

Abstract
Inflammatory bowel diseases (IBD) result from dysregulated immune responses toward microbial and perhaps other luminal antigens in a genetically susceptible host, and are associated with altered composition and diversity of the intestinal microbiota. The interleukin 10-deficient (IL-10−/−) mouse has been widely used to model human IBD; however the specific alterations that occur in the intestinal microbiota of this mouse model during the onset of colonic inflammation have not yet been defined. The aim of our study was to define the changes in diversity and composition that occur in the intestinal microbiota of IL-10−/− mice during the onset and progression of colonic inflammation. We used high throughput sequencing of the 16S rRNA gene to characterize the diversity and composition of formerly germ-free, wild-type and IL-10−/− mice associated with the same intestinal microbiota over time. Following two weeks of colonization with a specific pathogen-free (SPF) microbiota we observed a significant increase in the diversity and richness of the intestinal microbiota of wild-type mice. In contrast, a progressive decrease in diversity and richness was observed at three and four weeks in IL-10−/− mice. This decrease in diversity and richness was mirrored by an increase in Proteobacteria and Escherichia coli in IL-10−/− mice. An increase in E. coli was also observed in conventionally raised IL-10−/− mice at the point of colonic inflammation. Our data reports the sequential changes in diversity and composition of the intestinal microbiota in an immune-mediated mouse model that may help provide insights into the primary vs. secondary role of dysbiosis in human IBD patients.
Keywords: inflammatory bowel diseases, intestinal microbiota, Interleukin 10-deficient mouse
Introduction
Ulcerative colitis (UC) and Crohn disease (CD), collectively known as inflammatory bowel diseases (IBD), are prevalent in the United States (US) affecting 1.4 million individuals,1 are associated with reduced quality of life2 and a heavy economic burden that is estimated to be $6.3 billion annually in the US.3 The chronic nature, high rate of recurrence and lack of safe and curative medical treatments for IBD underscore the need for alternate therapeutic approaches for these complex diseases. Although, the precise pathophysiology of IBD remains unclear, it is widely accepted that the pathogenesis of IBD involves dysregulated immune responses toward microbial and other luminal antigens in a genetically susceptible host.4-7 Environmental factors also play an important role in the initiation and reactivation of inflammation in IBD.4,8 An altered composition of the intestinal microbiota (dysbiosis) has been reported for both UC and CD,9-11 however this association is not as profound in recent pediatric studies.12,13 Moreover, the primary vs. secondary nature of an intestinal microbial dysbiosis in IBD remains unknown. Thus, more in depth characterization of sequential enteric microbial changes during early and later phases of the inflammatory process may enable the development of better therapeutic strategies for these diseases.
Among the various rodent models for IBD, interleukin-10 deficient (IL-10−/−) mice are widely used for mechanistic studies investigating the pathogenesis of spontaneous, immune-mediated, intestinal inflammation.14-16 IL-10−/− mice maintained in germ-free (GF) conditions do not develop intestinal inflammation. However, once colonized with conventional or specific pathogen free (SPF) microbiota, IL-10−/− mice develop chronic intestinal inflammation as early as one week following colonization with an SPF microbiota.17 Although alterations in the intestinal microbiota in subsets of IBD patients with established disease compared with healthy controls have been reported,9,10,18 early changes in the composition and diversity of this complex microbial community at the onset of disease cannot be studied in the human intestinal tract, as it is impossible to predict who will develop disease.8,19 Currently, little is known about the changes in composition and diversity of the enteric microbiota of IL-10−/− mice during the onset of intestinal inflammation. Molecular methods are effectively used to characterize the intestinal microbiota due to the limitations of culture-based methods. A study using 16S rRNA-based fingerprinting techniques (denaturing gradient gel electrophoresis [DGGE] and repetitive DNA element-based PCR) reported compositional changes in the intestinal microbiota in IL-10−/− mice over time;20,21 however due to the limitations in technology used for microbiota analysis, these studies provided limited data regarding the abundances of specific taxa and the diversity of the microbiota. Thus, we conducted the current study to characterize the intestinal microbiota of IL-10−/− mice during the progression of colitis in comparison with wild-type (WT) mice. We controlled for variations in the composition of the microbiota using previously GF IL-10−/− and WT mice colonized with a microbiota from the same donor. We also characterized changes in diversity and composition of the intestinal microbiota in mice from two genetic backgrounds.
Results
16S rRNA gene sequences
The V1–3 regions of the 16S rRNA gene were amplified from all fecal DNA samples (n = 30). Following high throughput sequencing, four samples (three from week two of the IL-10−/− group and one from week one of the WT group) yielded sequence numbers that were too low (< 350 16S rRNA sequences per sample) to be included in our analyses. An average of 8,012 16S rRNA sequences per sample were obtained from the remaining 26 samples with the following ranges: WT week one (n = 4) 5647‒10502 sequences; WT week two (n = 5) 6927‒11490 sequences; IL-10−/− week one (n = 5) 1115‒8116 sequences; IL-10−/− week two (n = 2) 9666‒12789 sequences; IL-10−/− week three (n = 5) 6162‒8467 sequences; IL-10−/− week four (n = 5) 1340‒23189 sequences. To determine the numbers and abundances of different bacterial groups in each sample we used 97% similarity between 16S rRNA gene sequences as an indicator of a “species level” operational taxonomic unit (OTU). Using this procedure we found a total of 479 OTUs in our data set.
