

Abstract
The dual specific phosphatase, MAP kinase phosphatase-2 (MKP-2) has recently been demonstrated to negatively regulate macrophage arginase-1 expression, while at the same time to positively regulate iNOS expression. Consequently, MKP-2 is likely to play a significant role in the host interplay with intracellular pathogens. Here we demonstrate that MKP-2−/− mice on the C57BL/6 background have enhanced susceptibility compared with wild-type counterparts following infection with type-2 strains of Toxoplasma gondii as measured by increased parasite multiplication during acute infection, increased mortality from day 12 post-infection onwards and increased parasite burdens in the brain, day 30 post-infection. MKP-2−/− mice did not, however, demonstrate defective type-1 responses compared with MKP-2+/+ mice following infection although they did display significantly reduced serum nitrite levels and enhanced tissue arginase-1 expression. Early resistance to T. gondii in MKP-2+/+, but not MKP-2−/−, mice was nitric oxide (NO) dependent as infected MKP-2+/+, but not MKP-2−/− mice succumbed within 10 days post-infection with increased parasite burdens following treatment with the iNOS inhibitor L-NAME. Conversely, treatment of infected MKP-2−/− but not MKP-2+/+ mice with nor-NOHA increased parasite burdens indicating a protective role for arginase-1 in MKP-2−/− mice. In vitro studies using tachyzoite-infected bone marrow derived macrophages and selective inhibition of arginase-1 and iNOS activities confirmed that both iNOS and arginase-1 contributed to inhibiting parasite replication. However, the effects of arginase-1 were transient and ultimately the role of iNOS was paramount in facilitating long-term inhibition of parasite multiplication within macrophages.
Author Summary
Toxoplasma gondii is a protozoan (single cell) parasite that can be transmitted to humans via infection stages in cat feces or from eating under-cooked meat containing parasite cysts. The disease, which infects some 30% of the world population is a major cause of congenital infection and abortion and can be fatal in immune compromised hosts. Control of parasite growth has been shown to be largely, but not wholly, dependent on the production of nitric oxide (NO) as a result of the enzymatic activity of inducible nitric oxide synthase (iNOS) on the substrate L-arginine. It has been suggested that arginase-1 by depleting the L-arginine store can promote parasite growth. We have recently shown MAP kinase phosphatase-2 (MKP-2) to increase iNOS and decrease arginase-1 levels and mice lacking this gene were found to be more susceptible to T. gondii. Inhibition of iNOS activity in genetically intact mice demonstrated it was of paramount importance in controlling parasite growth. Surprisingly, however, inhibition of arginase-1 in MKP-2−/− mice demonstrated arginase-1 was also protective. Significantly T. gondii cannot make its own arginine and requires this from the host cell and consequently iNOS and arginase-1 can work together to starve the parasite of this essential metabolite.
Introduction
Toxoplasma gondii is an obligate intracellular protozoan parasite of significant public health importance, being a major cause of congenital infection and abortion as well as a significant and often fatal infection in immune compromised hosts. The early acute stage of infection is characterized by widespread tachyzoite dissemination and tissue damage. The rapid onset of immunity, initiated in large part by the well characterized T. gondii pathogen associated molecular patterns (PAMPS) controls parasite replication [1]–[4], and results in the life-long chronic stage of infection associated with encystment of the parasites in skeletal muscle and the central nervous system [reviewed in 5]. Protection against acute disease is mediated primarily by the interaction of neutrophils, dendritic cells, macrophages and natural killer (NK) cells that as part of the innate response not only limits parasite growth, but initiates an effective cytotoxic CD8+ T cell response that is responsible for long-term protection and prevention of encephalitis via IFN-γ production [6]–[9]. The mechanisms by which IFN-γ, the major effector cytokine mediating resistance during T. gondii infection, promotes anti-toxoplasma activity are not yet fully clear and vary between host species studied. Several IFN-γ-regulated genes including iNOS [6], [10], indoleamine 2,3 dioxygenase (IDO) [11], [12], and more recently, p47 GTPases, have been implicated in playing significant roles in mediating these protective responses [13]–[21].
Despite being essential to control parasite replication, an overactive type-1 response and overproduction of IFN-γ, TNF-α and NO can result in severe pathology and death. Consequently, protective immunity to T. gondii that needs to effectively control parasite proliferation without excessive inflammation is dependent on the regulation of type-1 responses by Th2 cells and Treg cells [22]–[29]. An overactive type-2 response could equally promote enhanced parasite proliferation and host death and indeed alternative macrophage activation has been shown to promote parasite growth [30]. Interestingly, the classic hallmark indicator of alternative macrophage activation, arginase-1, can be induced innately by T. gondii apparently via both STAT-6 dependent [30]–[32] and independent mechanisms [30], [33]. Paradoxically, arginase-1 has been associated with enhancing infection with T. gondii by competing with iNOS for their common substrate L-arginine [33] and promoting parasite replication by providing the polyamines needed for cell division [32]. Conversely arginase-1 might also limit parasite growth by starving the parasite of the L-arginine it requires for this process [31], [34].
A recent study has demonstrated that the dual specific phosphatase, MKP-2 is not only a negative regulator of macrophage arginase-1 but also a positive regulator of iNOS expression [35]. Furthermore MKP-2−/− C57BL/6 mice have been found to display enhanced susceptibility to the intracellular parasite Leishmania mexicana. Susceptibility in MKP-2−/− mice was in large part due to enhanced parasite growth that could be reversed by inhibiting arginase-1 activity. Given the importance of NO production as well as the apparently contradictory roles of arginase-1 in murine T. gondii infections it is likely that the role of MKP-2 in T. gondii infection would be one of major significance. We consequently studied the course of T. gondii infection in MKP-2−/− and MKP-2+/+ C57BL/6 mice. We identified that MKP-2 deficiency results in increased susceptibility to T. gondii infection and that this correlated strongly with impaired iNOS activity in vivo. Conversely we demonstrated an arginase-1 dependent mechanism responsible for control of parasite growth that functioned to partially protect the host independently of iNOS mediated effects.
Materials and Methods
Ethics statement
All animal procedures conformed to guidelines from The Home Office of the UK Government. All work was covered by two Home Office licences: PPL60/3929, “mechanism of control of parasite infection” and PPL60/3439,“genetic models of cancer and inflammation” with approval by the University of Strathclyde ethical review panel.
Mice
MKP-2+/+ and MKP-2−/− mice on a C57BL/6 background were bred and maintained and all experiments carried out in house at the Strathclyde Institute of Pharmacy and Biomedical Sciences, Glasgow, UK. Six to eight week old, male mice were used for infection and aged matched within each experiment.
Maintenance and infection of Toxoplasma gondii Beverly strain
Beverley cysts were maintained in vivo by intraperitoneal (i.p.) passage of infective brain tissue homogenates through outbred CD1 albino mice. For experimental infections MKP-2+/+ and MKP-2−/− mice were infected i.p. with 10 tissue cysts in 200 µl sterile PBS.
Maintenance of transfected Toxoplasma gondii Prugniaud strains
Tachyzoites were routinely maintained in confluent human foreskin fibroblasts (HFFs) grown in DMEM complete medium comprising; Dulbecco's Modified Eagle Medium (DMEM) containing L-glutamine (Invitrogen, UK), 10% foetal calf serum (PAA, UK), 100 U/ml penicillin (Cambrex Bioscience, Veniers, Belgium), 100 µg/ml streptomycin (Cambrex Bioscience, Veniers, Belgium) and 50 U/ml amphotericin B (Cambrex Bioscience, Veniers, Belgium) at 37°C in 5% CO2.
