) are identical when comparing Sort1+/+ and Sort1−/− mice on the PDAPP background by Western blotting (C) and densitometric scanning of replicate blots (D; n = 3 per genotype). E, Levels of sAPPα, sAPPβ, and Aβ40 were determined by ELISA in brain extracts of PDAPP wild-type mice (PDAPP+/−; Sort1+/+; white bars) or mice homozygous for the Sort1 null allele (PDAPP+/−; Sort1−/−; black bars) (n = 3–7 per group). F, G, Aβ40 levels in hippocampus (Hip) and cortex (Ctx) of mice of the indicated genotypes at 5 months (F; n = 3–5 per group) and 9 months (G; n = 8–10 per group) of age. Values are mean ± SEM.
Figure 2.
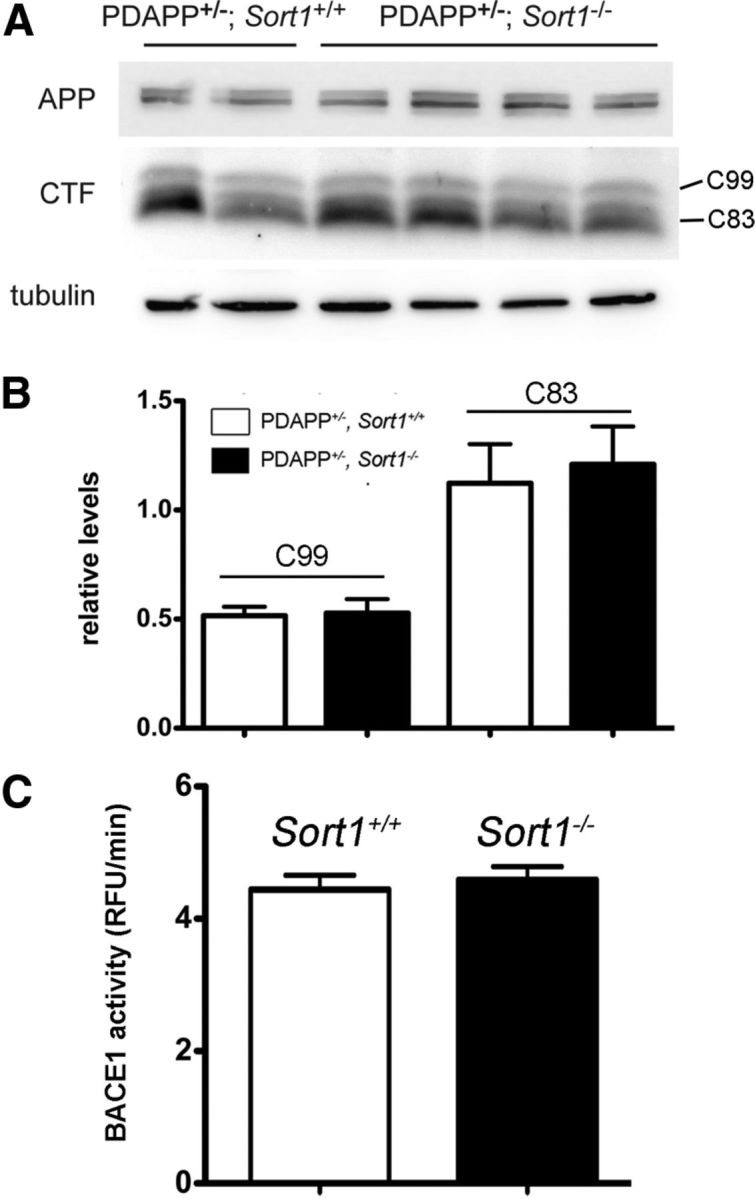
Sortilin deficiency does not impact α- and β-secretase activities. A, B, Quantitative analysis of C-terminal fragments (CTF) C99 and C83 in total brain extracts of Sort1+/+ and Sort1−/− mice on the PDAPP background using Western blotting (A) and densitometric scanning of replicate blots (B; n = 7–11 per genotype). Detection of APP and tubulin served as loading controls in A. C, Activity of BACE1 in mice of the indicate genotypes using FRET assay as described in Materials and Methods (n = 4 per group). RFU, Relative fluorescent unit.
The effect of sortilin deficiency on amyloidogenic processing was confirmed in 5xFAD mice, another commonly used AD model (Oakley et al., 2006). Thus, elevated levels of Aβ40 were seen in 5xFAD mice lacking sortilin (5xFAD; Sort1−/−) compared with control mice (5xFAD; Sort1+/+) at 2 months of age (Fig. 3A) and resulted in an aggravated plaque burden as documented by thioflavin S staining of hippocampal sections (Fig. 3B,C). At 10 months of age, the aggressive plaque burden observable in the 5xFAD model masked the consequences of sortilin deficiency on plaque deposition (Fig. 3B,C). This finding suggested an effect of receptor activity on time of onset and early progression of amyloidosis. Together, quantitative analyses of APP metabolism in vivo demonstrated an increase in Aβ concentrations in Sort1−/− animals that accelerated onset of senile plaques deposition. Because levels of soluble or membrane-bound APP processing products or β-secretase activity were not altered in these mice, our data argued for a role of sortilin in catabolism of Aβ peptides in the brain.
Loss of sortilin impairs neuronal catabolism of APOE
Several mechanisms have been shown to affect the half-life of Aβ peptides in brain parenchyma, including shunt into the circulation or enzymatic breakdown in the extracellular space (for review, see Kurz and Perneczky, 2011). The plasma levels of Aβ40 were identical in PDAPP+/−; Sort1−/− and control animals (Fig. 4A). Also, the levels of the Aβ-degrading enzymes CD10 (neprilysin) (Iwata et al., 2001) and insulin degrading enzyme (Farris et al., 2003) were not altered in the brain of receptor-deficient mice as shown by Western blot analyses (Fig. 4B). Thus, neither of the two mechanisms seemed likely responsible for the increase in Aβ levels in Sort1−/− mice. Conceivably, brain pathology as a consequence of altered (pro)-neurotrophin signaling may secondarily increase Aβ levels in sortilin-deficient mice. However, no signs of overt neuropathology were observable in histological brain sections from receptor null mice stained with thionine or antibodies directed against GFAP or NeuN, markers of glia cells and neurons, respectively (Fig. 5). Also, immunostaining for F4/80 failed to reveal any signs of reactive microglia in this mouse line (data not shown).
