

Abstract
A well-known mechanism through which new protein-coding genes originate is by modification of pre-existing genes, e.g. by duplication or horizontal transfer. In contrast, many viruses generate protein-coding genes de novo, via the overprinting of a new reading frame onto an existing (“ancestral”) frame. This mechanism is thought to play an important role in viral pathogenicity, but has been poorly explored, perhaps because identifying the de novo frames is very challenging. Therefore, a new approach to detect them was needed. We assembled a reference set of overlapping genes for which we could reliably determine the ancestral frames, and found that their codon usage was significantly closer to that of the rest of the viral genome than the codon usage of de novo frames. Based on this observation, we designed a method that allowed the identification of de novo frames based on their codon usage with a very good specificity, but intermediate sensitivity. Using our method, we predicted that the Rex gene of deltaretroviruses has originated de novo by overprinting the Tax gene. Intriguingly, several genes in the same genomic region have also originated de novo and encode proteins that regulate the functions of Tax. Such “gene nurseries” may be common in viral genomes. Finally, our results confirm that the genomic GC content is not the only determinant of codon usage in viruses and suggest that a constraint linked to translation must influence codon usage.
Author Summary
How does novelty originate in nature? It is commonly thought that new genes are generated mainly by modifications of existing genes (the “tinkering” model). In contrast, we have shown recently that in viruses, numerous genes are generated entirely de novo (“from scratch”). The role of these genes remains underexplored, however, because they are difficult to identify. We have therefore developed a new method to detect genes originated de novo in viral genomes, based on the observation that each viral genome has a unique “signature”, which genes originated de novo do not share. We applied this method to analyze the genes of Human T-Lymphotropic Virus 1 (HTLV1), a relative of the HIV virus and also a major human pathogen that infects about twenty million people worldwide. The life cycle of HTLV1 is finely regulated – it can stay dormant for long periods and can provoke blood cancers (leukemias) after a very long incubation. We discovered that several of the genes of HTLV1 have originated de novo. These novel genes play a key role in regulating the life cycle of HTLV1, and presumably its pathogenicity. Our investigations suggest that such “gene nurseries” may be common in viruses.
Introduction
Modification of existing genes, such as by duplication or fusion, is a common and well-understood mechanism by which protein-coding genes originate [1], [2]. In contrast, we have shown that viruses generate many proteins de novo (hereafter called “de novo proteins”) [3], [4]. Preliminary observations indicate that these proteins play an important role in the pathogenicity of viruses [3], [5], for instance by neutralizing the host interferon response [6] or antagonizing the host RNA interference [7]. Strikingly, p19, the only de novo protein characterised both structurally and functionally, has both a previously unknown structural fold and a previously unknown mechanism of action [7]. Thus, protein innovation seems to be a significant, but poorly understood part of the evolutionary arms race between hosts and their pathogens [5], 8,9.
Studying de novo proteins should thus greatly enhance our understanding of host-pathogen co-evolution and our knowledge of the function and structure of viral proteins [3], [10]–[14]. However, a major bottleneck that prevents the study of such proteins is their identification, which is very challenging. Finding that a viral protein has no detectable sequence homolog does not reliably indicate that it has originated de novo, because viral proteins evolve so fast that they can easily diverge in sequence beyond recognition. To circumvent this problem, in our previous work [3], [4] and in the current study, we focused on a special case of de novo proteins: those generated by overprinting. Overprinting is a process in which mutations in a protein-coding reading frame allow the expression of a second reading frame while preserving the expression of the first one (Figure 1), leading to an overlapping gene arrangement [10]. It is thought that most overlapping genes evolve by this mechanism, and that consequently each gene overlap contains one ancestral frame and one originated de novo [10]. Because overlapping genes are particularly abundant in viruses [15]–[17], they constitute a rich source of de novo proteins.
Figure 1. Rationale for our approach.

Identifying which frame is ancestral and which one is de novo (the “genealogy” of the overlap) can be done, in principle, by examining their phylogenetic distribution (the frame with the most restricted distribution is assumed to be the de novo one). One can exclude the possibility that the phylogenetically restricted frame is in fact present in other genomes but has diverged beyond recognition, by checking that outside of its clade, the ancestral frame is not overlapped by any reading frame [4]. This approach is simple and reliable [3], [4], but is not applicable to cases where the homologs of both frames have an identical phylogenetic distribution. For instance, it could identify the de novo frame in only a minority (40%) of overlaps in our previous study [3]. Therefore, a new method is needed to identify the de novo proteins in most overlapping genes.
The approach we investigated is based on the hypothesis that the ancestral frame should have a pattern of codon usage (i.e. which synonymous codon(s) is preferred to encode each amino acid [18]) closer to that of the rest of the viral genome than the de novo frame [10]. Indeed, analyses of plant RNA viruses and animal DNA viruses [19], [20] have shown that, within a given viral genome, genes generally have a similar pattern of codon usage, which is thought to depend on the overall GC content of the genome [19]–[21]. In overlapping genes, the ancestral frame, which has co-evolved over a long period with the other viral genes, is expected to have a codon usage similar to that of the rest of the genome (Figure 1). On the other hand, the de novo frame, at birth, will have a codon usage in effect randomized by the shift and thus unlikely to be close to that of the genome. In addition, constraints imposed by the ancestral frame might prevent the de novo frame from adopting, later, the typical genomic codon usage. Consequently, the de novo frame is expected to have a codon usage less similar to that of the viral genome than the ancestral frame (Figure 1). This approach has been empirically used to try and identify the de novo frame in a number of cases, as have been related methods which rely on the frequency on nucleotides at some or all codon positions [10], [22]–[29]. However, the reliability or accuracy of these methods has never been tested. Here we gathered a reference (“benchmark”) dataset composed of overlaps with known genealogy, and used it to answer the following questions: do de novo frames have a codon usage distinguishable from ancestral frames? If yes, can codon usage be used to identify the de novo frame? What is the reliability of the method and its sensitivity? Finally, we applied this method to overlapping genes whose genealogy was undeterminable by the phylogenetic method.
Results
A benchmark dataset of overlapping genes with known genealogy
As described in Material and Methods, we assembled a dataset of 27 independent, experimentally proven overlapping genes longer than 140 nt (Table 1). 16 of them have been described previously [3], as indicated by an asterisk in Table 1, and 11 additional overlaps were collected for this study. The 27 overlaps come from 25 genera, distributed in 16 viral families covering a wide range of viruses (Table 1). 18 overlaps involve one gene being completely overlapped by the other, while in 9 the overlap is partial (e.g. Figure 2). All overlapping genes are in the same orientation, i.e. there are no antiparallel overlapping genes in the dataset. To identify the genealogy of the overlaps, we used the same stringent criterion as in our previous study [3], selecting only cases in which one frame, predicted ancestral, had a much wider taxonomic distribution than the other frame, predicted de novo. To be confident about the taxonomic distribution of each frame, we carried out extensive searches involving the most up to date similarity search tools, supplemented by in-depth manual searches using contextual information (see Material and Methods). The taxonomic distribution of each frame, and the corresponding evidence, are presented in Supplementary Table S1. Our predictions of ancestry are supported by functional data: almost all proteins encoded by a frame identified as ancestral have a function central to the viral cycle (such as capsid or replication), while most proteins identified as de novo have a “secondary” function related to pathogenicity (such as silencing suppressor or apoptotic factor) (Table 1). Thus, the predicted genealogy of the overlapping genes of the dataset is highly reliable.
Table 1. Benchmark dataset of 27 overlapping genes with known genealogy.
| Viral family and nature of the genome | Genus and genome accession number(1) , (2) | Species | Ancestral frame [function(3)] | De novo frame [function(3)] | Length of overlapping regions (nt) | Length of non- overlapping regions (nt) | Host organism | Reference |
| Alphaflexiviridae ssRNA(+) | Betatetravirus (1) NC_001990 | Nudaurelia capensis beta virus | Capsid protein [capsid] | Replicase, C-term domain | 1833 | 3951 | Insect | [106] |
| Alphaflexiviridae ssRNA(+) | Mandarivirus (1) NC_003093 | Indian citrus ringspot virus | Capsid protein [capsid] | NABP, N-term domain | 303 | 7011 | Plant | [107] |
| Alphaflexiviridae ssRNA(+) | Potexvirus (1) NC_001658 | Cassava common mosaic virus | TGBp2 [viral Movement] | TGBp3, N-term domain | 153 | 5991 | Plant | [108] |
| Betaflexiviridae ssRNA(+) | Capillovirus (1) NC_001749 | Apple stem grooving virus | MP [viral Movement] | Polyprotein | 966 | 5352 | Plant | [109] |
| Betaflexiviridae ssRNA(+) | Trichovirus (1) NC_001409 | Apple stem leaf spot virus | Coat protein [capsid] | MP, C-term domain | 318 | 6804 | Plant | [110] |
| Birnaviridae dsRNA | Aquabirnavirus (1) NC_001915 | Infectious pancreatic necrosis virus | VP2 [capsid] | VP5 [apoptotic factor] | 396 | 5112 | Fish | [111] |
| Bunyaviridae ssRNA(−) | Orthobunyavirus (1) NC_001927 | Bunyamwera virus | N [Nucleoprotein] | NSs [blocks interferon and cellular transcription] | 309 | 11412 | Insect | [112] |
| Caliciviridae ssRNA(+) | Norovirus (2) NC_008311 | Murine norovirus | Capsid protein [capsid] | VF1 [virulence factor] | 642 | 6639 | Mammal | [113] |
| Circoviridae ssDNA | Gyrovirus (2) NC_001427 | Chicken anemia virus | VP2 [phosphatase?] | Apoptin [apoptotic factor] | 366 | 1275 | Bird | [114] |
| Dicistroviridae ssRNA(+) | Aparavirus (2) NC_009025 | Israel acute paralysis virus | Capsid Protein [capsid] | Pog | 315 | 8115 | Insects | [115] |
| Geminiviridae ssDNA | Begomovirus (2) NC_001467 | African cassava mosaic virus | AL1 [rolling circle replication initiator] | AC4 [silencing suppressor] | 426 | 2928 | Plant | [116] |
| Hepadnaviridae Retroid | Orthohepadnavirus (1) NC_003977 | Human hepatitis B virus | Pol, central domain [reverse transcriptase] | L [envelope glycoprotein] | 708 | 1578 | Mammal | [117] |
| Hepadnaviridae Retroid | Orthohepadnavirus (1) NC_003977 | Human hepatitis B virus | Pol, C-term domain [RNAse H] | X [virulence factor] | 252 | 1578 | Mammal | [118] |
| Luteoviridae ssRNA(+) | Luteovirus (2) NC_003680 | Barley yellow dwarf virus | P5 [capsid] | P4 [viral Movement] | 468 | 4311 | Plant | [119] |
| Parvoviridae ssDNA | Brevidensovirus (2) NC_004285 | Aedes albopictus densovirus | NS1 [rolling circle replication initiator] | NS2(4) [virulence factor] | 1122 | 2142 | Insect | [120] |
| Parvoviridae ssDNA | Densovirus (2) NC_005041 | Blattella germanica densovirus | NS1 [rolling circle replication initiator] | NS2(4) [virulence factor] | 792 | 4005 | Insect | [121] |
| Parvoviridae ssDNA | Dependovirus (2) NC_001401 | Adeno-associated virus 2 | VP2 [capsid] | AAP [capsid assembly co-factor] | 618 | 3429 | Mammal | [122] |
| Parvoviridae ssDNA | Parvovirus (2) NC_001718 | Porcine parvovirus | Capsid protein [capsid] | SAT [virulence factor] | 210 | 3639 | Mammal | [123] |
| Carmotetraviridae ssRNA(+) | Alphacarmotetravirus (2) NC_014126 | Providence virus | p104(5) [polymerase] | p130 | 2682 | 3312 | Insect | [31] |
| Tetraviridae ssRNA(+) | Omegatetravirus (1) NC_005899 | Dendrolimus punctatus tetravirus | Capsid protein [capsid] | p17 | 384 | 6591 | Insect | [124] |
| Tombusviridae ssRNA(+) | Carmovirus (1) NC_003608 | Hibiscus chlorotic ringspot virus | p28 [replicase cofactor] | p23 [virulence factor] | 633 | 2163 | Plant | [125] |
| Tombusviridae ssRNA(+) | Carmovirus (1) NC_003608 | Hibiscus chlorotic ringspot virus | Capsid protein [capsid] | p25 [viral movement] | 678 | 2163 | Plant | [125] |
| Tombusviridae ssRNA(+) | Tombusvirus (1) NC_003532 | Cymbidium ringspot virus | p22 [viral movement] | p19 [silencing suppressor] | 522 | 3648 | Plant | [126] |
| Tombusviridae ssRNA(+) | Machlomovirus (2) NC_003627 | Maize chlorotic mottle virus | Capsid protein(5) [capsid] | p31 | 453 | 2625 | Plant | [127] |
| Tymoviridae ssRNA(+) | Tymovirus (1) NC_004063 | Turnip yellow mosaic virus | Replicase [Methyltransferase-Guanylyltransferase] | MP [viral movement] | 1881 | 4230 | Plant | [128] |
| Virgaviridae ssRNA(+) | Hordeivirus (1) NC_003481 | Barley stripe mosaic virus | TGBp2 [viral movement] | TGBp3, N-term domain | 192 | 8796 | Plant | [129] |
| Unassigned ssRNA(+) | Umbravirus (1) NC_004366 | Tobacco bushy top virus | ORF4 [viral movement] | ORF3 [viral movement] | 699 | 2619 | Plant | [130] |
gene overlaps described previously (see reference [3]).