Bioluminescent imaging
In vivo parasite burden was assessed using bioluminescent imaging using type II Prugniaud T. gondii transfected with the firefly luciferase [36]. The light data was quantified using Living Image software (Caliper Life Science)
Briefly, intracellular FLUC T. gondii was harvested from in vitro culture. Culture media was removed and the HFF monolayer washed once with sterile PBS (Lonza, UK) prior to disruption with a cell scraper and harvesting in 10 ml sterile PBS. The intracellular parasites were then released from the host cells by passage through a 21 gauge needle and centrifuged at 1200 rpm for 10 minutes. The bioluminescent activity of the FLUC T. gondii was quality controlled for each experiment prior to infection using a standard curve in a black-walled 24 well plate by in vitro imaging using the IVIS Spectrum (Caliper Life Sciences) (Figure S1). Mice were then infected with 20,000 tachyzoites i.p. in a volume of 400 µl sterile PBS. FLUC T. gondii infected mice were imaged using the IVIS Spectrum (Caliper Life Sciences) to determine parasite burden. Mice to be imaged were given 150 mg/kg of D-luciferin potassium salt solution i.p. prior to anaesthesia with isoflurane. For optimal imaging, 1 minute exposures were taken with medium binning, 20 minutes post luciferin injection. For ex vivo imaging of the brain, mice were injected with 150 mg/kg of D-luciferin potassium salt solution. After 10 minutes the mice were sacrificed by CO2 inhalation. The brains were removed and soaked in 15 mg/ml D-luciferin potassium salt, dissolved in warmed RMPI 1640, for a further 10 minutes before being imaged.
Purification of Toxoplasma lysate antigen
RH tachyzoites where harvested from acutely infected BALB/c mice, by intra-peritoneal washout and lysate antigen (TLA) was prepared as described previously [37].
Cytokine ELISA
Spleen cell suspensions from infected mice were prepared as described previously [24]. For re-stimulation, 2×105 cells were incubated with 5 µg TLA in a total volume of 200 µl for 72 h at 37°C in 5% CO2. Supernatants were then used for IFN-γ, IL-5, IL-6 and IL-10 ELISA, using paired antibodies from BD Bioscience. Absorbance was the read at 405 nm using a microtitre plate reader (Spectramax 190, Molecular Devices, USA). Concentrations were determined against standard curves for each cytokine (R&D Systems).
Serum nitric oxide
Serum nitric oxide levels were determined by Griess assay. Blood was collected by cardiac puncture and cells removed by centrifugation at 13,000 rpm for 10 minutes. Protein was removed from the serum by adding ZnSO4 to a final concentration of 15 mg/ml, vortexing thoroughly and centrifuged at 13,000 rpm for 10 minutes. The supernatant was retained for the Griess assay. Griess reagent (equal volumes of 2% sulphanilamide in 5% H3PO4 and 0.2% Napthylene diamine HCL in ddH2O) was added to samples and standards in a 96 well plate and incubated in the dark for 10 minutes. Absorbance was read at 540 nm and serum nitrite concentrations were calculated against a standard curve.
In vivo iNOS and Arginase-1 inhibition
To inhibit iNOS and Arginase-I in vivo, mice received intraperitoneal doses of N (G)-nitro-L-arginine methyl ester (200 mg/kg, L-NAME, Sigma UK) or Nω-hydroxy-nor-Arginine (100 µg nor-NOHA, Merck, UK), respectively [38], [39]. Treatment commenced one day prior to infection.
Generation of murine macrophages
Bone marrow derived macrophages were cultured by flushing the femurs and tibiae of 6 week old MKP-2+/+ or MKP-2−/− mice with DMEM. Cells were then cultured in DMEM containing 20% heat inactivated FCS, 30% L-cell conditioned medium, 5 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin and incubated at 37°C for 10 days [35]. After this time adherent cells were harvested and then seeded into either 12 well plates (1×106 cells/ml) or black-walled 96 well plates (0.5×105).
Macrophage infection with T. gondii
For infection, 5×104 bone marrow derived MKP-2+/+ or MKP-2−/− macrophages were seeded into black-walled 96 well plates in complete phenol red free RPMI 1640 medium. The cells were infected with 5×104 type II Prugniaud strain T. gondii tachyzoites, expressing YFP (Donated by Marcus Meissner, University of Glasgow). Wells were made up to a final volume of 200 ml. Macrophages were treated with LPS (100 ng/ml), IFN-γ (100 U/ml), 50 µM nor-NOHA or 5 mM L-NAME as appropriate. Levels of YFP expression were assayed at 24, 48 and 72 h using the transillumination feature of the IVIS Spectrum (Caliper Lifescience). An excitation wavelength of 500 nm and emission wave of 540 nm was used. The light data was quantified using Living Image software (Caliper Lifescience) using the uninfected macrophages as background controls.
Flow cytometry
For re-stimulation, 1×106 splenocytes were incubated with 5 mg/ml TLA, ionomycin and phorbol-13-myristate-12-acetate (PMA, Sigma, UK) (0.5 µg/ml and 10 ng/ml, respectively) for 6 h at 37°C and 5% CO2. After 3 h incubation Brefeldin A (Sigma) was added for a further 3 h at a final concentration of 10 ng/ml. Cells were then harvested for staining and incubated with Fc-Block (5 µg/ml αCD16/CD32, BD Bioscience, 1% mouse serum in RPMI) for 10 minutes at room temperature, cells were stained for 1 hour at 4°C with αCD3-PerCP, αCD4-APC-H7 (BD Biosciences) and αCD8-Alexa488 (ebioscience). Cells were fixed using Fix & Perm (Life Technologies) following manufacturer's instructions. Intracellular cytokine staining was performed using αTNF-PE and αIFN-γ-APC (eBioscience). A total of 500,000 events per sample were acquired on a BD FACSCanto and data analysis was carried out using FlowJo software.
Natural killer or NKT cells from intraperitoneal exudates (1×106) from infected mice were surface stained with αPanNK-APC and αCD3-PerCP-eFluor710 before fixing and permeabilisation. Intracellular staining for IFN-γ was performed using αIFN-γ-PE or the respective isotype control, rat IgG1,κ-PE (all eBioscience). A total of 200,000 events was recorded and analysed using Kaluza software (Beckman Coulter). All values were normalized to their respective isotype control.
Measurement of Arginase-1 expression and activity
Cell lysates (20 µg protein/lane) were separated by 10% SDS-PAGE and transferred onto a nitrocellulose membrane and probed for Arginase-1 as previously described [35]. Arginase activity from murine BMD-macrophages was measured using an assay based on a reaction with α-isonitrosopropiophenon (ISPF) as described previously [35].
Preparation of cDNA
Peritoneal exudates from mice were counted and 1×106 cells from each sample taken for RNA extraction using RNeasy Mini Kit (Qiagen, UK) following manufacturer's instructions. For each reaction, 2 µg of total RNA and 300 ng of random primers (Promega) were incubated at 65°C for 5 minutes. Following a 10 minute incubation at room temperature 2 µl of AffinityScript reverse transcriptase (RT) buffer (Agilent Technologies), 10 mM dithiothreitol, 4 mM dNTP mix (Promega) and 1 µl of AffinityScript multiple RT (Agilent Technologies) were added to each sample. These were then incubated for 10 minutes at 25°C, 1 hour at 50°C and 15 minutes at 70°C.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR experiments were conducted on the Stratagene Mx3000p system (Stratagene, Agilent). Each reaction consisted of 5 µl Brilliant III Ultra-fast SYBR Green QPCR Master Mix (Agilent Technologies), 3.5 µl of molecular grade water (Life Technologies), 25 pmol of the forward and reverse primers (Table S1), and 1 µl of the cDNA template. PCR reactions were carried out with the following thermoprofile: one cycle at 95°C for 10 minutes, 40 cycles of 20 seconds at 95°C, 20 seconds at 64°C and 30 seconds at 70°C. Relative gene expression was calculated based on the ΔCT for each gene, normalized to that of the housekeeping gene (TBP).
