

Abstract
Many proteins cannot fold without the assistance of chaperonin machines like GroEL and GroES. The nature of this assistance, however, remains poorly understood. Here we demonstrate that unfolding of a substrate protein by GroEL enhances protein folding. We first show that capture of a protein on the open ring of a GroEL–ADP–GroES complex, GroEL’s physiological acceptor state for non-native proteins in vivo, leaves the substrate protein in an unexpectedly compact state. Subsequent binding of ATP to the same GroEL ring causes rapid, forced unfolding of the substrate protein. Notably, the fraction of the substrate protein that commits to the native state following GroES binding and protein release into the GroEL–GroES cavity is proportional to the extent of substrate-protein unfolding. Forced protein unfolding is thus a central component of the multilayered stimulatory mechanism used by GroEL to drive protein folding.
Many proteins cannot fold spontaneously in the concentrated environment of a cell1–3. These proteins typically show slow and complex folding behavior that renders them susceptible to misfolding and aggregation4,5. Aggregation of an indispensable cellular protein, and the loss of a key activity, is typically fatal. However, misfolding and aggregation of even nonessential proteins can result in cellular dysfunction and disease6–9. Several large families of specialized proteins, known as molecular chaperones, have evolved to prevent protein misfolding and aggregation10–13. In many cases, molecular chaperones seem to serve primarily as passive aggregation inhibitors, selectively binding aggregation-prone folding intermediates and blocking the self-association reactions that lead to aggregate formation10,11. In other cases, it has been suggested that molecular chaperones actively facilitate protein folding by using the energy of ATP hydrolysis to alter the conformations accessible to a non-native protein14–16.
Among the molecular chaperones, the chaperonins or Hsp60s use a distinct physical mechanism, capturing and confining non-native folding intermediates inside an isolated cavity17–21. The chaperonin system of Escherichia coli, comprising GroEL and its smaller co-chaperonin GroES, is perhaps the best-studied example of this molecular chaperone family13,14. GroEL is a large, homo-oligomer constructed from 14 subunits of 57 kDa arranged in a double ring complex, with 7 subunits in each ring22. Each GroEL ring possesses a large central cavity that, in the absence of ATP and GroES, is lined with hydrophobic amino acids that capture poorly folded non-native substrate proteins22,23. These hydrophobic binding sites are located on the inner surface of the GroEL apical domain at the open ends of each ring. The equatorial domain at the base of each GroEL subunit forms the interface between the two GroEL rings and contains the catalytic ATPase site that powers the GroEL reaction cycle24–26. A GroEL-mediated folding reaction begins with the capture of a non-native substrate protein on an open GroEL ring27–30. Subsequent binding of ATP to the same protein-loaded ring triggers the large-scale rearrangement of the GroEL apical domains, allowing GroES to bind to the GroEL ring18,31. GroES binding results in the encapsulation and release of the non-native substrate protein into the isolated GroEL–GroES cavity (a cis complex)17,19–21. Hydrolysis of ATP within the cis complex followed by ATP binding to the other GroEL ring (the trans ring) then causes GroES to be released and the substrate protein to be ejected into free solution, whether or not it has achieved a folded conformation that is committed to the native state17,26,27,30. Substrate-protein molecules that fail to fold must then be recaptured for another passage through the GroEL reaction cycle.
Precisely how the capture and encapsulation of a non-native protein within the GroEL–GroES cavity leads to stimulated protein folding remains poorly understood. It was initially suggested that, as do many other molecular chaperones, GroEL functions as a passive aggregation inhibitor, with the GroEL–GroES cavity serving to simply sequester aggregation-prone folding intermediates32,33. Because aggregation of a substrate protein would be prevented by encapsulation in the GroEL–GroES cavity, this model implied that GroEL’s primary role is to ‘infinitely’ dilute substrate proteins and allow them to fold at their intrinsic spontaneous rates within the GroEL–GroES chamber32. Although simple binding of non-native proteins by GroEL does indeed inhibit aggregation, a considerable body of evidence suggests that, for many substrate proteins, GroEL is much more than a passive box and is actively involved in facilitating protein folding (for review, see refs. 13,14). On the basis of both experimental and theoretical studies, it has been suggested that confinement of a non-native protein within the GroEL–GroES cavity enhances folding by altering the substrate protein’s folding landscape34–40. In essence, this model suggests that the restricted volume, and perhaps the surface character, of the GroEL–GroES cavity inhibits formation of expanded and otherwise misfolded conformational states that slow or block folding of the protein in free solution. At the same time, collapsed conformations that are more likely to find the native state are favored, resulting in an overall smoothing of the substrate protein’s folding landscape34–36,38,39.
A third model of GroEL action, based on the idea that GroEL stimulates productive protein folding through unfolding, has also been proposed (for review see refs. 14,15). The most general form of this model posits that substrate proteins that strictly depend upon GroEL for their folding (so-called ‘stringent’ substrate proteins) are those most likely to fall into deep energetic traps that ambient thermal fluctuations cannot reverse41. These quasi-stable, misfolded states are highly prone to irreversible aggregation. In principle, a simple way to stimulate the productive folding of such a protein would be to partially disrupt the nascent structure of the trapped protein conformation, giving the substrate protein a fresh opportunity to fold from a higher point on its free energy landscape41,42. Although a large body of evidence suggests that capture of a substrate protein by GroEL can indeed result in protein unfolding, the magnitude and impact of this effect has remained unclear14. In addition, unfolding by the full GroEL–GroES system has not been directly linked to enhanced folding of any stringent substrate protein to date. A variation on the unfolding model has also been proposed, in which it is suggested that GroEL delivers a mechanical unfolding force to a substrate protein in the course of GroES binding and substrate-protein encapsulation42–44. Although an early tritium-amide exchange study was consistent with this mechanism, termed ‘forced’ unfolding, subsequent and more detailed deuterium-amide exchange experiments were not44–46. Thus, the existence and importance of protein unfolding by GroEL has remained controversial14,36,45.
Here we demonstrate that unfolding of a stringent substrate protein by GroEL substantially enhances productive protein folding. We first examine the conformation of a non-native substrate protein bound to the open trans ring of a GroEL–ADP–GroES complex, GroEL’s physiological acceptor state for non-native proteins in vivo. Unexpectedly, we find that the non-native substrate protein is considerably more compact when bound to a trans ring than when bound to the GroEL tetradecamer alone. We then show that the captured folding intermediate is subjected to a rapid unfolding force when ATP binds to the loaded trans ring. Notably, we find that the fraction of the substrate protein that commits to the native state upon release into the GroEL–GroES cavity is directly proportional to the extent of substrate unfolding. Forced protein unfolding is thus a central component of the multilayered stimulatory mechanism employed by GroEL to drive protein folding.
RESULTS
Non-native Rubisco bound to a GroEL trans ring
The capture of a non-native substrate protein on an open GroEL ring is a necessary first step in GroEL-mediated protein folding. Because protein unfolding could, in principle, have a profound impact on productive folding, it remains cucially important to determine whether or not substrate-protein capture by a GroEL ring causes substantial protein unfolding. However, our current understanding of protein capture by GroEL is principally based on studies involving double-ring GroEL in the absence of nucleotide and GroES (referred to here as apoGroEL)11,14,47. Under in vivo conditions, by contrast, the open trans ring of an asymmetric GroEL–ADP–GroES complex (an ‘ADP bullet’) is the acceptor state for non-native substrate proteins (Fig. 1a and refs. 21,27,48). Whether or not the conformation of a substrate protein bound to the trans ring of an ADP bullet is the same as that populated on an apoGroEL ring has remained unclear.