additional overlaps collected for this study.
The function is that of the overlapping region of the protein; if it is not known, the field is left blank.
The NS2 proteins of brevidensoviruses and that of densoviruses are not homologous (they are encoded in different frames relative to NS1).
The alphacarmotetravirus polymerase and machlomovirus capsid have originated by horizontal transfer and thus the two corresponding overlaps are not part of the benchmark dataset, although we perform the same analyses on them than on other overlaps(see text).
Abbreviations: AAP, assembly-activating protein; dsRNA, double-stranded RNA; C-term, C-terminal; L, large envelope protein; MP, movement protein; NABP, nucleic-acid binding protein; NS, non-structural protein; NSs, non-structural protein of the small RNA segment; N-term, N-terminal; Pog, predicted overlapping gene; Pol, Polymerase; SAT, small alternatively translated protein; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA (+, positive or −, negative); TGBp2, Triple Gene Block protein 2; TGBp3, Triple Gene Block protein 3; VP, viral protein.
Figure 2. Definition used for overlapping regions and non-overlapping regions of a viral genome.

If a viral genome contains other overlapping genes than those under study (e.g. the genes to the right), we only considered non-overlapping regions of these genes; their overlapping regions (in grey) were excluded from the analysis.
We needed to exclude from the dataset ancestral frames that have entered their genome by distant horizontal transfer since these frames are not expected to have the same codon usage as that of their new viral genome, and are thus not suitable for codon usage analysis. Performing a detailed recombination analysis on all ancestral frames of the dataset was out of the scope of this article, and thus we simply detected cases of taxonomic incongruence (see Material and Methods). We detected two cases in which the ancestral frame had originated from another viral genome by distant horizontal transfer. The ancestral protein p104 of Providence virus (genus alphacarmotetravirus, family Carmotetraviridae) has statistically significant similarity with the replicase of viruses from a different family, Tombusviridae. Also, the capsid protein of Maize chlorotic virus (genus machlomovirus, family Tombusviridae) has significant similarity to that of sobemoviruses, an unassigned genus unrelated to Tombusviridae [30]. We established that horizontal transfer took place towards alphacarmotetravirus and machlomovirus from the other families by analysing the phylogenetic distribution of homologs of the ancestral proteins (not shown). Our results agree with previously reported findings that Providence virus has originated through recombination between a Tombusviridae-like and a Tetraviridae-like virus [31], and that the machlomovirus capsid protein is taxonomically incongruent [32]. We excluded these two cases from our analyses, and the final benchmark dataset is thus composed of 25 overlaps (Table 1).
Ancestral frames have a codon usage closer to that of the genome than de novo frames
As a measure of codon usage similarity between a given frame and the rest of the viral genome, we used the Spearman's rank correlation coefficient (rs) between the number of occurrences of each codon in that frame and in the viral genome (see Materials and Methods). Accordingly, the higher the rs of a frame, the more similar its codon usage is to that of the genome. For all gene overlaps of the benchmark dataset, we evaluated rsA, rsN (the rs of the ancestral and the de novo frame, respectively), and the difference (d 21) between rsA and rsN (d 21 = rsA−rsN). They are listed in the left moiety of Table 2, ranked by decreasing value of t-Hotelling. rsA is higher than rsN in 21 cases (i.e. d21>0) and lower (i.e. d21<0) in only 4 cases. This distribution is not random (P<0.001, in accordance to the binomial proportion test), suggesting that ancestral frames have a codon usage closer to their genome than de novo frames. This conclusion is supported quantitatively, since the median rsA (0.42) is significantly (P<0.01) higher than the median rsN (0.19) according to the Wilcoxon signed rank test [33]. These findings support the hypothesis that codon usage can, in principle, be used to determine the ancestral frame.
Table 2. Analysis of the codon usage of overlapping frames from the benchmark dataset.
| Calculations performed on actual frames | Calculations performed on simulated frames | |||||||||||||
| Genus | Ancestral frame | De novo frame | NA | NN | rsA | rsN | d21 = rsA−rsN | t-Hotelling | P< | rsA | rsN | d21 = rsA−rsN | P< | Agreement between t-Hotelling and simulation |
| Omegatetra | Capsid | p17 | 49 | 48 | 0.70 | 0.04 | 0.66 | 4.26 | 0.001 | 0.252 | 0.239 | 0.012 | 0.001 | Yes |
| Noro | Capsid | VF1 | 59 | 57 | 0.68 | 0.23 | 0.45 | 3.57 | 0.001 | 0.304 | 0.222 | 0.083 | 0.002 | Yes |
| Dependo | VP2 | AAP | 59 | 57 | 0.61 | 0.19 | 0.42 | 3.03 | 0.005 | 0.312 | 0.255 | 0.056 | 0.001 | Yes |
| Carmo | Replicase | p23 | 59 | 57 | 0.42 | −0.10 | 0.52 | 3.00 | 0.005 | 0.305 | 0.245 | 0.060 | 0.001 | Yes |
| Aquabirna | VP2 | VP5 | 53 | 57 | 0.62 | 0.24 | 0.38 | 2.74 | 0.005 | 0.285 | 0.239 | 0.046 | 0.004 | Yes |
| Luteo | P5 | P4 | 59 | 52 | 0.44 | 0.01 | 0.43 | 2.72 | 0.005 | 0.314 | 0.284 | 0.030 | 0.0005 | Yes |
| Tymo | Replicase | MP | 57 | 59 | 0.79 | 0.59 | 0.20 | 2.65 | 0.01 | 0.294 | 0.292 | 0.002 | 0.073 | No |
| Capillo | MP | Replicase | 59 | 59 | 0.65 | 0.40 | 0.25 | 2.30 | 0.025 | 0.357 | 0.288 | 0.069 | 0.080 | No |
| Mandari | Capsid | NABP | 37 | 44 | 0.58 | 0.22 | 0.36 | 2.22 | 0.025 | 0.229 | 0.247 | −0.019 | 0.011 | Yes |
| Carmo | Capsid | p25 | 59 | 59 | 0.35 | −0.05 | 0.40 | 2.13 | 0.025 | 0.316 | 0.227 | 0.089 | 0.003 | Yes |
| Betatetra | Capsid | Replicase | 59 | 59 | 0.40 | 0.11 | 0.29 | 1.88 | 0.05 | 0.306 | 0.290 | 0.017 | 0.003 | Yes |
| Gyro | VP2 | Apoptin | 59 | 51 | 0.29 | −0.01 | 0.30 | 1.78 | 0.05 | 0.259 | 0.217 | 0.042 | 0.020 | Yes |
| Potex | TGBp2 | TGBp3 | 20 | 33 | 0.42 | 0.03 | 0.39 | 1.75 | 0.05 | 0.237 | 0.206 | 0.031 | 0.015 | Yes |
| Parvo | VP2 | SAT | 29 | 35 | 0.47 | 0.12 | 0.35 | 1.69 | 0.10 | 0.234 | 0.224 | 0.010 | 0.063 | Yes |
| Tombus | p22 | p19 | 59 | 59 | 0.33 | 0.13 | 0.20 | 1.24 | 0.15 | 0.310 | 0.289 | 0.021 | 0.051 | Yes |
| Apara | Capsid | Pog | 37 | 49 | 0.25 | 0.02 | 0.23 | 1.04 | 0.20 | 0.276 | 0.262 | 0.014 | 0.061 | Yes |
| Denso | NS1 | NS2 | 59 | 57 | 0.36 | 0.19 | 0.17 | 1.04 | 0.20 | 0.278 | 0.303 | −0.024 | 0.083 | Yes |
| Orthohepadna | Pol | L | 59 | 55 | 0.42 | 0.29 | 0.13 | 0.98 | 0.20 | 0.241 | 0.252 | −0.011 | 0.146 | Yes |
| Umbra | ORF4 | ORF3 | 59 | 55 | 0.40 | 0.23 | 0.17 | 0.97 | 0.20 | 0.280 | 0.279 | 0.001 | 0.101 | Yes |
| Begomo | Replicase | AC4 | 53 | 55 | 0.18 | 0.10 | 0.08 | 0.49 | 0.50 | 0.317 | 0.281 | 0.036 | 0.377 | Yes |
| Hordei | TGBp2 | TGBp3 | 29 | 27 | 0.24 | 0.36 | −0.12 | 0.39 | 0.50 | 0.195 | 0.209 | −0.014 | 0.297 | Yes |
| Orthohepadna | Pol | X | 48 | 44 | 0.06 | 0.10 | −0.04 | 0.24 | 0.50 | 0.221 | 0.217 | 0.004 | 0.438 | Yes |
| Brevidenso | NS1 | NS2 | 59 | 59 | 0.62 | 0.63 | −0.01 | 0.09 | 0.50 | 0.348 | 0.353 | −0.005 | 0.556 | Yes |
| Orthobunya | N | NSs | 55 | 55 | 0.28 | 0.26 | 0.02 | 0.06 | 0.50 | 0.308 | 0.235 | 0.073 | 0.655 | Yes |
| Tricho | Capsid | MP | 43 | 41 | 0.31 | 0.32 | −0.01 | 0.03 | 0.50 | 0.313 | 0.278 | 0.035 | 0.426 | Yes |
| Recombinant: | ||||||||||||||
| Alphacarmotetra | Replicase | p130 | 59 | 59 | 0.00 | 0.51 | −0.51 | 2.94 | 0.005 | 0.279 | 0.303 | −0.024 | 0.001 | Yes |
| Machlomo | Capsid | p31 | 43 | 41 | 0.34 | 0.18 | 0.16 | 1.01 | 0.20 | 0.273 | 0.230 | 0.044 | 0.189 | Yes |
Abbreviations are the same as in Table 1. The last two overlaps have entered their genome by horizontal transfer (see text).
rsA is the Spearman rank correlation coefficient rs between the codon usage of the ancestral frame and that of its genome. rsN is the equivalent coefficient for the de novo frame. NA and NN are the number of codons on which rsA and rsN were calculated. The first row indicates whether calculations are presented for the actual overlapping frames or for the corresponding simulated frames. The calculation of P for the actual frames is based on Hotelling's t-test, whereas for simulated frames P is based on the distribution of the simulated d21 (see text). Agreement between t-Hotelling and simulation is calculated on the basis of whether corresponding P-values are both <0.05 or >0.05.