Intestinal microbial diversity decreases over time in formerly GF IL-10−/− mice
In our initial investigation we sought to characterize changes in diversity in the intestinal microbiota that arise over time in formerly GF WT and IL-10−/− mice. The microbiota was characterized in fecal samples collected weekly for two weeks in WT mice and for four weeks in IL-10−/− mice following association with an SPF microbiota. Based on IL-12 p40 secretion from colonic tissue and composite histology scores, we found that WT mice did not develop significant inflammation whereas IL-10−/− mice developed moderate intestinal inflammation four weeks following association with an SPF microbiota (Fig. 1A and B).

Figure 1. Inflammatory changes in the colons of formerly GF mice over time. Levels of IL-12 p40 secreted by colonic tissues from formerly GF WT and IL-10−/− mice (A) and SPF raised WT and IL-10−/− mice (C). The levels of IL-12 p40 in IL-10−/− mice are significantly higher compared with WT mice in both formerly GF (p = 0.008) and SPF (p = 0.0001) environments. Histological scores for formerly GF WT and IL-10−/− mice (B) and SPF WT (n = 7, as one tissue sample degraded during processing) and IL-10−/− mice (D). The degree of histological inflammation is significantly higher in IL-10−/− compared with WT mice in formerly GF (p = 0.008) and SPF (p = 0.001) environments.
We calculated UniFrac distances for all time points for each group. We found that average weighted and un-weighted UniFrac distances significantly increased (p = 0.003 and p = 8.5 × 10−5, respectively) over the two week observation period in the WT group (Fig. 2C). We investigated the weighted and un-weighted UniFrac distances of the microbiota in fecal samples obtained one, two, three and four weeks from formerly GF IL-10−/− mice following association with an SPF microbiota. We found a significant decrease in average weighted UniFrac distances at the three week (p = 0.005) and four week (p = 0.005) time points compared with the 1 week time point (Fig. 2A and B).

Figure 2. Changes in weighted and un-weighted average UniFrac distances in formerly GF wild-type (WT) and IL-10−/− mice over time. (A) Average weighted UniFrac distances of the intestinal microbiota significantly decrease in IL-10−/− mice three and four weeks following colonization with a specific-pathogen free (SPF) microbiota. (B) Average un-weighted UniFrac distances of the intestinal microbiota do not significantly alter in the IL-10−/− mice following colonization with an SPF microbiota. (C) Average weighted and un-weighted UniFrac distances significantly increase in WT mice between one and two weeks following colonization with an SPF microbiota.
Intestinal microbial richness decreases over time in formerly GF IL-10−/− mice
We determined the richness of the intestinal microbiota in all groups using rarefaction analysis. A significant increase (p = 0.004) in the number of observed microbial species was found in WT mice two weeks following association with an SPF microbiota when compared with the one week time point (Fig. 3B). In contrast, a significant decrease in the number of observed microbial species was found in IL-10−/− mice three (p = 0.02) and four (p = 0.009) weeks following association with an SPF microbiota when compared with the one week time point (Fig. 3A).
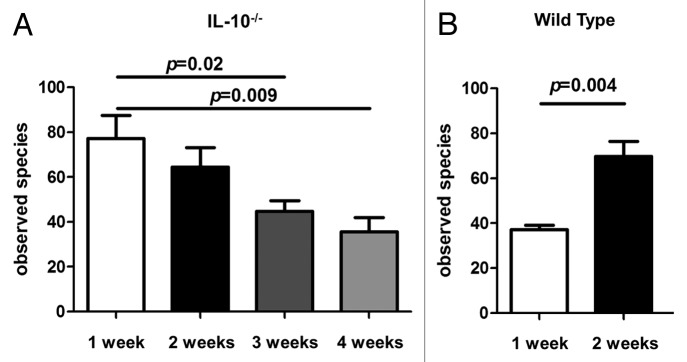
Figure 3. Microbial richness of 16S rRNA data. (A) The number of observed bacterial species in formerly GF IL-10−/− mice decreases between one and four weeks following colonization with an SPF microbiota. (B) The number of observed bacterial species in formerly GF WT mice significantly increases between one and two weeks following colonization with an SPF microbiota.