Figure 1.

Non-native Rubisco is more compact when bound to the trans ring of an ADP bullet than when bound to apoGroEL. (a) The conformation of a substrate protein bound to either apoGroEL or the trans ring of an ADP bullet can be investigated with intramolecular FRET, where the distance between two exogenous fluorescent probes attached to different parts of the substrate protein (indicated schematically as colored circles labeled ‘A’ and ‘D’) is examined. (b) Steady-state FRET measurement of the distance between the N- and C-terminal domains of a non-native Rubisco monomer (100 nM) bound to an ADP-bullet trans ring and apoGroEL (120 nM in each case). The C-terminal domain of a previously described Rubisco variant35 was labeled with a donor fluorophore and the N-terminal domain was labeled with an acceptor fluorophore. For these measurements, the donor probe was 5-(2-acetamidoethyl) aminonaphthalene-1-sulfonate (AEDANS; 454-ED) and the acceptor was fluorescein (58-F). The donor-emission spectra of donor-only (labeled apo-D and trans-D, respectively) and donor-acceptor (labeled apo and trans, respectively) samples are shown. (c) Time-resolved donor-intensity decays of the samples described in b. The donor-emission decay of donor-only and donor-acceptor samples, labeled as above, and the instrument response function (IRF) are shown. The average intraprobe distance was quantified from both steady-state and time-resolved FRET measurements (b, inset).
We therefore examined whether the conformation of a substrate protein bound to the trans ring of an ADP bullet is the same as that populated on an apoGroEL ring. We used Rubisco from Rhodospirillum rubrum as a model substrate protein for this analysis, because it is one of the most stringently dependent GroEL substrate proteins known29. We also used a mixing protocol that yields a nearly homogeneous population of asymmetric ADP bullets (see Methods). The ADP bullets are stable for at least an hour when maintained in low concentrations of ADP (5–15 μM; Supplementary Fig. 1 online). Binding of non-native Rubisco to the trans ring of an ADP bullet is comparable, in both rate and strength, to apoGroEL binding (ref. 27 and data not shown).
For these studies, we used an intramolecular fluorescence resonance energy transfer (FRET) assay that reports on changes in Rubisco conformation. In earlier work, we developed a mutagenesis and labeling protocol that allows the specific attachment of exogenous fluorescent probes to particular sites on the Rubisco monomer35. The fluorescent dyes have no detectable effect on the stability, enzymatic activity or GroEL-mediated folding of this substrate protein. By coupling distinct probes to the amino- and carboxyterminal domains of the Rubisco monomer, we demonstrated that the average expansion of the non-native Rubisco monomer could be monitored by FRET35. Binding of non-native Rubisco to an apoGroEL ring results in a near total loss of detectable FRET between the labeled regions of the Rubisco monomer (Fig. 1b,c and ref. 35). This observation suggests that, upon binding to an apoGroEL ring, the Rubisco monomer is subjected to a dramatic stretching or conformational expansion35. By contrast, when the Rubisco monomer is bound to the trans ring of an ADP bullet, we observe a much higher level of energy transfer, as evidenced by the considerable quenching of the donor fluorescence emission and decrease in the donor lifetime (Fig. 1b,c). These observations indicate that the Rubisco monomer populates a distinct conformational state or family of states when captured by a trans ring.
The large FRETsignal of trans ring–bound Rubisco suggests that the non-native monomer is more compact when bound to the ADP-bullet trans ring than when bound to an apoGroEL ring. Quantitative analysis of both steady state and time-resolved FRET data support this conclusion, indicating that the labeled segments of the non-native Rubisco monomer are, on average, approximately 25 Å closer together when the protein is bound to an ADP-bullet trans ring (Fig. 1b, inset). Binding of Rubisco to an ADP-bullet trans ring thus results in an expansion of no more than 4–6 Å of the monomer relative to a kinetically trapped and misfolded state of the protein35. By contrast, non-native Rubisco binding to an apoGroEL ring results in an expansion of 30 Å or more35. Because these FRET observations probe only the dimensions of the Rubisco monomer along a single spatial axis, we examined the overall conformational properties of GroEL-bound Rubisco with two more methods. A more collapsed Rubisco monomer would be expected to show (i) reduced susceptibility to proteolytic fragmentation; and (ii) decreased reactivity of buried cysteine residues toward soluble thiol alkylating agents. Consistent with our FRET observations, non-native Rubisco bound to the trans ring of an ADP bullet is substantially less susceptible to protease digestion than when it is bound to apoGroEL (with chymotrypsin, Fig. 2a,b; with trypsin, Supplementary Fig. 2 online). In addition, trans ring–bound Rubisco shows digestion patterns with both proteases that are distinct from those observed with apoGroEL. We also observe changes in the chemical reactivity of several endogenous Rubisco cysteine residues in trans ring–bound Rubisco (Fig. 2c). These cysteine residues are fully buried and nonreactive to small fluorescent probes in native Rubisco, but they are exposed and highly reactive when non-native Rubisco binds to apoGroEL (Fig. 2c). The same cysteine residues, however, show intermediate reactivity when the non-native Rubisco monomer is bound to the trans ring of an ADP bullet, consistent with the internal structure of the protein being considerably less exposed to solvent. Overall, these observations strongly suggest that the non-native Rubisco monomer bound to an ADP-bullet trans ring is more compact and is present in a different conformation than when it is bound to an apoGroEL ring.
Figure 2.
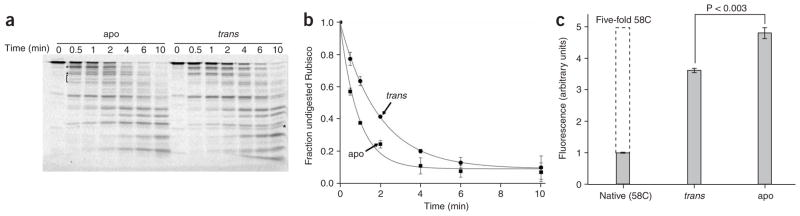
Non-native Rubisco bound to the trans ring of an ADP bullet is less susceptible to protease digestion and shows reduced chemical modification of internal cysteine residues. (a) Fluorescently labeled Rubisco (58-F; 100 nM) was denatured and bound to either ADP bullets or apoGroEL (labeled trans and apo, respectively; 120 nM in each case), treated with chymotrypsin for the indicated times and then analyzed by SDS-PAGE and laser-excited, fluorescence gel scanning. Partial digestion fragments that appear uniquely well represented in either the apo or trans digestion experiments are indicated by a square bracket and asterisks. (b) The amount of full-length, fluorescent Rubisco was quantified for three independent protease experiments, and the average of these replicates is plotted as a function of digestion time (error bars show one s.d.). The t1/2 for the disappearance of full-length Rubisco bound to apoGroEL and an ADP-bullet trans ring are 0.6 min and 1.3 min, respectively. (c) The internal structure of Rubisco monomers bound to apoGroEL and the ADP-bullet trans ring was probed with a small, highly reactive coumarin maleimide dye (CPM) that selectively modifies exposed cysteine residues60. Only a single surface-exposed cysteine residue at position 58 (out of a total of five cysteine residues in wild-type Rubisco) is modified in native Rubisco (labeled Native (58C); 500 nM). The graph shows the total level of CPM fluorescence detected in the Rubisco sample following analysis by reverse-phase HPLC. When non-native Rubisco (500 nM) is bound to either apoGroEL (apo; 600 nM) or an ADP-bullet trans ring (trans; 600 nM), the internal Rubisco cysteine residues become highly reactive toward CPM. However, the reactivity of internal cysteine residues in Rubisco bound to apoGroEL is significantly greater than the trans ring–bound sample (P < 0.003 based on a paired t-test with n = 3 replicates; error bars show one s.d.). The maximum level of internal CPM incorporation expected under the solution conditions used is also shown (indicated by a dashed line, labeled five-fold 58C) and was estimated by assuming that the internal Rubisco cysteine residues in unfolded Rubisco would be as exposed and reactive as the surface-exposed 58C site.
