

Abstract
The coordinated assembly of the DNA polymerase (gp43), the sliding clamp (gp45), and the clamp loader (gp44/62) to form the bacteriophage T4 DNA polymerase holoenzyme is a multistep process. A partially opened toroid-shaped gp45 is loaded around DNA by gp44/62 in an ATP-dependent manner. Gp43 binds to this complex to generate the holoenzyme in which gp45 acts to topologically link gp43 to DNA, effectively increasing the processivity of DNA replication. Stopped-flow fluorescence resonance energy transfer was used to investigate the opening and closing of the gp45 ring during holoenzyme assembly. By using two site-specific mutants of gp45 along with a previously characterized gp45 mutant, we tracked changes in distances across the gp45 subunit interface through seven conformational changes associated with holoenzyme assembly. Initially, gp45 is partially open within the plane of the ring at one of the three subunit interfaces. On addition of gp44/62 and ATP, this interface of gp45 opens further in-plane through the hydrolysis of ATP. Addition of DNA and hydrolysis of ATP close gp45 in an out-of-plane conformation. The final holoenzyme is formed by the addition of gp43, which causes gp45 to close further in plane, leaving the subunit interface open slightly. This open interface of gp45 in the final holoenzyme state is proposed to interact with the C-terminal tail of gp43, providing a point of contact between gp45 and gp43. This study further defines the dynamic process of bacteriophage T4 polymerase holoenzyme assembly.
DNA replication requires the coordinated assembly of many proteins to form a replisome responsible for copying an organism's genome. The generation of two holoenzymes, one for leading- and one for lagging-strand synthesis, proceeds stepwise with many intermediates. The bacteriophage T4 DNA polymerase holoenzyme is derived from the polymerase (gp43), the clamp (gp45), and the clamp–loader complex (gp44/62) (1, 2). Analogous proteins are found in both the Escherichia coli holoenzyme, consisting of the DNA polymerase III, the β clamp, and the clamp-loading γ complex, and in the eukaryotic holoenzyme, consisting of DNA polymerase δ, the proliferating cell nuclear antigen (PCNA) clamp, and the clamp-loading RF–C complex (3–7). The sliding clamp, gp45, of bacteriophage T4 is a ring-shaped protein trimer (Fig. 1A) that is an essential component of the holoenzyme, which acts by conferring the property of processivity on the DNA polymerase. X-ray crystallography has shown the clamps from the various systems to be similar in overall structure but different in oligomeric structure, with gp45 (8, 9) and PCNA (10) formed as trimers and the β clamp (11) as a dimer. The clamp–loader complex, gp44/62, is a 4:1 complex of gp44 and gp62 that sequentially hydrolyzes two sets of two molecules of ATP while loading gp45 onto DNA and also facilitates the gp45-gp43 interaction (12–14). The polymerase, gp43, incorporates nucleotides in a 5′ to 3′ direction complementary to a DNA template. The coordinated actions of these proteins in bacteriophage T4 result in a highly efficient model system for studying DNA replication.
Figure 1.

(A) X-ray crystal structure of gp45 showing the interdomain connecting loop and the subunit interface. (B) In-plane model of opening of gp45 showing the location of the mutations: V163C in blue, S158C in green, and T168C in pink, with the donor tryptophan in orange. Each mutation is shown only once for clarity. (C) Out-of-plane model of opening.
Without the crystal structure of gp44/62 or other structural information, an exact model of the interaction between gp45, gp44/62, gp43, and the holoenzyme waits to be determined. However, the structures of individual components of the holoenzyme have been elucidated as well as specific points of interaction between the proteins. The x-ray crystal structure of gp43 from bacteriophage RB69 has been solved (15) and shares 74% sequence similarity with the bacteriophage T4 gp43 (16). Only one region of gp43 from T4 lacks significant sequence homology to gp43 from RB69, and this area has been implicated in the dimerization of the polymerases (17). Photocrosslinking has determined that gp44/62 and gp43 interact with gp45 on the same “rough” face of the clamp (18–20). Recently, we have shown that the C-terminal tail of gp43 crosslinks to amino acid residues within the subunit interface of gp45 (21) and is absolutely required for holoenzyme assembly (22).
A variety of steady-state and presteady-state techniques have been used to investigate the kinetic assembly of the holoenzyme in bacteriophage T4 (23–25) and in E. coli (26–30) Both systems are assembled stepwise to form their holoenzymes. In bacteriophage T4, gp44/62 hydrolyzes two molecules of ATP to open gp45 and then two more molecules of ATP on interaction with DNA (14, 23, 24, 31). In E. coli, the γ complex hydrolyzes two to three molecules of ATP (32) on interaction with DNA. It has not been unequivocally determined whether this hydrolysis is sequential or simultaneous because of conflicting results (26, 30). In either case, the structural changes that occur from ATP hydrolysis in the clamp or clamp–loader complex have not been identified.
In the present experiments, we investigated the directionality of gp45 subunit interface opening and closing during holoenzyme assembly by using presteady-state fluorescence resonance energy transfer (FRET) techniques. Initially, we discovered that one of the three subunit interfaces of gp45 is open to a distance of 35–38 Å, and the other two interfaces are closed with a distance of 19 Å (as measured between W91 on one subunit interface and a fluorophore on the opposite interface) (33). By creating three site-specific mutants of gp45 (V163C, S158C, and T168C) singly labeled with a fluorophore, energy transfer was detected from a single endogenous tryptophan (W91) during holoenzyme assembly. These fluorophores are located across the subunit interface from W91 and provide a means of assessing the gp45 conformation. From those experimental FRET values, the distance between a fluorophore acceptor and a tryptophan donor was calculated and provided a model for the opening and closing of gp45 during the formation of the holoenzyme. We propose two limiting models for the opening and closing of gp45: the first in which gp45 opens within the plane of the ring and a second in which gp45 opens out-of-plane of the ring (Fig. 1 B and C). The actual mechanism of opening and closing during holoenzyme assembly may be a combination of these two models. The site-specific mutants of gp45 were chosen because the location of the residues in the subunit interface relative to the fixed tryptophan would distinguish between the in-plane and out-of-plane model of opening.
We had previously used the V163C mutant to develop a complex kinetic mechanism of holoenzyme assembly (23). This mechanism proceeded through 10 steps and 7 conformational changes in gp45. As the clamp-loading process proceeded, we discovered that interaction of gp45 with gp44/62 and ATP powers an opening of gp45 separating the fluorophore pair by a distance greater than 45 Å. The binding to DNA leads to spontaneous ring closure of gp45, which left a partial separation of the subunit interface in the final complex, allowing for an insertion of C terminus of gp43. Our present measurements confirm and further define the magnitude and directionality of these stepwise events.
