

Abstract
Background
Vibrio parahaemolyticus AphA and OpaR are the two master quorum sensing (QS) regulators that are abundantly expressed at low cell density (LCD) and high cell density (HCD), respectively, with a feature of reciprocally gradient production of them with transition between LCD and HCD. The type VI secretion system 2 (T6SS2) gene cluster can be assigned into three putative operons, namely VPA1027-1024, VPA1043-1028, and VPA1044-1046. T6SS2 contributes to adhesion of V. parahaemolyticus to host cells.
Methodology/Principal Findings
OpaR box-like sequences were found within the upstream promoter regions of all the above three operons, while none of AphA box-like elements could be identified for them. The subsequent primer extension, LacZ fusion, electrophoretic mobility shift, and DNase I footprinting assays disclosed that OpaR bound to the promoter regions of these three operons to stimulate their transcription, while AphA negatively regulated their transcription most likely through acting on OpaR. This regulation led to a gradient increase of T6SS2 transcription with transition from LCD to HCD.
Conclusions/Significance
V. parahaemolyticus OpaR and AphA positively and negatively regulate T6SS2 expression, respectively, leading to a gradient elevation of T6SS2 expression with transition from LCD to HCD. T6SS2 genes are thus assigned as the QS regulon members in V. parahaemolyticus.
Introduction
Quorum sensing (QS) systems are widely distributed in Vibrio species and act though complex signal transduction cascades involving cell density-dependent synthesis, release, and detection of signal molecules called autoinducers [1,2]. AphA and HMR [an abbreviation of high cell density (HCD) master regulator] are the two master QS regulators that are abundantly expressed at low cell density (LCD) and HCD, respectively, and a reciprocally gradient production of these two regulators has been recorded with transition between LCD and HCD [3–6] (see also Figure 1). Notably, HMR has distinct names in different Vibrio species, e.g. OpaR in V. parahaemolyticus [7], LuxR in V. harveyi [8], HapR in V. cholerae [9], and SmcR in V. vulnificus [10].
Figure 1. Action of V. parahaemolyticus QS systems.
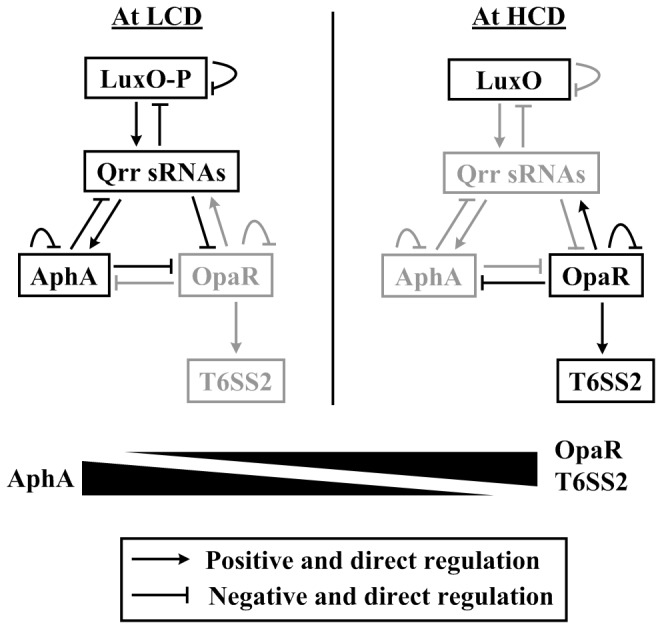
The regulatory associations between LuxO, Qrr sRNAs, AphA, and OpaR were summarized with the integration of relevant observations in V. parahaemolyticus [4,5] and closely related V. harveyi [3,32–40]. The grey fonts denoted the inhibited production of relevant proteins or the cease of relevant regulatory cascades. At LCD, low concentrations of autoinducers lead to phosphorylation of LuxO (LuxO-P), and LuxO-P activates expression of Qrr sRNA genes [32,33]. Redundant Qrr sRNAs promote AphA translation and, meanwhile, inhibit OpaR translation [34–36]. AphA further represses opaR transcription [3,5]. Overproduced AphA feeds back to inhibit transcription of qrr2-3 and its own gene [3,5]. In addition, over-production of Qrr sRNAs and LuxO-P triggers three additional feedback regulatory loops: i) LuxO-P represses transcription of its own gene, ii) Qrr sRNAs inhibits luxO translation, and iii) Qrr sRNAs repress translation of luxMN encoding the membrane-anchoring autoinducer-binding receptor protein LuxM and its cognate receptor LuxN [37–39]. The above feedbacks will contribute to control the LuxO-P, Qrr and AphA levels within the physiological states. At HCD, high concentrations of autoinducers reverse the phosphate flow in the circuit, leading to dephosphorylation of LuxO. Dephosphorylated LuxO is inactive as a regulator, leading to cessation of Qrr sRNA production; thus, there is no production of AphA but OpaR translation occurs [34,35]. OpaR in turns represses aphA transcription [4,40] but stimulates T6SS2 transcription (this study), and it also feeds back to inhibit its own expression [4,40]. OpaR is also able to activate the qrr2-4 transcription, leading to rapid down-regulation of opaR [4,40]; this OpaR-qrr feedback dramatically accelerates transition from HCD to LCD, but it has no effect on QS behaviors at steady-state LCD or HCD [41]. Taken together, there is the reciprocal gradients of cellular AphA and OpaR levels during transition between LCD and HCD, and AphA and OpaR act as the master QS regulators at LCD and HCD, respectively. In addition, T6SS2 transcription enhances in a gradient manner with transition from LCD to HCD, which is coordinately controlled by AphA and OpaR.