Statistics
Statistical analysis was performed using GraphPad Prism Program (Version 5.0, GraphPad Software, California). All results are shown as standard error of the mean (SEM). Statistically significant differences were determined using students t-test for parametric and Mann Whitney U test for non-parametric data, respectively. P values equal or below 0.05 were considered significant.
Results
MKP-2 deficiency results in increased susceptibility following infection with T. gondii
Intraperitoneal infection with 20 cysts of the Beverley (type-II) strain of T. gondii resulted in significant mortality, typically between 80–100%, 15–25 days post-infection in MKP-2−/−, but not MKP-2+/+ mice (Figure 1A). In order to determine whether this was associated with any inability of the MKP-2−/− mice to control parasite growth, MKP-2−/− and MKP-2+/+ mice were infected i.p. with 20,000 type-II strain Prugniaud-FLUC tachyzoites and parasite burdens measured at 2 day intervals by bioluminescent imaging (Figure 1B and C). At days 8 (Figure 1B) and 10 the bioluminescent intensity and consequently parasite burdens were significantly greater (p<0.031 and p<0.0079 respectively) in MKP-2−/− compared with MKP-2+/+ mice (Figure 1C). Greater parasite numbers in infected MKP-2−/− as opposed to MKP-2+/+ mice during acute infections were confirmed by qRT-PCR (Figure S2) The increased susceptibility of MKP-2−/− mice infected with Prugniaud-FLUC tachyzoites compared with their wild-type counterparts was also evident during the chronic phase of the disease: mortality was typically 40–60% in MKP-2−/− mice and by day 30 post-infection the excised brains of MKP-2−/− mice were revealed to have significantly higher parasite burdens (p<0.02) than similarly infected MKP-2+/+ animals (Figure 1D and E). Increased parasite burdens in infected MKP-2−/− compared with MKP-2+/+ mice were also evident in other tissues at this late stage (Liver: MKP-2−/− 4.76×105±4.7×103p/s, MKP-2+/+ 3.85×105±4.6×104p/s; Kidney: MKP-2−/− 1.31×106±4×104p/s, MKP-2+/+ 4.76×105±1.3×105p/s p<0.05).
Figure 1. MKP-2 deficiency results in increased susceptibility to T. gondii infection.

Mice were infected with 10 Beverley tissue cysts intraperitoneally and mortality monitored (A). For in vivo bioluminescent imaging and parasite quantification, mice were infected with 20,000 type II Prugniaud tachyzoites, expressing firefly luciferase. Mice were imaged as outlined in the methods (B). Parasite burdens were determined by measuring whole body Total Flux (Photons/second) using Living Image 4.0 (C). Each value represents the mean of 5 animals per experimental group ± SEM. On day 30 post-infection brains were imaged ex vivo as outlined in the methods (D). Chronic parasite burden was determined by measuring Total Flux (photons/second) from each brain (E). Each value represents the mean of four animals per experimental group ± SEM. All experiments were carried out on at least two occasions.
MKP-2−/− mice infected with T. gondii do not have an impaired immune response
In order to determine whether the increased susceptibility of MKP-2−/− mice infected with T. gondii was the result of an impaired adaptive immune response spleens were removed and splenocytes stimulated with TLA at days 10, 20 and 30 post-infection and IFN-γ, IL-4, IL-5 and IL-10 production measured in the supernatants. There were generally no differences between the ability of splenocytes from MKP-2−/− or MKP-2+/+ mice infected with T. gondii to produce cytokines when stimulated with parasite lysate antigen. This was consistent through both acute and chronic infection for the primary mediator of parasite control IFN-γ (Figure 2). There was also no difference in the absolute number of cytokine producing cells between MKP-2−/− and MKP-2+/+ mice (Figure S3). Flow cytometry analysis (Figure 3) of splenocytes from MKP-2−/− and MKP-2+/+ mice day 10 post-infection either under resting conditions, following antigen stimulation, or following treatment with ionomycin and PMA revealed no differences in the overall numbers of CD4+ or CD8+ T cells. In addition no differences in the frequencies of CD4+ (Figure 3B) or CD8+ (Figure 3C) T cells producing IFN-γ, or TNF-α, or both IFN-γ and TNF-α (Figure 3) were noted. In addition, while no IFN-γ was detected in NK cells equivalent amounts of NKT cells producing IFN-γ were detected from both infected MKP-2−/− and MKP-2+/+ mice (Figure S4). Consequently, there was little evidence that MKP-2−/− mice were limited in their ability to mount a type-1 response. Furthermore we examined the expressions of IFN-γ dependent GTPases by qRT-PCR and found these to be comparable in both infected MKP-2−/− and MKP-2+/+ mice (Figure S2).
Figure 2. MKP-2−/− and MKP-2+/+ splenocyte IFN-γ production was similar through the acute and chronic stages of infection.
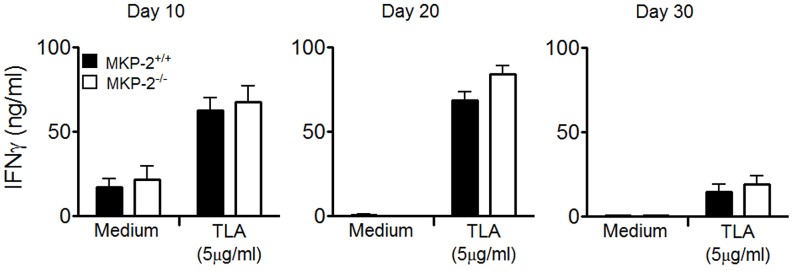
Splenocytes from T. gondii infected mice were stimulated with TLA (5 µg/ml) and the supernatants assessed for IFN-γ by ELISA. Each value represents the mean of four animals per experimental group ± SEM. All experiments were carried out on at least two occasions.
Figure 3. MKP-2 deletion does not impair T cell responses during infection with T. gondii.

T cell responses were determined by flow cytometry. Cells were surface-stained for CD3, CD4 and CD8 and intracellular for IFN-γ and TNF-α. Live cells were gated on forward (FSC) versus side scatter (SSC). T cells were first gated for CD3 and then sub-gated for either CD4 or CD8 and subsequently their respective antigen-specific intracellular cytokine production, following stimulation with TLA (10 µg/ml). The specificity of the intracellular staining was ensured by analysing the respective isotype controls and normalizing samples accordingly (A). Populations of CD3+ CD4+ T cells (B) and CD3+ CD8+ T cells (C) single or double positive for IFN-γ and TNF-α were determined using FlowJo software. Each value represents the mean of four animals per experimental group ± SEM. All experiments were carried out on at least two occasions.