Figure 4.

Sortilin activity does not affect levels of Aβ in plasma or levels of Aβ peptide degrading enzymes in the brain. A, Levels of Aβ40 in plasma of PDAPP+/−; Sort1+/+ and PDAPP+/−; Sort1−/− mice are given. Values are mean ± SEM (n = 4–5 per group). B, Expression levels of CD10 (neprilysin) and insulin degrading enzyme (IDE) in hippocampus (Hip) and cortex (Ctx) mice of the indicated genotypes were determined by Western blotting. Detection of tubulin served as loading control.
Figure 5.
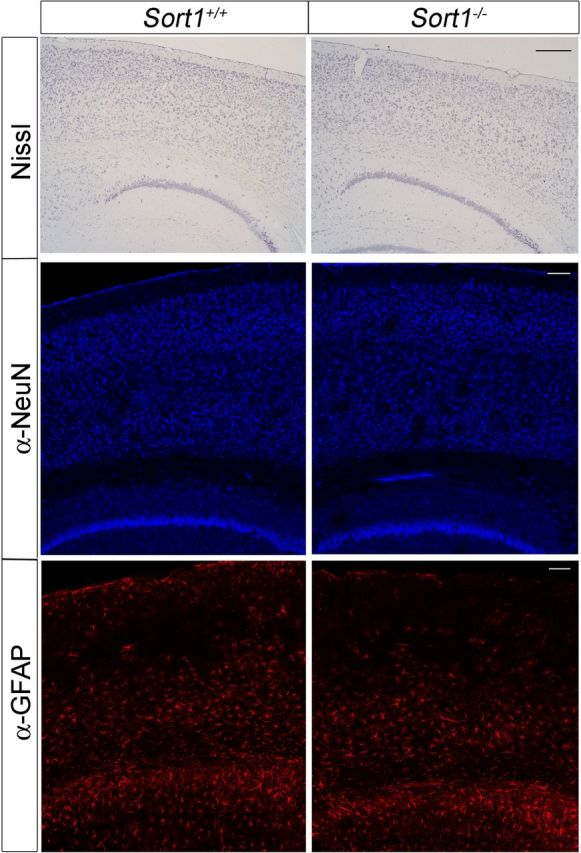
Histological analyses of sortilin-deficient mouse brains. Histological sections of the brain cortex from mice of the indicated genotypes stained with thionine or with antisera directed against the neuronal marker NeuN or the glial marker GFAP. Scale bars, 400 μm.
Another pathway implicated in brain turnover of Aβ is clearance by APOE receptors when the peptide is bound to APOE (Koistinaho et al., 2004; Morikawa et al., 2005; Jiang et al., 2008). Interestingly, a twofold increase in the concentration of APOE in hippocampus and cortex of sortilin null mice was evident when determining protein levels by Western blot (Fig. 6A,B). Elevated levels of APOE were not attributable to an increased rate of transcription as demonstrated by quantitative RT-PCR of Apoe transcripts in hippocampal and cortical tissues of the respective animals (Fig. 6C). Also, the rate of secretion of APOE from astrocytes, the main source of this APO in the brain, was not affected by the absence of the receptor as shown by metabolic labeling experiments in primary astrocyte cultures (Fig. 7). Thus, our data argued for impaired cellular uptake of APOE as the cause of Aβ accumulation and deposition in the brain of sortilin-deficient mice.
Figure 6.
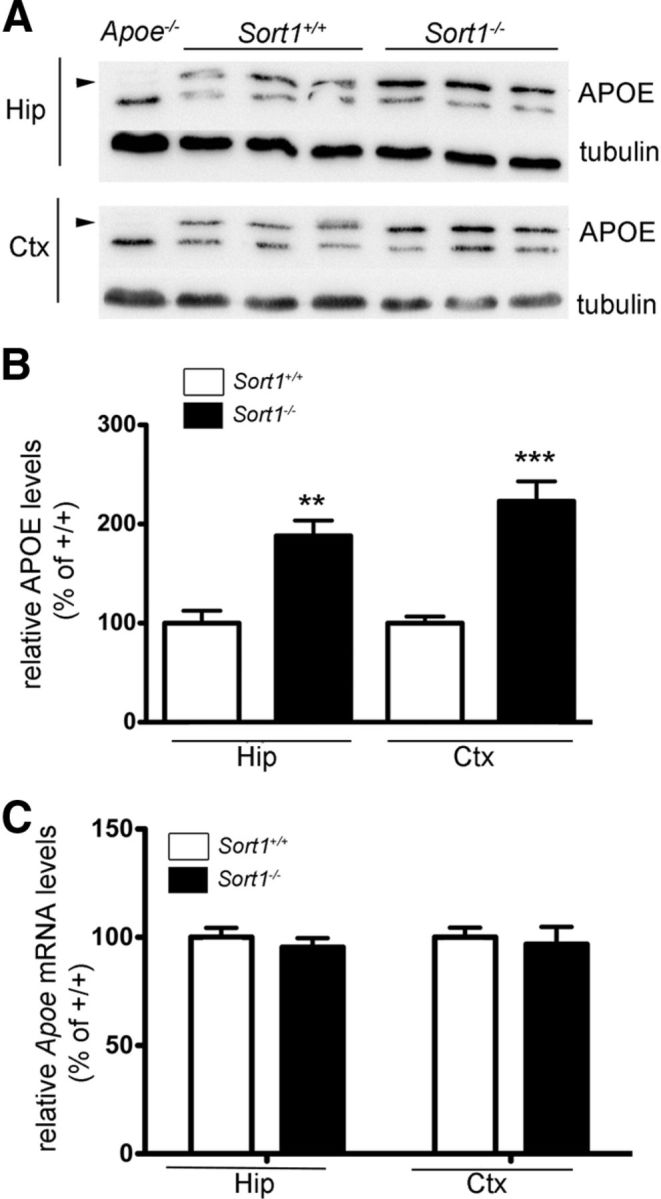
Accumulation of APOE in the brain of sortilin-deficient mice. A, B, Levels of APOE in hippocampus (Hip) and cortex (Ctx) of wild-type (Sort1+/+) and sortilin-deficient (Sort1−/−) mice at 3 months of age were determined by Western blot analyses (A) and quantified by densitometric scanning of the respective membranes (n = 6 per group) (B). In A, detection of tubulin served as loading control. Extracts from mice genetically deficient for APOE (Apoe−/−) were used as negative control for specificity of the APOE immunoreactivity (indicated by arrowheads). A similar difference in APOE levels was also seen in mice at 9 months of age (Sort1+/+, 100.0 ± 10.19%, n = 6; Sort1−/−, 176.1 ± 10.3%, n = 7). C, Quantitative RT-PCR analyses of Apoe mRNA levels in hippocampus and cortex of Sort1+/+ and Sort1−/− animals at 2 months of age (n = 4 per group). All values are mean ± SEM and are given as percentage of the wild-type control (mean set at 100%). Significant differences between genotypes are indicated (**p < 0.01, ***p < 0.001).