V. parahaemolyticus is a natural inhabitant of estuarine and marine environments. A small proportion of V. parahaemolyticus isolates, which harbor one or more key virulence genes [11,12], are pathogenic to human beings. V. parahaemolyticus is a worldwide cause of food-borne gastroenteritis which is usually self-limited and lasts within several days, but severe V. parahaemolyticus-caused diseases may occur in persons with weakened immune systems [12,13]. V. parahaemolyticus can also cause a skin infection if the bacterium gets in an open sore [12,13].
Type VI secretion system (T6SS) is a bacterial protein injection machinery with roles in virulence, symbiosis, interbacterial interaction, anti-pathogenesis, and environmental stress responses. There are two separate T6SS gene clusters, namely T6SS1 (VP1386-1414) and T6SS2 (VPA1024-1046), in chromosomes I and II of V. parahaemolyticus, respectively [14]. T6SS2 harbors 23 consecutive genes that can be assigned into three putative operons, VPA1027-1024, VPA1043-1028, and VPA1044-1046. T6SS1 is present in the majority of clinical isolates but only in a very low fraction of environmental isolates, while T6SS2 is universally present in the V. parahaemolyticus populations [15]. T6SS1 is predominantly expressed and active under high-salt marine-like conditions, while elevated secretion of T6SS2 effectors occurs under low salt conditions [16]. T6SS1 rather than T6SS2 has an anti-bacterial activity and, thus, is responsible for interbacterial competition under warm marine conditions, suggesting a role of T6SS1 in enhancing fitness of clinical isolates in marine environments [16]. Both T6SS1 and T6SS2 contribute to adhesion of V. parahaemolyticus to host cells [15].
QS-dependent expression of T6SS has be characterized in a bit of bacterial species such as Vibrio cholerae [5,11], V . alginolyticus [17], Pseudomonas aeruginosa [4,18], Yersinia pseudotuberculosis [19], and Aeromonas hydrophila [20]. It has been also previously shown that V. parahaemolyticus OpaR is a positive regulator of T6SS2 and a negative regulator of T6SS1 [16,21,22], but detailed mechanism of action of OpaR is unclear. These observations encourage us to hypothesize cell density- and QS-dependent expression of T6SS2 in V. parahaemolyticus. This study reported that OpaR bound to the promoter regions of VPA1027-1024, VPA1043-1028, and VPA1044-1046 to stimulate their transcription, while AphA negatively regulated their transcription most likely through acting on OpaR (Figure 1). In addition, this regulation led to a gradient increase of T6SS2 transcription with transition from LCD to HCD (Figure 1).
Materials and Methods
Bacterial strains
The wild-type (WT) V. parahaemolyticus strain RIMD 2210633 is isolated from a patient with traveler’s diarrhea in Japan in 1996 [14]. The base pairs (bps) 2 to 516 of coding region of aphA or the entire coding region of opaR were deleted from WT in our previous studies [4,5], generating the ΔaphA or ΔopaR mutant, respectively. The non-polar deletion of aphA or opaR was verified in Figure S1. The empty and recombinant pHRP309 plasmids tested were transformed into WT, ΔaphA, and ΔopaR for LacZ fusion assays (see below). All the primers used in the present work were listed in Table 1.