Materials and Methods
Materials and Their Sources.
Oligonucleotide primers and substrates were prepared as previously described (14, 33, 34). The forked bio62/34/36mer primer-template blocked with streptavidin (34) was used as the DNA substrate in all experiments. 7-Diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM) was obtained from Molecular Probes. ATP was from Boehringer–Mannheim. Deep Vent DNA polymerase and all restriction enzymes were from New England Biolabs. Steptavidin was from Sigma. All other chemicals were of analytical grade or better.
Cloning, Protein Purification, and Labeling.
The following PCR primers were used in preparation of the mutants in this manuscript: (A) 5′-GCG GAA TTC CAT ATG AAA CTG TCT AAA GAT; (B) 5′-GCG GAA TTC GGA TTC CTA TTA AAA ATC GTG GGT; (C) 5′-GGT TTT AAT AAA GTA GAA GAT TGC GCA CTG ACC CGT GTT AAA TAT; (D) 5′-ATA TTT AAC ACG GGT CAG TGC GCA ATC TTC TAC TTT ATT AAA ACC; (E) 5′-GTT AAA TAT TCT TTG TGC CTA GGT GAT TAT GAT; (F) 5′-ATC ATA ATC ACC TAG GCA CAA AGA ATA TTT AAC; (G) 5′-CGC TCA ACA ATT TTT TTC GCG GCG GCC GAT CCG AGT ACA; and (H) 5′-TGT ACT CGG ATC GGC CGC CGG GAA AAA AAT TGT TGA GCG. Fragment AD was prepared by PCR by using primers A and D and pET26b-W199F (33) as a template. Fragment BC was created similarly by using PCR. Overlap extension PCR by using fragments AD and BC yielded the mutant gp45 gene S158C/W199F that included a new FspI site. The full-length fragment was digested with NdeI and BamHI and ligated into pET26b similarly digested to yield pET26b-S158C/W199F. Likewise, pET26b-T168C/W199F was created by using fragments AF and BE for overlap extension and included a new AvrII site. pET26b-W92F/S158C/W199F and pET26b-W92F/T168C/W199F were then created from the respective mutants above by using fragments AH and BG in the overlap extension reaction yielding a new BsiEI site. Purification and labeling with CPM were performed as described previously (12, 33). Mutations were confirmed by DNA sequencing. Protein concentrations of CPM-labeled gp45 mutants were determined by a spectrometric Bradford protein assay (Bio-Rad) by using wild-type gp45 as a standard. Wild-type gp45 and other protein concentrations were determined by UV spectroscopy at 280 nm by using the appropriate extinction coefficients. The ability of all mutants, in both the unlabeled and CPM-labeled forms, to stimulate ATPase activity was verified (Table 1), as described earlier (33). Wild-type gp45, gp44/62, and exonuclease-deficient gp43 were purified as previously described (35–37).
Table 1.
Activities of wild-type and mutant forms of gp45
| gp45 species | ATPase, nM s−1*
|
||
|---|---|---|---|
| gp45 and gp44/62 | gp45, gp44/62, and DNA | gp45, gp44/62, DNA, and gp43 | |
| Wild type | 19 | 303 | 23 |
| S158C/W199F | 13 | 316 | 17 |
| S158C/W199F-CPM | 12 | 101 | 12 |
| T168C/W199F | 19 | 263 | 27 |
| T168C/W199F-CPM | 19 | 318 | 45 |
| W92F/S158C/W199F | 25 | 297 | 28 |
| W92F/S158C/W199F-CPM | 23 | 196 | 26 |
| W92F/T168C/W199F | 22 | 304 | 35 |
| W92F/T168C/W199F-CPM | 14 | 266 | 28 |
Verification of the ability of the mutants to simulate ATPase activity in the presence of DNA. Shutdown rates of ATPase activity with the addition of gp43 were very close to the basal rate with gp45 and gp44/62 alone. The ability to stimulate and shut down ATPase activity is a sufficient test for the formation of a functional holoenzyme. Experimental error and reproducibility is 10%.
Steady-State Fluorescence Spectroscopy.
Steady-state fluorescence experiments were performed on an ISA (Edison, NJ) FluoroMax-2 spectrofluorometer thermostated to 25°C. Fluorescence experiments were carried out as described previously (33). Polarization experiments were performed on the same instrument equipped with automated polarizers. Distances between the tryptophan donor and the CPM acceptor-labeled mutants can be calculated (Eq. 1) by using the following relationships for tryptophan quenching (Eq. 2) and acceptor sensitization (Eq. 3) (38):
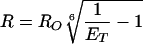 |
1 |
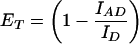 |
2 |
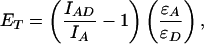 |
3 |
where ET is the transfer efficiency of the FRET process, and IAD and ID are the fluorescent intensities of the tryptophan donor in the presence and absence, respectively, of the CPM acceptor. IA is the fluorescence intensity of the CPM acceptor in the absence of the tryptophan donor. ɛA and ɛD are the extinction coefficients of the CPM acceptor and the tryptophan donor at 290 nm and are equal to 3,340 M−1⋅cm−1 and 4,100 M−1⋅cm−1, respectively (23). RO is the Förster distance at which the transfer efficiency is 50%, and R is the distance between the donor and acceptor. RO was calculated by using Eq. 4 (39) and determined to be 29 Å for both S158C/W199F and T168C/W199F mutants, which is consistent with previous values (33, 40).
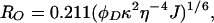 |
4 |
where φD is the quantum yield of the donor, κ2 is the orientation factor, η is the refractive index of the medium, and J is the overlap integral between the fluorescence spectrum of the donor and the absorption spectrum of the acceptor. κ2 relates the relative orientation of the donor and acceptor pair and has a value between 0 and 4. For a freely rotating probe, κ2 is assumed to be 2/3 (38). The rotational freedom of the probe is measured by using steady-state fluorescence polarization spectroscopy. Both W92 and CPM in the gp45-labeled mutants have anisotropy values less than 0.3 when exciting at 290 and 390 nm, respectively, which is consistent with a freely rotating probe (41). By using our previous assumption that RO values measured for the CPM-labeled mutants in complex buffer are appropriate for the intermediate states of gp45 during holoenzyme assembly (23, 33), polarization values less than 0.3 (23) will result in errors in the R values being less than 10% (41). IAD and IA for the gp45 mutants in complex buffer were determined by steady-state fluorescence as before (23) to obtain the starting point distance between W91 and CPM-labeled mutants in solution.