Figure 7.

Sortilin activity does not impact APOE release from primary astrocytes. APOE was immunoprecipitated from cell lysate and conditioned media of primary cultures of astrocytes of newborn Sort1+/+ and Sort1−/− animals. The levels of APOE in lysate and media at the indicated time points were determined by autoradiography (A) and quantified by densitometric scanning of replicate blots (B). APOE levels were normalized with respect to the sample with the highest level of APOE in lysate or media (set at 100%), respectively. Bar graphs in B show mean ± SD value of three separate experiments.
Sortilin acts as endocytic receptor for APOE
The ability of sortilin to bind APOB100 has been reported previously (Kjolby et al., 2010; Musunuru et al., 2010). However, a possible function as receptor for APOE has not been documented so far. Thus, we applied surface plasmon resonance analysis to test the ability of sortilin to interact with the latter APO. As test ligands, we used proteoliposomes enriched with recombinant human APOE2, APOE3, and APOE4 (Heeren et al., 1999). As shown in Figure 8A–C, the extracellular domain of sortilin bound all three APOE isoforms (Kd = 117 nm for APOE2; Kd = 44 nm for APOE3; Kd = 114 nm for APOE4). Binding was significantly reduced by NT, a neuropeptide that binds to the VPS10P domain and blocks interaction with ligands that target this receptor domain (Mazella et al., 1998; Quistgaard et al., 2009). In contrast, poor binding to the sortilin ectodomain was seen for isolated Aβ40 (Fig. 8D; Kd = 0.8 μm).
Figure 8.

Sortilin interacts with all human APOE isoforms. A–C, Surface plasmon resonance analyses of ligand binding to sortilin immobilized on the BIAcore chip surface. Binding of APOE-free liposomes and of liposomes complexed with recombinant APOE2 (A), APOE3 (B), and APOE4 (C) was tested in the presence and absence of 20 μm NT. D, Surface plasmon resonance analyses of binding of a concentration series of Aβ40 to sortilin immobilized on the BIAcore chip surface.
To further substantiate a role for sortilin as APOE receptor, we tested its ability to mediate endocytic uptake of this APO into cells. To do so, we generated a Chinese hamster ovary (CHO) cell line that stably overexpresses murine sortilin (CHO–mSort). The receptor mainly localized to intracellular compartments in CHO–mSort cells, in line with what has been observed previously in other cell types (Fig. 9A) (Nielsen et al., 2001). Despite a predominant intracellular localization, sortilin efficiently internalized ligands as shown for GST–pro, a surrogate ligand composed of the pro-domain of proNGF (the sortilin binding site) fused to GST (Nykjaer et al., 2004) (Fig. 9A). When replicate layers of CHO–mSort cells were incubated with conditioned media from HEK293 cells secreting lipidated APOE2, APOE3, or APOE4, avid internalization of all three human APOE isoforms was seen by immunofluorescence microscopy. Little APOE uptake was observed in parental CHO cells lacking the receptor (Fig. 9B). Efficient uptake of APOE in CHO–mSort but not in parental cells was substantiated by Western blot analysis using recombinant APOE4 lipidated with APOE-free HDL in vitro (Fig. 9C). Similar findings were obtained with recombinant APOE2 and APOE3 (data not shown). To directly document the ability of sortilin to act as a surface receptor for APOE, we immunostained CHO–mSort cells incubated with the APO in the absence of detergent. Colocalization of receptor and ligands at the cell surface of nonpermeabilized cells was readily visible for both APOE3 (Fig. 9D) and APOE4 (Fig. 9E).
Figure 9.