ATP binding causes forced protein unfolding
The more compact state of the Rubisco monomer indicates that the trans ring of an ADP bullet cannot induce the same level of binding-driven protein unfolding as an apoGroEL ring35. We refer to this type of unfolding as ‘passive unfolding’ because it depends on only simple, physical contact between the non-native Rubisco monomer and the GroEL ring. Although passive unfolding has also been observed with other substrate proteins, a direct linkage between this effect and GroEL-stimulated folding has been difficult to establish14. Furthermore, although passive unfolding of a substrate protein on an apoGroEL ring can be extensive, it is also slow. With Rubisco, much of the passive unfolding seen with apoGroEL proceeds much more slowly (t1/2 = ~5–8 min at 25 °C) than GroEL’s functional reaction cycle (t1/2 = ~15–20 s at 25 °C)25,27,35,41. In this context, our observation of limited passive unfolding of Rubisco by the ADP-bullet trans ring raises a key question: how much unfolding, if any, is necessary for productive folding? The modest level of passive unfolding on an ADP-bullet trans ring might represent a maximally effective level of substrate unfolding. Alternatively, passive unfolding on the ADP-bullet trans ring might represent only the first phase of substrate-protein unfolding. A bound Rubisco monomer might be subjected to further unfolding before encapsulation within the GroEL–GroES cavity. In particular, the dramatic rearrangements of the GroEL apical domains caused by ATP binding, movements that precede and are required for GroES binding, might well induce further alterations in the bound substrate protein42,43.
To address this question, we examined conformational changes in the trans ring–bound Rubisco by using a FRET-based rapid mixing assay (Fig. 3a and ref. 35). Notably, upon ATP and GroES binding to the Rubisco-occupied trans ring of a wild-type GroEL bullet, we observed two initial phases of conformational alteration in the non-native Rubisco monomer, based on the occurrence of distinct, time-dependent changes in intramolecular FRET: an initial rapid drop in FRET efficiency followed a slower increase in FRET efficiency (Fig. 3b). The same overall changes in FRET signal are also seen with a second pair of FRET dyes, as well as with a distinct set of dye-attachment sites, indicating that the conformational changes we observe occur across multiple spatial axes of the Rubisco monomer (Supplementary Fig. 3 online). The rising phase of the FRET signal is reminiscent of previous observations and probably reflects the compaction of the folding intermediate upon GroES binding35. This conclusion is supported by the fact that the rate of the rising phase is dependent on the concentration of GroES in a classically bimolecular fashion (Fig. 3c). A third phase of FRET signal change is also detectable when experiments are conducted under conditions that allow extended observation of the stable complex (manual mixing and rapid enzymatic depletion of ambient ATP; Supplementary Fig. 4 online). Under these conditions, the initial drop and subsequent rise in FRET efficiency are followed by a much slower decrease in FRET efficiency, consistent with previous observations35, and this corresponds to substrate-protein release and folding inside the GroEL–GroES cis complex.
Figure 3.

GroES and ATP binding to a Rubisco-occupied trans ring results in forced conformational expansion, followed by compaction, of the non-native protein. (a) Schematic of a FRET experiment designed to examine conformational changes in a non-native substrate protein upon encapsulation beneath GroES on the trans ring of an ADP bullet. (b) GroES binding and substrate encapsulation results in an initial rapid expansion of the non-native Rubisco intermediate, followed by a compaction. Substrate-bound complexes in the presence of excess GroES were rapidly mixed in a stopped-flow apparatus (60 nM) with ATP (1 mM). For these measurements, the donor probe was fluorescein and the acceptor was rhodamine. Each FRET trace is the average of n = 10 replicates of matched experimental pairs, calculated from donor-only (Rubisco 454-F) and donor-acceptor (Rubisco 454-F/58-R) samples. The inset shows the FRET change over the first second of data. The rising phase in the observed FRET signal reflects GroES binding and substrate-protein encapsulation and compaction35. (c) The observed rate constants for the initial rapid drop in FRET efficiency (kobs) and the subsequent rise upon GroES binding (kES) for a series of experiments at different concentrations of GroES are shown. In each case, a fixed concentration of the Rubisco-bound GroEL ADP bullets (60 nM) and ATP (1 mM) was used, whereas the total concentration of GroES was varied.
It is important that the rising FRET signal indicative of GroES binding is preceded by a rapid decrease in FRET efficiency (Fig. 3b). This initial falling phase reflects a rapid increase in the distance between the labeled segments of the Rubisco monomer and, therefore, a conformational expansion or partial unfolding of the captured Rubisco intermediate. Because the observed rate of this expansion phase shows no dependence on the concentration of GroES (Fig. 3c), this unfolding event seems to occur independently of and before GroES binding. The observation of rapid, partial unfolding of the Rubisco intermediate before GroES binding suggests that ATP binding alone, and the attendant structural shifts of the GroEL ring, are sufficient to drive a forced conformational rearrangement of the bound substrate protein. We tested this idea by repeating a series of experiments similar to those shown in Fig. 3, but under conditions in which GroES was prevented from binding to the Rubisco-occupied trans ring (excess SR1, a single-ringed GroEL variant, added as a GroES trap21). Under these conditions, the consequences of ATP binding alone to the substrate-occupied trans ring can be observed (Fig. 4a). Binding of ATP to the trans ring of a wild-type GroEL bullet induces a rapid drop in the Rubisco FRET efficiency (Fig. 4b), similar to the rapid expansion that precedes GroES binding in Figure 3. The same overall changes were also observed with a second set of FRET probes (Supplementary Fig. 3). The partial unfolding event is well characterized by a single exponential rate law across a 20-fold range of ATP concentration, and the observed rate constant shows classical saturation behavior (Fig. 4c). The fitted rate constant of the fast phase in the absence of free GroES seems to be slower than the fast FRET change observed in the presence of GroES (Fig. 4c). However, this disparity is probably an artifact of the competing change in FRET efficiency, induced by the binding of GroES, artificially increasing the apparent rate of the initial falling phase in the presence of GroES.
Figure 4.