We now needed a method to infer, given any pair of overlapping frames, whether one frame had a codon usage significantly closer to the rest of the viral genome than does the other frame. In principle, a suitable method to assess the significance of the difference between the rs coefficients of each frame is Hotelling's t-test [34], [35]. However, Hotelling's t-test is applicable to correlation coefficients estimated from independent data, whereas our data are clearly not independent (see Material and Methods). Therefore, we assessed whether Hotelling's t-test was robust to the violation of the non-independence of data by comparing the results of Hotelling's t-test with simulated codon usage data (see Material and Methods). Values of rsA, rsN and d21 for simulated frames corresponding to each overlap are presented in the right moiety of Table 2. We performed a McNemar test [33], which indicated that both methods provide equivalent results (McNemar chi-square = 0.6; P = 0.50). Therefore, Hotelling's t-test is reasonably robust to violation of independence and is applicable to our problem.
Codon usage can predict the ancestral frame with a high specificity but intermediate sensitivity
Having established the validity of Hotelling's t-test, we used it to predict the ancestral frame (and consequently the de novo frame) in our dataset. Given two overlapping frames 1 and 2, a frame (for instance frame 2) was predicted ancestral only if it matched the following two criteria:
its codon usage was significantly closer to that of the genome than the other frame, i.e. r s2>r s1 and the difference d 21 = r s2−r s1 was statistically significant (P<0.05) according to Hotelling's t-test;
its codon usage was positively correlated to that of the genome, i.e. r s2>0.
The first criterion corresponds to our main biological hypothesis, whereas the second criterion avoids a scenario in which the first criterion gives results that are mathematically significant but not biologically meaningful. For instance, if one frame had an r s of −0.7 and the overlapping frame had an r s of −0.1, the difference would be significant. However, it would be unjustified to return a prediction that the second frame is ancestral, because the negative value of its r s contrasts with our central hypothesis that the ancestral frame has conserved traces of the genome's codon usage.
The overlaps are listed in Table 3 by decreasing value of t-Hotelling. We found that both criterions were fulfilled for 13 of our 25 overlaps, and in all these cases the ancestral frame prediction was correct, i.e. concordant with that established by phylogeny (Table 3). Consequently, the specificity of the codon usage approach is high (specificity = 1.0, 95% confidence interval [CI] 0.77–1.00), but its sensitivity is moderate (sensitivity = 0.52, 95% CI 0.31–0.72).
Table 3. Prediction of the ancestral frame in overlapping genes from the benchmark dataset.
| Genus | rsA | rsN | d21 = rsA−rsN | t-Hotelling | P< | Predicted ancestral frame | Prediction correct? |
| Omegatetra | 0.70 | 0.04 | 0.66 | 4.26 | 0.001 | Capsid | Yes |
| Noro | 0.68 | 0.23 | 0.45 | 3.57 | 0.001 | Capsid | Yes |
| Dependo | 0.61 | 0.19 | 0.42 | 3.03 | 0.005 | VP2 | Yes |
| Carmo (replicase/p23) | 0.42 | −0.10 | 0.52 | 3.00 | 0.005 | Replicase | Yes |
| Aquabirna | 0.62 | 0.24 | 0.38 | 2.74 | 0.005 | VP2 | Yes |
| Luteo | 0.44 | 0.01 | 0.43 | 2.72 | 0.005 | P5 | Yes |
| Tymo | 0.79 | 0.59 | 0.20 | 2.65 | 0.01 | Replicase | Yes |
| Capillo | 0.65 | 0.40 | 0.25 | 2.30 | 0.025 | MP | Yes |
| Mandari | 0.58 | 0.22 | 0.36 | 2.22 | 0.025 | Capsid | Yes |
| Carmo (capsid/p25) | 0.35 | −0.05 | 0.40 | 2.13 | 0.025 | Capsid | Yes |
| Betatetra | 0.40 | 0.11 | 0.29 | 1.88 | 0.05 | Capsid | Yes |
| Gyro | 0.29 | −0.01 | 0.30 | 1.78 | 0.05 | VP2 | Yes |
| Potex | 0.42 | 0.03 | 0.39 | 1.75 | 0.05 | TGBp2 | Yes |
| Parvo | 0.47 | 0.12 | 0.35 | 1.69 | 0.10 | - | - |
| Tombus | 0.33 | 0.13 | 0.20 | 1.24 | 0.15 | - | - |
| Apara | 0.25 | 0.02 | 0.23 | 1.04 | 0.20 | - | - |
| Denso | 0.36 | 0.19 | 0.17 | 1.04 | 0.20 | - | - |
| Orthohepadna (pol/L) | 0.42 | 0.29 | 0.13 | 0.98 | 0.20 | - | - |
| Umbra | 0.40 | 0.23 | 0.17 | 0.97 | 0.20 | - | - |
| Begomo | 0.18 | 0.10 | 0.08 | 0.49 | 0.50 | - | - |
| Hordei | 0.24 | 0.36 | −0.12 | 0.39 | 0.50 | - | - |
| Orthohepadna (pol/X) | 0.06 | 0.10 | −0.04 | 0.24 | 0.50 | - | - |
| Brevidenso | 0.62 | 0.63 | −0.01 | 0.09 | 0.50 | - | - |
| Orthobunya | 0.28 | 0.26 | 0.02 | 0.06 | 0.50 | - | - |
| Tricho | 0.31 | 0.32 | −0.01 | 0.03 | 0.50 | - | - |
| Recombinant(1): | - | - | |||||
| Alphacarmotetra | 0.00 | 0.51 | −0.51 | 2.94 | 0.005 | p130 | No(1) |
| Machlomo | 0.34 | 0.18 | 0.16 | 1.01 | 0.20 | - | - |
The last two overlaps have entered their genome by horizontal transfer and are not taken into account for calculations of specificity and sensitivity of the method.
Abbreviations and conventions are the same as in Table 2. A frame is predicted ancestral if its rs is positive and significantly higher than the rs of the other frame (P<0.05, corresponding to t-Hotelling >1.70). If no prediction is possible, the field is left blank. Numerical values are the same as in Table 3 for actual frames, but are reproduced here for clarity.
Excluding possible confounding factors: genome segmentation, GC content, age, amino acid composition, relative frame, and recombination
We examined several factors that could influence the ability to predict the de novo frame by its codon usage.
A first factor is genome segmentation: five overlaps of the dataset belong to viruses with segmented genomes (Aquabirnavirus, Begomovirus, Hordeivirus, Omegatetravirus, Orthobunyavirus). The calculations above were done by considering all genomic segments of such viruses as their “genome”. However, considering only the segment encoding the overlap under study yielded the same predictions, suggesting that genome segmentation is not a confounding factor.
Second, an extreme GC content could also, in principle, confound codon usage analysis. However, the GC contents of the genomes we analysed here are in a moderate range (35–57%), and thus are probably not a source of bias.
Third, in principle, the relative frame (+1 or +2) of the de novo region with respect to the ancestral region could influence the power of codon usage analysis to distinguish their genealogy. As can be seen in Supplementary Table S2, 16 de novo coding regions are in the +1 frame relative to the ancestral region they overlap, while the remaining 9 de novo regions are in the +2 frame. Among the 13 overlaps for which there was a significant difference in codon usage between the two overlapping regions, in 9 cases the de novo region was in the +1 frame relative to the ancestral region, while in 4 cases it was in the +2 frame (Supplementary Table S2). A chi-square test (chi-square = 0.023; P = 0.90) indicates that the sensitivity of our method does not change depending on the relative frame of the de novo region with respect to the ancestral region, and thus that the relative frame is not a confounding factor.
A fourth factor is the age of overlaps: as de novo proteins age, they may progressively impose increased constraints on the ancestral frames, which may change their codon usage, and make it difficult or impossible to distinguish them from de novo frames [4]. Precisely estimating the age of overlaps is not possible given the state of our knowledge of viruses. However, one can use the taxonomical distribution of de novo frames as a heuristic to obtain a very approximate idea of their relative ages. For instance, a de novo frame found in a single species of viral family A has almost certainly appeared more recently than a de novo frame found in a whole family B (provided there is a good sequencing coverage in both families). We have applied this idea to infer the age of overlaps of the benchmark dataset.
De novo frames found only in one species were considered “young” (provided there are several species in the genus considered); overlaps found in more than one species but less than one genus were considered of “Intermediate” age, and overlaps found in more than one genus were considered “old”. We have indicated these estimated relative “ages” in Supplementary Table S2 (the exact taxonomic distribution of de novo frames is in Supplementary Table S1).
There is insufficient taxonomic coverage to estimate the age of overlaps in two genera, for which only a single species is known (betatetravirus and mandarivirus). The remaining 23 overlaps cluster in the following way: 3 young, 13 medium, and 7 old (supplementary Table S2). By codon usage analysis we have (correctly) predicted the genealogy of 3 young, 6 medium and 2 old overlaps (supplementary Table S2). We have analysed these data by the chi-square contingency table test. The Chi-square value was 1.95 (P = 0.30). Therefore, the predictive power of codon usage to identify the de novo frames does not seem to be dependent on their taxonomic distribution, and by extension, on their relative ages.
A fifth potential confounding factor is that some de novo frames have a biased amino acid (aa) composition. This raises the possibility that the aa composition of de novo frames could be the major explanatory factor of our results, and that differences in codon usage would be a consequence of this biased aa composition. To empirically determine whether aa composition contains more information about frame ancestry than codon usage, we carried out a correlation analysis of the aa composition of overlapping frames with the same statistical test as for codon usage, and compared the predictive power of both methods. We performed the same analysis as on codon usage data but on the frequency of the 18aas that have a degree of codon-degeneracy >1. The median value of the Spearman correlation between the aa composition of the ancestral frame and that of non-overlapping regions was 0.62, while the median value of the Spearman correlation between the aa. composition of the de novo frame and that of non-overlapping regions was 0.50. Unlike for codon usage (see above), the difference was not significant (P = 0.35 in accordance to the Wilcoxon signed rank test). Therefore, aa composition does not have as much predictive power regarding the genealogy of overlaps as codon usage, and our results are unlikely to be explained by the difference in aa composition between ancestral and de novo frames.
Finally, to study whether recombination could be a confounding factor, we examined codon usage in the two cases in which the ancestral frame had arisen by recombination (see above), excluded from the above statistics. For machlomovirus, the difference between r sN and r sA was not significant (Table 3, bottom, t-Hotelling = 1.01, P<0.20). On the other hand, in the case of Providence virus (Alphacarmotetravirus), r sN (0.51) was significantly higher than r sA (0.00) (t-Hotelling = 2.94; P<0.005), and positive. Thus, ignoring the recombination event would lead to the erroneous prediction that the replicase is the de novo frame. It would be interesting to determine whether the codon usage of the Providence virus replicase gene is similar to that of its original genome. However, we could not find the species from which the recombination had occurred, since a similarity search based on the nucleotide sequence of the replicase found no similar viral (or cellular) sequence.
Application of our method to cases irresolvable by the phylogenetic approach
We applied the codon usage method defined above to seven pairs of overlapping genes (gathered from the literature), in which both frames have the same phylogenetic distribution. Table 4 presents the codon usage values for these overlaps by decreasing value of t-Hotelling, and the corresponding predictions of ancestry. The codon usage of overlapping frames was significantly different in only two cases (deltaretrovirus Tax/Rex and alphanodavirus replicase/B2). Deltaretrovirus Tax and the betanodavirus replicase, respectively, had a codon usage significantly closer to that of the viral genome than the other frames, suggesting that they are the ancestral frames and that the de novo frames are Rex and B2. We discuss these two overlaps in more depth below (case studies number 1 and 2).