Ecological succession of bacterial taxa in formerly GF IL-10−/− mice over time
In order to determine the dominant bacterial groups that are altered over time in WT and IL-10−/− mice following colonization with an SPF intestinal microbiota we summarized the bacterial taxa identified by our 16S rRNA sequences at the phylum level (Fig. 4). In WT mice we found no significant changes in bacterial phyla between one and two weeks post association with an SPF microbiota. In the IL-10−/− group we observed a significant decrease in the levels of Bacteroidetes (20.06%‒6.30%, FDR = 0.01) and Verrucomicrobia (0.74%‒0.17%, false discovery rate [FDR] = 0.02) at three weeks compared with the one week time point. These changes were further reduced by four weeks: Bacteroidetes (20.06–9.5%, FDR = 0.003) and Verrucomicrobia (0.74%‒0.09%, FDR = 0.02). At four weeks we observed a significant increase in the levels of Proteobacteria (0.17%‒7.71%, FDR = 1.5 × 10−5) and Tenericutes (39.21%‒70.66%, FDR = 0.07) compared with the one week time point. In tandem, at four weeks we observed a decrease in the levels of Actinobacteria (1.27%‒0.53%, FDR = 0.07) and Firmicutes (38.48%‒11.46%, FDR = 0.03) compared with the one week time point. Additionally, we observed significant changes in the abundances of genus-level taxa over time in IL-10−/− mice (Table 1) and no significant changes in bacterial genera in WT mice over time.

Figure 4. Bacterial taxa alterations over time in formerly GF WT and IL10−/− mice.
Table 1. Changes in the abundances of genus-level taxa over time in formerly GF IL-10−/− mice.
| Taxon | % week 1 | % week 3 | % week 4 | p | FDR* |
|---|---|---|---|---|---|
|
Coprobacillus |
0.86 |
0.06 |
|
0.004 |
0.07 |
|
Akkermansia |
0.75 |
0.17 |
|
0.007 |
0.06 |
|
Lactobacillus |
1.20 |
12.83 |
|
0.008 |
0.05 |
|
Staphylococcus |
0.50 |
0.00 |
|
0.020 |
0.10 |
|
Parabacteroides |
9.15 |
4.05 |
|
0.023 |
0.10 |
|
Raoultella |
0.00 |
|
0.08 |
0.002 |
0.03 |
|
Akkermansia |
0.75 |
|
0.09 |
0.003 |
0.02 |
|
Clostridium |
0.02 |
|
0.00 |
0.006 |
0.04 |
|
Enterobacter |
0.10 |
|
5.14 |
0.009 |
0.04 |
| Oscillospira | 0.83 | 0.04 | 0.010 | 0.04 |
False discovery rate (FDR) < 0.1 = statistically significant.
Escherichia coli concentrations increase in formerly GF IL-10−/− mice over time
Given that E. coli is a member of the Proteobacteria phylum and has been established as a pro-inflammatory microbe in the context of IBD,22 we used species-specific qPCR to determine the levels of this bacterial species in fecal samples from the IL-10−/− group of mice. We found that the levels of E. coli in the intestinal microbiota of IL-10−/− mice increased over time and became significantly higher at the four week time point when compared with the week one time point (Fig. 5). The stepwise increase of E. coli over time in the IL-10−/− group closely paralleled the increase in Proteobacteria in the same mice, confirming results by two different molecular methods (Fig. 5).
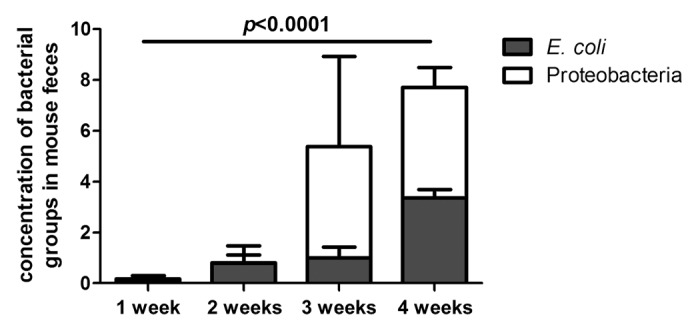
Figure 5. Change in levels of Proteobacteria and Escherichia coli in formerly GF IL-10−/− mice over time. Proteobacteria are expressed as the percentage of total 16S rRNA sequences. E. coli are expressed as fold increase with respect to baseline (WT at week 1). *The levels of Proteobacteria and E. coli are significantly higher at week 4 post colonization compared with week 1 (E. coli, p = 0.0001; Proteobacteria, FDR = 1.5 × 10−5).
Alterations in the abundance of E. coli in formerly GF IL-10−/− mice over time are mirrored in SPF IL-10−/− mice
In order to determine whether inflammation associated alterations of specific taxa in formerly GF IL-10−/− mice are also altered in a gut that has developed naturally, we investigated the levels of E. coli, Lactobacillus species and Akkermansia muciniphila in WT and IL-10−/− mice raised in an SPF environment. This group of IL-10−/− mice developed significant intestinal inflammation at 10 weeks of age compared with WT controls that did not develop inflammation (Fig. 1C and D). Similarly to formerly GF mice the abundance of E. coli was significantly higher in IL-10−/− mice at week ten compared with WT mice (Fig. 6A). Akkermansia muciniphila was significantly higher in IL-10−/− mice compared with WT mice at both eight and ten weeks (Fig. 6B). We did not observe any significant differences in the abundance of Lactobacillus species in IL-10−/− mice compared with WT mice at either time point (Fig. 6C).