Stopped-Flow Fluorescence Spectroscopy.
Stopped-flow fluorescence experiments were performed on an Applied Photophysics (Surrey, U.K.) SX.18MV stopped-flow reaction analyzer in fluorescence mode at the constant temperature of 25°C. The samples were excited at 290 or 390 nm, as explained previously (23). A 420-nm cutoff filter was used to detect only CPM fluorescence. The excitation path length was 2 mm. Single or split time bases were used as appropriate, with 2,000 data points collected. Excitation at 290 nm gave FRET between W92 and S158C-CPM or T168C-CPM, whereas excitation at 390 nm allowed for direct excitation of CPM without any contribution from tryptophan. The relative changes in fluorescence (F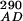 ) were corrected for changes in fluorophore environment and interprotein FRET (F
) were corrected for changes in fluorophore environment and interprotein FRET (F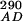 and F
and F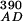 and converted to fluorescence intensities (IAD and IA), as described previously (23).
and converted to fluorescence intensities (IAD and IA), as described previously (23).
The energy transfer values (ET) obtained experimentally are an average of the distances between the tryptophan donor and the CPM acceptor at all three subunit interfaces. We have assumed before that two of the subunit interfaces are closed (EC) and do not change, whereas the third interface is opened (EO) during the holoenzyme formation process (23, 33). Using this assumption, we can calculate the amount of EO from the open interface when EC is known for the closed interfaces by using Eq. 5
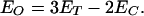 |
5 |
Molecular Modeling Calculations.
Molecular modeling was performed by using insight ii (ver. 98, Molecular Simulations, Waltham, MA) running on a Silicon Graphics O2 12000 (Mountain View, CA). The crystal structure of gp45 (Protein Data Bank accession no. 1CZD) (8) was modified by introducing two new bonds (A1166:O—C3089:O and C3166:O—B2089:O) to connect the subunits. Closed subunit distances for mutants W199F/S158C and W199F/T168C were modeled on the basis of the experimental ET value of 0.95 for the crosslinked A42C/V163C/W199F/A214C gp45 mutant described previously (33). EC values of the closed subunit interfaces were then calculated to be 0.59 and 0.60 for the W199F/S158C and W199F/T168C mutants, respectively. Torsion angles, φ between residues K105 and P106 and ψ of residue P108 in the interdomain-connecting loop opposite the subunit interface, were modified in the x-ray crystal structure to open and close gp45 in either an in-plane or out-of-plane orientation.
Determination of the Kd Values.
The Kd values were determined by fitting the plot of FAD or FA versus protein concentration to the quadratic equation, as explained previously (23), by using kaleidagraph (Synergy Software, Reading, PA).
Results
Steps in the Formation of the T4 Holoenzyme.
We have used two gp45 mutants, S158C/W199F and T168C/W199F, along with a previous mutant, V163C/W199F (23), to triangulate the direction of opening and closing of gp45 during the formation of the bacteriophage T4 polymerase holoenzyme. With the aid of the x-ray crystal structure, sites for the attachment of an acceptor fluorophore were chosen on the basis of the distances across the subunit interface from an inherent tryptophan (W92) in one subunit to various residues on an adjacent subunit (Fig. 1). The other tryptophan (W199) in gp45 was mutated to a nonfluorescent phenylalanine to simplify FRET measurements. Previous measurements using the V163C/W199F mutant were consistent with a model solution structure for gp45 in which one subunit interface has a measured distance (between the W91 donor and the V162C-CPM acceptor) of 36 Å with the other two interfaces closed (33). Changes in the FRET emanating from the W91 donor-V162C-CPM acceptor pair on interaction with other holoenzyme components were used previously to determine a 10-step kinetic scheme for assembly (Scheme S1) (23). In this study, we tracked the direction of gp45 opening and closing by using three site-specific fluorescent mutants of gp45. The experimental distances were then integrated to provide a global fit of the movements of gp45 to either an in-plane or out-of-plane model.
Scheme 1.

Steady-State Fluorescence of gp45 Alone in Solution.
The distance between subunits of gp45A (see Scheme S1 for a diagram of states A–K, designated by superscripts) alone in solution was measured with acceptor-sensitization or tryptophan-quenching techniques to determine the distance between the fluorescent tryptophan donor and the CPM acceptor. Excitation at 290 and 390 nm resulted in an acceptor-sensitization ratio IAD/IA for S158C/W199F of 1.502 and for T168C/W199F of 1.508. ET values determined from Eq. 3 were 0.409 for S158C/W199F and 0.414 for T168C/W199F (Table 2). ET values for acceptor sensitization were consistent with ET values of 0.479 for S158C/W199F and 0.482 for T168C/W199F determined by tryptophan quenching. Calculation of the distance between the tryptophan donor and CPM acceptor for the open subunit in gp45A is 48 Å for both S158C/W199F and T168C/W199F.
Table 2.
Steady-state fluorescence data during holoenzyme assembly
| Assembly state | S158C/W199F
|
T168C/W199F
|
||
|---|---|---|---|---|
| ET | Rave, Å* | ET | Rave, Å* | |
| gp45A | 0.409 | 30.9 | 0.414 | 30.4 |
| gp45D, gp44/62, ATP | 0.352 | 32.2 | 0.384 | 31.1 |
| gp45H, gp44/62, ATP, DNA | 0.237 | 35.4† | 0.453 | 29.6† |
| gp45K, gp44/62, ATP, DNA, gp43 | 0.379 | 31.6 | 0.506 | 28.6 |
Distances reported as an average for all subunit interfaces.
Addition of DNA to the assembly state caused a decrease in ET and therefore an increase in distance in S158C/W199F, whereas the same change in the assembly state caused an increase in ET and therefore an decrease in distance in T168C/W199F.
Steady-state fluorescence measurements were also taken at each step of the holoenzyme assembly for each of the mutants (Table 2). Decreases in ET were observed with addition of gp44/62 and ATP for both mutants, suggesting an opening of the gp45D interface. When DNA was added to gp45D, the ET decreased for the S158C/W199F mutant and increased for the T168C/W199F mutant. The apparent difference in the ET values for each mutant suggests that CPM labels are moving in different directions from the donor tryptophan. This can be explained by an out-of-plane movement in gp45H in which the fluorophore in S158C/W199F increased its distance from the donor, whereas that in T168C/W199F decreased its distance from the donor. Increases in ET were then observed for both mutants with the addition of gp43 to the assembly, which is explained by a closing of the subunit interface of gp45K in plane. Further experiments were then done to determine the changes in FRET and associated distances, along with the evaluation of presteady-state rates by using stopped-flow fluorescence spectroscopy.