MKP-2−/− mice infected with T. gondii have decreased systemic nitrite levels and increased tissue arginase-1 expression
Serum nitrite levels were significantly higher (p<0.0028) in MKP-2+/+ mice compared with MKP-2−/− at day 10 post-infection coincident with the noted inverse differences in T. gondii parasite burdens between MKP-2−/− and MKP-2+/+ mice (Figure 4A). Nitrite levels were below the level of detection in both non-infected MKP-2−/− and MKP-2+/+ mice. At the same time spleen arginase-I expression was higher in MKP-2−/− compared with MKP-2+/+ mice in both, the uninfected as well as in the T. gondii infected groups (Figure 4B). Infection with T. gondii in general increased splenic arginase-I levels in all mice independent of the genotype (Figure 4B).
Figure 4. Systemic serum nitrite levels are reduced and tissue arginase-1 expression is enhanced in MKP-2−/− compared with MKP-2+/+ mice infected with T. gondii.
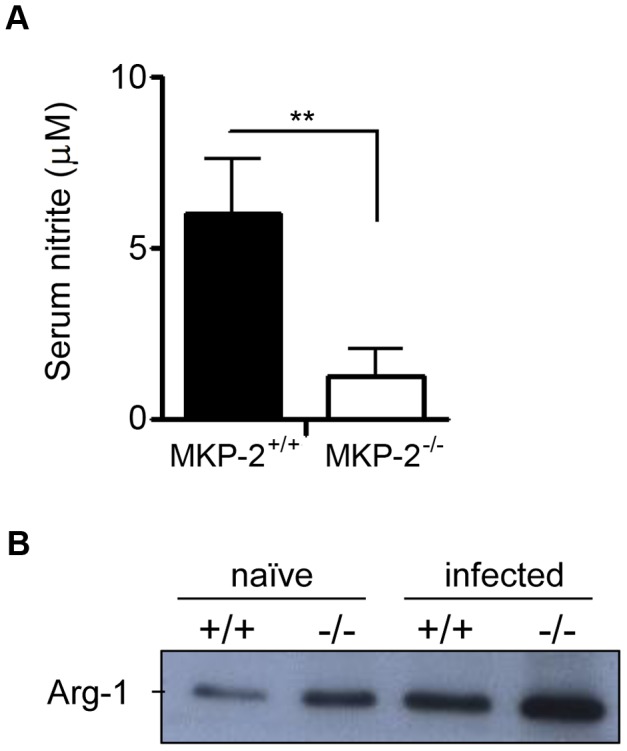
Serum from infected animals was assessed for its nitrite content by Griess assay. Each value represents the mean from ten animals per experimental group ± SEM (A). Splenocyte cell lysates were prepared and assayed for Arginase-1 by western blot. Cells were lysed in sample buffer and protein concentrations measured for each mouse. Equal amounts/animal were pooled and 20 µg loaded for each lane (B). **P<0.005. All experiments were carried out on at least two occasions.
Inhibition of NO production by L-NAME enhances the susceptibility of MKP-2+/+ but not MKP-2−/− mice to T. gondii infection
In order to determine whether the enhanced NO production by T. gondii infected MKP-2+/+ mice over their MKP-2 deficient counterparts was responsible for their increased resistance to parasite growth and survival in vivo, NO production was inhibited in infected mice by intraperitoneal injection with the iNOS inhibitor L-NAME. While L-NAME treatment of infected MKP-2+/+ mice resulted in 100% mortality by day 10 post-infection, surprisingly the majority of L-NAME treated infected MKP-2−/− mice survived until day 12 post-infection (Figure 5A). No nitrite was detectable in L-NAME treated mice. All non-treated infected MKP-2−/− and MKP-2+/+ mice survived until day 12 (Figure 5A) and in agreement with previous studies at day 10 there were significantly more parasites in MKP-2−/− mice (p<0.03). Measurement of bioluminescence (Figure 5B) allowed quantification of parasite burdens and demonstrated a significant increase in parasite growth at days 6 and 8 post-infection (day 6 p = 0.017, day 8 p = 0.005) in L-NAME treated as opposed to non-treated MKP-2+/+ mice (Figure 5C). By comparison treatment of infected MKP-2−/− mice with L-NAME did not significantly alter parasite growth (Figure 5C).
Figure 5. NO inhibition by L-NAME enhances susceptibility of MKP-2+/+ but not MKP-2−/− mice to T. gondii infection.

Mice were pre-treated with L-NAME and subsequently treated with L-NAME (100 mg per mouse) daily following infection with 20,000 Prugniaud tachyzoites expressing firefly luciferase. Mortality was measured over 12 days (A). Mice were imaged every second day. (B) Represents day 8 post-infection. The parasite burden was determined by measuring the total flux (photons/second) for each group (C). Each value represents the mean of five mice per group ± SEM. *P<0.05. All experiments were carried out on at least two occasions.
Inhibition of arginase-1 activity by nor-NOHA enhances the susceptibility of MKP-2−/− but not MKP-2+/+ mice to T. gondii infection
Unlike their wild-type counterparts, inhibition of NO production did not increase parasite growth or the early mortality of MKP-2−/− mice infected with T.gondii. This implied an alternative mechanism of controlling early infection in the absence of MKP-2. As T. gondii is an L-arginine auxotroph [40] we suspected that over expression of arginase-1 in MKP-2−/− mice could also be playing a protective role. To determine the possibility that arginase-1 expression might arrest parasite growth through arginine depletion, infected MKP-2−/− and MKP-2+/+ mice were treated with the arginase-1 inhibitor nor-NOHA. Nor-NOHA did not significantly increase mortality in either MKP-2−/− nor MKP-2+/+ mice infected with T. gondii compared with non-drug treated controls (Figure 6A). However, measurement of bioluminescence (Figure 6B) demonstrated significantly greater (p = 0.02 parasite burden day 8 post-infection in nor-NOHA treated as opposed to non-treated MKP-2−/− mice (Figure 6C). This is despite nor-NOHA treatment up-regulating iNOS activity in MKP-2−/− mice as measured by increased nitrite production by as much as ×2. By comparison nor-NOHA treatment had little effect on parasite burden during early T. gondii infection in MKP-2+/+ mice (Figure 6C).
Figure 6. Arginase-1 inhibition by nor-NOHA enhances susceptibility of MKP-2−/− but not MKP-2+/+ mice to T. gondii infection.

Mice were pre-treated with nor-NOHA (200 mg/kg) and treated daily following infection with 20,000 FLUC Prugniaud tachyzoites (A). Mice were imaged every second day post-infection. (B) Represents day 8 post-infection. Total flux (photons/second) was determined for each animal to determine parasite burden (C). Each value represents the mean of five mice per group ± SEM. *P<0.05. All experiments were carried out on at least two occasions.