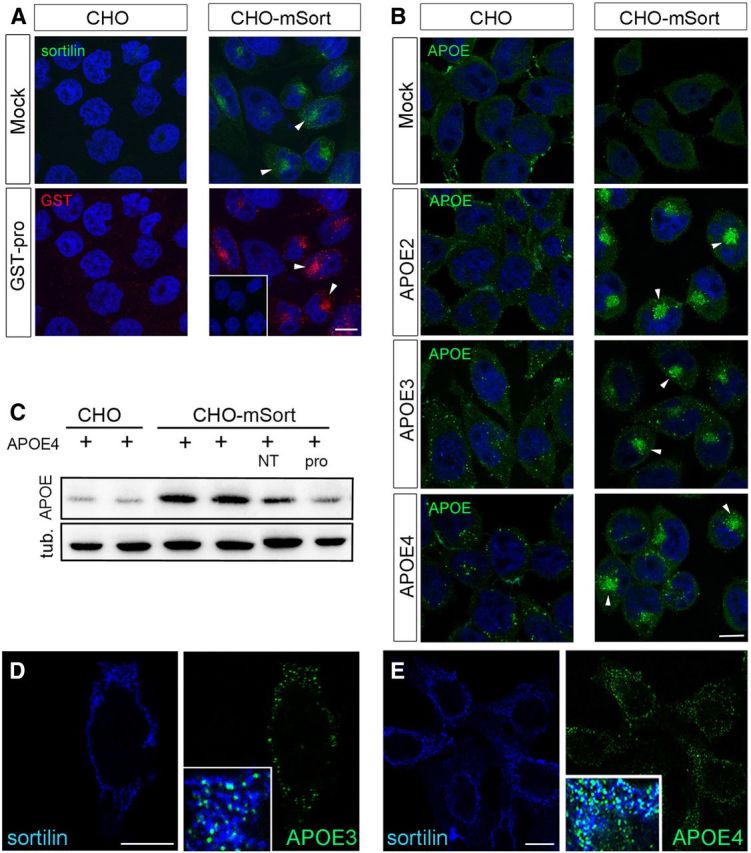
Sortilin mediates cellular uptake of human APOE. A, B, Replicate layers of parental CHO cells or CHO cells overexpressing murine sortilin (CHO–mSort) were incubated with blank medium (mock), with medium containing 500 nm of the pro-domain of NGF fused to GST (GST–pro), or with conditioned media from HEK293 cells transfected with expression constructs for human APOE2, APOE3, or APOE4. Receptor-dependent uptake of ligands was determined by immunodetection of sortilin (A), GST–pro (A), as well as APOE (B). Arrowheads highlight the localization of receptor and ligands in the perinuclear region of the cells. The inset in A indicates lack of ligand uptake in cells treated with GST instead of GST–pro. Scale bars, 4 μm. C, Detection of APOE and tubulin (loading control) in extracts of parental CHO or CHO–mSort cells incubated for 30 min with lipidated recombinant APOE4. When indicated, the medium also contained 10 μm NT or GST–Pro (pro). D, E, Detection of sortilin and APOE3 (D) and sortilin and APOE4 (E) on the surface of nonpermeabilized CHO–mSort cells by immunocytochemistry. Insets in D and E show the merged pictures, with white color indicating colocalization of sortilin (blue) and APOE (green) on the cell surface. Scale bars, 4 μm.
Sortilin mediates cellular uptake of Aβ bound to APOE
Next, we confirmed the ability of sortilin to mediate cellular uptake of Aβ peptides when bound to APOE. Thus, we incubated fluorescent-labeled Aβ40 (HiLyte–Aβ) with a 1:1 mixture of APOE-free HDL and recombinant APOE3 or APOE4. This ratio of APOE to HDL protein content is characteristic of lipoproteins in the CSF (Koch et al., 2001). The APOE/Aβ complexes were applied in a final concentration of 5–10 μg of APOE per milliliter to the culture medium. Similar concentrations of APOE have been measured in human CSF (5 μg/ml) (Hesse et al., 2000). As a negative control, HiLyte–Aβ was mixed with APOE-free HDL only. In line with our hypothesis, efficient uptake of Aβ40 was seen in cells expressing sortilin when the amyloid peptide was complexed with lipidated APOE3 or APOE4 (CHO–mSort; Fig. 10A). In contrast, no sortilin-mediated uptake of HiLyte–Aβ was seen in APOE-free HDL. Also, no uptake of free or APOE-bound HiLyte–Aβ40 was detectable in cells lacking receptor expression (CHO; Fig. 10A). Sortilin-dependent uptake of Aβ40 was also confirmed using antiserum 4G8 (Fig. 10A, inset). Quantitative analysis by ELISA demonstrated an approximate fivefold increase in uptake of APOE3-bound Aβ40 in CHO–mSort compared with parental CHO cells (Fig. 10B). Furthermore, threefold less uptake was seen in CHO–mSort cells incubated with free Aβ40 compared with APOE3-bound Aβ40, substantiating the ability of APOE to promote sortilin-dependent clearance of the peptide (Fig. 10B). Cellular uptake of APOE-bound Aβ40 was reduced by the receptor antagonists GST–pro and NT (Fig. 10C). Internalized Aβ40 and APOE molecules colocalized with sortilin in intracellular compartments of the cells (Fig. 11A). Colocalization of Aβ40 with the marker cathepsin B suggested predominant lysosomal targeting of the internalized peptides (Fig. 11B). To exclude any effect of sortilin on cellular Aβ40 production, we transiently transfected replicate HEK293 cell layers with expression constructs for human APP and either mSort or empty control vector. Determination of Aβ40 levels in cell supernatants after 48 h gave identical results for parental HEK293 (121.7 ± 13.62 pg Aβ/ng APP; n = 11) and HEK293–mSort cells (99.87 ± 6.260 pg Aβ/ng APP; n = 11; p = 0.39 by Mann–Whitney U test).
Figure 11.

Sortilin delivers Aβ to lysosomes. A, Replicate layers of CHO–mSort cells were incubated with HiLyte–Aβ40 complexed with lipidated APOE4 for 1 h. Subsequently, the subcellular localization of sortilin, APOE4, and Aβ40 was documented by immunofluorescence microscopy. B, Immunocytochemical detection of HiLyte–Aβ40 and lysosomal marker cathepsin B in CHO–mSort. The inset depicts the merged micrographs. Scale bars, 4 μm.