Figure 6. Change in levels of E. coli(A),Akkermansia muciniphila(B) and Lactobacillus species (C) in SPF WT and IL-10−/− mice over time.
Discussion
Based on the established association of the intestinal microbiota with IBD and the frequent use of the IL-10−/− mouse as a model of spontaneous, immune-mediated colonic inflammation, we characterized the diversity and composition of the intestinal microbiota in this mouse model during the progression of experimental colitis. Previous studies characterizing the intestinal microbiota of IL-10−/− mice focused on viable bacteria or used molecular techniques that characterize a limited number of bacterial taxa.20,21 Our study used a current molecular technique that provided a comprehensive characterization of the changes in microbial composition and diversity in WT and IL-10−/− mice over time. Additionally, the use of formerly GF WT and IL-10−/− mice colonized with the same fecal microbiota provided a highly controlled environment to study these changes.
In our investigations we observed an increase in microbial richness (as determined by rarefaction of bacterial species) and diversity (as determined by UniFrac distances) in formerly GF WT mice two weeks following association with an SPF microbiota. This finding suggests that the intestinal microbiota in formerly GF WT mice may still be establishing a homeostatic balance two weeks after colonization. UniFrac distances can be calculated in a weighted (relative abundance of taxa) or un-weighted (presence or absence of taxa) manner. Low average UniFrac distances indicate higher similarity in the composition of the microbiota within a group of samples, whereas high average UniFrac distances indicate more dissimilarity within a group of samples. We observed an increase in both weighted and un-weighted UniFrac distances two weeks post colonization in WT mice, suggesting that the rise in diversity values at this time point is due to an increase in both high and low abundance bacterial species. A limitation of our study is that we did not investigate the microbial composition and diversity of fecal samples in formerly GF WT mice at three and four weeks following conventionalization with an intestinal microbiota. Thus we cannot conclude whether the intestinal microbiota has reached equilibrium at this point. Interestingly, it has been reported that the microbial composition and diversity of cecal contents in formerly GF WT mice, from a different genetic background (C57BL/6), exhibited stability seven days post-inoculation.23
We subsequently characterized the richness and diversity of the intestinal microbiota in formerly GF IL-10−/− mice up to four weeks following colonization with an SPF microbiota to investigate alterations in the intestinal microbiota during the progression of colitis. Indeed, we observed moderate inflammation at four weeks post colonization in IL-10−/− mice. A progressive decrease in weighted UniFrac distances was observed three and four weeks post colonization in comparison with one week values. In addition, the richness of the microbiota was significantly decreased in this cohort of mice at these later time points. Interestingly, we did not observe a significant decrease in un-weighted UniFrac distances at the three and four week time points compared with one week in IL-10−/− mice. These data suggest that the composition of newly introduced complex microbial communities in formerly GF IL-10−/− mice become more alike during the progression of colonic inflammation and that high abundance taxa are responsible for this change. In the absence of an established fecal marker indicative of intestinal inflammation we are unable to conclude whether the alterations in the richness and diversity of the enteric microbiota in IL-10−/− mice occur before, during or after the onset of colonic inflammation. In previous experiments we find that mild inflammation occurs in formerly GF IL-10−/− mice on a 129 SvEv background two weeks following microbial colonization.17 Our current study shows that enteric microbial richness and diversity dramatically changes at three weeks post association with an SPF microbiota. Thus, we speculate that decreases in enteric microbial richness and diversity in IL-10−/− mice occur after the onset of colonic inflammation in this mouse model.