ATP binding alone to a Rubisco-occupied trans ring drives a forced conformational expansion of the non-native protein. (a) Schematic of a FRET experiment designed to follow conformational changes in a non-native substrate protein upon ATP binding alone to the substrate-loaded trans ring of an ADP bullet. In this case, a large excess of a single-ringed GroEL variant SR1 (ref. 21) was added as a GroES trap. SR1 contains a set of equatorial mutations that prevent assembly of a double-ringed complex (indicated by red crosses). SR1 binds ATP and GroES, executes a single round of ATP hydrolysis and then stalls, failing to release GroES and ADP21. (b) The allosteric transition triggered by ATP alone results in the forced conformational expansion of the non-native protein. Stopped-flow FRET experiments were conducted in essentially the same manner as in Figure 3, except that SR1 was present in a 20-fold excess (1.2 μM) to prevent GroES binding to the substrate-loaded GroEL ring. (c) The observed rate of Rubisco expansion on the trans ring of an ADP bullet at different concentrations of ATP is shown. Each data point in the plot represents a single exponential fit to the observed change in FRET efficiency of the labeled Rubisco calculated from the average of n = 10 experimental replicates. The solid line shows a fit of the data to the Hill equation with kobs = 2.49 ± 0.06 s−1, K1/2 = 82.6 ± 3.7 μM and nH = 1.9 ± 0.2.
Unfolding enhances productive Rubisco folding
We thus observe two phases of Rubisco unfolding by GroEL: (i) slow passive unfolding of the protein following capture by an open GroEL ring; and (ii) rapid, ATP-driven unfolding. Although these observations are highly suggestive, the ultimate significance of substrate-protein unfolding by GroEL rests on whether unfolding is linked to productive folding. In principle, substrate-protein unfolding provides a simple means of repositioning a kinetically trapped folding intermediate to a higher point on its free energy landscape, potentially opening efficient and otherwise inaccessible routes to the substrate protein’s native state14,15. This model predicts that the fraction of a non-native substrate protein that commits to the native state should be correlated with the extent of GroEL-induced unfolding (Fig. 5). Unfortunately, testing this prediction on the trans ring of an ADP bullet is difficult, given the speed with which ATP-driven unfolding proceeds. A powerful alternative approach, however, is provided by the much slower rate of passive unfolding induced by the single-ringed GroEL variant SR1 (ref. 35).
Figure 5.
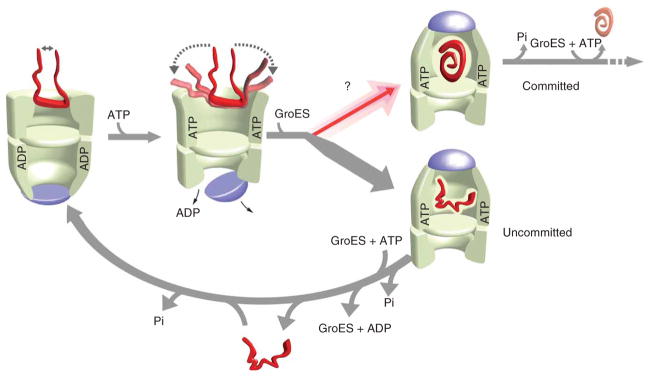
A functional model for substrate-protein unfolding by GroEL. Partial substrate unfolding by GroEL occurs in two phases: (i) passive unfolding upon protein capture by a GroEL ring; and (ii) forced unfolding upon ATP binding to a substrate-loaded trans ring. Although substrate confinement within the GroEL–GroES cavity enhances folding, the GroEL–GroES cavity exists for only a short period of time (~15–20 s at 25 °C) during a functional GroEL cycle17,19,20,25,27,34–36. Substrate unfolding provides, in principle, a method for driving a non-native substrate protein to a higher level of its folding free energy landscape, opening efficient folding paths (committed) that are not readily accessible to more compact states (uncommitted)14,15. Substrate-protein unfolding before encapsulation might substantially enrich the fraction of protein that commits to a productive folding path with each round of the GroEL cycle. ATP binding to the trans ring of the asymmetric complex, once hydrolysis of ATP within the GroEL–GroES cavity is complete (Pi), induces disassembly of the GroEL–ADP–GroES complex and ejection of the substrate protein, folded or not17,26,27,30. Uncommitted substrate must be recaptured on the trans ring of an ADP bullet for another round of unfolding and encapsulation (lower arrow). Two full rounds of ATP hydrolysis, one in each ring, followed by a round of substrate capture on the trans ring, returns the asymmetric ATP-containing complex to the starting ADP-bullet state.
Upon binding to an SR1 ring, Rubisco is subjected to a substantial level of passive unfolding35 (Fig. 6a). SR1 also binds ATP and GroES and initiates a fully productive folding reaction inside a long-lived SR1–GroES complex17,19,20. Importantly, however, SR1 supports only a minimal level of ATP-driven unfolding. In previous experiments, we detected a weak, ATP-driven expansion of the FRET-labeled Rubisco intermediate with SR1 (ref. 35). The magnitude of this apparent unfolding was small, and it was not detectable in the presence of GroES. However, these experiments were conducted under conditions (a 20-min preincubation of the SR1–Rubisco binary complex) in which passive unfolding on the SR1 ring is complete (Fig. 6a). SR1 could fail to support ATP-driven unfolding under these conditions because the long preincubation time allows maximal expansion of the substrate protein through passive unfolding. We therefore examined the ability of SR1 to cause ATP-driven Rubisco unfolding at different points along the SR1 passive-unfolding curve (Fig. 6b). Binary complexes between labeled Rubisco samples were formed and rapidly loaded into the syringe of a stopped flow apparatus. The preincubation time in this experiment takes into account the time (~15–20 s) required for complex formation and loading. When ATP is mixed with an SR1–Rubisco complex at preincubation times for which passive unfolding is less extensive, SR1 shows the same, low level of ATP-driven unfolding that is observed at longer times (Fig. 6b,c). Additionally, SR1 seems incapable of maintaining even this reduced level of ATP-driven expansion of the Rubisco monomer. Within 3–4 s, the SR1-bound Rubisco relaxes back to a conformational state similar to the state it populated before ATP addition (Fig. 6b). By contrast, the more extensive ATP-forced expansion of the Rubisco monomer on an ADP-bullet trans ring is stable for at least 7–10 s (Fig. 4b). SR1 thus shows a considerably reduced ability to support ATP-driven substrate unfolding. As a consequence, the slow passive unfolding of Rubisco on an SR1 ring provides a straightforward way to control the extent of substrate unfolding (Fig. 7a,b).
Figure 6.