Investigation of gp45 Opening and Closing by Using Stopped-Flow Fluorescence Spectroscopy.
Presteady-state fluorescence values were measured for each state of the holoenzyme assembly following Scheme S1. Steps 1, 4, and 8 and thus gp45 states B, E, and I are fluorescently silent and correspond to binding events (23). FRET measurements were made by exciting double or triple mutants of gp45 at either 290 or 390 nm. Excitation of the double mutants of gp45 at 290 nm results in observed FRET changes because of intramolecular FRET (W92 to gp45-CPM), intermolecular FRET (tryptophans in other interacting proteins), and changes in CPM environment. Excitation of the triple mutants of gp45 at 290 nm results in observed FRET changes because of intermolecular FRET and CPM environmental changes. Excitation of the triple mutants of gp45 at 390 nm result in observed FRET changes because of CPM environmental changes. By using both double and triple mutants of gp45 and excitation at 290 or 390 nm, intermolecular FRET values and CPM environmental changes can be subtracted to yield only changes in intramolecular FRET resulting from the opening and closing of a subunit interface of gp45.
Interaction of gp45 and gp44/62 in the Presence of ATP.
The first step in the holoenzyme assembly process is the interaction between gp45A and gp44/62 followed by hydrolysis of ATP and opening of the clamp. Solutions containing 2 μM of either S158C/W199F-CPM or T168C/W199F-CPM in complex buffer were mixed with 2 μM gp44/62 in complex buffer and 2 mM ATP. Excitation at 290 nm showed two increases in CPM-associated fluorescent amplitude for both mutants (gp45C and gp45D), with the first increase larger than the second. S158C/W199F-CPM yielded a larger positive amplitude change than T168C/W199F-CPM (Fig. 2 and Tables 4 and 5, which are published as supplementary material on the PNAS web site, www.pnas.org). Crosslinking data previously identified gp44/62 as interacting on the “rough” face of gp45, which is the same face where S158 is located (19, 20, 42). Consequently, the amplitude of the fluorescent change is much larger in the S158C/W199F-CPM mutant because of a much larger contribution from intermolecular FRET. Altering the gp44/62 concentration did not change the observed rate constants of 6.3 ± 0.1 s−1 and 0.77 ± 0.01 s−1 for S158C/W199F-CPM and 2.1 ± 0.1 s−1 and 0.28 ± 0.02 s−1 for T168C/W199F-CPM for steps 2 and 3. The normalized fluorescent amplitude, however, increased with increasing concentration of gp44/62 for both mutants, yielding a KD for S158C/W199F-CPM of 0.020 ± 0.038 μM and a KD for T168C/W199F-CPM of 0.036 ± 0.020 μM for the dissociation of gp45C⋅gp44/62 to gp45A and gp44/62.
Figure 2.

Relative stopped-flow fluorescence values for the interaction of gp45A with gp44/62 and ATP. S158C/W199F data are in green, and T168C/W199F data are in pink.
When the experiment was repeated by using W92F/S158C/W199F-CPM or W92F/T168C/W199F-CPM and exciting at either 290 or 390 nm, two increases in fluorescent amplitude were also observed, with the first change larger than the second for both labeled mutants (Tables 4 and 5). By using these triple mutants, any fluorescence because of interprotein FRET can be calculated and subtracted from the 290-nm excitation value above to yield exclusively changes in intramolecular FRET. The observed rate constants at 290 nm excitation for W92F/S158C/W199F-CPM were 6.0 ± 0.1 s−1 and 0.47 ± 0.01 s−1 and for W92F/T168C/W199F-CPM were 4.1 ± 0.1 s−1 and 1.0 ± 0.1 s−1. The observed rate constants at 390 nm excitation for W92F/S158C/W199F-CPM were 5.9 ± 0.1 s−1 and 0.62 ± 0.01 s−1 and for W92F/T168C/W199F-CPM were 2.3 ± 0.1 s−1 and 0.74 ± 0.05 s−1. These observed rates compared well with the above rates for the double mutants. The extent of this opening cannot be measured accurately with the donor and acceptor pair used in these experiments because of the extreme distance that gp45 opens on addition of 44/62 and ATP, therefore neither limiting model can be suggested at this stage.
Formation of a gp45-gp44/62–DNA Complex.
Solutions containing 2 μM of the gp45D–gp44/62 complex formed in the presence of 2 mM ATP and 2 μM of the DNA substrate in complex buffer were mixed and gave rise to three changes in fluorescent amplitude for each of the double mutants of gp45 when excited at 290 nm. The S158C/W199F-CPM mutant showed one increase and two decreases, whereas the T168C/W199F-CPM mutant showed an increase, a decrease, and another increase in normalized fluorescence (Fig. 3 and Tables 4 and 5). The observed rate constants for S158C/W199F-CPM were 49 ± 1 s−1, 8.9 ± 0.4 s−1, and 0.72 ± 0.07 s−1 and for T168C/W199F-CPM were 42 ± 1 s−1, 4.4 ± 0.2 s−1, and 1.06 ± 0.02 s−1 compared well with previous results (23). The first rate constant depended on DNA concentration, suggesting that DNA binding was rate limiting in the first step. Doubling the DNA concentration increased the rate to 67 ± 2 s−1 for S158C/W199F-CPM and 72 ± 1 s−1 for T168C/W199F-CPM for step 5. Halving the DNA concentration decreased the rate to 26 ± 1 s−1 for S158C/W199F-CPM and 25 ± 1 s−1 for T168C/W199F-CPM. The rate constants of the second and third changes in amplitude did not depend on DNA concentrations, indicating that these changes are first-order processes.
Figure 3.

Relative stopped-flow fluorescence values for the interaction of gp45D and gp44/62 and ATP with DNA. S158C/W199F data are in green, and T168C/W199F data are in pink.