In tandem with altered enteric microbial richness and diversity in the intestinal microbiota of IL-10−/− mice over time, we observed changes in the relative abundances of specific bacterial phyla. Most notable was a step-wise increase in the levels of Proteobacteria that mirrored a significant increase of E. coli during the onset of colonic inflammation. Elevated Proteobacteria levels have been reported in UC and CD patients.9 Moreover, adherent, invasive E. coli strains are associated with ileal CD.22,24 A significant depletion in the concentrations of the Bacteroidetes and Firmicutes phyla were observed over time in IL-10−/− mice, which also reflect changes reported in the intestinal microbiota of human IBD.9,10 Thus, our data demonstrate that the IL-10−/− mouse model of colitis exhibits alterations in specific members of the intestinal microbiota that parallel those of human IBD. We also observed significant decreases in the levels of the Actinobacteria and Verrucomicrobia phyla, which have not been reported for human IBD and may be a consequence of enteric inflammation. It is interesting to note that Bifidobacterium species are encompassed within Actinobacteria phylum and are considered probiotic microbes. Alterations in the Verrucomicrobia phylum may be yet unreported components of a human IBD dysbiosis, or alternatively, they may be unique to this mouse model. However, it has been reported that Akkermansia muciniphila (a member of the Verrucomicrobia phylum) is depleted in the mucus of IBD patients.25 Indeed, in support of this finding, our 16S rRNA sequence data revealed a significant decrease in the Akkermansia genus in IL-10−/− mice over time. As bacterial richness and diversity decreased in IL-10−/− mice over time we observed a profound increase in the levels of the Tenericutes phylum. This phylum encompasses bacteria that lack a cell wall and are thus Gram negative. Tenericutes have been reported to be significantly elevated in fecal samples from colonic Crohn disease patients but decreased in fecal samples from ileal Crohn disease and ulcerative colitis patients.10 From our data set the dominant genus within the Tenericutes phylum is Allobaculum. However, although taxonomies were assigned to our 16S rRNA sequences using the Greengenes database26 it is interesting to note that the RDP database27 classifies the Allobaculum genus as belonging to the Firmicutes phylum, which would make the Firmicutes the dominant phylum in our data set. Nevertheless, it has been reported that the Greengenes database, which contains the largest and most diverse set of 16S rRNA sequences, is superior to other databases when classifying sequences at the phylum level, particularly with respect to the Tenericutes.28 It has been reported that GF mice have abnormal mucosal and immunologic maturation resulting in increased morbidity in the oxazolone-induced mouse model of colitis when compared with SPF mice.29 Thus, we determined the abundances of specific bacterial groups in cohorts of WT and IL-10−/− mice that were raised with a normal intestinal microbiota. Even though the mice used were of a different genetic background (C57BL/6) to the mice used in our GF experiments we observed an increase in the abundance of E. coli in IL-10−/− mice compared with WT mice, suggesting a strong association of this bacterial species with the development of intestinal inflammation in this mouse model over time. We also found that although abundances of A. muciniphila and Lactobacillus species appeared to decrease over time in IL-10−/− mice compared with WT mice, these decreases were not significant. Thus, it is possible that the alterations in the enteric microbiota during the development of intestinal inflammation that we observed in formerly GF IL-10−/− mice are influenced by host genetics and an abnormal gut physiology associated with GF mice.
Our data reports the changes in diversity and composition of the intestinal microbiota in the IL-10−/− mouse model of spontaneous, immune-mediated colitis over time. The reduction in diversity and most changes in specific bacterial taxa in the intestinal microbiota of this mouse model reflect the changes in this complex microbial community observed in human IBD patients. Thus, our study validates the use of this mouse model for studies relating to the intestinal microbiota and immune-mediated colonic inflammation. The progressive loss of diversity, of dominant commensal microbiota and expansion of Proteobacteria, notably E. coli, with increasing experimental inflammation suggests that dysbiosis is secondary to the immune response in this model. Despite the postulated secondary nature of the observed changes, the altered microbiota profiles may still be responsible for perpetuation and amplification of mucosal immune responses and inflammation, since we have documented enteric bacterial-specific TH1 and TH17 responses in this model.30
Materials and Methods
Mice
Adult WT and IL-10−/− 129 SvEv mice were maintained in a GF state at the National Gnotobiotic Rodent Resource Center at UNC-Chapel Hill, NC USA. One group of WT (n = 5) and one group of IL-10−/− (n = 5) mice were used in this study. Formerly GF mice were inoculated with fecal slurry obtained from a common SPF WT 129 SvEv donor mouse via oral and rectal swabbing (see Fig. 7 for a schematic design of the study). Each experimental group (WT and IL-10−/−) consisted of mice from the same litter that were housed in separate cages. Fresh fecal pellets were obtained weekly from all mice. All fecal pellets collected were flash frozen immediately in liquid nitrogen to retain the integrity of the fecal microbiota.
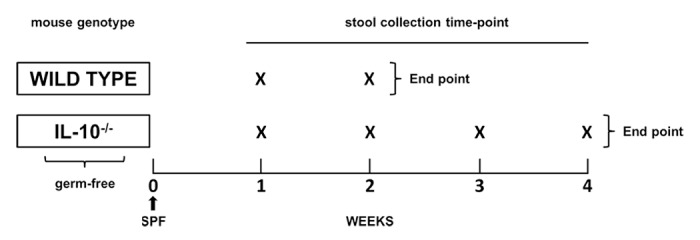
Figure 7. Schematic outline of experimental design. Adult WT and IL-10−/− 129 SvEv GF mice were inoculated with an SPF microbiota from a single donor (black arrow). Fresh fecal pellets were obtained from the WT group (n = 5) at 1 and 2 weeks following SPF inoculation. Fecal pellets were obtained from the IL-10−/− group (n = 5) at 1, 2, 3 and 4 weeks following association with an SPF intestinal microbiota.
In order to translate our findings from GF animals to mice that were raised from birth with a normal microbiota, we investigated WT (n = 8) and IL-10−/− (n = 9) C57BL/6 mice housed in a specific pathogen-free (SPF) environment. Each experimental group (WT and IL-10−/−) consisted of mice from the same litter that were housed in separate cages. Fecal samples were collected from all mice at eight and 10 weeks of age.