ATP binding to a Rubisco-loaded SR1 ring does not cause extensive forced unfolding. (a) SR1 and the trans ring of an ADP bullet drive passive unfolding of Rubisco at a similar rate. The passive unfolding of Rubisco by SR1 and the ADP-bullet trans ring was monitored by FRET using fluorescein as the donor and rhodamine as the acceptor. The time-dependent drops in FRET efficiency for each complex are shown. Matched samples of donor-only (454-F) and donor-acceptor (454-F/58-R) Rubisco were denatured in acid-urea and rapidly diluted 50-fold (100 nM final) into buffer containing either SR1 (400 nM final) or ADP bullets (120 nM final) at 25 °C. Under the conditions used for this experiment, binding is complete within 5 s27. In both cases, mixing was conducted by rapid manual mixing into a thermally jacketed stirring cuvette in a standard fluorimeter to allow stable long-time observation of the sample. (b) SR1 does not support extensive forced unfolding of non-native Rubisco. Acid-urea denatured Rubisco was diluted to a final concentration of 100 nM into buffer containing 400 nM SR1. At different times over the course of a passive unfolding curve (main plot), aliquots of the SR1–Rubisco complex were rapidly mixed with ATP in a stopped-flow apparatus, and changes in the FRET efficiency of the labeled Rubisco sample were monitored for ~10 s. The insets show representative stopped-flow traces (red) taken at early (left) and late (right) points in the passive-unfolding process. For reference, the progress of passive unfolding over the same time window is also shown in each inset. The stopped-flow FRET traces are the average of n = 3 replicates of matched experimental pairs, calculated from donor-only (Rubisco 454-F) and donor-acceptor (Rubisco 454-F/58-R) samples. (c) The maximum amplitude of forced Rubisco expansion by SR1 upon ATP binding was measured for a series of stopped-flow experiments over the full passive-unfolding curve and is plotted as a function of time. The level of preexisting passive unfolding does not affect the weak forced unfolding supported by SR1 upon ATP binding.
Figure 7.

The fraction of non-native Rubisco that commits to the native state in a single round of GroES encapsulation increases in proportion to the extent of unfolding. (a) The slow, passive unfolding of Rubisco on SR1 can be used to examine the linkage between productive folding and substrate-protein unfolding. The progress curve of passive Rubisco unfolding by SR1, monitored by FRET at 25 °C, is shown schematically. For comparison, a schematic of the progress curve of Rubisco unfolding on the trans ring of an ADP bullet, and the extent of forced unfolding driven by ATP binding to the trans ring, are also shown. (b) Schematic of the experiment. Denatured Rubisco bound to excess SR1 was incubated for different preincubation times at 25 °C (indicated by red circles and letters a–d) to allow passive unfolding before the addition of GroES and ATP. Because SR1 shows a dramatically reduced capacity to support forced unfolding upon ATP binding (Fig. 6), the preincubation time primarily sets the level of unfolding imposed on the bound Rubisco monomer. Following addition of ATP and GroES, the encapsulated Rubisco is allowed to fold within the enclosed SR1–GroES cavity. (c) Rubisco folding is monitored as a function of time following GroES and ATP addition for several different preincubation times. Although the overall refolding kinetics are similar, the fraction of Rubisco that commits to the native state in the first few seconds following ATP and GroES binding increases considerably at longer preincubation times. (d) The extent to which unfolding on SR1 enhances the initial commitment of Rubisco to the native state is quantified by extrapolating refolding curves like those in c to zero preincubation time for a series of identical experiments (n = 3; error bars show one s.d.). The inset in d shows the final native Rubisco yield (following 30 min of folding within the SR1–GroES complex) observed at each preincubation point.
We therefore used SR1 to test whether unfolding stimulates productive Rubisco folding. We conducted a series of refolding experiments in which non-native Rubisco was bound to SR1 and incubated for various times before the addition of ATP and GroES (Fig. 7b). Productive folding was assessed by tracking the regain of enzymatic activity as a function of time (Fig. 7c). Whereas the overall refolding curves at each preincubation time are similar, the zero-time preincubation point (that is, the y-intercept in Fig. 7c) is directly proportional to the extent of SR1-mediated unfolding up to the point (~150 s) at which passive unfolding on the SR1 ring is complete (Fig. 7d). Thus, the fraction of non-native Rubisco that commits to the native state in the first few seconds following GroES encapsulation on SR1 increases in proportion to the amount of time that passive unfolding is allowed to proceed on the SR1 ring. Importantly, the final yield of native Rubisco following extended incubation within the SR1–GroES cavity is independent of the preincubation time (Fig. 7d, inset), demonstrating that the effect of a short preincubation time is not the result of incomplete Rubisco binding or encapsulation. Our results with SR1 thus strongly suggest that substrate unfolding by GroEL leads directly to improved partitioning of the substrate protein between more and less efficient folding routes.
DISCUSSION
Although several mechanisms have been proposed to explain how GroEL facilitates protein folding, the role of substrate-protein unfolding has remained controversial. Here we show that GroEL-induced unfolding can have a profound and direct influence on the productive folding of the stringent substrate protein Rubisco. Using GroEL’s physiological acceptor state for non-native proteins, the ADP bullet, we show that non-native Rubisco is substantially more compact when bound to a trans ring than when bound to an apoGroEL ring. This observation suggests that the trans ring of an ADP bullet is much less capable of passive protein unfolding, although precisely why this is the case remains unclear. Passive unfolding is probably caused by the high density of hydrophobic surface on the inner apical face of an open GroEL ring, where non-native proteins like Rubisco make multiple, simultaneous binding contacts49. The high local concentration of hydrophobic surface on the interior of an open GroEL ring probably catalyzes the rearrangement and partial unfolding of the protein, perhaps in a manner analogous to surface-mediated denaturation50,51. The observation that the trans ring of an ADP bullet is less capable of passive unfolding suggests that either a trans ring cannot engage a substrate protein across multiple apical domains in the same way that an apoGroEL ring can, or that the trans ring is not capable of dynamically adjusting its structure in response to substrate-protein binding in the same manner as the less constrained apoGroEL ring52. Whatever the ultimate reason, our observations suggest that, at least for substrate proteins like Rubisco, the conformation of a substrate protein bound to the trans ring of an ADP bullet can be substantially different from that observed with an apoGroEL ring.
Although the extent of passive unfolding on the ADP-bullet trans ring is limited, it is only the first stage of substrate-protein unfolding by GroEL. Here we show that the binding of ATP to a Rubisco-loaded trans ring results in the rapid conformational expansion of the bound protein. This ATP-driven stretching of the GroEL-bound folding intermediate is highly consistent with forced protein unfolding42–44. In the original forced-unfolding model, extensive destabilization of a substrate protein’s internal structure was directly linked to substrate-protein encapsulation within the GroEL–GroES cavity, requiring the binding of both ATP and GroES. By contrast, the forced conformational change we observe seems to depend only on ATP binding, and it actually precedes GroES binding. Furthermore, detailed hydrogen-deuterium exchange experiments with malate dehydrogenase (MDH) and Rubisco failed to detect evidence for the extensive, global unfolding that was suggested by the original forced-unfolding model45,46. These studies, however, left open the possibility that less extensive and longer-range structural alteration of a misfolded protein, including disruption of stabilizing tertiary contacts not readily observed by hydrogen-deuterium exchange, could be important for initiating productive folding. Our observations are consistent with the forced disruption of this kind of residual structure in a GroEL-bound folding intermediate and provide the best current evidence that the ATP-driven allosteric shift of a GroEL ring can cause partial, forced unfolding of a stringent substrate protein.