Sortilin is a major APOE receptor in neurons
A number of potential APOE receptors has been identified in neurons and shown to affect turnover of APOE-bound Aβ (for review, see Holtzman et al., 2012). One receptor discussed in this context is the low-density lipoprotein receptor-related protein 1 (LRP1), a member of the low-density lipoprotein (LDL) receptor gene family (Liu et al., 2010). In addition, other receptors of this gene family (including the LDL receptor itself) or VPS10P-domain receptors, such as SORLA or SORCS1, SORCS2, and SORCS3, may contribute to APO clearance in the brain. Comparative quantitative RT-PCR analysis revealed transcript levels for Sort1 and Sorl1 (the gene encoding SORLA) in the murine cortex that were 10- and 20-fold higher than those for Lrp1, arguing for high levels of sortilin (and SORLA) expression in the brain (Fig. 12A). Levels of transcripts encoding SORLA, LRP1, LDL receptor, very-low-density lipoprotein receptor, LRP8, or SORCS1, SORCS2, and SORCS3 were unchanged in the cortex of sortilin-deficient compared with wild-type mice as documented by quantitative RT-PCR analyses (Fig. 12B). This finding argued against reduced expression of any of these APOE receptors as the underlying cause of APOE accumulation in sortilin null mice. In support of this conclusion, no accumulation of APOE was seen in mice genetically deficient for Lrp1 or Sorl1 in this study. As shown in the inset to Figure 12D, crossing mice carrying a loxP-modified Lrp1 gene with animals expressing Cre recombinase under control of the neuron-specific enolase promoter (Nse–creCK1) resulted in efficient elimination of receptor expression from the brain. Still, levels of APOE in hippocampus and cortex of the LRP1-deficent mice were not altered compared with matched controls as shown by Western blotting (Fig. 12C) and by densitometric scanning thereof (Fig. 12D). Also, mice carrying a targeted disruption of Sorl1 (Andersen et al., 2005) displayed similar levels of APOE in cortex as wild-type controls (Fig. 12E,F).
Figure 12.

Functional expression of APOE receptors in the murine brain. A, Quantitative RT-PCR of levels of Sort1, Sorl1, and Lrp1 transcripts in the cortex of wild-type mice of the indicated ages (n = 3 per group). B, Quantitative RT-PCR of levels of the indicated receptor transcripts in the cortex of wild-type and sortilin-deficient mice (n = 4 per group). C, D, Levels of APOE in hippocampus (Hip) and cortex (Ctx) of wild-type mice (Lrp1 wt) or animals with neuron-specific Lrp1 gene defect (Lrp1 ko) were determined by Western blotting (C) and quantified by densitometric scanning of the respective blots (n = 3–4) (D). Detection of tubulin served as loading control. Extracts from mice genetically deficient for APOE (Apoe−/−) were used as negative control for specificity of the APOE immunoreactivity (APOE indicated by arrowheads). The inset in D demonstrates robust expression of LRP1 in brain extracts of wild-type mice but not of animals with neuron-specific Lrp1 deletion. E, F, Levels of APOE in cortex of wild-type (Sorl1+/+) or SORLA-deficient (Sorl1−/−) mice were determined by Western blotting (E) and quantified by densitometric scanning of the respective blots (n = 6) (F). Detection of tubulin served as loading control.
To better appreciate the quantitative importance of sortilin over other endocytic receptors for neuronal catabolism of Aβ, we determined the uptake of APOE4-bound Aβ40 in primary neurons from sortilin-deficient mice compared with control mice. Despite normal expression of LRP1 and SORLA in sortilin-deficient neurons (Fig. 13A), these cells exhibited a 70–80% reduction in uptake of exogenously added APOE/Aβ40 complexes compared with wild-type cells (Fig. 13B). Similar to transfected HEK293 cells discussed above, the quantitative difference in cellular handling of Aβ was dependent on the presence of APOE in the medium as documented by testing the levels of endogenous amyloidogenic products without addition of APOE. Under these experimental conditions, primary neurons produce very low levels of the endogenous APO (Harris et al., 2004). In the absence of exogenous APOE, the concentrations of sAPPα and sAPPβ (Fig. 13C) or of Aβ40 and Aβ42 (Fig. 13D) were not changed in sortilin-deficient compared with control neurons. Thus, sortilin activity does not impact the rate of Aβ production or clearance in neurons when APOE is absent.
Lipid profiles in sortilin-deficient mice recapitulate features of murine APOE deficiency
So far, our data indicated a major role for sortilin as APOE receptor in neurons. To further substantiate this model, we analyzed the brain lipid metabolism in sortilin-deficient mice. One of the main effects of altered APOE levels in mice is a change in brain sulfatide levels. Sulfatides are sulfated galactosylceramides synthesized by oligodendrocytes and incorporated into the myelin sheath of axons. APOE-containing lipoproteins extract sulfatides from the myelin sheath and deliver them to neurons for uptake and catabolism via APOE receptors (Han et al., 2003; Cheng et al., 2010). Accordingly, loss of APOE expression in Apoe−/− mice causes an approximate twofold increase in brain sulfatide levels, whereas overexpression of human APOE3 and APOE4 reduces the concentration of these lipids in mice (Han et al., 2003). At 5 months of age, sortilin-deficient animals exhibited a twofold increase in levels of the major brain sulfatide variant N24:1 in the hippocampus. A significant increase in brain sulfatides was also confirmed at 10 months of age (Fig. 14). In contrast to the situation with sortilin, deficiency for LRP1 did not affect the brain levels of sulfatides in mice with conditional inactivation of Lrp1 in neurons at 5 months of age (Fig. 14). Because of the premature lethality of mice with brain-specific LRP1 deficiency (May et al., 2004), sulfatide levels could not be evaluated at later stages in this line.
In conclusion, our findings have identified a hitherto unknown function for sortilin as neuronal APOE receptor in the brain, and they uncovered APOE-dependent clearance as a molecular mechanism of receptor action in AD. Absence of this receptor recapitulates features seen in mice lacking APOE (such as accumulation of brain sulfatides) despite higher than normal levels of the APO in the CNS. Because other neuronal APOE receptors seemed properly expressed in sortilin-deficient mice, our data suggest that sortilin represents a pathway of major quantitative importance for neuronal clearance of APOE-containing lipoproteins and APOE/Aβ complexes.