Table 4. Prediction, by codon usage, of the ancestral frame in overlapping reading frames with identical phylogenetic distribution.
| Phylogenetic distribution | Genome accession number | Species | Frame 1 [function] | Frame 2 [function] | Length of overlap (nt) | Length of non-overlapping regions (nt) | rs1 | rs2 | t-Hotelling | P< | Predicted ancestral frame | Predicted de novo frame |
| Genus Alphanodavirus | NC_004142 | Boolarra virus | Replicase A, C-term domain | B2 [silencing suppressor] | 300 | 3930 | 0.62 | 0.16 | 2.74 | 0.005 | Replicase A | B2 |
| Genus Deltaretrovirus | HTU19949 | Human T-Cell lymphotropic virus 1 | Rex [post-transcriptional regulator] | Tax [transcription activator] | 510 | 6021 | 0.32 | 0.58 | 2.06 | 0.025 | Tax | Rex |
| Genus Betanodavirus | NC_003448 | Striped Jack nervous necrosis virus | Replicase A, C-term domain | B2 | 231 | 3744 | 0.04 | 0.47 | 1.59 | 0.10 | - | - |
| Genus Ilarvirus | NC_003842 | Tobacco streak virus | Replicase, C-term domain | 2b [silencing suppressor] | 276 | 7338 | 0.32 | 0.26 | 0.37 | 0.50 | ||
| Genus Polerovirus | NC_001747 | Potato leafroll virus | P0 [silencing suppressor] | P1, N-term domain | 612 | 3789 | 0.30 | 0.24 | 0.38 | 0.50 | - | - |
| Genus Polerovirus | NC_001747 | Potato leafroll virus | P1 | Replicase, N-term domain | 456 | 3789 | 0.35 | 0.33 | 0.10 | 0.50 | - | - |
| Genus Cucumovirus | NC_002035 | Cucumber mosaic virus | Replicase, C-term domain | 2b [silencing suppressor] | 243 | 6900 | 0.14 | 0.11 | 0.12 | 0.50 | - | - |
Conventions are the same as in Table 3. A frame is predicted ancestral if its rs is positive and significantly higher than the rs of the other frame (P<0.05, corresponding to t-Hotelling>1.70).
In the five other overlaps analyzed in Table 4, both frames had a comparable codon usage, preventing prediction of the de novo frame.
Case study 1: De novo origin of three deltaretrovirus accessory genes
We examined in more detail the deltaretrovirus genome, which contains a complex pattern of overlapping genes at its 3′ end, in the pX region [36]–[39]. In addition to Tax and Rex, the pX region of Human T-lymphotropic virus 1 (HTLV1) encodes two other proteins in the sense strand, p12 and p30, and a fifth protein, HBZ, from the antisense strand [36], [37], [40] (Figure 3). The resulting arrangement has two long (>80 aa) triple overlaps: the N-terminus of p30 overlaps both p12 and the N-terminus of HBZ, while the C-terminus of p30 overlaps the N-termini of both Tax and Rex (Figure 3). The phylogenetic distribution of the overlapping genes in deltaretroviruses is summarized in Figure 4. P30 is expressed only in HTLV1 [36]. p12 has only been reported in HTLV1 [36], and its coding sequence is interrupted by a stop codon in HTLV2, while it has no equivalent in bovine leukemia virus. HBZ is present in HTLV1 but also in HTLV2, 3 and 4, where it is called respectively APH2, APH3 and APH4 (these proteins have statistically significant similarity with HBZ, indicative of homology). In the bovine leukemia virus genome, no equivalent of HBZ is expressed from the antisense strand in the region between the Env and Tax genes (Luc Willems, personal communication); instead microRNAs are expressed from the sense strand [41], [42]. Taking into account this phylogenetic distribution, and our codon usage predictions, the most likely evolutionary scenario (Figure 4) is that HBZ has originated in the common ancestor of HTLV1 to 4, after its divergence from bovine leukemia virus; p12 has originated de novo in HTLV1 by overprinting HBZ; and p30 has originated de novo in HTLV1 by overprinting both HBZ (in the N-terminus of p30) and Tax and Rex (in the C-terminus of p30). It is not possible to conclude whether p30 or p12 originated first, nor how Tax or HBZ originated (de novo or by horizontal gene transfer).
Figure 3. A “gene nursery”: the pX region of deltaretroviruses.

The pX region of HTLV1 encodes five genes unique to deltaretroviruses by a complex pattern of alternative splicing and leaky scanning [36], [39]. The initial exons of these genes are very short and have not been represented, nor have been shorter versions of p12 and p30 expressed alternatively. Only the 3′ end of the Env gene is represented. The figure is approximately to scale. Ancestral regions in red and de novo regions in blue. Frame numbering is as in [45], with the Tax frame taken as “0”. Protein regions with unusually low sequence complexity are indicated by dashed, grey lines.
Figure 4. Presumed evolution of the deltaretrovirus pX region.

The deltaretrovirus phylogeny is shown as a cladogram. Conventions are the same as in Figure 3.
We made two additional observations regarding codon usage. First, the fact that Tax and Rex are involved in a triple overlap with a short region of p30 (Figure 3) constitutes a potential confusing factor in our prediction of ancestry by codon usage above. Nevertheless, the region of p30 overlapping Tax and Rex has a codon usage significantly more distant to that of the genome than that of Tax (t-Hotelling = 2.16; P<0.025). Therefore, the codon usage of Tax is close to that of the genome over the entire length of its overlapping region, which further suggests that Tax is the ancestral gene. Second, genes expressed from an antisense strand are not expected to have a similar codon usage to genes from the sense strand. Accordingly, the codon usage of HBZ is not correlated to that of the rest of the genome (rs = 0.00 for the entire HBZ gene, rs = 0.06 for the region of HBZ overlapping p30).
The existence of triple overlaps poses severe constraints on the sequence of the proteins encoded by the pX region, and we thus examined whether they had an unusual sequence composition, or were predicted to be structurally disordered [3] (see Material and Methods). We found that all proteins encoded by the pX region, with the exception of Tax, contained long regions with low sequence complexity (as defined by SEG [43]) over most of their length (dashed lines in Figure 3; see Supplementary Table S3), indicating that they were unlikely to adopt a typical globular structure [43], [44]. Tax has no specific region of low sequence complexity, but both its N-terminus, overlapping Rex and p30, and its non-overlapping C-terminus have a highly biased composition, being enriched in hydrophobic residues (P<0.005) and depleted in polar and charged residues (P<0.005).
In addition, HBZ and Rex were predicted to be mostly disordered, at least in the absence of binding partners, while p30 contained several long regions predicted disordered (see Supplementary Table S3). Only p12 and Tax were predicted to be mostly ordered. These results suggest that sequence constraints imposed by triple overlaps may lead to proteins with a highly biased sequence composition and/or structurally disordered [3], and further highlight the fact that Tax is different from the other proteins encoded by the pX region.
Finally, it may seem extraordinary that triple overlaps exist at all, given the sequence constraints they impose; in that light, we note that the relative frame arrangement that would impose the highest constraint, called “−2” [45], is not used for the overlap involving HBZ. (In this arrangement, codon positions 1 and 2 of a frame overlap respectively codon positions 2 and 1 of the antisense frame, with the result that the sequences of each frame are almost fixed by each other). As can be seen in Figure 3, the frame that is in the −2 arrangement relative to HBZ is the non-coding frame 0, rather than the p12 or p30 frames.
Case study 2: The B2 proteins of Nodaviridae, a complex evolutionary history
In the second case, the codon usage of alphanodavirus B2 (a suppressor of RNA silencing [46]) suggests that it has originated de novo by overprinting the disordered C-terminal extension of the polymerase domain (Table 4). However, several observations cast a doubt on the reliability of this prediction. A similar genomic arrangement occurs in a closely related genus, betanodavirus (though there is no detectable sequence similarity between either the C-terminal extensions of the replicases or the B2 proteins of both genera) (Figure 5). A parsimonious scenario would demand that the overlaps of both genera have the same origin and thus presumably a similar codon usage pattern. Yet this is not the case: in betanodavirus it is B2 that has a codon usage closer to that of the genome (though not significantly so). This discrepancy might be due to horizontal transfer (see below).
Figure 5. A genomic hotspot of origination of silencing suppressors in plus-strand RNA viruses.

The replicases of Nodaviridae and Bromoviridae contain C-terminal extensions predicted disordered (thin boxes) downstream of their homologous polymerase (RdRP) domain. These extensions encode structurally unrelated suppressors of RNA silencing, B2 and 2b (PDB accession codes respectively 2AZ2 and 2ZI0) in different reading frames. Neither the C-terminal extensions nor the suppressors of RNA silencing have detectable sequence similarity, even between closely related genera. Which region is ancestral in each overlap could not be determined (see text).
Intriguingly, a very similar arrangement occurs in two genera (ilarviruses and cucumoviruses) of another family of positive-strand RNA viruses, Bromoviridae, in which a silencing suppressor called 2b overlaps a C-terminal extension of the polymerase (Figure 5). Like in Nodaviridae, neither the overlapping regions of the replicases nor the 2b proteins of the two genera have any similarity. The codon usage of the 2b proteins of ilarviruses and cucumoviruses is indistinguishable from that of the region of the replicase they overlap (Table 4), making a prediction of ancestry impossible. In fact, the 2b proteins of ilarviruses might have a different origin from those of cucumoviruses, since these genera do not form a monophyletic clade [47]. Despite their similar genomic location, size and function, alphanodavirus B2 and cucumovirus 2b have different structural folds and different modes of binding to RNA, both previously unknown [46], [48]–[50], clearly indicating an independent origin. We think that together, these observations indicate that the overlaps have a complex evolutionary origin; the ancestral protein could differ in each genus (for instance it might be the C-terminal extension of the replicase in alphanodaviruses and the B2 protein in betanodaviruses), and in some genera the ancestral proteins might have entered their genome by horizontal transfer (see below).
Discussion
Our method provides a quantitative improvement on previous empirical approaches
We have shown that de novo frames originated by overprinting have a pattern of codon usage distinguishable from ancestral frames, which can be used to predict the de novo frame with a good specificity but intermediate sensitivity (working in around half the cases).
How do our results compare with previous empirical studies of codon usage? The codon usage of six of the overlaps presented here has been studied previously using a different method, the “codon similarity index” [4]. The qualitative trends reported were similar to the ones we observe. Four of the overlaps presented here were also analysed previously, by Pavesi et al [24] who studied their information content and their codon usage. Again, the numerical values they reported for codon usage are in very good agreement with those obtained here, as are their general conclusions. However, our improved statistical analysis allowed us to draw more powerful conclusions. For instance, Pavesi et al reported that both the tymovirus replicase and movement genes had a codon usage correlated with that of their genome, and concluded that it was consequently not possible to determine the ancestral gene [24]. In the present article, the use of Hotelling's t-test to compare two dependent correlation coefficients [51] allowed us to determine that the replicase gene had a codon usage significantly closer to its genome than the movement gene (Table 3), indicating (correctly) that it was the ancestral frame. Another study, on the VP2/VP5 overlap of avibirnavirus (homologous to the aquabirnavirus overlap studied herein), showed that VP5 had an unusual nucleotide usage and predicted that it was the de novo frame [27]. Our quantitative analysis is in agreement with these findings: VP2 has a codon usage significantly closer to the viral genome than VP5 (Table 3). Finally, a previous analysis of the cucumovirus replicase/2b overlap predicted that 2b was the de novo frame, based on its uridine content at the third codon position [22]. In contrast, our analysis detects no statistically significant difference between the codon usage of the overlapping region of the replicase and that of 2b (Table 4).