Our observations also provide direct experimental evidence that protein unfolding by GroEL is mechanistically linked to enhanced protein folding. By exploiting the slow rate of passive Rubisco unfolding by SR1, we show that unfolding directly enhances the efficiency with which Rubisco commits to the native state upon substrate-protein release into the SR1–GroES cavity. At the same time, however, encapsulation of Rubisco within the GroEL–GroES cavity also dramatically stimulates folding under conditions in which spontaneous folding is not possible17,19,20,35,36. Several mechanisms have been proposed to explain how simple confinement of a non-native folding intermediate within the spatially confined GroEL–GroES cavity could lead to enhanced folding (for review, see ref. 14). Notably, even when unfolding by an SR1 ring is reduced, long-term confinement within the SR1–GroES cavity results in fully productive folding34–36 (Fig. 7). This raises an important question: if confinement within a GroEL–GroES cavity can be fully efficient, to what extent can substrate-protein unfolding actually contribute to productive folding?
The answer to this question rests, at least in part, with the limited lifetime of the GroEL–GroES cavity during a physiologically relevant GroEL reaction cycle. Although long-term confinement within an isolated SR1–GroES cavity supports nearly complete Rubisco folding, Rubisco unfolding results in a maximal stimulation of ~10% for any given release event (Fig. 7d). However, in the case of Rubisco folding by SR1, the Rubisco monomer is bound and ejected only once into the productive SR1–GroES cavity, where the protein then spends the total remainder of the experimental observation time in an optimized folding environment. In the case of the cycling GroEL–GroES system, however, the relatively short lifetime (15–20 s) of the GroEL–GroES cavity during the course of a typical reaction cycle25,27 limits the capacity of cis cavity confinement to drive folding. During a typical GroEL reaction cycle, a non-native Rubisco monomer must spend a substantial fraction of its time in one of several states that have little or no capacity to fold. These states include: (i) kinetically trapped, non-productive states that are populated after the protein is ejected into free solution with each turn of the GroEL cycle and that must then be recaptured; (ii) bound to an open GroEL ring, waiting for ATP and GroES to bind; and (iii) stretched on a GroEL ring as GroES binds and encloses the protein. In other words, a protein transiting the cycling GroEL–GroES system has access to only a fraction of the in-cavity folding time of a protein confined within the artificially stable SR1–GroES cavity. A several-fold improvement in folding efficiency caused by substrate-protein unfolding during each turn of the GroEL cycle will, therefore, have a considerable impact.
The ability of GroEL to unfold kinetically trapped folding intermediates also provides a simple explanation for how GroEL and GroES drive folding under conditions in which encapsulation of a substrate protein is not possible. In several cases, large substrate proteins that cannot be accommodated within the GroEL–GroES cavity have been shown to gain a considerable folding benefit from binding to the trans ring of a GroEL–GroES complex, followed by direct ejection of the protein into free solution53,54. Indeed, trans-only GroEL–GroES complexes that are incapable of protein encapsulation can substantially enhance the folding Rubisco and another classical GroEL substrate protein, MDH55. Partial unfolding of these proteins on the open GroEL–GroES trans ring before their ejection into free solution provides a simple and elegant explanation for these observations.
The ATP-driven, forced expansion of a folding intermediate by GroEL can be viewed, then, as a means to rapidly complete the process of substrate unfolding that is initiated by the much slower step of passive, binding-driven unfolding. The combined steps of passive and forced protein unfolding serve to disrupt misfolded and inhibitory conformations of a substrate protein, providing the protein with a fresh opportunity at productive folding14,15. Confinement of the non-native protein within the constricted space of the GroEL–GroES cavity then enhances and amplifies the probability that a protein finds a productive folding path34–36. In folding Rubisco, GroEL thus makes use of a multilayered folding mechanism, involving both passive and forced partial unfolding, in combination with brief confinement within the GroEL–GroES cavity. GroEL’s ability to use multiple physical methods to stimulate Rubisco folding may also explain how this chaperonin machine assists the folding of diverse substrate proteins that possess a range of physical and energetic constraints on their productive folding.
METHODS
Proteins
Wild-type GroEL, cysteine-free GroEL and SR1 were expressed and purified using a combination of previously described methods17,27,56 (Supplementary Methods). Wild-type GroES, GroES98C, wild-type Rubisco and the various Rubisco cysteine mutants were expressed and purified as previously described17,27,35.
Labeling of Rubisco with fluorescent dyes
Labeling of Rubisco variants was carried out as previously described27,35. The thiol-reactive dyes used in this study were: 5-iodoacetamidofluorescein (F), 5-(2-acetamidoethyl) aminonaphthalene-1-sulfonate (AEDANS), tetratmethylrhodamine-5-iodoacetamide (R) and 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM). All reactive dyes were obtained from Invitrogen (Molecular Probes) and were prepared fresh from dry powder in anhydrous dimethyl formamide (DMF) immediately before use. The extent and specificity of dye conjugation was confirmed by (i) determination of the protein:dye ratio using a combination of dye absorption and protein quantitation (Bradford assay; BioRad); (ii) denaturing ion-exchange chromatography of labeled proteins using anion-exchange chromatography in 6 M urea57; (iii) quantitation of the residual free thiol content of proteins following dye conjugation using a thiol-detection assay (Invitrogen, Molecular Probes); and (iiii) analysis of dye-labeled peptides following trypsin digestion, reverse-phase (C18) chromatography and either in-line fluorescence detection57 or MS. Unless otherwise stated, the extent of dye conjugation to specific cysteine residues was between 95% and 100%. The labeling efficiency of ES98F was ~15% (~1 dye per heptamer). Exposure of Rubisco cysteine residues was examined with a short pulse (10 min) of the coumarin-derived, maleimide-reactive dye CPM. Dye incorporation was examined by reverse-phase HPLC analysis. See Supplementary Methods for further details.
Refolding and enzymatic assays
The refolding of Rubisco with SR1 was assayed essentially as previously described17,35. Rubisco was denatured in acid-urea and mixed with excess SR1 for the indicated times at 25 °C before the addition of GroES and ATP.
Asymmetric GroEL–GroES complex formation and protease protection
The binding of Rubisco to the trans ring of a GroEL–GroES complex was conducted by first creating an asymmetric GroEL–ADP–GroES complex. Samples of GroEL (7 μM), GroES (24.5 μM) and ATP (250 μM) were mixed in 50 mM HEPES (pH 7.6), 5 mM potassium acetate, 10 mM magnesium acetate and 2 mM DTT (buffer A) and incubated for 10 min at 25 °C. The asymmetric complex was diluted to the desired concentration and supplemented with acid-urea denatured Rubisco, and the sample was then incubated for 10 min at 25 °C to allow trans ring binding. For protease protection experiments, the asymmetric GroEL–ADP–GroES complex was mixed (120 nM GroEL, 350 nM GroES and 14 μM ADP final) with acid-urea denatured, fluorescein-labeled Rubisco (58-F, 100 nM). Binary complexes between apoGroEL and denatured, fluorescent Rubisco were created by mixing 100 nM denatured, fluorescent Rubisco with apoGroEL (120 nM). GroES (350 nM) and ADP (14 μM) were then added in order to match the total protein content of each reaction. Following a 30 min incubation at 25 °C, samples were supplemented with either trypsin (0.2 μg ml−1; Sigma) or chymotrypsin (0.3 μg ml−1; Sigma) and incubated at 25 °C. Aliquots were removed at various times, treated with 0.2 mg ml−1 PMSF and then subjected to SDS-PAGE. The level of undigested Rubisco was determined by quantifying the amount of intact, fluorescent Rubisco using a Molecular Dynamics Storm 860 system.