MKP-2 deficiency does not render macrophages more susceptible to infection with T. gondii
We next determined whether the increased susceptibility of MKP-2−/− mice to infection with T. gondii was a result of their macrophages being more permissive to parasite growth. MKP-2−/− and MKP-2+/+ bone marrow derived macrophages were infected with YFP expressing Prugniaud T. gondii tachyzoites at a 1∶1 ratio. In addition the macrophages were either treated with LPS and IFN-γ with or without L-NAME or nor-NOHA. Parasite growth was then determined by assessing YFP fluorescence. At 24 hours, through to 72 hours post-infection parasite growth was similar in non-stimulated MKP-2−/− and MKP-2+/+ macrophages (Figure 7A). In addition, following LPS/IFN–γ stimulation parasite growth was significantly, and equally, reduced over the non-treated macrophages (p = 0.0001) and this was irrespective of whether the macrophages were derived from MKP-2+/+ or MKP-2−/− mice (Figure 7A). Treatment of LPS/IFN–γ activated MKP-2−/− and MKP-2+/+ macrophages with L-NAME partially ablated their ability to control parasite replication at 24 hours and totally ablated their ability to control parasite growth through 48 hours and 72 hours post-infection (p<0.0001) (Figure 7A). Conversely, treatment of LPS/IFN-γ activated MKP-2−/− and MKP-2+/+ macrophages with nor-NOHA partially ablated their ability to control parasite growth at 24 hours (MKP-2+/+ p = 0.005 and MKP-2−/− p = 0.006 respectively) but not at 48 or 72 hours post-infection (Figure 7A). While LPS/IFN-γ activation stimulated NO production, as measured by nitrite production 72 hours post-infection in the supernatants of T. gondii infected MKP-2−/− and MKP-2+/+ macrophages, this was significantly higher in the infected MKP-2+/+ culture supernatants (p<0.0001) (Figure 7B). Treatment of LPS/IFN-γ activated macrophages with L-NAME, but not nor-NOHA, largely ablated NO production by both MKP-2−/− and MKP-2+/+ macrophages (p<0.0001) (Figure 7B). T. gondii tachyzoite infection was found to enhance macrophage arginase-1 expression (Figure 8A) although this was consistently higher in MKP-2−/− than in MKP-2+/+ macrophages up to 24 hours post-infection. Increased expression of arginase-1 following macrophage infection with T. gondii (Figure 8A) was also reflected in up-regulated enzyme activity at 24 hours post-infection in both MKP-2−/− (p<0.05) and MKP-2+/+ macrophages (Figure 8B). Arginase-1 activity was significantly greater in MKP-2−/− than in MKP-2+/+ infected macrophages (p<0.001 for non-activated macrophages and p<0.0001 for LPS/IFN–γ activated macrophages) but was effectively and equally inhibited by nor-NOHA in both MKP-2−/− and MKP-2+/+ macrophages. Interestingly while L-NAME treatment significantly increased arginase-1 activity in non-activated MKP-2−/− and MKP-2+/+ macrophages (p<0.02 for both) and LPS/IFN-γ activated MKP-2−/− cultures (p<0.01) this was not noted in activated MKP-2+/+ macrophages (Figure 8B).
Figure 7. MKP-2 deficient macrophages display deficient iNOS activity but do not display an increased susceptibility to infection.

BMD macrophages were treated as appropriate with L-NAME, nor-NOHA, LPS and IFN-γ and subsequently infected with Prugniaud tachyzoites expressing YFP. After 24 and 72 h parasite burdens was determined by measuring YFP fluorescence (A). Supernatants from cultures were assayed for nitrite content by Griess assay (B). Each value represents four replicates ± SEM. ***P<0.005. All experiments were carried out on at least two occasions.
Figure 8. MKP-2 deficient macrophages display enhanced arginase-1 expression and activity.

BMD macrophage cell lysates were examined for arginase-1 expression following infection (A). BMD macrophages were treated as appropriate with L-NAME, nor-NOHA, LPS and IFN-γ and subsequently infected with Prugniaud tachyzoites expressing YFP (B). At 24 hours post-infection supernatants were assayed for arginase-1 activity as described in Methods. Each value represents four replicates ± SEM. **P<0.01 ***P<0.005.
Discussion
The present study demonstrates an important role for MKP-2 in controlling infection with T. gondii as infected MKP-2−/− C57BL/6 mice were found to be less able to control parasite growth during both acute and chronic infection as well as displaying increased mortality compared with their wild-type counterparts. The enhanced susceptibility of MKP-2−/− mice was associated with increased tissue arginase-1 expression, generally associated with Th2 responses, and at the same time lowered serum nitrite levels, generally associated with type-1 responses. Nevertheless, increased susceptibility was not associated with any significant modifications of the adaptive immune response between MKP-2 deficient and wild-type mice and the type-1 responses generated in infected MKP-2−/− mice were at least as strong as in their MKP-2+/+ counterparts. Rather, the recently identified unique feature of MKP-2 as a negative regulator of macrophage arginase-1 expression and a positive regulator of macrophage iNOS expression [35] would appear to underlie the important role of this member of the dual specific phosphatase family in controlling infection with T. gondii. While highlighting the importance of iNOS and NO production in controlling T. gondii infection, the present study also uncovered a protective role for arginase-1 in controlling parasite multiplication that can compensate for NO deficiency during early infection.
Generally protective immunity against T. gondii is associated with a type-1 response where IFN-γ synergizes with TNF-α to activate macrophages and induce the expression of inducible nitric oxide synthase (iNOS) that catalyzes the formation of nitric oxide (NO) from L-Arginine. While NO can directly kill the parasites [6], [10] some studies have also shown that NO promotes tachyzoite conversion to the much slower dividing bradyzoite form of the parasite through inhibition of mitochondrial and nuclear enzymes essential for parasite respiration [41]. Although iNOS seems to be the predominant pathway used by classically activated macrophages to control T. gondii proliferation in tissue culture, the role of NO during in vivo infection is less clear. Studies using iNOS-deficient mice have shown that mice lacking iNOS are able to survive and control tachyzoite growth during the acute stage of infection via an IFN-γ dependent mechanism and only succumb during chronic toxoplasmosis [42]. Death was associated with uncontrolled proliferation of tachyzoites in the brain, suggesting that the protective anti-toxoplasmic effect in the brain was iNOS-dependent [42], [43]. Nevertheless, the observation that iNOS deficient mice were able to survive acute infection in an IFN-γ mediated manner suggested that there were alternative pathways other than NO production mediating anti-toxoplasma resistance in vivo [44]. In addition, a further IFN-γ and NO independent control of T. gondii has been identified in IFN regulatory factor-1 gene deficient mice [45].
In the present study we found that L-NAME treatment of wild-type mice infected with 20,000 type-II Prugniaud-FLUC tachyzoites but not MKP-2−/− mice resulted in enhanced parasite burden and mortality early in infection. This indicated that an NO independent mechanism was playing a protective role in MKP-2−/− mice and controlling parasite growth under conditions where NO generation was being inhibited. Previously induction of the IFN-γ inducible gene IDO has been implicated in mediating some of the IFN-γ dependent NO independent anti-Toxoplasma activity [11]. IDO catalyzes the degradation of the essential amino acid L-tryptophan through the kynurenine pathway, thereby depriving the parasite of this essential amino acid [12]. Interestingly the relative contributions of iNOS and IDO to parasite control appear to be tissue specific [12]. More recently immunity-related GTPases (IRGs) have emerged as potent effectors of T gondii killing in mice [13], [14]. Thus, murine astrocytes have been shown to have the ability to kill intracellular T. gondii independently of iNOS and IDO via IFN-γ inducible IRGs [18]–[20]. Different IRGs have been shown to play roles in controlling acute and chronic infection [16], [17] although the mechanisms through which p47 GTPases confer resistance to T. gondii infection have not been determined [21]. However, as no difference in IFN-γ production was noted between infected MKP-2+/+ and MKP-2−/− mice infected with T. gondii this would suggest GTPase production was not associated with the NO independent resistance demonstrated by MKP-2−/− mice. We have now examined qRT-PCR the expression of the IFN-γ inducible genes LRG47 and IGTP GTPase in infected MKP-2−/− and MKP-2+/+ mice and indeed found them comparable. Consequently, this would suggest that an IFN-γ independent mechanism was operating to protect MKP-2−/− mice in the absence of NO production.