Repeating these experiments with the triple mutants showed similar trends. Excitation at 290 nm yielded three changes in fluorescent amplitude, with observed rate constants of 33 ± 1 s−1, 19 ± 1 s−1, and 0.64 ± 0.10 s−1 for W91F/S158C/W199F-CPM and 30 ± 1 s−1, 8.9 ± 0.2 s−1, and 0.70 ± 0.02 s−1 for W91F/T168C/W199F-CPM. Excitation at 290 nm showed an increase and two decreases for W91F/S158C/W199F-CPM, and an increase, a decrease, and another increase for W91F/T168C/W199F-CPM (Tables 4 and 5). Excitation at 390 nm resulted in an increase and two decreases for both W91F/S158C/W199F-CPM and W91F/T168C/W199F-CPM (Tables 4 and 5). Observed rate constants at 390 nm excitation were 45 ± 1 s−1, 20 ± 2 s−1, and 1.7 ± 0.1 s−1 for W91F/S158C/W199F-CPM and 39 ± 1 s−1, 5.2 ± 0.1 s−1, and 0.98 ± 0.02 s−1 for W91F/T168C/W199F-CPM. Distances at gp45 states F, G, and H were still too large to be measured accurately for S158C/W199F-CPM, but for T168C/W199F-CPM, the donor/acceptor pair distance in assembly states G and H can be measured accurately at 42 and 32 Å, respectively. These distances, along with the previously determined distances of 35, 33, and 35 Å for states F, G, and H of V163C/W199F-CPM mutant (23), suggest that the donor/acceptor pair decreases in distance for two of the mutants while remaining unchanged for the other mutant. Modeling of these experimental distances revealed gp45 to be in an in-plane conformation at step D, but after binding of DNA, gp45D proceeds through three steps, with the first being an in-plane closing of the clamp through step G, sliding outwards to an out-of-plane conformation at step H. Interestingly, modeling of gp45H with DNA shows this out-of-plane conformation orthogonal to the pitch of the DNA duplex, with gp45H open wide enough to hover over the surface of the DNA without making significant contacts in either the major or minor groove.
Formation of the Holoenzyme.
The final state of the holoenzyme was observed by mixing 2 μM of the gp45H-gp44/62–DNA complex formed in the presence of 2 mM ATP with 2 μM of gp43 in complex buffer. Excitation at 290 nm showed two increases in amplitude for both S158C/W199F-CPM and T168C/W199F-CPM, leading to gp45 states J and K (Fig. 4 and Tables 4 and 5), with observed rate constants of 21 ± 1 s−1 and 0.50 ± 0.01 s−1 for S158C/W199F-CPM and 47 ± 1 s−1 and 1.9 ± 0.1 s−1 for T168C/W199F-CPM. Again, S158C/W199F-CPM has a larger positive amplitude because the polymerase (gp43) interacts on the same face of gp45 as gp44/62 does (18). Therefore, the interprotein FRET caused by tryptophans in gp43 increases the total fluorescence detected for the S158C/W199F-CPM mutant.
Figure 4.

Relative stopped-flow fluorescence values for the interaction of gp45H and gp44/62 and ATP and DNA with gp43. S158C/W199F data are in green, and T168C/W199F data are in pink.
The triple mutants also yielded two changes in fluorescent amplitude and similar observed rate constants to the double mutants. Excitation at 290 nm resulted in two increases in fluorescent amplitude for both mutants, whereas excitation at 390 nm resulted in an increase and a decrease in fluorescent amplitude for W91F/S158C/W199F-CPM and two increases in fluorescent amplitude for W91F/T168C/W199F-CPM (Tables 4 and 5). Observed rate constants at 290 nm excitation were 20 ± 1 s−1 and 0.40 ± 0.01 s−1 for S158C/W199F-CPM and 20 ± 1 s−1 and 1.2 ± 0.1 s−1 for T168C/W199F-CPM. Observed rate constants at 390 nm excitation were 47 ± 2 s−1 and 0.32 ± 0.01 s−1 for S158C/W199F-CPM and 36 ± 1 s−1 and 0.45 ± 0.01 s−1 for T168C/W199F-CPM. These changes resulted in a closing of both mutants to donor/acceptor pair distances of 36 and 28 Å for S158C/W199F-CPM and T168C/W199F-CPM, respectively, for gp45 state K. Triangulation and computer modeling by using these experimental distances shows gp45K to be closed further in an in-plane orientation. These final distance measurements in the formation of the holoenzyme show that gp45K is closed further than initially in solution (gp45A) but not as fully as seen in the crystal structure. This final state of gp45K in holoenzyme assembly has an open interface distance of about 11 Å. It has been suggested previously that the C terminus of gp43 is inserted into the subunit interface of gp45 (21). Although the C-terminal tail of gp43 is largely unstructured, interaction with gp45 could allow the tail to become more structured when it interacts with gp45 in the subunit interface.
Discussion
We have used stopped-flow FRET to monitor the directionality of the opening and closing of the gp45 clamp during holoenzyme assembly. The location of a tryptophan donor in the subunit interface as well as three mutants of gp45 that are individually labeled with a CPM acceptor at precise locations across the subunit interface were used in these experiments. Global triangulation of experimental distances calculated from the three mutants of gp45 was performed to create a model of clamp opening and closing. T4 holoenzyme assembly is an ordered process (2, 23, 24, 43) that proceeds by the sequential addition of the various components of the holoenzyme through a 10-step mechanism (Scheme S1). The magnitude of the fluorescent amplitude (Tables 4 and 5) for each of the gp45 mutants was used to determine the distances (Table 3) between the tryptophan and CPM-labeled cysteine in the subunit interface according to Eqs. 1, 3, and 5. By using computer modeling, experimental distances were integrated to provide a unique description of the direction and extent of clamp opening by modifying torsion angles within the interdomain connecting loop on the opposite side of the subunit interface. Gp45 opens in-plane in the presence of gp44/62 and ATP, closes out-of-plane on the addition of DNA, and finally closes in-plane with the addition of gp43 (Fig. 5). The final gp45K state features a partially open subunit interface that would allow for the C-terminal tail of gp43 to be inserted into the subunit interface of gp45. This proposed protein–protein interaction could be important in stabilizing the holoenzyme and conferring processivity to the bacteriophage T4 DNA replication complex.
Table 3.