Discussion
APOE is the most important genetic risk factor for sporadic AD (Corder et al., 1993; Strittmatter et al., 1993). In the CNS, this 34 kDa protein is mainly produced by microglia and astrocytes (Pitas et al., 1987) and delivers cholesterol-rich lipoproteins to neurons to support synaptogenesis and maintenance of synaptic connections (Mauch et al., 2001). Many hypotheses have been advanced concerning the contribution of APOE to neurodegeneration, including effects on Aβ clearance or deposition, on cell signaling, neuroinflammation, synaptic transmission, or neurotoxicity (for review, see Mahley et al., 2006; Holtzman et al., 2012). One common hypothesis states that APOE sequesters soluble Aβ in the extracellular space, resulting in cellular uptake and catabolism of this neurotoxic peptide. This hypothesis is supported by many in vitro studies (Koistinaho et al., 2004; Morikawa et al., 2005; Jiang et al., 2008). Data from transgenic mouse models produced more conflicting results with impaired plaque deposition seen in mice lacking murine APOE (Bales et al., 1997; Holtzman et al., 2000) but also in animals overexpressing the human APOE3 or APOE4 isoforms (Holtzman et al., 1999). Still, recent findings that pharmacological induction of Apoe expression in mice ameliorates Aβ production and plaque deposition strongly supports an inverse correlation of APOE concentration with brain Aβ levels (Cramer et al., 2012).
Although the multiple (and perhaps opposing) effects of APOE on Aβ degradation versus deposition still warrant clarification, it is well appreciated that cellular APOE receptors likely play critical roles in these processes. The two main receptors discussed in this context are the LDL receptor and LRP1. Whereas the LDL receptor mainly mediates uptake of APOE into microglia and astrocytes (Kim et al., 2009), LRP1 constitutes an uptake pathway for this APO in neurons (for review, see Holtzman et al., 2012). Experimental support for involvement of LDL receptors and LRP1 in brain APOE catabolism stems from the fact that inactivation of either Ldlr or Lrp1 causes accumulation of APOE in the brain of mice (Fryer et al., 2005; Liu et al., 2007). Conversely, overexpression of the LDL receptor (Kim et al., 2009) or of LRP1 mini receptors reduces systemic levels of APOE in the CNS (Zerbinatti et al., 2006). Now, our studies identified sortilin as another APOE receptor to be considered in this context. Sortilin binds and mediates cellular uptake of all human APOE isoforms (Figs. 9–11). Loss of receptor expression in gene-targeted mice coincides with an increase in systemic APOE levels that parallels increases seen in the LDL receptor null mice (100% in Sort1−/− vs 60% in Ldlr−/− lines) (Fig. 6A,B) (Fryer et al., 2005). The rate of Apoe transcription (Fig. 6C) or of APOE release from astrocytes (Fig. 7) is unchanged in sortilin-deficient animals, suggesting impaired clearance as the cause of APOE buildup. Most importantly, sortilin mediates cellular uptake of Aβ bound to APOE (Fig. 10) and receptor deficiency results in accumulation of Aβ peptides and in increased plaque burden in two AD mouse models (Figs. 1, 3). In contrast to Aβ, the levels of other soluble or membrane-tethered APP processing products are not affected by receptor deficiency (Figs. 1, 2, 13C). Although our data substantiate an important role for sortilin in amyloidogenic processes, they disagree with previous studies in cellular overexpression systems that argued for a direct role of this receptor in APP processing through regulation of β-secretase trafficking (Finan et al., 2011).
Recently, sortilin was identified as a genetic risk factor in cardiovascular disease associated with high plasma cholesterol levels and occurrence of myocardial infarction in the human population (Kathiresan et al., 2009). The underlying mechanism was traced to the ability of sortilin to act as intracellular sorting receptor for APOB100 and to modulate release of nascent APOB100-containing lipoproteins from the liver (Kjolby et al., 2010; Musunuru et al., 2010). Intriguingly, our studies now uncovered the relevance of this receptor pathway in control of lipoprotein homeostasis in the brain as well. Support for a critical role for sortilin in systemic APOE metabolism stems from changes in lipid profiles in sortilin-deficient mice that recapitulate features shown for Apoe−/− animals previously (Han et al., 2003). Thus, although total levels of cholesterol remain unaltered (data not shown), loss of sortilin coincides with elevated hippocampal levels of sulfatides that are in the same order of magnitude as in APOE-deficient animals (twofold) (Fig. 14) (Han et al., 2003). Although the molecular pathways involved in APOE-dependent turnover of brain sulfatides are poorly understood, levels of the APO are typically inversely correlated with levels of these lipids (Han et al., 2003). Sortilin-deficient animals exhibit elevated sulfatide concentrations albeit at higher than normal levels of APOE. These data strongly argue for a defect in neuronal clearance of APOE-containing lipoproteins (and associated sulfatides) in mice lacking this receptor.
In the brain, endocytic pathways in glia cells (likely the LDL receptor) and in neurons are implicated in cellular catabolism of APOE/Aβ complexes (Koistinaho et al., 2004; Jiang et al., 2008; Holtzman et al., 2012). Because expression of sortilin in the CNS is restricted to neurons (Sarret et al., 2003), we assessed the quantitative contribution of this receptor pathway to neuronal APOE/Aβ catabolism. Surprisingly, APOE-dependent uptake of Aβ was impaired in primary neurons lacking sortilin despite normal levels of LRP1 and SORLA (Fig. 13). Also, transcript levels of members of the LDL receptor and VPS10P-domain receptor gene families were not affected by sortilin deficiency (Fig. 12B), strongly arguing for a direct effect of sortilin on APOE and Aβ turnover in vivo. In support of our model, no accumulation of APOE was seen in mice lacking SORLA or LRP1 in this study (Fig. 12C–F). In addition, accumulation of brain sulfatides was seen in sortilin-deficient but not in LRP1-deficient animals in this study (Fig. 14). In line with this notion, lower (rather than higher) sulfatide levels were observed in another mouse model with neuron-specific LRP1 deficiency previously (Liu et al., 2010). Finally, neuron-specific inactivation of Lrp1 was shown to decrease brain Aβ levels (Liu et al., 2007), whereas overexpression of an LRP1 mini receptor was shown to increase brain Aβ levels (Zerbinatti et al., 2006). Obviously, many of these data are inconsistent with a simple role for LRP1 in neuronal APOE/Aβ clearance. Likely, the multiple alternative functions assigned to this receptor in AD, including intracellular APP transport (Ulery et al., 2000; Pietrzik et al., 2002), blockade of APP signaling (Kinoshita et al., 2003), or Aβ shunt across the blood–brain barrier (Deane et al., 2004) complicate interpretation of the available experimental data.