Butcher and colleagues (2011) have recently demonstrated that the type-1 strain T. gondii parasites deficient in ROP16 have enhanced growth in macrophages, and in vivo infection results in increased parasite multiplication and dissemination in the host. This has been associated with the inability of ROP16 deficient parasites to induce STAT-6 mediated arginase-1 expression resulting in maintenance of host cell L-arginine which is exploited by the parasite. It has previously been demonstrated that T. gondii is an L-arginine auxotroph [40] and parasite multiplication in the host cell is L-arginine dependent. Previously studies have demonstrated that polymorphisms in ROP-16 from type-2 parasites result in their inability or greatly reduced ability to activate STAT3/6 [46], [47]. However, in the course of the present study these type-2 parasites were shown to increase arginase-1 expression and activity in both MKP-2+/+ and MKP-2−/− macrophages. While often thought of as a Th2 associated product of alternative macrophage activation, innate macrophage activation via TLR-4 ligation [48], [49] has also been shown to induce arginase-1 expression. As T gondii has been demonstrated to have a number of TLR-4 ligands such as GPI anchors [50] and HSP70 [51], [52] this is the likely source of the STAT-6 independent induction of arginase-1 by type-2 strain parasites as demonstrated in the present study. In vivo treatment with nor-NOHA to inhibit arginase-1 activity during the course of the present study resulted in enhanced parasite multiplication in MKP-2−/− mice. This indicated that the enhanced expression of arginase-1 expression in these mice could in some part compensate for iNOS deficiency compared with MKP-2+/+ animals.
Many studies suggest that macrophage killing of parasites via iNOS catalyzed NO production is the main mechanism by which T. gondii parasite multiplication is controlled [33], [53] and that L-arginine depletion by arginase-1 counter-regulates the effectiveness of iNOS and facilitates parasite growth. Our in vitro studies utilizing classically activated BMDM clearly demonstrate that iNOS catalyzed NO production plays the major role in controlling parasite growth and this could be reversed by treatment of cultures with L-NAME. This was irrespective of whether MKP-2−/− or MKP-2+/+ macrophages were examined. Interestingly despite producing less NO than activated MKP-2+/+ macrophages, activated MKP-2−/− macrophages were equally able to control parasite growth. Indeed inhibition of arginase-1 activity by nor-NOHA treatment did not facilitate parasite growth inhibition in activated MKP-2−/− or MKP-2+/+ macrophages, but in fact reversed inhibition of parasite growth early under conditions of activation. This clearly indicates a role for arginase-1 in protection against T. gondii. Surprisingly no differences in intracellular parasite growth in MKP-2−/− versus MKP-2+/+ macrophages in vitro were noted, suggesting the differential weighting of the alternative control mechanisms were compensating for each other over the course of the experiment. Nevertheless, the in vivo consequences of MKP-2 deficiency are significant in terms of parasite growth and long-term survival, and this would be in keeping with NO playing a role post-acute infection. It is also likely that cells other than macrophages are contributing to the dysregulated iNOS/arginase-1 expression bias in vivo in infected MKP-2−/− mice. Likely candidates would be neutrophils: these have previously been shown to play a significant role in early T. gondii infections [54] and have been shown to be major sources of arginase-1 activity in vivo and influence the disease process in humans [55]. This is currently under investigation in the murine model.
Our studies on the consequences of MKP-2 deficiency have revealed some fascinating insights into the control of T. gondii infection. Firstly NO induction is ultimately of paramount significance in controlling parasite multiplication and host survival. However, in the absence of NO production enhanced arginase-1 is able to in part compensate for this deficiency presumably by starving the parasite of L-arginine as previously demonstrated [31]. Previously it has been suggested that arginase-1 stimulates parasite growth by converting L-arginine to the polyamines needed by the parasite [32] or inhibiting iNOS activity by L-arginine depletion [33]. We could find no evidence for this. Rather we would propose that arginase-1 and iNOS work together to control parasite multiplication by a combination of L-arginine starvation (arginase-1 and iNOS) and NO killing (iNOS).
Overall our results demonstrate that MKP-2 through its ability to reciprocally modulate arginase-1 and iNOS expression is a key regulator in L-arginine metabolism and consequently this has clear consequences for the control of intracellular parasites. Furthermore as arginase-1 has also been shown to have potent T cell modulatory effects [55], [56] MKP-2 influences are likely to have significant consequences for inflammatory disease and cancers where arginase-1 and iNOS have already been identified as key players [57]–[59]. These observations identify manipulation of MKP-2 expression or activity as a significant target for future therapeutic strategies.
Supporting Information
In vitro assessment of bioluminescence of T. gondii prior to murine infection and assessment of optimal in vivo imaging time. Prior to infections being carried out T. gondii tachyzoites were assessed for their ability to luminesce (A). This was carried out as a quality control step prior to every infection. The optimal time to image mice following luciferin injection (B) was determined. MKP-2+/+ mice infected with 20,000 FLUC tachyzoites were imaged for 1 minute, every 5 minutes post luciferin injection. A consistent maximum signal was evident from 15–35 minutes post luciferin injection. 20 minutes was therefore chosen for all future experiments.
(TIF)
MKP-2−/− mice have an increased parasite burden and display no differences in immune regulated GTPases associated with control of T. gondii infection. On day 7 post infection, peritoneal exudates from MKP-2+/+ and MKP-2−/− mice infected with FLUC T. gondii were assessed for parasite burden by quantitative real-time PCR (qRT-PCR) for parasitic SAG1 (A) There were no parasites detected by qRT-PCR in the tissues at this stage but by day 10 post-infection increased parasite burdens were noted in the tissues including the lungs of MKP-2−/− mice (B). Splenocytes from infected MKP-2+/+ and MKP-2−/− were analysed by qRT-PCR for expression of murine LRG47 and IRGP (C). Each value represents 4 replicates ±SEM. * P<0.05. Results are from 2 independent experiments
(TIF)
Total numbers of cytokine producing T cells. Splenocytes from T. gondii infected mice were re-stimulated and analysed using flow cytometry. Cells were surface-stained for CD3, CD4 and CD8 and intracellular for IFN-γ and TNF-α. Populations of CD3+ CD4+ T cells (A) and CD3+ CD8+ T cells (B) single or double positive for IFN-γ and TNF-α were determined and normalized to their respective isotype control. Total numbers of cytokine producing cells were calculated by multiplying the percentage value with the total cell number for the individual spleen. Each bar represents the mean of four animals per experimental group ± SEM. All experiments were carried out on at least two occasions.
(TIF)
Infected MKP-2+/+ and MKP-2−/− mice have similar numbers of IFN-γ producing NKT cells. Intraperitoneal exudates from T. gondii infected mice were surface stained for CD3 and Pan-NK and intracellular for IFN-γ and analysed by flow cytometry. Viable, Pan-NK positive cells were analysed for the presence of CD3 and distinguished between CD3+, Pan-NK+ (NKT cells) and CD3−, Pan-NK+ (NK cells) populations and values for IFN-γ positive cells were normalized to their respective isotype control. Since NK cells did not produce any IFN-γ only NKT cells are presented. Shown are the %IFN-γ+ cells from all PanNK+ cells (A) and %IFN-γ+ NKT cells from the total cell population (B). Each bar represents the mean of four animals per experimental group ± SEM.
(TIF)
Primer sequences. Oligonucleotide sequences of primers used for analysis of gene expression by Real-Time PCR.