Limitations of our study
Why are ancestral and de novo frames distinguishable by their codon usage in only half of the overlaps? We examined in the Results several confounding factors, such as the relative frame of the overlapping regions, their sequence composition, and the estimated age of overlaps. They did not appear to have a significant impact on the predictive power of codon usage analysis. One note of caution is that we used a very crude estimate of the relative ages of overlaps (i.e. their taxonomic distribution). We could not use a more precise estimate, unlike a previous study [4], because our dataset contains both RNA and DNA viruses, which have no protein in common that could be used as a molecular clock, and because the proteins we studied often have more than 50% sequence divergence, preventing the determination of reliable phylogenies.
Potential avenues for future research
During the revision of this manuscript, following the suggestion of a reviewer, we tested a distance measure based on information theory approaches: the modified Kullback-Leibler (KL) distance [52]. Since dinucleotide frequency is an important genome signature [53], we have re-analysed our dataset by calculating the KL distance (based on the frequency of the 16 dinucleotides at codon positions 1-2, 2-3, and 3-1) between the ancestral frame and the non-overlapping coding regions of the genome (KLA), and between the de novo frame and the non-overlapping coding regions of the genome (KLN). The ancestral frame had a KL distance to non-overlapping regions lower than that of the de novo frame in 23 out of 25 overlaps. In contrast, in our approach, the rs of the de novo frame was lower than the rs of the ancestral frame in 21 out of 25 overlaps.
We could not evaluate by analytical methods whether the KL distance between the ancestral frame and the non-overlapping regions (KLA) was significantly smaller than that of the KL distance between the novel frame and the non-overlapping regions (KLN), because KL distances are gamma-distributed [52] and there is no generic analytical solution for the distribution of the difference in gamma distributed variables. Therefore, instead, we performed, on each pair of overlapping genes from our dataset, a permutation test to estimate whether the observed (KLN−KLA) was significantly higher than the null distribution of (KLN−KLA) derived from 10,000 random permutations of the nucleotide sequence of the ancestral and the novel frame. We found that, on our dataset, this permutation test on the KL distance has the same specificity as the t-Hotelling test (1.00) and a slightly better sensitivity (0.60) than the t-Hotelling test (0.52), although the performance of the two methods is not significantly different (McNemar chi-square = 0.16; P = 0.70). We hope that our publicly available dataset of overlapping genes with known genealogy (expected to grow) will encourage others to continue exploring these methods and others.
Deltaretrovirus Rex has probably originated de novo by overprinting Tax
Our new method allowed us to make predictions of ancestry for two overlaps in which both frames have the same phylogenetic distribution (Table 4). In the alphanodavirus replicase/B2 overlap (case study 2), several elements suggest that horizontal transfer might have taken place and thus that the codon usage prediction is not reliable. In the deltaretrovirus Tax/Rex overlap (case study 1), our prediction that Rex has originated de novo by overprinting Tax is consistent with the function of Tax, which occurs upstream of that of Rex in the viral cycle [37], [39], [54]. It is also coherent with the fact that Tax has a higher sequence complexity than Rex or p30, and is under stronger selection pressure than Rex [55], [56], which is generally the case of ancestral frames [3], [4]. Our prediction is in agreement with that of a previous work, reached by analyzing the substitution rates of Tax and Rex, their nucleotide composition and their amino acid composition [55]. Tax and Rex are encoded by the same mRNA but have different start codons [57] and thus Rex presumably originated by the appearance of a new ATG upstream of Tax. Both Tax and Rex are present in all deltaretroviruses and only in those viruses, which suggests that Tax originated first in the common ancestor of deltaretroviruses, and that Rex originated by overprinting it “rapidly” afterwards (by biological timescales), before the divergence of deltaretroviruses. Rex must have then undergone a rapid functionalization, since it is indispensable for the viral cycle, like Tax [37], [39], [54].
An alternative scenario is possible but appears much less parsimonious: Rex might have originated in another organism with a different codon usage, and entered the genome of the ancestor of deltaretroviruses by horizontal transfer. It would then have diverged in sequence beyond recognition, and have been overprinted by Tax (which would have a codon usage similar to that of the genome by coincidence).
Hotspots of de novo origination or of horizontal transfer (“gene nurseries”) in viral genomes?
The pX region encodes five genes unique to deltaretroviruses [36]–[39], at least three of which (p12, p30 and Rex) have originated de novo, while the two others (Tax and HBZ) have either also originated de novo too (although earlier), or by horizontal transfer (Figure 3). The pX region thus constitutes a hotspot of gene origination, or gene “nursery” [58]. Strikingly, the two genes that have overprinted Tax, Rex and p30, play roles that are respectively complementary and antagonistic to Tax [38], [39], [59], while HBZ plays a role antagonistic to that of Tax [60], [61]. This suggests that the function of Tax was gradually controlled and refined by the appearance of new genes encoded in the same genomic location. Interestingly, other gene nurseries are found in a similar genomic position in other Retroviridae, such as lentiviruses or spumaviruses [62]. As seen above, the 3′ end of the replicase gene of some positive-strand viruses may also favour the origination of gene encoding silencing suppressors (Figure 5).
Such hotspots of origination of genes coding for proteins involved in the same pathways, and combining horizontal transfer and de novo origin, may be common in viruses. For instance, the movement proteins of Alphaflexiviridae and Betaflexiviridae are encoded in the same genomic position (downstream of the replicase gene) but belong either to the Triple Gene Block type [63], [64] or to the 30K type [65], indicating that at least one or possibly both types of movement proteins have entered these families by horizontal transfer [66].
The mechanisms that presumably favour the appearance of gene nurseries are unknown, but obviously of great interest. In the case of the deltaretrovirus pX region, we note that the high constraints imposed by the triple overlaps severely restrict the evolution of p12, p30 and Rex, and that consequently their present-day sequence composition is probably rather similar to the one they had when they first originated. We speculate that the pattern of origination seen in the pX region, in which de novo genes regulate the function of an ancestral protein, may be facilitated by the fact that low sequence complexity (and/or structural disorder) is compatible with a range of regulatory functions [67]–[69]. Thus, at birth, despite having a very “simple” sequence not honed by natural selection, these proteins may have had, by chance, a regulatory function and provided the virus with a fitness advantage that lead to their fixation.
The need to annotate the genomes of Retroviridae, and to look for triple overlaps
Retroviridae encode numerous short, accessory genes, often accessed by alternative splicing or complex mechanisms leading to partially overlapping genes, and no doubt many remain to be discovered [62]. Yet at the time this article was submitted, almost none of these genes were annotated in the NCBI reference genomes [70]. This poor annotation is prejudicial to the study of these viruses. It is important that researchers who discover, or have discovered new genes, contact the NCBI viral genomes team to ensure that they are annotated properly.
Another, more general implication for genome annotation is that long, triple overlaps may have the potential to yield functional proteins relatively easily. Therefore, triple overlaps might be more abundant than previously thought (we know only two triple overlaps outside of deltaretroviruses, involving the P, V, and D or W proteins in Paramyxovirinae [71]–[74]). We thus recommend re-investigating known overlapping gene pairs to find whether in some cases a third overlapping frame might be expressed.
A virus-specific evolutionary constraint that operates on top of GC mutational bias and influences codon usage
It has been proposed that the GC content of a genome was the main, though not the only, determinant of codon usage [19]–[21]. Our results confirm that it cannot be the unique determinant, otherwise the de novo and ancestral frames (which have the same GC content) would necessarily have a similar codon usage. Therefore, a significant evolutionary constraint(s) on codon usage must operate in addition to the GC content, and this constraint must be greater on ancestral frames than on de novo frames. Belalov et al. recently reported that the frequency of the dinucleotide CpG was an important factor of viral codon usage, and that CpG was significantly rarer at codon positions 2-3 than at positions 3-1 [75]. CpG is known to be underrepresented in RNA viruses [76], perhaps to avoid recognition from an antiviral CpG sensor [77]. However, the difference in frequency of CpG at different codon positions suggests that a second type of pressure exists, imposed by the translational apparatus. The authors thus suggested the existence of an evolutionary constraint on the genome deriving from a hypothetical cellular CpG sensor coupled (by an unknown mechanism) to the translational machinery. The existence of such a constraint would be coherent with our results, and we speculate that it might cause the difference in codon usage between ancestral and de novo frames.
Conclusion
Very little is known about de novo protein origination, although it is by now clear that this mechanism plays an important role in viral pathogenicity. Our method should allow the identification of more de novo proteins, and thus enhance our understanding of host-pathogen co-evolution. It will be of particular interest to apply it to gene “nurseries” such as the ones we have identified here, and to elucidate the pressures that shape them. Finally, we note that recent experimental and computational reports suggest that de novo origination of genes by overprinting may not be confined to viruses but on the contrary, be a much wider phenomenon than previously thought, both in eukaryotic [78]–[82] and bacterial genomes [83].
Materials and Methods
Sequence analyses
We retrieved all sequences from the NCBI viral genome database [84]. We used MAFFT [85] for multiple sequence alignment, HHpred [86] and HHblits [87] for remote homology detection, Phylogeny.fr [88] for phylogenetic analyses, and METAPRDOS [89] for prediction of protein structural disorder, respecting the guidelines of [44]. We used Composition Profiler [90] for analyses of protein global compositional bias with respect to Swiss-Prot (release 51), and SEG for analyses of protein local compositional bias [43]. SEG analyses were obtained from the web server ANNIE [91] with parameters 45/3.75/3.4 in order to identify long regions with a composition bias indicative of non-globular proteins [44].
Assembly of a benchmark dataset of overlapping genes with known genealogy
We searched the NCBI genome database [84] for viruses that infected eukaryotes, with a genome shorter than 30,000 nucleotides, and which contained overlapping genes longer than 120 nucleotides. The cut-off of 30,000 nucleotides was chosen because curation of larger genomes is impractical [3]. We included an overlapping gene into the benchmark dataset only when two criteria were fulfilled: 1) the expression of both overlapping reading frames was experimentally verified; 2) the genealogy of the overlapping reading frames could be determined with good support by using the very stringent criterion described previously [3], regarding the taxonomic distribution of both overlapping frames. According to this criterion, one reading frame can be considered ancestral only if it has homologs in at least two viral families whereas the other, overlapping frame had in at most one viral family. Since viral proteins diverge very fast, identifying viral proteins conserved in at least two families requires powerful similarity search techniques, which are described below. The final dataset, presented in Table 1, contains 27 independent (non-homologous) overlapping genes whose genealogy is reliably established. The dataset contains no antiparallel overlapping genes because we could not find any whose existence had been convincingly proven experimentally in the genomes of short or medium size considered (<30 kb).
Definition of the boundaries of overlapping regions
We used the following conventions to define the precise boundaries of the overlapping regions on which we performed calculations of codon usage. There are two types of overlaps: in internal overlaps, one overlapping gene is contained entirely within the other gene whereas terminal overlaps involve only the 3′ end of one gene and the 5′ end of another [92]. In the case of internal overlaps, for the longest frame, the first codon counted as overlapping was the most upstream codon that overlaps the start codon of the internal frame, and the last codon counted as overlapping was the most downstream codon that overlaps the stop codon of the internal frame. In the case of terminal overlaps, for the upstream frame, the first codon counted as overlapping was the most upstream codon that overlaps the other frame, and for the downstream frame the last codon counted as overlapping was the most downstream codon that overlaps the stop codon of the other frame.