Assessment of intestinal inflammation
Sections of fixed (10% neutral buffered formalin) colons were embedded in paraffin and stained with hematoxylin and eosin. Using a well-validated scale17,31 the severity of inflammation was blindly assessed. Histological scores (0 to 4) were based on the degree of lamina propria and submucosal mononuclear cellular infiltration, crypt hyperplasia, goblet cell depletion and architectural distortion. A composite histology score was generated by the aggregate scores from the cecum and proximal and distal colon.
Colonic explant cultures were prepared as previously described.17 Briefly, colonic tissue was thoroughly irrigated with phosphate-buffered saline (PBS), shaken at room temperature in Roswell Park Memorial Institute Medium (RPMI) containing 1% penicillin and streptomycin (GIBCO, Grand Island, NY) for 30 min at 280 rpm, cut into 1-mm fragments and weighed. Intestinal tissue fragments were then distributed (0.05 g per well) into 24-well plates and incubated in 1 mL of RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% antibiotic/antimycotic (penicillin/streptomycin) for 22 h at 37°C. Supernatants were collected and stored at -20°C before use for cytokine quantification. Commercially available monoclonal anti-mouse IL-12 p40 capture and detection reagents (BD Biosciences PharMingen, San Diego, CA) in an enzyme-linked immunosorbent assay (ELISA) were used to measure the levels of IL-12 p40 secreted constitutively in colonic explant cultures.17,31 IL-12 p40 levels were measured in supernatants and compared with standard curves generated by using recombinant murine IL-12 p40.
Isolation of DNA
Bacterial DNA was isolated from all fecal pellets (n = 47) using a phenol/chloroform extraction method combined with physical disruption of bacterial cells and a DNA clean-up kit (Qiagen DNeasy® Blood and Tissue extraction kit) as previously described.32 Briefly, 100 mg of frozen feces were suspended in 750 µL of sterile bacterial lysis buffer (200 mM NaCl, 100 mM Tris [pH 8.0], 20 mM EDTA, 20 mg mL−1 lysozyme) and incubated at 37°C for 30 min. Next, 40 µL of proteinase K (20 mg mL−1) and 85 µL of 10% SDS were added to the mixture and incubated at 65°C for 30 min. 300 mg of 0.1 mm zirconium beads (BioSpec Products) were then added and the mixture was homogenized in a bead beater (BioSpec Products) for 2 min. The homogenized mixture was cooled on ice and centrifuged at 14,000 rpm for 5 min. The supernatant was transferred to a new 1.5 ml microfuge tube and fecal DNA was further extracted by phenol/chloroform/iso-amyl alcohol (25:24:1) and chloroform/iso-amyl alcohol (24:1). Following extraction the supernatant was precipitated by absolute ethanol at -20°C for 1 h. The precipitated DNA was suspended in DNase free H2O and cleaned using the Qiagen DNeasy® Blood and Tissue extraction kit per the manufacturer’s instructions.
454 pyro-sequencing of 16S rRNA genes
Bacterial community composition in isolated DNA samples was characterized by amplification of the V1–3 (forward, 8f:5′-AGAGTTTGATCMTGGCTCAG-3′; reverse 518r: 5′-ATTACCGCGGCTGCTGG-3′) variable regions of the 16S rRNA gene by polymerase chain reaction (PCR) as previously described.33 Forward primers were tagged with 10 bp unique barcode labels at the 5′ end along with the adaptor sequence (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3′) to allow multiple samples to be included in a single 454 Genome Sequencer (GS) FLX Titanium sequencing plate as previously described.34,35 16S rRNA PCR products were quantified, pooled and purified for the sequencing reaction. 454 GS FLX Titanium sequencing was performed on a 454 Life Sciences GS FLX machine (Roche, Florence, SC) at the Microbiome Core at UNC-Chapel Hill (http://www.med.unc.edu/microbiome).