All evidence in this study is consistent with a model whereby sortilin represents a major neuronal receptor pathway for APOE-containing lipoproteins that counteracts Aβ accumulation in the brain. Sortilin-mediated endocytosis likely results in lysosomal degradation of the Aβ peptides (Fig. 11B). A related role for sortilin as neuronal clearance receptor has been shown recently for progranulin, a protein implicated in frontotemporal lobar degeneration (Hu et al., 2010). In this study, we observed low-affinity binding of Aβ to sortilin in BIAcore (Fig. 8) and some residual uptake of the free peptide in CHO–mSort cells (Fig. 10A,B). Thus, we cannot exclude that sortilin may also clear free Aβ in vivo. However, one has to keep in mind that the concentration of Aβ applied to cells (0.7 μg/ml) is significantly higher than the amount of total Aβ detected, for example, in the PDAPP+/− mouse brain (<3 ng/ml) (Castellano et al., 2012). Also, Deane et al. (2004) documented an affinity for Aβ binding to LRP1 (Kd = 0.57 nm) that is a 1000-fold higher than the one determined for sortilin. Data obtained so far also failed to identify obvious differences in cellular handling of APOE3 versus APOE4 by sortilin. However, we noticed a twofold lower Kd for binding of APOE3 (44 nm) versus APOE4 (114 nm) (Fig. 8). Also, NT blocked receptor interaction with APOE3 much more efficiently than with APOE4 (80 vs 30% inhibition, respectively) (Fig. 8). These observations point toward an isoform-specific interaction of sortilin with human APOE. Whether this distinction may explain differences in risk of AD associated with these two APOE variants remains to be resolved. Regardless of possible differences in handling of APOE3 versus APOE4, the ability of sortilin to modulate brain lipid and APOE homeostasis strongly implicates this receptor pathway in pathophysiological mechanisms whereby alterations in brain lipoprotein metabolism affect the risk of sporadic AD.
In conclusion, our studies have identified the pro-neurotrophin receptor sortilin as a major neuronal pathway involved in brain APOE and Aβ metabolism. Thus, this receptor represents a pathway in which pro-neurotrophin signaling, Aβ catabolism, and brain lipoprotein metabolism converge. Additional studies are likely to uncover exciting details of this protein critical to several important concepts in neurodegeneration.
Footnotes
This work was funded by grants from the Helmholtz Association and the European Commission (Memories) (T.E.W.), the American Health Assistance Foundation (M.R.), and the Federal Ministry of Education and Research (S.K. and G.M.). We are indebted to T. Breiderhoff for providing expressions constructs, Z. Cseresnyes for help with microscopy, and T. Pantzlaff, M. Schmeisser, J. Diesbach, and S. Ehret for expert technical assistance.
The authors declare no competing financial interests.
References
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, Bales KR, Cappai R, Masters CL, Gliemann J, Mufson EJ, Hyman BT, Paul SM, Nykjaer A, Willnow TE. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc Natl Acad Sci U S A. 2012;109:15502–15507. doi: 10.1073/pnas.1206446109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A. 2010;107:12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Zhou Y, Holtzman DM, Han X. Apolipoprotein E mediates sulfatide depletion in animal models of Alzheimer's disease. Neurobiol Aging. 2010;31:1188–1196. doi: 10.1016/j.neurobiolaging.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer's disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan GM, Okada H, Kim TW. BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin. J Biol Chem. 2011;286:12602–12616. doi: 10.1074/jbc.M110.170217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Demattos RB, McCormick LM, O'Dell MA, Spinner ML, Bales KR, Paul SM, Sullivan PM, Parsadanian M, Bu G, Holtzman DM. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J Biol Chem. 2005;280:25754–25759. doi: 10.1074/jbc.M502143200. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, Mcconlogue L, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Han X, Cheng H, Fryer JD, Fagan AM, Holtzman DM. Novel role for apolipoprotein E in the central nervous system. Modulation of sulfatide content. J Biol Chem. 2003;278:8043–8051. doi: 10.1074/jbc.M212340200. [DOI] [PubMed] [Google Scholar]
- Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang S, Wyss-Coray T, Mahley RW, Huang Y. Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer's disease. J Biol Chem. 2004;279:3862–3868. doi: 10.1074/jbc.M309475200. [DOI] [PubMed] [Google Scholar]
- Heeren J, Weber W, Beisiegel U. Intracellular processing of endocytosed triglyceride-rich lipoproteins comprises both recycling and degradation. J Cell Sci. 1999;112:349–359. doi: 10.1242/jcs.112.3.349. [DOI] [PubMed] [Google Scholar]
- Hesse C, Larsson H, Fredman P, Minthon L, Andreasen N, Davidsson P, Blennow K. Measurement of apolipoprotein E (apoE) in cerebrospinal fluid. Neurochem Res. 2000;25:511–517. doi: 10.1023/a:1007516210548. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Herz J, Bu G. Apolipoprotein e and apolipoprotein e receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Jansen P, Giehl K, Nyengaard JR, Teng K, Lioubinski O, Sjoegaard SS, Breiderhoff T, Gotthardt M, Lin F, Eilers A, Petersen CM, Lewin GR, Hempstead BL, Willnow TE, Nykjaer A. Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat Neurosci. 2007;10:1449–1457. doi: 10.1038/nn2000. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myocardial Infarction Genetics Consortium. Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand S, Engert JC, Samani NJ, Schunkert H, Erdmann J, Reilly MP, Rader DJ, Morgan T, Spertus JA, Stoll M, Girelli D, McKeown PP, Patterson CC, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A, Shah T, Tangredi MM, Strickland DK, Hyman BT. The intracellular domain of the low density lipoprotein receptor-related protein modulates transactivation mediated by amyloid precursor protein and Fe65. J Biol Chem. 2003;278:41182–41188. doi: 10.1074/jbc.M306403200. [DOI] [PubMed] [Google Scholar]