Remote homology detection
In order to obtain a highly reliable genealogy of the overlaps, we needed to identify as distant homologs as possible for each protein of the dataset. However, not all homologs of a protein can be detected by conventional sequence similarity searches even if they have retained some sequence identity with the query, for a number of reasons [93], including the fact that databases of protein domains are underrepresented for viruses (our observations). We thus exploited “contextual” information available for viral proteins, such as taxonomy and genome organisation, to identify distant homologs overlooked by conventional searches [94]. We proceeded in the following way (the procedure is the same as in our previous article [3] but had not been described in detail). We first identified “straightforward” homologs of the query protein in the NCBI nr database (release 1st April 2012), by using HHpred [95] and HHblits [87] and selecting hits whose E-value was below the standard cut-off of 10−3. We then examined subsignificant hits (i.e. hits with an E-value superior to 10−3) up to E-values of 1000, looking for viral proteins that came from a taxonomically related virus, and/or occurred in the same position of the genome. Such subsignificant hits, which have weak similarity with the query protein and occur in a similar genomic context, constitute potential homologs. In order to test whether they were actually homologous with the query, we gathered homologs of these subsignificant hits (with E≤10−3), and used HHalign [96] to compare homologs of the query protein (obtained above) with homologs of the subsignificant hits. We considered that an HHalign E-value inferior to 10−3 indicated homology between the subsignificant hit and the query, but performed additional checks, such as verifying that the secondary structure and function (when available) of the hits were compatible with that of the query.
Whenever the structure of a protein from the dataset was available, we also performed structural similarity searches to identify structural homologs, using DALI [97] and FATCAT [98].
Because overlapping genes are not systematically recognised [16], [99] there is a theoretical possibility that some homologs of an overlapping frame might exist in related genomes but not be annotated, and therefore missed by similarity searches. For each overlap, we thus systematically checked that the genomes of other taxonomically related viruses did not contain conserved, unannotated open reading frames, as in [4].
We present in Supplementary Table S1 the taxonomic distribution of the homologs detected by our searches, together with the corresponding PFAM family (or clan) identified in the process.
Detection of horizontal transfer events
Genes that have entered their genome by horizontal transfer can be identified by the fact that their phylogeny is discordant with the rest of the genome. A robust measure of this discordance is taxonomic incongruence, e.g. the existence of close homologs in a distant taxon. To detect taxonomic incongruence, we collected homologs of the protein products of each ancestral reading frame using blastp [100] on the Refseq database [101] with a cutoff E-value of 10−3. Hits to proteins from a different viral family than that of the query indicated taxonomic incongruence. To infer the direction of horizontal transfer, we analysed the phylogenetic distribution of homologs of the ancestral protein, both from the same family and from the distant taxon detected, and applied a parsimony criterion: the clade that has the wider phylogenetic distribution of the gene was most likely to be the clade of origin.
Calculation of codon usage
In the genetic code, 18 amino acids (aas) are degenerate, e.g. encoded by more than one codon, and they are encoded by 59 “synonymous” codons in total. For each viral genome sequence, we measured the number of occurrences of the 59 synonyms in the non-overlapping coding regions and in each of the two overlapping reading frames (Figure 2). For clarity we will refer to the ensemble of the numbers of occurrences of the 59 synonymous codons of a given reading frame as its “codon usage”. The codon usage of non-overlapping regions will be called the “codon usage of the genome”.
In some overlapping reading frames (generally short, i.e. less than 400 nucleotides), the number of occurrences of the synonymous codons for a given aa was smaller than the degree of degeneracy of this aa (for instance only 3 synonyms for arginine, a 6-fold degenerate aa). In these cases, we restricted the analysis to synonymous codons whose number of occurrences was at least equal to the degree of degeneracy of the encoded aa. We indicated in Table 2 the number of synonymous codons on which the analysis was carried out.
Preliminary tests on canonical methods of codon usage analysis
We wanted to utilize codon usage as a method to predict the genealogy of overlapping genes, and not simply to characterise the behaviour of overlapping genes. Therefore, we needed a method to assess whether the codon usage of ancestral frames was closer to the rest of the genome that the codon usage of de novo frames, and to assess whether this difference was statistically significant.
We have examined various canonical methods to evaluate codon usage bias: the Effective number of codons (ENC), [102] Codon Adaptation Index (CAI) [103], and Dmean index [104]. We found that ENC and Dmean had poor predictive power on the genealogy of overlaps (not shown). Initial tests suggested that CAI may have been more sensitive, but we could not easily test the statistical significance of the difference between the observed CAI distances. Therefore, we developed a new method, that had a good predictive power and that could yield estimates of statistical significance.
A measure of the similarity in codon usage between two reading frames
Our hypothesis was that, in overlapping reading frames, the ancestral frame could be identified by having a codon usage that was more similar to the codon usage of the genome than that of the other frame. Thus we designed a measure of the similarity of codon usage of each frame with that of the genome, and a method to assess whether one frame had a codon usage significantly closer to that of the genome than the other frame.
In order to quantify the similarity between the codon usages of two given reading frames, we used as a measure the Spearman's rank correlation coefficient (r s) [33] between the number of occurrences of the 59 synonymous codons of these two frames (i.e. between their “codon usages”, see above). Each viral genome was divided into three sets: a) the overlapping region of the reading frame 1; b) the overlapping region of the reading frame 2, and c) non-overlapping regions of the genome, composed of the sequences of non-overlapping genes, and, in cases where some genes of the genome partially overlapped, of their non-overlapping regions (Figure 2). For viruses with segmented genomes, all segments were included in the calculations. For simplicity, the codon usage of the third set, i.e. non-overlapping regions, will be referred to as the “codon usage of the genome”. In all viral genomes, we calculated the r s between the codon usage of the genome and that of each of the two overlapping frames under consideration (r s1 and r s2). The reason we collected the non-overlapping coding regions of a virus genome into an integrated set (as opposed to studying individual non-overlapping genes and analyzing their variance) is because the individual non-overlapping genes (or their non-overlapping regions, in cases of genes that partially overlap) are often short, which would have restricted correlation analysis to 2 or 3 dozens of synonyms.
Assessing whether one of the two overlapping frames has a codon usage significantly closer to that of the genome
Determining if a given frame “1” has a codon usage closer to that of the genome than the other frame “2” is equivalent to determining whether rs1 is significantly greater than rs2, i.e. whether the correlation between the codon usage of the first frame and that of the genome is significantly greater than the correlation between the codon usage of the second frame and that of the genome. This comparison involves two correlations coefficients that refer to a common variable (the codon usage of the genome), a situation categorized as “dependent correlation” [51] or as the study of “correlated correlation coefficients”, which can be addressed by the Hotelling t-test [34], [35]. The conventional Hotelling formula involves comparing Pearson correlation coefficients r
p, but can be used with Spearman's correlation coefficients r
s by converting them into their Pearson equivalents: 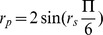 [105].
[105].
The Hotelling t-value was calculated as follows:
 |
where n is the number of the compared codon frequencies, rp1 and rp2 are respectively the Pearson equivalents of rs1 and r s2, and rp12 is the Pearson equivalent of rs12 (codon usage correlation between the overlapping frames). We assess the Hotelling t-value according to the one-tailed Student's t-test.
Evaluation of the robustness of Hotelling's t-test to non-independence, by simulation
The Hotelling's t-test is designed for correlation coefficients estimated from independent data. However, the data we examine in this study (the number of occurrences of synonymous codons) are clearly not independent, since the sum of the numbers of synonymous codons encoding a given aa is fixed. Consider, for example, a reading frame containing 28 Glutamine codons (an aa encoded by two synonyms, CAA and CAG). If the number of occurrences of CAA is 11, that of CAG will inevitably be 17 (i.e. 28−11), i.e. the number of occurrences of CAA and CAG are not independent. Therefore, we assessed whether Hotelling's t-test was robust to non-independence of data by comparing it with a simulation-based exact test. For each pair of overlapping frames of the dataset, we generated two simulated overlapping frames with an aa composition identical to that of the two original frames, and used the actual non-overlapping regions of the genome as a reference set.
One round of simulation was performed as follows: we randomly generated a number (n) of occurrences for each of the 59 codons encoding the 18 degenerate aas, keeping the sum of the occurrences of codons encoding each aa equal to that of the original frame (e.g. if there were 28 Glutamine codons in the original frame, the simulated frame could have any number of CAA and CAG totalling 28). We calculated the correlation coefficients rs 1 and rs 2 between the number of occurrences of all synonyms in both simulated frames and that of the actual genome. We repeated the same process 10,000 times, thus simulating the distribution of d 21 expected assuming that the reading frames are randomly generated and that codon usage is not related to ancestry (i.e. the null distribution). We then tested whether the observed d21 (Table 2) was significantly larger than this null distribution.
Finally, we used the McNemar's non-parametric test [33] to determine whether the Hotelling's t-test and the simulation provide equivalent results (which would indicate that the Hotelling's t-test is robust to non-independence of data).
Supporting Information
Taxonomic distribution of the ancestral and de novo frames of the benchmark dataset.
(DOC)
Examination of possible confounding factors of codon usage analysis: relative frame of de novo genes compared to ancestral genes, and relative age of de novo frames. (1) We used taxonomic distribution as a very approximate, empirical proxy to estimate comparative ages of the overlaps. De novo frames found only in one species are considered “young” (provided there are several species in the genus considered, see note 2 below); overlaps found in more than one species but less than one genus are considered of “Intermediate” age, and overlaps found in more than one genus are considered “old”. The taxonomic distribution of de novo frames is taken from Supplementary Table S1. (2) We excluded cases where there was insufficient taxonomic sampling, such as the betatetravirus overlap, since the betatetravirus genus comprises only ones species.
(DOCX)
Low complexity and predicted structural disorder in proteins encoded by overlapping genes in human T-lymphotropic virus 1 (HTLV1). (1) Predictions of low sequence complexity were made with SEG with parameters 45/3.75/3.4. (2) Predictions of structural disorder were made with MetaPrDOS (see Material and Methods).
(DOC)
Acknowledgments
We thank A. Firth for suggesting to perform a recombination analysis, J. Turpin for providing the precise boundaries of HTLV-1 genes, C. Journo and JM Mesnard for advice on deltaretroviruses, and R. Belshaw and A. Vianelli for critical reading of the manuscript.