Comparisons of open interface distance measurements for gp45 mutants
| Assembly state | V163C/W199F*
|
S158C/W199F
|
T168C/W199F
|
|||
|---|---|---|---|---|---|---|
| ET | RO, Å | ET | RO, Å | ET | RO, Å | |
| A | 0.692 | 40 | 0.409 | 48 | 0.414 | 48 |
| B | 0.692 | 40 | 0.409 | 48 | 0.414 | 48 |
| C | 0.688 ± 0.010 | 41 | 0.353 ± 0.001 | >45 | 0.412 ± 0.001 | >45 |
| D | 0.622 ± 0.005 | >45 | 0.296 ± 0.001 | >45 | 0.389 ± 0.001 | >45 |
| E | 0.622 ± 0.005 | >45 | 0.296 ± 0.001 | >45 | 0.389 ± 0.001 | >45 |
| F | 0.741 ± 0.016 | 35 | 0.265 ± 0.002 | >45 | 0.389 ± 0.002 | >45 |
| G | 0.767 ± 0.010 | 33 | 0.224 ± 0.002 | >45 | 0.431 ± 0.002 | 42 |
| H | 0.745 ± 0.010 | 35 | 0.170 ± 0.002 | >45 | 0.512 ± 0.003 | 32 |
| I | 0.745 ± 0.010 | 35 | 0.170 ± 0.002 | >45 | 0.512 ± 0.003 | 32 |
| J | 0.800 ± 0.013 | 31 | 0.269 ± 0.003 | >45 | 0.525 ± 0.003 | 31 |
| K | 0.809 ± 0.011 | 30 | 0.462 ± 0.006 | 36 | 0.576 ± 0.003 | 28 |
Previous experimental values determined by Alley et al. (23).
Figure 5.

Molecular models of gp45 were created on the basis of values from Table 3. V163 is blue, S158 is green, T168 is pink, and W91 is purple. (A) Gp45A open in-plane in solution. (B) Gp45D in the presence of gp44/62 and ATP open further in-plane. (C) Gp45H in the presence of gp44/62, ATP, and DNA closes out-of plane. (D) Gp45K in the presence of gp44/62, ATP, DNA, and gp43 closes further in-plane.
The proposed in-plane open gp45A state in Fig. 5A is different from the closed crystal structure (8). A possible explanation for this discrepancy is that gp45 is in equilibrium between two states in solution, and only one state crystallizes in the more symmetric closed state. Efforts are underway to determine whether an equilibrium exists between these states by using time-resolved fluorescence spectroscopy. Further gp45 states in Scheme S1 represent changes in distances across the subunit interface from state A. These states are calculated by using stopped-flow FRET and are described as a single state derived from gp45A. We have this broken symmetry in gp45A by assuming one interface of the three to be partially open and the other two to be closed (33).
The gp44/62 complex sequentially hydrolyzes two sets of two molecules of ATP in the loading of gp45 onto DNA and its interaction with gp43 (14, 24, 31). We have previously assigned steps 3 and 7 as the ATP hydrolysis steps because of: (i) the need for a hydrolyzable form of ATP in these steps; (ii) results from kinetic simulations implicating an irreversible step; and (iii) the similarity of the rate constant for these steps to those previously determined rates measured during the sequential ATP hydrolysis events by gp44/62 in the presence of gp45 (23, 24). These studies also concluded that ATP hydrolysis was associated with the opening of the clamp in step 3 and conformational changes in gp44/62 in step 7 (23). By measuring changes in the donor/acceptor distances for the three mutants of gp45 for each step, a deeper insight was obtained for the role of ATP hydrolysis in the formation of the holoenzyme. The hydrolysis of ATP in step 3 opens the clamp in an in-plane conformation (Fig. 5B). On the other hand, the hydrolysis of ATP in step 7 is also responsible for movements that close the clamp in the out-of-plane direction (Fig. 5C). With only the previous single gp45 mutant (W199F/V162C-CPM), the closing of the clamp in the out-of-plane direction through the hydrolysis of ATP was not detected because a similar donor/acceptor pair distance was maintained for both gp45G and gp45H. It is noteworthy that gp45H is in an out-of -plane conformation opposite to the groove of DNA, giving an inner diameter of 38 Å for gp45, which is slightly larger than double-stranded DNA, allowing the clamp protein to slide over the DNA. Positively charged residues in the interior of gp45 provide an electrostatic attraction to the negatively charged DNA backbone (11, 44). Gp45 is not a sequence-specific DNA-binding protein and does not make significant contacts within the DNA grooves. Therefore, the out-of-plane closing of the clamp on DNA provides a favorable conformation to interact with gp43.
When gp43 is added, gp45K returns to an in-plane conformation with the interface still open about 11 Å (Fig. 5D), sufficient to accommodate the C terminus of gp43. A crystal structure of the C-terminal peptide of gp43 bound to gp45 of bacteriophage RB69 shows that the peptide binds away from the subunit interface in a hydrophobic pocket close to the interdomain connecting loop (9). This mode of interaction is inconsistent with the experimental solution data, which would suggest the gp43 interaction to be in the subunit interface of gp45. The C-terminal tail of gp43 is required for holoenzyme assembly (22) and interacts with residues in the subunit interface of gp45 (21). This proposed protein–protein interaction, where gp45 “bites” down on the C-terminal tail of gp43, is an important contact point partially responsible for the highly processive DNA replisome.
Many other proteins also used in DNA replication and recombination have been identified as toroid-shaped molecules (45). Circular sliding clamps have been found in species varying from bacteriophages to humans, and they all have the same basic three-dimensional structure (8, 11, 44, 46). Hexameric helicase structures have also been determined by x-ray crystallography and electron microscopy (47–50), and all are circularly shaped proteins thought to encircle either single-stranded or double-stranded DNA and to unwind the DNA ahead of the polymerase. DNA recombination proteins including: bacteriophage λ exonuclease (51) and β protein (52), eukaryotic Rad52 (53) and a RecA homologue DMC1 (54), as well as topoisomerases (55–57), likewise form toroidal oligomeric structures with central channels large enough to accommodate single- or double-stranded DNA. Various mechanisms are apparently used to load these proteins onto DNA. The sliding clamps use a clamp-loader complex to guide them onto DNA. The similarity in structural and biochemical properties would predict that many would assemble like gp45. Hexameric helicases seem to self-associate in the presence of nucleoside triphosphates (58), which suggests that an alternate dimer association pathway is used to assemble helicases onto DNA (59, 60), possibly with the help of accessory factors such as gp59 in bacteriophage T4 (61) or DnaC protein in E. coli (62). Many of these circular DNA metabolism proteins may have DNA threading through the center channel of the proteins, but that remains to be determined. The mechanism of interaction of these toroid-shaped proteins with DNA may be vastly different from the sliding clamps, but the detailed conformational changes of gp45 described here provide one possible model for assembly.