- Kjolby M, Andersen OM, Breiderhoff T, Fjorback AW, Pedersen KM, Madsen P, Jansen P, Heeren J, Willnow TE, Nykjaer A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12:213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Koch S, Donarski N, Goetze K, Kreckel M, Stuerenburg HJ, Buhmann C, Beisiegel U. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42:1143–1151. [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Kurz A, Perneczky R. Amyloid clearance as a treatment target against Alzheimer's disease. J Alzheimers Dis. 2011;24(Suppl 2):61–73. doi: 10.3233/JAD-2011-102139. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhou J, Li Y, Kim KW, Hensley LL, Baker SJ, Parada LF. Neuron-specific enolase-cre mouse line with cre activity in specific neuronal populations. Genesis. 2006;44:130–135. doi: 10.1002/gene.20197. [DOI] [PubMed] [Google Scholar]
- Lane RF, Raines SM, Steele JW, Ehrlich ME, Lah JA, Small SA, Tanzi RE, Attie AD, Gandy S. Diabetes-associated SorCS1 regulates Alzheimer's amyloid-beta metabolism: evidence for involvement of SorL1 and the retromer complex. J Neurosci. 2010;30:13110–13115. doi: 10.1523/JNEUROSCI.3872-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, Herz J, Muglia L, Bu G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56:66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, Han X, Weeber EJ, Bu G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci. 2010;30:17068–17078. doi: 10.1523/JNEUROSCI.4067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- May P, Rohlmann A, Bock HH, Zurhove K, Marth JD, Schomburg ED, Noebels JL, Beffert U, Sweatt JD, Weeber EJ, Herz J. Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol Cell Biol. 2004;24:8872–8883. doi: 10.1128/MCB.24.20.8872-8883.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazella J, Zsürger N, Navarro V, Chabry J, Kaghad M, Caput D, Ferrara P, Vita N, Gully D, Maffrand JP, Vincent JP. The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. J Biol Chem. 1998;273:26273–26276. doi: 10.1074/jbc.273.41.26273. [DOI] [PubMed] [Google Scholar]
- Morikawa M, Fryer JD, Sullivan PM, Christopher EA, Wahrle SE, DeMattos RB, O'Dell MA, Fagan AM, Lashuel HA, Walz T, Asai K, Holtzman DM. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol Dis. 2005;19:66–76. doi: 10.1016/j.nbd.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Morris NJ, Ross SA, Lane WS, Moestrup SK, Petersen CM, Keller SR, Lienhard GE. Sortilin is the major 110-kDa protein in GLUT4 vesicles from adipocytes. J Biol Chem. 1998;273:3582–3587. doi: 10.1074/jbc.273.6.3582. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Wuu J, Counts SE, Nykjaer A. Preservation of cortical sortilin protein levels in MCI and Alzheimer's disease. Neurosci Lett. 2010;471:129–133. doi: 10.1016/j.neulet.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MS, Madsen P, Christensen EI, Nykjaer A, Gliemann J, Kasper D, Pohlmann R, Petersen CM. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 2001;20:2180–2190. doi: 10.1093/emboj/20.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykjaer A, Willnow TE. Sortilin: a receptor to regulate neuronal viability and function. Trends Neurosci. 2012;35:261–270. doi: 10.1016/j.tins.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, Hempstead BL, Petersen CM. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CM, Nielsen MS, Nykjaer A, Jacobsen L, Tommerup N, Rasmussen HH, Roigaard H, Gliemann J, Madsen P, Moestrup SK. Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J Biol Chem. 1997;272:3599–3605. doi: 10.1074/jbc.272.6.3599. [DOI] [PubMed] [Google Scholar]
- Pietrzik CU, Busse T, Merriam DE, Weggen S, Koo EH. The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. EMBO J. 2002;21:5691–5700. doi: 10.1093/emboj/cdf568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setäl ä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- Quistgaard EM, Madsen P, Grøftehauge MK, Nissen P, Petersen CM, Thirup SS. Ligands bind to Sortilin in the tunnel of a ten-bladed beta-propeller domain. Nat Struct Mol Biol. 2009;16:96–98. doi: 10.1038/nsmb.1543. [DOI] [PubMed] [Google Scholar]
- Rellin L, Heeren J, Beisiegel U. Recycling of apolipoprotein E is not associated with cholesterol efflux in neuronal cells. Biochim Biophys Acta. 2008;1781:232–238. doi: 10.1016/j.bbalip.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Rohe M, Synowitz M, Glass R, Paul SM, Nykjaer A, Willnow TE. Brain-derived neurotrophic factor reduces amyloidogenic processing through control of SORLA gene expression. J Neurosci. 2009;29:15472–15478. doi: 10.1523/JNEUROSCI.3960-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlmann A, Gotthardt M, Willnow TE, Hammer RE, Herz J. Sustained somatic gene inactivation by viral transfer of Cre recombinase. Nat Biotechnol. 1996;14:1562–1565. doi: 10.1038/nbt1196-1562. [DOI] [PubMed] [Google Scholar]
- Sarret P, Krzywkowski P, Segal L, Nielsen MS, Petersen CM, Mazella J, Stroh T, Beaudet A. Distribution of NTS3 receptor/sortilin mRNA and protein in the rat central nervous system. J Comp Neurol. 2003;461:483–505. doi: 10.1002/cne.10708. [DOI] [PubMed] [Google Scholar]
- Schmidt V, Sporbert A, Rohe M, Reimer T, Rehm A, Andersen OM, Willnow TE. SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J Biol Chem. 2007;282:32956–32964. doi: 10.1074/jbc.M705073200. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulery PG, Beers J, Mikhailenko I, Tanzi RE, Rebeck GW, Hyman BT, Strickland DK. Modulation of beta-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP). Evidence that LRP contributes to the pathogenesis of Alzheimer's disease. J Biol Chem. 2000;275:7410–7415. doi: 10.1074/jbc.275.10.7410. [DOI] [PubMed] [Google Scholar]
- Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006;281:36180–36186. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]