Steady-state and time-resolved FRET
Steady-state fluorescence emission spectra were acquired with a PTI photon-counting spectrofluorometer equipped with a temperature-jacketed cuvette holder. Donor excited state lifetimes were measured in the time domain using a Photon Technologies International TimeMaster Fluorescence Lifetime Spectrometer. The lifetimes of donor-only and donor-plus-acceptor samples were measured using a pulsed, N2 laser-pumped dye laser coupled to an optical boxcar detector58. The average distance between donor and acceptor probes (〈r〉) was calculated from the average FRET efficiency 〈E〉 as59:
A value of R0 was calculated for each donor-acceptor pair in each state examined. The value of κ2 was assumed to be 2/3 for all distance measurements. See ref. 35 for more detailed descriptions of instrument settings and energy transfer calculations. In all cases, steady-state and time-resolved measurements of the GroEL–Rubisco complexes were taken only after a 10–20 min incubation of the complex at 25 °C to allow the conformational rearrangements of the Rubisco monomer to come to completion.
Stopped-flow fluorescence and data analysis
Stopped-flow experiments were carried out essentially as described previously35, using an SFM-400 rapid mixing unit (BioLogic) equipped with a custom-designed, two-channel fluorescence detection system. The time-dependent change in donor-side FRET efficiency of the labeled Rubisco monomer was extracted from matched sets of donor-only and donor-acceptor stopped-flow experiments as described previously35. Fitting of experimental data was accomplished with either Igor Pro (Wavemetrics) or Origin (OriginLab).
Supplementary Material
Acknowledgments
We would like to thank F. Hughson, T. Silhavy and C. Carr for comments on the manuscript. This work was supported by a grant from the US National Institutes of Health (GM065421).
Footnotes
AUTHOR CONTRIBUTIONS
Z.L. and D.M. contributed equally to the experimental design, execution of the experiments and the analysis and interpretation of the data; H.S.R. contributed to the experimental design, analysis and interpretation of the data and was primarily responsible for writing the manuscript.
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Chapman E, et al. Global aggregation of newly translated proteins in an Escherichia coli strain deficient of the chaperonin GroEL. Proc Natl Acad Sci USA. 2006;103:15800–15805. doi: 10.1073/pnas.0607534103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kerner MJ, et al. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell. 2005;122:209–220. doi: 10.1016/j.cell.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 3.Houry WA, Frishman D, Eckerskorn C, Lottspeich F, Hartl FU. Identification of in vivo substrates of the chaperonin GroEL. Nature. 1999;402:147–154. doi: 10.1038/45977. [DOI] [PubMed] [Google Scholar]
- 4.Dobson CM. The structural basis of protein folding and its links with human disease. Phil Trans R Soc Lond B. 2001;356:133–145. doi: 10.1098/rstb.2000.0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grantcharova V, Alm EJ, Baker D, Horwich AL. Mechanisms of protein folding. Curr Opin Struct Biol. 2001;11:70–82. doi: 10.1016/s0959-440x(00)00176-7. [DOI] [PubMed] [Google Scholar]
- 6.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 7.Horwich A. Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions. J Clin Invest. 2002;110:1221–1232. doi: 10.1172/JCI16781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 9.Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–810. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 10.Ellis RJ, van der Vies SM. Molecular chaperones. Annu Rev Biochem. 1991;60:321–347. doi: 10.1146/annurev.bi.60.070191.001541. [DOI] [PubMed] [Google Scholar]
- 11.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 12.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 13.Horwich AL, Fenton WA, Chapman E, Farr GW. Two families of chaperonin: physiology and mechanism. Annu Rev Cell Dev Biol. 2007;23:115–145. doi: 10.1146/annurev.cellbio.23.090506.123555. [DOI] [PubMed] [Google Scholar]
- 14.Lin Z, Rye HS. GroEL-mediated protein folding: making the impossible, possible. Crit Rev Biochem Mol Biol. 2006;41:211–239. doi: 10.1080/10409230600760382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thirumalai D, Lorimer GH. Chaperonin-mediated protein folding. Annu Rev Biophys Biomol Struct. 2001;30:245–269. doi: 10.1146/annurev.biophys.30.1.245. [DOI] [PubMed] [Google Scholar]
- 16.Slepenkov SV, Witt SN. The unfolding story of the Escherichia coli Hsp70 DnaK: is DnaK a holdase or an unfoldase? Mol Microbiol. 2002;45:1197–1206. doi: 10.1046/j.1365-2958.2002.03093.x. [DOI] [PubMed] [Google Scholar]
- 17.Rye HS, et al. Distinct actions of cis and trans ATP within the double ring of the chaperonin GroEL. Nature. 1997;388:792–798. doi: 10.1038/42047. [DOI] [PubMed] [Google Scholar]
- 18.Xu Z, Horwich AL, Sigler PB. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 19.Mayhew M, et al. Protein folding in the central cavity of the GroEL-GroES chaperonin complex. Nature. 1996;379:420–426. doi: 10.1038/379420a0. [DOI] [PubMed] [Google Scholar]
- 20.Weissman JS, Rye HS, Fenton WA, Beechem JM, Horwich AL. Characterization of the active intermediate of a GroEL-GroES-mediated protein folding reaction. Cell. 1996;84:481–490. doi: 10.1016/s0092-8674(00)81293-3. [DOI] [PubMed] [Google Scholar]
- 21.Weissman JS, et al. Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES. Cell. 1995;83:577–587. doi: 10.1016/0092-8674(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 22.Braig K, et al. The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature. 1994;371:578–586. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- 23.Fenton WA, Kashi Y, Furtak K, Horwich AL. Residues in chaperonin GroEL required for polypeptide binding and release. Nature. 1994;371:614–619. doi: 10.1038/371614a0. [DOI] [PubMed] [Google Scholar]
- 24.Boisvert DC, Wang J, Otwinowski Z, Horwich AL, Sigler PB. The 2.4 crystal structure of the bacterial chaperonin GroEL complexed with ATP γS. Nat Struct Biol. 1996;3:170–177. doi: 10.1038/nsb0296-170. [DOI] [PubMed] [Google Scholar]
- 25.Burston SG, Ranson NA, Clarke AR. The origins and consequences of asymmetry in the chaperonin reaction cycle. J Mol Biol. 1995;249:138–152. doi: 10.1006/jmbi.1995.0285. [DOI] [PubMed] [Google Scholar]
- 26.Todd MJ, Viitanen PV, Lorimer GH. Dynamics of the chaperonin ATPase cycle: implications for facilitated protein folding. Science. 1994;265:659–666. doi: 10.1126/science.7913555. [DOI] [PubMed] [Google Scholar]
- 27.Rye HS, et al. GroEL-GroES cycling: ATP and nonnative polypeptide direct alternation of folding-active rings. Cell. 1999;97:325–338. doi: 10.1016/s0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- 28.Ranson NA, Dunster NJ, Burston SG, Clarke AR. Chaperonins can catalyse the reversal of early aggregation steps when a protein misfolds. J Mol Biol. 1995;250:581–586. doi: 10.1006/jmbi.1995.0399. [DOI] [PubMed] [Google Scholar]