(TIF)
Funding Statement
This work was supported by grants from the Wellcome Trust, Ref No. 089273 (http://www.wellcome.ac.uk) to RP and JA and the Rhona Reid Charitable Trust to CWR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS (2007) Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 110: 3234–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Denkers EY (2010) Toll-like receptor initiated host defence against Toxoplasma gondii . J Biomed Biotechnol 2010: 737125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Erridge C, Duncan SH, Bereswill S, Heimesaat MM (2010) The induction of colitis and ileitis in mice is associated with marked increases in intestinal concentrations of stimulants of TLRs 2, 4, and 5. PloS One 5: e9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pifer R, Yarovinsky F (2011) Innate responses to Toxoplasma gondii in mice and humans. Trends Parasitol 27: 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leiberman LA, Hunter CA (2002) The role of cytokines and their signaling pathways in the regulation of immunity to Toxoplasma gondii . Int Rev Immunol 21: 373–403. [DOI] [PubMed] [Google Scholar]
- 6. Gazzinelli RT, Thomas A, Sher A (1993) Acute Cerebral Toxoplasmosis is Induced by in vivo neutralization of TNF-α and correlates with the down-regulated expression of inducible nitric oxide synthase and other markers of macrophage activation. J Immunol 151: 3672–3681. [PubMed] [Google Scholar]
- 7. Suzuki Y, Remington JS (1988) Dual regulation of resistance against Toxoplasma gondii infection by Lyt-2+ and Lyt-1+, L3T4+ T cells in mice. J Immunol 140: 3943–3946. [PubMed] [Google Scholar]
- 8. Parker SJ, Roberts CW, Alexander J (1991) CD8 + T cells are the major lymphocyte subpopulation involved in the protective immune response to Toxoplasma gondii in mice. Clin Exp Immunol 84: 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Subauste CS, Koniaris AH, Remington JS (1991) Murine CD8+ cytotoxic T lymphocytes lyse Toxoplasma gondii infected cells. J Immunol 147: 3955–3959. [PubMed] [Google Scholar]
- 10. Jun CD, Kim SH, Kang SS, Chung HT (1993) Nitric Oxide mediates the toxoplasmastic activity of murine microglial cells in vitro. Immunol Invest 22: 487–501. [DOI] [PubMed] [Google Scholar]
- 11. Pfefferkorn ER, Guyre PM (1984) Inhibition of growth of Toxoplasma gondii in cultured fibroblasts by human recombinant gamma interferon. Infect Immun 44: 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fujigaki S, Saito K, Takemura M, Yamada Y, Wada H (2002) l-Tryptophan-l-kynurenine pathway metabolism accelerated by Toxoplasma gondii infection is abolished in gamma interferon-gene-deficient mice: cross-regulation between inducible nitric oxide synthase and pathway metabolism accelerated by Toxoplasma gondii . Infect Immun 70: 770–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taylor GA, Collazo CM, Yap GS, Nguyen K, Gregorio TA, et al. (2000) Pathogen-specific loss of host resistance in mice lacking the IFN-gamma-inducible gene IGTP. Proc Natl Acad Sci USA 97: 751–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collazo CM, Yap GS, Sempowski GD, Lusby KC, Tessarollo L, et al. (2001) Inactivation of LRG-47 and IRG-47 reveals a family of interferon gamma – inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med 194: 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Butcher BA, Greene RI, Henry SC, Annecharico KL, Weinberg JB, et al. (2005) p47 GTPases regulate Toxoplasma gondii survival in activated macrophages. Infect Immun 73: 3278–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, et al. (2005) Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog 1: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hunn JP, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, et al. (2008) Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii . EMBO J 27: 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Halonen SK, Chiu F, Weiss LM (1998) Effect of cytokines on growth of Toxoplasma gondii in murine astrocytes. Infect Immun 66: 4989–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Halonen SK, Taylor GA, Weiss LM (2001) Gamma interferon-induced inhibition of Toxoplasma gondii in astrocytes is mediated by IGTP. Infect Immun 69: 5573–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Melzer T, Duffy A, Weiss LM, Halonen SK (2008) The gamma interferon (IFN-gamma)-inducible GTP-binding protein IGTP is necessary for Toxoplasma vacuolar disruption and induces parasite egression in IFN-gamma-stimulated astrocytes. Infect Immun 76: 4883–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Howard JC, Hunn JP, Steinfeldt T (2011) The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii . Curr Opin Microbiol 14: 414–421. [DOI] [PubMed] [Google Scholar]
- 22. Denkers EY, Gazzinelli RT (1998) Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev 11: 569–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, et al. (1996) In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-γ, and TNF-α. J Immunol 157: 798–805. [PubMed] [Google Scholar]
- 24. Roberts CW, Ferguson DJ, Jebbari H, Satoskar A, Bluethmann H, et al. (1996) Different roles for interleukin-4 during the course of Toxoplasma gondii infection. Infect Immun 64: 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suzuki Y, Sher A, Yap G, Park D, Neyer E, et al. (2000) IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii . J Immunol 164: 5375–5382. [DOI] [PubMed] [Google Scholar]
- 26. Suzuki Y, Yang Q, Yang S, Nguyen N, Lim S (1996) IL-4 is protective against development of toxoplasmic encephalitis. J Immunol 157: 2564–2569. [PubMed] [Google Scholar]
- 27. Wilson EH, Wille-Reece U, Dzierszinski F, Hunter CA (2005) A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. J Neuroimmunol 165: 63–74. [DOI] [PubMed] [Google Scholar]
- 28. Couper KN, Lanthier PA, Perona-Wright G, Kummer LW, Chen W, et al. (2009) Anti-CD25 antibody-mediated depletion of effector T cell populations enhances susceptibility of mice to acute but not chronic Toxoplasma gondii infection. J Immunol 182: 3985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morampudi V, De Craeye S, Le Moine A, Detienne S, Braun MY, et al. (2011) Partial depletion of CD4(+)CD25(+)Foxp3(+) T regulatory cells significantly increases morbidity during acute phase Toxoplasma gondii infection in resistant BALB/c mice. Microbes Infect 13: 394–404. [DOI] [PubMed] [Google Scholar]
- 30. Marshall ES, Elshekiha HM, Hakimi MA, Flynn RJ (2011) Toxoplasma gondii peroxiredoxin promotes altered macrophage function, caspase-1-dependent IL-1β secretion enhances parasite replication. Vet Res 42: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Butcher B, Fox BA, Rommereim LM, Kim SG, Maurer KJ, et al. (2011) Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog 7: e1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jensen KDC, Wang Y, Wojno EDT, Shastri AJ, Hu K, et al. (2011) Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe 9: 472–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. El-Kasmi KC, Qualls JE, Pesce JT, Smith AM, Robert W, et al. (2008) Toll-like receptor–induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol 9: 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pfaff AW, Villard O, Klein JP, Mousli M, Candolfi E (2005) Regulation of Toxoplasma gondii multiplication in BeWo trophoblast cells: cross-regulation of nitric oxide production and polyamine biosynthesis. Int J Parasitol 35: 1569–1576. [DOI] [PubMed] [Google Scholar]
- 35. Al-Mutairi MS, Cadalbert LC, McGachy HA, Shweash M, Schroeder J, et al. (2010) MAP kinase phosphatase-2 plays a critical role in response to infection by Leishmania mexicana . PloS Pathog 6: e1001192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim SK, Karasoc A, Boothroyd JC (2007) Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infect Immun 75: 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jones LA, Roberts F, Nickdel MB, Brombacher F, McKenzie ANJ, et al. (2010) IL-33 receptor (T1/ST2) signalling is necessary to prevent the development of encephalitis in mice infected with Toxoplasma gondii . Eur J Immunol 40: 426–36. [DOI] [PubMed] [Google Scholar]