The results from this study show that in bacteriophage T4, a partially open trimer of gp45 is loaded onto DNA in the presence of gp44/62 and ATP and interacts with gp43 to form a holoenzyme through 10 discrete steps. Gp45 opens in-plane by gp44/62 and ATP, closes out-of-plane in the presence of DNA, and closes further in-plane with the addition of gp43. ATP hydrolysis steps are responsible for first opening the clamp in the presence of gp44/62 and then closing the clamp out-of-plane on DNA. Gp44/62 likely undergoes significant structural changes as well during the holoenzyme loading process, although a dynamic analysis such as that described here would be difficult with this large protein complex. We have shown that the bacteriophage T4 DNA polymerase holoenzyme assembly process is extremely dynamic and well coordinated. Solution-phase FRET analysis was critical in defining a detailed structural model to track this process by using gp45 as a reference for this assembly.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health Grants GM19492 (S.C.A.) and GM13306 (S.J.B.).
Abbreviations
- CPM
7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin
- FRET
fluorescence resonance energy transfer
Footnotes
This paper results from the National Academy of Sciences colloquium, “Links Between Recombination and Replication: Vital Roles of Recombination,” held November 10–12, 2000, in Irvine, CA.
References
- 1.Nossal N G. FASEB J. 1992;6:871–878. doi: 10.1096/fasebj.6.3.1310946. [DOI] [PubMed] [Google Scholar]
- 2.Sexton D J, Berdis A J, Benkovic S J. Curr Opin Chem Biol. 1997;1:316–322. doi: 10.1016/s1367-5931(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 3.Stillman B. Cell. 1994;78:725–728. doi: 10.1016/s0092-8674(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 4.Waga S, Stillman B. Annu Rev Biochem. 1998;67:721–751. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- 5.Baker T A, Bell S P. Cell. 1998;92:295–305. doi: 10.1016/s0092-8674(00)80923-x. [DOI] [PubMed] [Google Scholar]
- 6.Keck J L, Berger J M. Chem Biol. 2000;7:R63–R71. doi: 10.1016/s1074-5521(00)00094-6. [DOI] [PubMed] [Google Scholar]
- 7.Benkovic S J, Valentine A M, Salinas F. Annu Rev Biochem. 2001;70:181–208. doi: 10.1146/annurev.biochem.70.1.181. [DOI] [PubMed] [Google Scholar]
- 8.Moarefi I, Jeruzalmi D, Turner J, O'Donnell M, Kuriyan J. J Mol Biol. 2000;296:1215–1223. doi: 10.1006/jmbi.1999.3511. [DOI] [PubMed] [Google Scholar]
- 9.Shamoo Y, Steitz T A. Cell. 1999;99:155–166. doi: 10.1016/s0092-8674(00)81647-5. [DOI] [PubMed] [Google Scholar]
- 10.Krishna T S, Fenyo D, Kong X P, Gary S, Chait B T, Burgers P, Kuriyan J. J Mol Biol. 1994;241:265–268. doi: 10.1006/jmbi.1994.1495. [DOI] [PubMed] [Google Scholar]
- 11.Kong X P, Onrust R, O'Donnell M, Kuriyan J. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 12.Kaboord B F, Benkovic S J. Curr Biol. 1995;5:149–157. doi: 10.1016/s0960-9822(95)00036-4. [DOI] [PubMed] [Google Scholar]
- 13.Kaboord B F, Benkovic S J. Biochemistry. 1996;35:1084–1092. doi: 10.1021/bi9520747. [DOI] [PubMed] [Google Scholar]
- 14.Berdis A J, Benkovic S J. Biochemistry. 1996;35:9253–9265. doi: 10.1021/bi952569w. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Sattar A K, Wang C C, Karam J D, Konigsberg W H, Steitz T A. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 16.Wang C C, Yeh L S, Karam J D. J Biol Chem. 1995;270:26558–26564. doi: 10.1074/jbc.270.44.26558. [DOI] [PubMed] [Google Scholar]
- 17.Salinas F, Benkovic S J. Proc Natl Acad Sci USA. 2000;97:7196–7201. doi: 10.1073/pnas.97.13.7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latham G J, Bacheller D J, Pietroni P, von Hippel P H. J Biol Chem. 1997;272:31685–31692. doi: 10.1074/jbc.272.50.31685. [DOI] [PubMed] [Google Scholar]
- 19.Latham G J, Bacheller D J, Pietroni P, von Hippel P H. J Biol Chem. 1997;272:31677–31684. doi: 10.1074/jbc.272.50.31677. [DOI] [PubMed] [Google Scholar]
- 20.Alley S C, Ishmael F T, Jones A D, Benkovic S J. J Am Chem Soc. 2000;122:6126–6127. [Google Scholar]
- 21.Alley S C, Jones A D, Soumillion P, Benkovic S J. J Biol Chem. 1999;274:24485–24489. doi: 10.1074/jbc.274.35.24485. [DOI] [PubMed] [Google Scholar]
- 22.Berdis A J, Soumillion P, Benkovic S J. Proc Natl Acad Sci USA. 1996;93:12822–12827. doi: 10.1073/pnas.93.23.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alley S C, Abel-Santos E, Benkovic S J. Biochemistry. 2000;39:3076–3090. doi: 10.1021/bi992377r. [DOI] [PubMed] [Google Scholar]
- 24.Sexton D J, Kaboord B F, Berdis A J, Carver T E, Benkovic S J. Biochemistry. 1998;37:7749–7756. doi: 10.1021/bi980088h. [DOI] [PubMed] [Google Scholar]
- 25.Sexton D J, Carver T E, Berdis A J, Benkovic S J. J Biol Chem. 1996;271:28045–28051. doi: 10.1074/jbc.271.45.28045. [DOI] [PubMed] [Google Scholar]
- 26.Bertram J G, Bloom L B, Hingorani M M, Beechem J M, O'Donnell M, Goodman M F. J Biol Chem. 2000;275:28413–28420. doi: 10.1074/jbc.M910441199. [DOI] [PubMed] [Google Scholar]