- 29.Goloubinoff P, Christeller JT, Gatenby AA, Lorimer GH. Reconstitution of active dimeric ribulose bisphosphate carboxylase from an unfolded state depends on two chaperonin proteins and Mg-ATP. Nature. 1989;342:884–889. doi: 10.1038/342884a0. [DOI] [PubMed] [Google Scholar]
- 30.Weissman JS, Kashi Y, Fenton WA, Horwich AL. GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell. 1994;78:693–702. doi: 10.1016/0092-8674(94)90533-9. [DOI] [PubMed] [Google Scholar]
- 31.Motojima F, Chaudhry C, Fenton WA, Farr GW, Horwich AL. Substrate polypeptide presents a load on the apical domains of the chaperonin GroEL. Proc Natl Acad Sci USA. 2004;101:15005–15012. doi: 10.1073/pnas.0406132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellis RJ. Molecular chaperones. Opening and closing the Anfinsen cage. Curr Biol. 1994;4:633–635. doi: 10.1016/s0960-9822(00)00140-8. [DOI] [PubMed] [Google Scholar]
- 33.Agard DA. To fold or not to fold. Science. 1993;260:1903–1904. doi: 10.1126/science.8100365. [DOI] [PubMed] [Google Scholar]
- 34.Park ES, Fenton WA, Horwich AL. Disulfide formation as a probe of folding in GroEL-GroES reveals correct formation of long-range bonds and editing of incorrect short-range ones. Proc Natl Acad Sci USA. 2007;104:2145–2150. doi: 10.1073/pnas.0610989104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin Z, Rye HS. Expansion and compression of a protein folding intermediate by GroEL. Mol Cell. 2004;16:23–34. doi: 10.1016/j.molcel.2004.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brinker A, et al. Dual function of protein confinement in chaperonin-assisted protein folding. Cell. 2001;107:223–233. doi: 10.1016/s0092-8674(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 37.Jewett AI, Baumketner A, Shea JE. Accelerated folding in the weak hydrophobic environment of a chaperonin cavity: creation of an alternate fast folding pathway. Proc Natl Acad Sci USA. 2004;101:13192–13197. doi: 10.1073/pnas.0400720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thirumalai D, Klimov DK, Lorimer GH. Caging helps proteins fold. Proc Natl Acad Sci USA. 2003;100:11195–11197. doi: 10.1073/pnas.2035072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betancourt MR, Thirumalai D. Exploring the kinetic requirements for enhancement of protein folding rates in the GroEL cavity. J Mol Biol. 1999;287:627–644. doi: 10.1006/jmbi.1999.2591. [DOI] [PubMed] [Google Scholar]
- 40.Takagi F, Koga N, Takada S. How protein thermodynamics and folding mechanisms are altered by the chaperonin cage: molecular simulations. Proc Natl Acad Sci USA. 2003;100:11367–11372. doi: 10.1073/pnas.1831920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Todd MJ, Lorimer GH, Thirumalai D. Chaperonin-facilitated protein folding: optimization of rate and yield by an iterative annealing mechanism. Proc Natl Acad Sci USA. 1996;93:4030–4035. doi: 10.1073/pnas.93.9.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stan G, Lorimer GH, Thirumalai D, Brooks BR. Coupling between allosteric transitions in GroEL and assisted folding of a substrate protein. Proc Natl Acad Sci USA. 2007;104:8803–8808. doi: 10.1073/pnas.0700607104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lorimer G. Protein folding. Folding with a two-stroke motor. Nature. 1997;388:720–721. 723. doi: 10.1038/41892. [DOI] [PubMed] [Google Scholar]
- 44.Shtilerman M, Lorimer GH, Englander SW. Chaperonin function: folding by forced unfolding. Science. 1999;284:822–825. doi: 10.1126/science.284.5415.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park ES, Fenton WA, Horwich AL. No evidence for a forced-unfolding mechanism during ATP/GroES binding to substrate-bound GroEL: no observable protection of metastable Rubisco intermediate or GroEL-bound Rubisco from tritium exchange. FEBS Lett. 2005;579:1183–1186. doi: 10.1016/j.febslet.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 46.Chen J, Walter S, Horwich AL, Smith DL. Folding of malate dehydrogenase inside the GroEL-GroES cavity. Nat Struct Biol. 2001;8:721–728. doi: 10.1038/90443. [DOI] [PubMed] [Google Scholar]
- 47.Fenton WA, Horwich AL. Chaperonin-mediated protein folding: fate of substrate polypeptide. Q Rev Biophys. 2003;36:229–256. doi: 10.1017/s0033583503003883. [DOI] [PubMed] [Google Scholar]
- 48.Sparrer H, Buchner J. How GroES regulates binding of nonnative protein to GroEL. J Biol Chem. 1997;272:14080–14086. doi: 10.1074/jbc.272.22.14080. [DOI] [PubMed] [Google Scholar]
- 49.Farr GW, et al. Multivalent binding of nonnative substrate proteins by the chaperonin GroEL. Cell. 2000;100:561–573. doi: 10.1016/s0092-8674(00)80692-3. [DOI] [PubMed] [Google Scholar]
- 50.Sharp JS, Forrest JA, Jones RA. Surface denaturation and amyloid fibril formation of insulin at model lipid-water interfaces. Biochemistry. 2002;41:15810–15819. doi: 10.1021/bi020525z. [DOI] [PubMed] [Google Scholar]
- 51.Swain JF, Gierasch LM. First glimpses of a chaperonin-bound folding intermediate. Proc Natl Acad Sci USA. 2005;102:13715–13716. doi: 10.1073/pnas.0506510102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elad N, et al. Topologies of a substrate protein bound to the chaperonin GroEL. Mol Cell. 2007;26:415–426. doi: 10.1016/j.molcel.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chaudhuri TK, Farr GW, Fenton WA, Rospert S, Horwich AL. GroEL/GroES-mediated folding of a protein too large to be encapsulated. Cell. 2001;107:235–246. doi: 10.1016/s0092-8674(01)00523-2. [DOI] [PubMed] [Google Scholar]
- 54.Paul S, Singh C, Mishra S, Chaudhuri TK. The 69 kDa Escherichia coli maltodextrin glucosidase does not get encapsulated underneath GroES and folds through trans mechanism during GroEL/GroES-assisted folding. FASEB J. 2007;21:2874–2885. doi: 10.1096/fj.06-7958com. [DOI] [PubMed] [Google Scholar]
- 55.Farr GW, et al. Folding with and without encapsulation by cis- and trans-only GroEL-GroES complexes. EMBO J. 2003;22:3220–3230. doi: 10.1093/emboj/cdg313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poso D, Clarke AR, Burston SG. A kinetic analysis of the nucleotide-induced allosteric transitions in a single-ring mutant of GroEL. J Mol Biol. 2004;338:969–977. doi: 10.1016/j.jmb.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 57.Rye HS. Application of fluorescence resonance energy transfer to the GroEL-GroES chaperonin reaction. Methods. 2001;24:278–288. doi: 10.1006/meth.2001.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.James DR, Siemiarczuk A, Ware WR. Stroboscopic optical boxcar technique for the determination of fluorescence lifetimes. Rev Sci Instrum. 1992;63:1710–1716. [Google Scholar]
- 59.Lakowicz JR. Principles of Fluorescence Spectroscopy. Kluwer Academic; Plenum Publishers; New York: 1999. Energy transfer; pp. 368–391. [Google Scholar]
- 60.Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego: 1996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.