- 38. Roberts F, Roberts CW, Ferguson DJP, McLeod R (2000) Inhibition of nitric oxide production exacerbates chronic ocular Toxoplasmosis. Parasite Immunol 22: 1–5. [DOI] [PubMed] [Google Scholar]
- 39. Kropf P, Fuentes JM, Fahnrich E, Arpa L, Herath S, et al. (2005) Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo . The FASEB J 19: 1000–2. [DOI] [PubMed] [Google Scholar]
- 40. Fox BA, Gigley JP, Bzik DJ (2004) Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int J Parasitol 34: 323–331. [DOI] [PubMed] [Google Scholar]
- 41. Bohne W, Heesemann J, Gross U (1993) Induction of bradyzoite-specific Toxoplasma gondii antigens in gamma interferon-treated mouse macrophages. Infect Immun 61: 1141–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scharton-Kersten TM, Yap G, Magram J, Sher A (1997) Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii . J Exp Med 185: 1261–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alexander J, Scharton-Kersten TM, Yap G, Roberts CW, Liew FY, et al. (1997) Mechanisms of innate resistance to Toxoplasma gondii infection. Philos Trans R Soc Lond B Biol Sci 352: 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khan IA, Matsuura T, Kasper LH (1998) Inducible nitric oxide synthase is not required for long-term vaccine-based immunity against Toxoplasma gondii . J Immunol 161: 2994–3000. [PubMed] [Google Scholar]
- 45. Khan IA, Matsuura T, Fonseka S, Kasper LH (1996) Production of nitric oxide (NO) is not essential for protection against acute Toxoplasma gondii infection in IRF-1−/− mice. J Immunol 156: 636–643. [PubMed] [Google Scholar]
- 46. Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, et al. (2007) Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445: 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, et al. (2009) A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med 206: 2747–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Menzies FM, Henriquez FL, Alexander J, Roberts CW (2011) Selective inhibition and augmentation of alternative macrophage activation by progesterone. Immunol 134: 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shweash M, McGachy AH, Schroeder J, Neamatallah T, Bryant CE, et al. (2011) Leishmania mexicana promastigotes inhibit macrophage IL-12 production via TLR-4 dependent COX-2, iNOS and arginase-1 expression. Mol Immunol 48: 1800–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yap GS, Shaw MH, Ling Y, Sher A (2006) Genetic analysis of host resistance to intracellular pathogens: lessons from studies of Toxoplasma gondii infection. Microbes Infect 8: 1174–1178. [DOI] [PubMed] [Google Scholar]
- 51. Debierre-Grockiego F, Campos MA, Azzouz N, Schmidt J, Bieker U, et al. (2007) Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii . J Immunol 179: 1129–1137. [DOI] [PubMed] [Google Scholar]
- 52. Mun H, Aosai F, Norose K, Mun H, Aosai F, et al. (2005) Toll-Like receptor 4 mediates tolerance in macrophages stimulated with Toxoplasma gondii- derived heat shock protein 70. Infect Immun 73: 4634–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li Z, Zhao Z-J, Zhu X-Q, Ren Q-S, Nie F-F, et al. (2012) Differences in iNOS and arginase expression and activity in the macrophages of rats are responsible for the resistance against T. gondii infection. PloS One 7: e35834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Egan CE, Sukhumavasi W, Bierly AL, Denkers EY (2008) Understanding the multiple functions of Gr-1(+) cell subpopulations during microbial infection. Immunol Res 40: 35–48. [DOI] [PubMed] [Google Scholar]
- 55. Munder M (2009) Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol 158: 638–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Modolell M, Choi B-S, Ryan RO, Hancock M, Titus RG, et al. (2009) Local suppression of T cell responses by arginase-induced L-arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis 3: e480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cao W, Sun B, Feitelson MA, Wu T, Tur-Kaspa R, et al. (2009) Hepatitis C virus targets over-expression of arginase I in hepatocarcinogenesis. Int J Cancer 124: 2886–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thanan R, Ma N, Iijima K, Abe Y, Koike T, et al. (2012) Proton pump inhibitors suppress iNOS-dependent DNA damage in Barrett's esophagus by increasing Mn-SOD expression. Biochem Bioph Res Commun 421: 280–285. [DOI] [PubMed] [Google Scholar]
- 59. Waha A, Felsberg J, Hartmann W, von dem Knesebeck A, Mikeska T, et al. (2010) Epigenetic downregulation of mitogen-activated protein kinase phosphatase MKP-2 relieves its growth suppressive activity in glioma cells. Cancer Res 70: 1689–1699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vitro assessment of bioluminescence of T. gondii prior to murine infection and assessment of optimal in vivo imaging time. Prior to infections being carried out T. gondii tachyzoites were assessed for their ability to luminesce (A). This was carried out as a quality control step prior to every infection. The optimal time to image mice following luciferin injection (B) was determined. MKP-2+/+ mice infected with 20,000 FLUC tachyzoites were imaged for 1 minute, every 5 minutes post luciferin injection. A consistent maximum signal was evident from 15–35 minutes post luciferin injection. 20 minutes was therefore chosen for all future experiments.
(TIF)
MKP-2−/− mice have an increased parasite burden and display no differences in immune regulated GTPases associated with control of T. gondii infection. On day 7 post infection, peritoneal exudates from MKP-2+/+ and MKP-2−/− mice infected with FLUC T. gondii were assessed for parasite burden by quantitative real-time PCR (qRT-PCR) for parasitic SAG1 (A) There were no parasites detected by qRT-PCR in the tissues at this stage but by day 10 post-infection increased parasite burdens were noted in the tissues including the lungs of MKP-2−/− mice (B). Splenocytes from infected MKP-2+/+ and MKP-2−/− were analysed by qRT-PCR for expression of murine LRG47 and IRGP (C). Each value represents 4 replicates ±SEM. * P<0.05. Results are from 2 independent experiments
(TIF)
Total numbers of cytokine producing T cells. Splenocytes from T. gondii infected mice were re-stimulated and analysed using flow cytometry. Cells were surface-stained for CD3, CD4 and CD8 and intracellular for IFN-γ and TNF-α. Populations of CD3+ CD4+ T cells (A) and CD3+ CD8+ T cells (B) single or double positive for IFN-γ and TNF-α were determined and normalized to their respective isotype control. Total numbers of cytokine producing cells were calculated by multiplying the percentage value with the total cell number for the individual spleen. Each bar represents the mean of four animals per experimental group ± SEM. All experiments were carried out on at least two occasions.
(TIF)
Infected MKP-2+/+ and MKP-2−/− mice have similar numbers of IFN-γ producing NKT cells. Intraperitoneal exudates from T. gondii infected mice were surface stained for CD3 and Pan-NK and intracellular for IFN-γ and analysed by flow cytometry. Viable, Pan-NK positive cells were analysed for the presence of CD3 and distinguished between CD3+, Pan-NK+ (NKT cells) and CD3−, Pan-NK+ (NK cells) populations and values for IFN-γ positive cells were normalized to their respective isotype control. Since NK cells did not produce any IFN-γ only NKT cells are presented. Shown are the %IFN-γ+ cells from all PanNK+ cells (A) and %IFN-γ+ NKT cells from the total cell population (B). Each bar represents the mean of four animals per experimental group ± SEM.
(TIF)
Primer sequences. Oligonucleotide sequences of primers used for analysis of gene expression by Real-Time PCR.
(TIF)