Analysis of 16S rRNA sequences using the QIIME pipeline
16S rRNA sequence data generated by the 454 GS FLX Titanium sequencer were processed by the quantitative insights into microbial ecology (QIIME) pipeline.36 Briefly, sequences that were less than 200 bp or greater than 1,000 bp in length, contained incorrect primer sequences or contained more than 1 ambiguous base were discarded. The remaining sequences were assigned to WT and IL-10−/− groups based on their unique nucleotide barcodes, including error-correction.34 Sequences were clustered into OTUs based on 97% sequence similarity (similar to species level) using BLAST37 with the Greengenes reference database.38 α-diversity (diversity within samples) was determined with ten iterations at a maximal sequence depth where all samples could be included. β-diversity (diversity between groups of samples) was calculated using weighted and un-weighted UniFrac distances.39-41 UniFrac distances are a β-diversity measure that utilizes phylogenetic information to compare environmental samples.39,40
Quantitative real-time PCR (qPCR)
qPCR assays were performed using the SYBR® Green PCR Master Mix (Applied Biosystems, Carlsbad, CA) with primers that amplify the genes encoding 16S rRNA from E. coli (forward, 5′- GTTAATACCTTTGCTCATTGA -3′; reverse, 5′- ACCAGGGTATCTAATCCTGTT -3′), Lactobacillus species (forward, 5′- AGCAGTAGGGAATCTTCCA -3′; reverse, 5′- CACCGCTACACATGGAG -3′), A. muciniphila (forward, 5′- CAGCACGTGAAGGTGGGGAC -3′; reverse, 5′- CCTTGCGGTTGGCTTCAGAT -3′) and all bacteria (forward, 5′-GTGSTGCAYGGYTGTCGTCA-3′; reverse, 5′- ACGTCRTCCMCACCTTCCTC-3′). qPCR assays were conducted in 96-well plates on an Eppendorf Realplex2 mastercycler thermocycler (Eppendorf, Hauppauge, NY). Each PCR was performed in a final volume of 25 µL and contained the following: 1 × SYBR Green Master Mix, 0.5 µM of each primer and 10 ng of purified fecal DNA. PCR conditions were as follows: 10 min at 95°C, followed by 40 cycles of 95°C for 15 sec, 20 sec at 50°C and 72°C for 30 sec. Each plate included duplicate reactions per DNA sample, the appropriate set of standards and a “no template” negative control for each primer set. qPCR standards were generated by PCR amplification of target sequences from genomic DNA of an appropriate positive control strain. Analysis of melting curves confirmed that the fluorescence signal originated from specific PCR products and not from primer-dimers or other artifacts.
Statistical analyses
Bacterial taxon percentages and average UniFrac value data sets were assessed for normality using the D’Agostino and Pearson omnibus normality test prior to making statistical. When a data set was identified as not having a normal distribution it was transformed by log10,42 and retested for normality. Normally distributed data sets were compared using a Student’s t-test. All statistical comparisons were performed using GraphPad Prism software (v4.0a; Prism). We used taxon and phylogenetic-based analyses to compare 16S rRNA gene sequences within WT and IL-10−/− groups over time. Taxon-based: the means and standard deviations of abundances of bacterial Phyla were calculated and compared between all time-points for each group using the Benjamini–Hochberg procedure43 to correct for multiple corrections. A p value of less than 0.05 and a FDR less than 0.1 was considered significant. Phylogenetic-based: phylogenetic trees for WT and IL-10−/− groups were generated using the QIIME pipeline.36 Each tree was subjected to un-weighted and weighted UniFrac analyses39,40 through the QIIME pipeline. UniFrac distances represent the fraction of branch length that is shared by any two samples’ communities in a phylogenetic tree built from 16S rRNA sequence data from all samples. Average UniFrac distances, which represent the similarity or dissimilarity of microbial communities within a group, were compared using a student’s t test.
For qPCR assays, the percentage of E. coli was determined in all fecal samples ([copies 16S rRNA gene for E. coli/copies of 16S rRNA gene for all bacteria] × 100). The concentration of E. coli group was expressed as a “fold change” with respect to the concentration in the control group (WT) at week 1 after colonization. qPCR data were compared between groups using a non-parametric Mann-Whitney test. Similarly, the concentrations of IL-12p40 secreted by colonic tissue (pg/mL/mg of tissue) were compared between groups using a non-parametric Mann-Whitney test.
Acknowledgments
The authors would like to thank UNC-CH Microbiome core (P30 DK 34987) and the National Gnotobiotic Rodent Resource Center (funded by USPHS P40 OD010995), P30 DK 34987–10 and Crohn and Colitis Foundation of America (CCFA) (R.B.S.) for their contribution to this study. This study was supported by the UNC-CH university research council (URC) small grant program (I.C.), the CCFA Research Fellowship Awards (N.M. and C.D.P.), the NIH T32 DK07737 institutinal research training grant (C.D.P.), the NIH T35 DK007386 short-term training grant [S.M. and J.S.], R01 DK 53347 (R.B.S.) and K01 DK 092330 (I.C.).