Funding Statement
This work was supported by the Wellcome Trust grant number [090005] to DK, and by a Marie Curie Research Fellowship and an MRC Clinician Scientist Fellowship to GM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Long M, Betran E, Thornton K, Wang W (2003) The origin of new genes: glimpses from the young and old. Nat Rev Genet 4: 865–875. [DOI] [PubMed] [Google Scholar]
- 2. Taylor JS, Raes J (2004) Duplication and divergence: the evolution of new genes and old ideas. Annu Rev Genet 38: 615–643. [DOI] [PubMed] [Google Scholar]
- 3. Rancurel C, Khosravi M, Dunker AK, Romero PR, Karlin D (2009) Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J Virol 83: 10719–10736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sabath N, Wagner A, Karlin D (2012) Evolution of viral proteins originated de novo by overprinting. Mol Biol Evol 29: 3768–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li F, Ding SW (2006) Virus counterdefense: diverse strategies for evading the RNA-silencing immunity. Annu Rev Microbiol 60: 503–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Knippenberg I, Carlton-Smith C, Elliott RM (2010) The N-terminus of Bunyamwera orthobunyavirus NSs protein is essential for interferon antagonism. J Gen Virol 91: 2002–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vargason JM, Szittya G, Burgyan J, Hall TM (2003) Size selective recognition of siRNA by an RNA silencing suppressor. Cell 115: 799–811. [DOI] [PubMed] [Google Scholar]
- 8. Dawkins R, Krebs JR (1979) Arms Races between and within Species. P Roy Soc B-Biol Sci 205: 489–511. [DOI] [PubMed] [Google Scholar]
- 9. Meyerson NR, Sawyer SL (2011) Two-stepping through time: mammals and viruses. Trends in Microbiology 19: 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keese PK, Gibbs A (1992) Origins of genes: “big bang” or continuous creation? Proc Natl Acad Sci U S A 89: 9489–9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaessmann H (2010) Origins, evolution, and phenotypic impact of new genes. Genome Res 20: 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abroi A, Gough J (2011) Are viruses a source of new protein folds for organisms? - Virosphere structure space and evolution. Bioessays 33: 626–635. [DOI] [PubMed] [Google Scholar]
- 13. Bornberg-Bauer E, Huylmans AK, Sikosek T (2010) How do new proteins arise? Curr Opin Struc Biol 20: 390–396. [DOI] [PubMed] [Google Scholar]
- 14. Keller J, Leulliot N, Soler N, Collinet B, Vincentelli R, et al. (2009) A protein encoded by a new family of mobile elements from Euryarchaea exhibits three domains with novel folds. Protein Science 18: 850–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sabath N, Price N, Graur D (2009) A potentially novel overlapping gene in the genomes of Israeli acute paralysis virus and its relatives. Virol J 6: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Firth AE, Brown CM (2006) Detecting overlapping coding sequences in virus genomes. BMC Bioinformatics 7: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chirico N, Vianelli A, Belshaw R (2010) Why genes overlap in viruses. Proc Biol Sci 277: 3809–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams MJ, Antoniw JF (2004) Codon usage bias amongst plant viruses. Arch Virol 149: 113–135. [DOI] [PubMed] [Google Scholar]
- 20. Shackelton LA, Parrish CR, Holmes EC (2006) Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J Mol Evol 62: 551–563. [DOI] [PubMed] [Google Scholar]
- 21. Jenkins GM, Holmes EC (2003) The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res 92: 1–7. [DOI] [PubMed] [Google Scholar]
- 22. Ding SW, Li WX, Symons RH (1995) A Novel Naturally-Occurring Hybrid Gene Encoded by a Plant Rna Virus Facilitates Long-Distance Virus Movement. Embo J 14: 5762–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pavesi A (2006) Origin and evolution of overlapping genes in the family Microviridae. J Gen Virol 87: 1013–1017. [DOI] [PubMed] [Google Scholar]
- 24. Pavesi A, De Iaco B, Granero MI, Porati A (1997) On the informational content of overlapping genes in prokaryotic and eukaryotic viruses. J Mol Evol 44: 625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lartey RT, Voss TC, Melcher U (1996) Tobamovirus evolution: gene overlaps, recombination, and taxonomic implications. Mol Biol Evol 13: 1327–1338. [DOI] [PubMed] [Google Scholar]
- 26. McVeigh A, Fasano A, Scott DA, Jelacic S, Moseley SL, et al. (2000) IS1414, an Escherichia coli insertion sequence with a heat-stable enterotoxin gene embedded in a transposase-like gene. Infect Immun 68: 5710–5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tan DY, Hair Bejo M, Aini I, Omar AR, Goh YM (2004) Base usage and dinucleotide frequency of infectious bursal disease virus. Virus Genes 28: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McGeoch DJ, Dolan A, Donald S, Rixon FJ (1985) Sequence determination and genetic content of the short unique region in the genome of herpes simplex virus type 1. J Mol Biol 181: 1–13. [DOI] [PubMed] [Google Scholar]
- 29. Lee S, Weon S, Kang C (2010) Relative codon adaptation index, a sensitive measure of codon usage bias. Evol Bioinform Online 6: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stuart GW, Moffett PK, Bozarth RF (2006) A comprehensive open reading frame phylogenetic analysis of isometric positive strand ssRNA plant viruses. Arch Virol 151: 1159–1177. [DOI] [PubMed] [Google Scholar]
- 31. Walter CT, Pringle FM, Nakayinga R, de Felipe P, Ryan MD, et al. (2010) Genome organization and translation products of Providence virus: insight into a unique tetravirus. J Gen Virol 91: 2826–2835. [DOI] [PubMed] [Google Scholar]
- 32. Dolja VV, Koonin EV (1991) Phylogeny of Capsid Proteins of Small Icosahedral Rna Plant-Viruses. J Gen Virol 72: 1481–1486. [DOI] [PubMed] [Google Scholar]
- 33.Sheskin DJ (2007) Handbook of parametric and nonparametric statistical procedures. Chapman & Hall/CRC. 1193p.
- 34. Hotelling H (1940) The selection of variates for use in prediction with some comments on the problem of nuisance parameters. Ann Math Stat 11: 271–283. [Google Scholar]
- 35.Dawson B, Trapp RG (2001) Basic and Clinical Biostatistics. 3rd edition. New York: McGraw-Hill. pp. 188–189.
- 36. Gessain A, Rua R, Betsem E, Turpin J, Mahieux R (2013) HTLV-3/4 and simian foamy retroviruses in humans: Discovery, epidemiology, cross-species transmission and molecular virology. Virology 435: 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nicot C, Harrod RL, Ciminale V, Franchini G (2005) Human T-cell leukemia/lymphoma virus type 1 nonstructural genes and their functions. Oncogene 24: 6026–6034. [DOI] [PubMed] [Google Scholar]
- 38. Edwards D, Fenizia C, Gold H, de Castro-Amarante MF, Buchmann C, et al. (2011) Orf-I and orf-II-encoded proteins in HTLV-1 infection and persistence. Viruses 3: 861–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baydoun HH, Bellon M, Nicot C (2008) HTLV-1 Yin and Yang: Rex and p30 master regulators of viral mRNA trafficking. AIDS Rev 10: 195–204. [PMC free article] [PubMed] [Google Scholar]
- 40. Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, et al. (2002) The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76: 12813–12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kincaid RP, Burke JM, Sullivan CS (2012) RNA virus microRNA that mimics a B-cell oncomiR. P Natl Acad Sci USA 109: 3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cullen BR (2012) MicroRNA expression by an oncogenic retrovirus. Proc Natl Acad Sci U S A 109: 2695–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wootton JC (1994) Nonglobular Domains in Protein Sequences - Automated Segmentation Using Complexity-Measures. Comput Chem 18: 269–285. [DOI] [PubMed] [Google Scholar]
- 44. Ferron F, Longhi S, Canard B, Karlin D (2006) A practical overview of protein disorder prediction methods. Proteins 65: 1–14. [DOI] [PubMed] [Google Scholar]
- 45. Firth AE, Brown CM (2005) Detecting overlapping coding sequences with pairwise alignments. Bioinformatics 21: 282–292. [DOI] [PubMed] [Google Scholar]
- 46. Chao JA, Lee JH, Chapados BR, Debler EW, Schneemann A, et al. (2005) Dual modes of RNA-silencing suppression by Flock House virus protein B2. Nat Struct Mol Biol 12: 952–957. [DOI] [PubMed] [Google Scholar]
- 47. Boulila M (2009) Recombination structure and genetic relatedness among members of the family Bromoviridae based on their RNAs 1 and 2 sequence analyses. Virus Genes 38: 435–444. [DOI] [PubMed] [Google Scholar]
- 48. Chen HY, Yang J, Lin C, Yuan YA (2008) Structural basis for RNA-silencing suppression by Tomato aspermy virus protein 2b. EMBO Rep 9: 754–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Korber S, Shaik Syed Ali P, Chen JC (2009) Structure of the RNA-binding domain of Nodamura virus protein B2, a suppressor of RNA interference. Biochemistry 48: 2307–2309. [DOI] [PubMed] [Google Scholar]
- 50. Lingel A, Simon B, Izaurralde E, Sattler M (2005) The structure of the flock house virus B2 protein, a viral suppressor of RNA interference, shows a novel mode of double-stranded RNA recognition. EMBO Rep 6: 1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nieuwenhuis S, Forstmann BU, Wagenmakers EJ (2011) Erroneous analyses of interactions in neuroscience: a problem of significance. Nat Neurosci 14: 1105–1107. [DOI] [PubMed] [Google Scholar]
- 52. Trifonov V, Rabadan R (2010) Frequency analysis techniques for identification of viral genetic data. MBio 1: e00156–00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karlin S, Burge C (1995) Dinucleotide relative abundance extremes: a genomic signature. Trends Genet 11: 283–290. [DOI] [PubMed] [Google Scholar]
- 54. Kfoury Y, Nasr R, Journo C, Mahieux R, Pique C, et al. (2012) The multifaceted oncoprotein Tax: subcellular localization, posttranslational modifications, and NF-kappaB activation. Adv Cancer Res 113: 85–120. [DOI] [PubMed] [Google Scholar]
- 55. McGirr KM, Buehuring GC (2006) Tax & rex: overlapping genes of the Deltaretrovirus group. Virus Genes 32: 229–239. [DOI] [PubMed] [Google Scholar]
- 56. Zhao X, McGirr KM, Buehring GC (2007) Potential evolutionary influences on overlapping reading frames in the bovine leukemia virus pXBL region. Genomics 89: 502–511. [DOI] [PubMed] [Google Scholar]
- 57. Nagashima K, Yoshida M, Seiki M (1986) A single species of pX mRNA of human T-cell leukemia virus type I encodes trans-activator p40x and two other phosphoproteins. J Virol 60: 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nahon JL (2003) Birth of ‘human-specific’ genes during primate evolution. Genetica 118: 193–208. [DOI] [PubMed] [Google Scholar]
- 59. Nicot C, Dundr M, Johnson JM, Fullen JR, Alonzo N, et al. (2004) HTLV-1-encoded p30(II) is a post-transcriptional negative regulator of viral replication. Nat Med 10: 197–201. [DOI] [PubMed] [Google Scholar]
- 60. Zhao T, Matsuoka M (2012) HBZ and its roles in HTLV-1 oncogenesis. Front Microbiol 3: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barbeau B, Mesnard JM (2011) Making sense out of antisense transcription in human T-cell lymphotropic viruses (HTLVs). Viruses 3: 456–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoye JP, Blomberg J, Coffin JM, Fan H, Hahn B, et al.. (2011) Retroviridae. In: King AM, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy - Ninth Report of the International Committee on Taxonomy of Viruses: Elsevier Academic Press. pp. 477–495.