- 27.Hingorani M M, O'Donnell M. J Biol Chem. 1998;273:24550–24563. doi: 10.1074/jbc.273.38.24550. [DOI] [PubMed] [Google Scholar]
- 28.Bertram J G, Bloom L B, Turner J, O'Donnell M, Beechem J M, Goodman M F. J Biol Chem. 1998;273:24564–24574. doi: 10.1074/jbc.273.38.24564. [DOI] [PubMed] [Google Scholar]
- 29.Bloom L B, Turner J, Kelman Z, Beechem J M, O'Donnell M, Goodman M F. J Biol Chem. 1996;271:30699–30708. doi: 10.1074/jbc.271.48.30699. [DOI] [PubMed] [Google Scholar]
- 30.Hingorani M M, Bloom L B, Goodman M F, O'Donnell M. EMBO J. 1999;18:5131–5144. doi: 10.1093/emboj/18.18.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young M C, Weitzel S E, von Hippel P H. J Mol Biol. 1996;264:440–452. doi: 10.1006/jmbi.1996.0652. [DOI] [PubMed] [Google Scholar]
- 32.Turner J, Hingorani M M, Kelman Z, O'Donnell M. EMBO J. 1999;18:771–783. doi: 10.1093/emboj/18.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alley S C, Shier V K, Abel-Santos E, Sexton D J, Soumillion P, Benkovic S J. Biochemistry. 1999;38:7696–7709. doi: 10.1021/bi9827971. [DOI] [PubMed] [Google Scholar]
- 34.Kaboord B F, Benkovic S J. Proc Natl Acad Sci USA. 1993;90:10881–10885. doi: 10.1073/pnas.90.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nossal N G. J Biol Chem. 1979;254:6026–6031. [PubMed] [Google Scholar]
- 36.Rush J, Lin T C, Quinones M, Spicer E K, Douglas I, Williams K R, Konigsberg W H. J Biol Chem. 1989;264:10943–10953. [PubMed] [Google Scholar]
- 37.Frey M W, Nossal N G, Capson T L, Benkovic S J. Proc Natl Acad Sci USA. 1993;90:2579–2583. doi: 10.1073/pnas.90.7.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selvin P R. Methods Enzymol. 1995;246:300–334. doi: 10.1016/0076-6879(95)46015-2. [DOI] [PubMed] [Google Scholar]
- 39.van der Meer B W, Coker G I, Chen S Y. Resonance Energy Transfer. New York: VCH; 1994. [Google Scholar]
- 40.Dunn B M, Pham C, Raney L, Abayasekara D, Gillespie W, Hsu A. Biochemistry. 1981;20:7206–7211. doi: 10.1021/bi00528a023. [DOI] [PubMed] [Google Scholar]
- 41.Hass E, Katchalski-Katzir E, Steinberg I Z. Biochemistry. 1978;17:5064–5070. doi: 10.1021/bi00616a032. [DOI] [PubMed] [Google Scholar]
- 42.Pietroni P, Young M C, Latham G J, von Hippel P H. J Biol Chem. 1997;272:31666–31676. doi: 10.1074/jbc.272.50.31666. [DOI] [PubMed] [Google Scholar]
- 43.Latham G J, Pietroni P, Dong F, Young M C, von Hippel P H. J Mol Biol. 1996;264:426–439. doi: 10.1006/jmbi.1996.0651. [DOI] [PubMed] [Google Scholar]
- 44.Kuriyan J, O'Donnell M. J Mol Biol. 1993;234:915–925. doi: 10.1006/jmbi.1993.1644. [DOI] [PubMed] [Google Scholar]
- 45.Hingorani M M, O'Donnell M. Nat Rev Mol Cell Biol. 2000;1:22–30. doi: 10.1038/35036044. [DOI] [PubMed] [Google Scholar]
- 46.Krishna T S, Kong X P, Gary S, Burgers P M, Kuriyan J. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 47.Egelman H H, Yu X, Wild R, Hingorani M M, Patel S S. Proc Natl Acad Sci USA. 1995;92:3869–3873. doi: 10.1073/pnas.92.9.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawaya M R, Guo S, Tabor S, Richardson C C, Ellenberger T. Cell. 1999;99:167–177. doi: 10.1016/s0092-8674(00)81648-7. [DOI] [PubMed] [Google Scholar]
- 49.Yu X, Jezewska M J, Bujalowski W, Egelman E H. J Mol Biol. 1996;259:7–14. doi: 10.1006/jmbi.1996.0297. [DOI] [PubMed] [Google Scholar]
- 50.Sato M, Gotow T, You Z, Komamura-Kohno Y, Uchiyama Y, Yabuta N, Nojima H, Ishimi Y. J Mol Biol. 2000;300:421–431. doi: 10.1006/jmbi.2000.3865. [DOI] [PubMed] [Google Scholar]
- 51.Kovall R, Matthews B W. Science. 1997;277:1824–1827. doi: 10.1126/science.277.5333.1824. [DOI] [PubMed] [Google Scholar]
- 52.Passy S I, Yu X, Li Z, Radding C M, Egelman E H. Proc Natl Acad Sci USA. 1999;96:4279–4284. doi: 10.1073/pnas.96.8.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stasiak A Z, Larquet E, Stasiak A, Muller S, Engel A, Van Dyck E, West S C, Egelman E H. Curr Biol. 2000;10:337–340. doi: 10.1016/s0960-9822(00)00385-7. [DOI] [PubMed] [Google Scholar]
- 54.Passy S I, Yu X, Li Z, Radding C M, Masson J Y, West S C, Egelman E H. Proc Natl Acad Sci USA. 1999;96:10684–10688. doi: 10.1073/pnas.96.19.10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mondragon A, DiGate R. Struct Folding Des. 1999;7:1373–1383. doi: 10.1016/s0969-2126(00)80027-1. [DOI] [PubMed] [Google Scholar]
- 56.Lima C D, Wang J C, Mondragon A. Nature (London) 1994;367:138–146. doi: 10.1038/367138a0. [DOI] [PubMed] [Google Scholar]
- 57.Redinbo M R, Stewart L, Kuhn P, Champoux J J, Hol W G. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 58.Dong F, Gogol E P, von Hippel P H. J Biol Chem. 1995;270:7462–7473. doi: 10.1074/jbc.270.13.7462. [DOI] [PubMed] [Google Scholar]
- 59.Lohman T M, Bjornson K P. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 60.Morris P D, Raney K D. Biochemistry. 1999;38:5164–5171. doi: 10.1021/bi9822269. [DOI] [PubMed] [Google Scholar]
- 61.Morrical S W, Hempstead K, Morrical M D. J Biol Chem. 1994;269:33069–33081. [PubMed] [Google Scholar]
- 62.Allen G C, Jr, Kornberg A. J Biol Chem. 1991;266:22096–22101. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.