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
These authors contributed equally to this study
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/25486
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Cohen RD. The quality of life in patients with Crohn’s disease. Aliment Pharmacol Ther. 2002;16:1603–9. doi: 10.1046/j.1365-2036.2002.01323.x. [DOI] [PubMed] [Google Scholar]
- 3.Kappelman MD, Rifas-Shiman SL, Porter CQ, Ollendorf DA, Sandler RS, Galanko JA, et al. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology. 2008;135:1907–13. doi: 10.1053/j.gastro.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Packey CD, Sartor RB. Interplay of commensal and pathogenic bacteria, genetic mutations, and immunoregulatory defects in the pathogenesis of inflammatory bowel diseases. J Intern Med. 2008;263:597–606. doi: 10.1111/j.1365-2796.2008.01962.x. [DOI] [PubMed] [Google Scholar]
- 5.Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis. 2009;22:292–301. doi: 10.1097/QCO.0b013e32832a8a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albenberg LG, Lewis JD, Wu GD. Food and the gut microbiota in inflammatory bowel diseases: a critical connection. Curr Opin Gastroenterol. 2012;28:314–20. doi: 10.1097/MOG.0b013e328354586f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shanahan F. The microbiota in inflammatory bowel disease: friend, bystander, and sometime-villain. Nutr Rev. 2012;70(Suppl 1):S31–7. doi: 10.1111/j.1753-4887.2012.00502.x. [DOI] [PubMed] [Google Scholar]
- 8.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–94. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 9.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–5. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–54, e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 11.Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJ. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2006;44:4136–41. doi: 10.1128/JCM.01004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellermayer R, Mir SA, Nagy-Szakal D, Cox SB, Dowd SE, Kaplan JL, et al. Microbiota separation and C-reactive protein elevation in treatment-naïve pediatric granulomatous Crohn disease. J Pediatr Gastroenterol Nutr. 2012;55:243–50. doi: 10.1097/MPG.0b013e3182617c16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hansen R, Russell RK, Reiff C, Louis P, McIntosh F, Berry SH, et al. Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn’s but not in ulcerative colitis. Am J Gastroenterol. 2012;107:1913–22. doi: 10.1038/ajg.2012.335. [DOI] [PubMed] [Google Scholar]
- 14.Büchler G, Wos-Oxley ML, Smoczek A, Zschemisch NH, Neumann D, Pieper DH, et al. Strain-specific colitis susceptibility in IL10-deficient mice depends on complex gut microbiota-host interactions. Inflamm Bowel Dis. 2012;18:943–54. doi: 10.1002/ibd.21895. [DOI] [PubMed] [Google Scholar]
- 15.Barnett MP, McNabb WC, Cookson AL, Zhu S, Davy M, Knoch B, et al. Changes in colon gene expression associated with increased colon inflammation in interleukin-10 gene-deficient mice inoculated with Enterococcus species. BMC Immunol. 2010;11:39. doi: 10.1186/1471-2172-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen JJ, Holt L, Sartor RB. Gene expression patterns in experimental colitis in IL-10-deficient mice. Inflamm Bowel Dis. 2009;15:890–9. doi: 10.1002/ibd.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–31. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–6. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140:859–70. doi: 10.1016/j.cell.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bibiloni R, Simon MA, Albright C, Sartor B, Tannock GW. Analysis of the large bowel microbiota of colitic mice using PCR/DGGE. Lett Appl Microbiol. 2005;41:45–51. doi: 10.1111/j.1472-765X.2005.01720.x. [DOI] [PubMed] [Google Scholar]
- 21.Peña JA, Li SY, Wilson PH, Thibodeau SA, Szary AJ, Versalovic J. Genotypic and phenotypic studies of murine intestinal lactobacilli: species differences in mice with and without colitis. Appl Environ Microbiol. 2004;70:558–68. doi: 10.1128/AEM.70.1.558-568.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–21. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 23.Gillilland MG, 3rd, Erb-Downward JR, Bassis CM, Shen MC, Toews GB, Young VB, et al. Ecological succession of bacterial communities during conventionalization of germ-free mice. Appl Environ Microbiol. 2012;78:2359–66. doi: 10.1128/AEM.05239-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wine E, Ossa JC, Gray-Owen SD, Sherman PM. Adherent-invasive Escherichia coli, strain LF82 disrupts apical junctional complexes in polarized epithelia. BMC Microbiol. 2009;9:180. doi: 10.1186/1471-2180-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Png CW, Lindén SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–8. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- 26.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35(Database issue):D169–72. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, et al. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012;6:94–103. doi: 10.1038/ismej.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–93. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Veltkamp C, Tonkonogy SL, De Jong YP, Albright C, Grenther WB, Balish E, et al. Continuous stimulation by normal luminal bacteria is essential for the development and perpetuation of colitis in Tg(epsilon26) mice. Gastroenterology. 2001;120:900–13. doi: 10.1053/gast.2001.22547. [DOI] [PubMed] [Google Scholar]
- 32.Gulati AS, Shanahan MT, Arthur JC, Grossniklaus E, von Furstenberg RJ, Kreuk L, et al. Mouse background strain profoundly influences Paneth cell function and intestinal microbial composition. PLoS One. 2012;7:e32403. doi: 10.1371/journal.pone.0032403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carroll IM, Ringel-Kulka T, Siddle JP, Ringel Y. Alterations in composition and diversity of the intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil. 2012;24:521–30, e248. doi: 10.1111/j.1365-2982.2012.01891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods. 2008;5:235–7. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A. 2008;105:17994–9. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 38.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–72. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576–85. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramette A. Multivariate analyses in microbial ecology. FEMS Microbiol Ecol. 2007;62:142–60. doi: 10.1111/j.1574-6941.2007.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–84. doi: 10.1016/S0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]