- 63. Morozov SY, Solovyev AG (2003) Triple gene block: modular design of a multifunctional machine for plant virus movement. J Gen Virol 84: 1351–1366. [DOI] [PubMed] [Google Scholar]
- 64. Verchot-Lubicz J, Torrance L, Solovyev AG, Morozov SY, Jackson AO, et al. (2010) Varied movement strategies employed by triple gene block-encoding viruses. Mol Plant Microbe Interact 23: 1231–1247. [DOI] [PubMed] [Google Scholar]
- 65. Melcher U (2000) The ‘30K’ superfamily of viral movement proteins. J Gen Virol 81: 257–266. [DOI] [PubMed] [Google Scholar]
- 66. Martelli GP, Adams MJ, Kreuze JF, Dolja VV (2007) Family Flexiviridae: a case study in virion and genome plasticity. Annu Rev Phytopathol 45: 73–100. [DOI] [PubMed] [Google Scholar]
- 67. Miller M (2009) The Importance of Being Flexible: The Case of Basic Region Leucine Zipper Transcriptional Regulators. Curr Protein Pept Sc 10: 244–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Garza AS, Ahmad N, Kumar R (2009) Role of intrinsically disordered protein regions/domains in transcriptional regulation. Life Sci 84: 189–193. [DOI] [PubMed] [Google Scholar]
- 69. Rado-Trilla N, Alba MM (2012) Dissecting the role of low-complexity regions in the evolution of vertebrate proteins. BMC Evol Biol 12: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brister JR, Bao Y, Kuiken C, Lefkowitz EJ, Le Mercier P, et al. (2010) Towards Viral Genome Annotation Standards, Report from the 2010 NCBI Annotation Workshop. Viruses 2: 2258–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lo MK, Harcourt BH, Mungall BA, Tamin A, Peeples ME, et al. (2009) Determination of the henipavirus phosphoprotein gene mRNA editing frequencies and detection of the C, V and W proteins of Nipah virus in virus-infected cells. J Gen Virol 90: 398–404. [DOI] [PubMed] [Google Scholar]
- 72. Pelet T, Curran J, Kolakofsky D (1991) The P-Gene of Bovine Parainfluenza Virus-3 Expresses All 3 Reading Frames from a Single Messenger-Rna Editing Site. Embo J 10: 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Durbin AP, McAuliffe JM, Collins PL, Murphy BR (1999) Mutations in the C, D, and V open reading frames of human parainfluenza virus type 3 attenuate replication in rodents and primates. Virology 261: 319–330. [DOI] [PubMed] [Google Scholar]
- 74. Kulkarni S, Volchkova V, Basler CF, Palese P, Volchkov VE, et al. (2009) Nipah Virus Edits Its P Gene at High Frequency To Express the V and W Proteins. J Virol 83: 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Belalov IS, Lukashev AN (2013) Causes and implications of codon usage bias in RNA viruses. PLoS One 8: e56642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Karlin S, Doerfler W, Cardon LR (1994) Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J Virol 68: 2889–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Greenbaum BD, Rabadan R, Levine AJ (2009) Patterns of Oligonucleotide Sequences in Viral and Host Cell RNA Identify Mediators of the Host Innate Immune System. PLoS One 4: e5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Michel AM, Choudhury KR, Firth AE, Ingolia NT, Atkins JF, et al. (2012) Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res 22: 2219–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chung WY, Wadhawan S, Szklarczyk R, Pond SK, Nekrutenko A (2007) A first look at ARFome: Dual-coding genes in mammalian Genomes. Plos Comput Biol 3: 855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lin MF, Kheradpour P, Washietl S, Parker BJ, Pedersen JS, et al. (2011) Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes. Genome Res 21: 1916–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Neme R, Tautz D (2013) Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genomics 14: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liang H, Landweber LF (2006) A genome-wide study of dual coding regions in human alternatively spliced genes. Genome Res 16: 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sharma V, Firth AE, Antonov I, Fayet O, Atkins JF, et al. (2011) A pilot study of bacterial genes with disrupted ORFs reveals a surprising profusion of protein sequence recoding mediated by ribosomal frameshifting and transcriptional realignment. Mol Biol Evol 28: 3195–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bao Y, Federhen S, Leipe D, Pham V, Resenchuk S, et al. (2004) National center for biotechnology information viral genomes project. J Virol 78: 7291–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Soding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33: W244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Remmert M, Biegert A, Hauser A, Soding J (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9: 173–175. [DOI] [PubMed] [Google Scholar]
- 88. Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, et al. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36: W465–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ishida T, Kinoshita K (2008) Prediction of disordered regions in proteins based on the meta approach. Bioinformatics 24: 1344–1348. [DOI] [PubMed] [Google Scholar]
- 90. Vacic V, Uversky VN, Dunker AK, Lonardi S (2007) Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics 8: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ooi HS, Kwo CY, Wildpaner M, Sirota FL, Eisenhaber B, et al. (2009) ANNIE: integrated de novo protein sequence annotation. Nucleic Acids Res 37: W435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Belshaw R, Pybus OG, Rambaut A (2007) The evolution of genome compression and genomic novelty in RNA viruses. Genome Res 17: 1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pearson WR, Sierk ML (2005) The limits of protein sequence comparison? Curr Opin Struct Biol 15: 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Aravind L (2000) Guilt by association: contextual information in genome analysis. Genome Res 10: 1074–1077. [DOI] [PubMed] [Google Scholar]
- 95. Hildebrand A, Remmert M, Biegert A, Soding J (2009) Fast and accurate automatic structure prediction with HHpred. Proteins 77 Suppl 9: 128–132. [DOI] [PubMed] [Google Scholar]
- 96. Biegert A, Mayer C, Remmert M, Soding J, Lupas AN (2006) The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res 34: W335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38 Suppl: W545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ye Y, Godzik A (2004) FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res 32: W582–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sabath N, Graur D (2010) Detection of functional overlapping genes: simulation and case studies. J Mol Evol 71: 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pruitt KD, Tatusova T, Brown GR, Maglott DR (2012) NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res 40: D130–D135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wright F (1990) The ‘effective number of codons’ used in a gene. Gene 87: 23–29. [DOI] [PubMed] [Google Scholar]
- 103. Sharp PM, Li WH (1987) The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Suzuki H, Saito R, Tomita M (2009) Measure of synonymous codon usage diversity among genes in bacteria. BMC Bioinformatics 10: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Myers L, Sirois MJ (2006) Spearman Correlation Coefficients, Differences between. Encyclopedia of Statistical Sciences. pp. 7901–7902.
- 106. Gordon KH, Williams MR, Hendry DA, Hanzlik TN (1999) Sequence of the genomic RNA of nudaurelia beta virus (Tetraviridae) defines a novel virus genome organization. Virology 258: 42–53. [DOI] [PubMed] [Google Scholar]
- 107. Rustici G, Milne RG, Accotto GP (2002) Nucleotide sequence, genome organisation and phylogenetic analysis of Indian citrus ringspot virus. Brief report. Arch Virol 147: 2215–2224. [DOI] [PubMed] [Google Scholar]
- 108. Verchot J, Angell SM, Baulcombe DC (1998) In vivo translation of the triple gene block of potato virus X requires two subgenomic mRNAs. J Virol 72: 8316–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yoshikawa N, Takahashi T (1992) Evidence for translation of apple stem grooving capillovirus genomic RNA. J Gen Virol 73 (Pt 5) 1313–1315. [DOI] [PubMed] [Google Scholar]
- 110. Sato K, Yoshikawa N, Takahashi T, Taira H (1995) Expression, subcellular location and modification of the 50 kDa protein encoded by ORF2 of the apple chlorotic leaf spot trichovirus genome. J Gen Virol 76 (Pt 6) 1503–1507. [DOI] [PubMed] [Google Scholar]
- 111. Mundt E, Beyer J, Muller H (1995) Identification of a novel viral protein in infectious bursal disease virus-infected cells. J Gen Virol 76 (Pt 2) 437–443. [DOI] [PubMed] [Google Scholar]
- 112. Fuller F, Bhown AS, Bishop DH (1983) Bunyavirus nucleoprotein, N, and a non-structural protein, NSS, are coded by overlapping reading frames in the S RNA. J Gen Virol 64 (Pt 8) 1705–1714. [DOI] [PubMed] [Google Scholar]
- 113. McFadden N, Bailey D, Carrara G, Benson A, Chaudhry Y, et al. (2011) Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog 7: e1002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chandratilleke D, O'Connell P, Schat KA (1991) Characterization of proteins of chicken infectious anemia virus with monoclonal antibodies. Avian Dis 35: 854–862. [PubMed] [Google Scholar]
- 115. Ren Q, Wang QS, Firth AE, Chan MM, Gouw JW, et al. (2012) Alternative reading frame selection mediated by a tRNA-like domain of an internal ribosome entry site. Proc Natl Acad Sci U S A 109: E630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rojas MR, Jiang H, Salati R, Xoconostle-Cazares B, Sudarshana MR, et al. (2001) Functional analysis of proteins involved in movement of the monopartite begomovirus, Tomato yellow leaf curl virus. Virology 291: 110–125. [DOI] [PubMed] [Google Scholar]
- 117. Peterson DL (1981) Isolation and characterization of the major protein and glycoprotein of hepatitis B surface antigen. J Biol Chem 256: 6975–6983. [PubMed] [Google Scholar]
- 118. Pfaff E, Salfeld J, Gmelin K, Schaller H, Theilmann L (1987) Synthesis of the X-protein of hepatitis B virus in vitro and detection of anti-X antibodies in human sera. Virology 158: 456–460. [DOI] [PubMed] [Google Scholar]
- 119. Dinesh-Kumar SP, Miller WA (1993) Control of start codon choice on a plant viral RNA encoding overlapping genes. Plant Cell 5: 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Azarkh E, Robinson E, Hirunkanokpun S, Afanasiev B, Kittayapong P, et al. (2008) Mosquito densonucleosis virus non-structural protein NS2 is necessary for a productive infection. Virology 374: 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Tijssen P, Li Y, El-Far M, Szelei J, Letarte M, et al. (2003) Organization and expression strategy of the ambisense genome of densonucleosis virus of Galleria mellonella. J Virol 77: 10357–10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sonntag F, Schmidt K, Kleinschmidt JA (2010) A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci U S A 107: 10220–10225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zadori Z, Szelei J, Tijssen P (2005) SAT: a late NS protein of porcine parvovirus. J Virol 79: 13129–13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hanzlik TN, Dorrian SJ, Johnson KN, Brooks EM, Gordon KH (1995) Sequence of RNA2 of the Helicoverpa armigera stunt virus (Tetraviridae) and bacterial expression of its genes. J Gen Virol 76 (Pt 4) 799–811. [DOI] [PubMed] [Google Scholar]
- 125. Huang M, Koh DC, Weng LJ, Chang ML, Yap YK, et al. (2000) Complete nucleotide sequence and genome organization of hibiscus chlorotic ringspot virus, a new member of the genus Carmovirus: evidence for the presence and expression of two novel open reading frames. J Virol 74: 3149–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Johnston JC, Rochon DM (1996) Both codon context and leader length contribute to efficient expression of two overlapping open reading frames of a cucumber necrosis virus bifunctional subgenomic mRNA. Virology 221: 232–239. [DOI] [PubMed] [Google Scholar]
- 127. Scheets K (2000) Maize chlorotic mottle machlomovirus expresses its coat protein from a 1.47-kb subgenomic RNA and makes a 0.34-kb subgenomic RNA. Virology 267: 90–101. [DOI] [PubMed] [Google Scholar]
- 128. Bozarth CS, Weiland JJ, Dreher TW (1992) Expression of ORF-69 of turnip yellow mosaic virus is necessary for viral spread in plants. Virology 187: 124–130. [DOI] [PubMed] [Google Scholar]
- 129. Zhou H, Jackson AO (1996) Expression of the barley stripe mosaic virus RNA beta “triple gene block”. Virology 216: 367–379. [DOI] [PubMed] [Google Scholar]
- 130. Kim SH, Ryabov EV, Kalinina NO, Rakitina DV, Gillespie T, et al. (2007) Cajal bodies and the nucleolus are required for a plant virus systemic infection. Embo J 26: 2169–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Taxonomic distribution of the ancestral and de novo frames of the benchmark dataset.
(DOC)
Examination of possible confounding factors of codon usage analysis: relative frame of de novo genes compared to ancestral genes, and relative age of de novo frames. (1) We used taxonomic distribution as a very approximate, empirical proxy to estimate comparative ages of the overlaps. De novo frames found only in one species are considered “young” (provided there are several species in the genus considered, see note 2 below); overlaps found in more than one species but less than one genus are considered of “Intermediate” age, and overlaps found in more than one genus are considered “old”. The taxonomic distribution of de novo frames is taken from Supplementary Table S1. (2) We excluded cases where there was insufficient taxonomic sampling, such as the betatetravirus overlap, since the betatetravirus genus comprises only ones species.
(DOCX)
Low complexity and predicted structural disorder in proteins encoded by overlapping genes in human T-lymphotropic virus 1 (HTLV1). (1) Predictions of low sequence complexity were made with SEG with parameters 45/3.75/3.4. (2) Predictions of structural disorder were made with MetaPrDOS (see Material and Methods).
(DOC)