Table 1. Oligonucleotide primers used in this study.
| Target |
Primers (forward/reverse, 5'–3')
|
||
|---|---|---|---|
| Primer extension | |||
| aphA | /GCTCTTACTGGCGCTTGAG | ||
| opaR | /ATCCATTTTCCTTGCCATTTG | ||
| VPA1027 | /CTGCATGCTAATCTCCTAGAGC | ||
| VPA1043 | /GATTTGAAGCTTTAATTATTAACAT | ||
| VPA1044 | /CCGCTATCGCTGCTATTT | ||
| LacZ fusion | |||
| VPA1027 | GCGCGTCGACTATTACCTTACTTGCCTCTCGG /GCGCGAATTCTGCTTCACGGTCCATTGC | ||
| VPA1043 | GCGCGTCGACTTTGTTGATAGGTGGTATTGTG /ATATGAATTCTGAGCGTCCGAAGGTTAC | ||
| VPA1044 | GCGCGTCGACGGGACAAAGCAAGCTCATTC /ATATGAATTCAGCGGAGTCTTGTTTATTAACG | ||
| Protein production | |||
| aphA | AGCGGGATCCATGTCATTACCACACGTAATC /AGCGAAGCTTTTAACCAATCACTTCAAGTTC | ||
| opaR | AGCGGGATCCATGGACTCAATTGCAAAGAG /AGCGAAGCTTTTAGTGTTCGCGATTGTAG | ||
| Complementation of mutant # | |||
| aphA | GATTCTAGAA G G A G G AATTCACCATGTCATTACCACACGTAATC/GACAAGCTTTTAACCAATCACTTCAAGTTC | ||
| opaR | GATTCTAGAA G G A G G AATTCACCATGGACTCAATTGCAAAGAG /GACAAGCTTTTAGTGTTCGCGATTGTAG | ||
| EMSA | |||
| VPA1027 | TATTACCTTACTTGCCTCTCGG/TGCTTCACGGTCCATTGC | ||
| VPA1043 | AGCGGAGTCTTGTTTATTAACG/CGAGAAAATCTAACCGAAG | ||
| VPA1044 | TTGTGGAAACTCGTTATGG/TTGACGGGTGAAAGTTTGAG | ||
| 16S rRNA | GACACGGTCCAGACTCCTAC/GGTGCTTCTTCTGTCGCTAAC | ||
| DNase I footprinting | |||
| VPA1027 | GAGTTGCTTCATAATAAC/GTTCCGCTGTCGCTTCAC TATTACCTTACTTGCCTCTCGG/CGTCTTACCATTAAGAATTGC | ||
| VPA1043 | AGCGGAGTCTTGTTTATTAACG/CGAGAAAATCTAACCGAAG | ||
| VPA1044 | TTGTGGAAACTCGTTATGG/TTGACGGGTGAAAGTTTGAG | ||
#, amplification of the aphA or opaR coding region together with a ribosome binding site consensus AGGAGG (underlined) and a spacer AATTCACC (bold and italic).
Bacterial growth conditions
For general V. parahaemolyticus cultivation and maintenance, bacteria were cultured in the HI broth [2.5% Bacto heart infusion (BD Bioscience)] or on the HI plate (2.5% Bacto heart infusion, and 1.5% bacteriological grade agar) at 37 °C. For long term storage, bacteria were stored in the HI broth with the addition of 30% glycerol at -85 °C. For the following gene regulation assays, we used a design of two-round precultivation of bacterial cells: firstly, the glyceric stocks of bacteria were inoculated into 15 ml of HI broth for growing at 37 °C with shaking at 200 rpm for 12 to 14 h to enter the stationary growth phase; secondly, the resulting cell cultures were 50-fold diluted into 15 ml of HI broth, and allowed to grow under the above conditions to reach an optical density at 600 nm (OD600) of 1.4 to 1.6 (at the mid-exponential growth phases), and then the cell cultures were diluted with the HI broth to an OD600 value of 1.4. The precultivated bacterial cells were 1,000-fold diluted into 15 ml of HI broth for a third-round growth under the above conditions for cell harvest at different cell densities.
RNA isolation and primer extension assay
Total bacterial RNAs were extracted using TRIzol Reagent (Invitrogen) [4,5]. RNA quality was monitored by agarose gel electrophoresis, and RNA quantity was determined by spectrophotometry. For primer extension assay [4,5], an oligonucleotide primer complementary to a portion of RNA transcript of each indicated gene was employed to synthesize cDNAs from RNA templates. Three to 10 µg of total RNA was annealed with 1 pmol of [γ-32P] end-labeled reverse primer using a Primer Extension System (Promega) according to the manufacturer’s instructions. For a single target gene, the same amount of total RNAs was used as starting materials to determine its relative mRNA levels in different isogenic strains grown at different cell densities. The same labeled primer was also used for sequencing with an AccuPower & Top DNA Sequencing Kit (Bioneer). The primer extension products and sequencing materials were concentrated and analyzed in a 6% polyacrylamide/8 M urea gel. The result was detected by autoradiography (Kodak film).
LacZ fusion and β-galactosidase assay
The promoter-proximal DNA region of each indicated gene was amplified by PCR with ExTaq™ DNA polymerase (Takara) using RIMD 2210633 genome DNA as the template. PCR fragments were then cloned between SalI and EcoRI sites of low-copy-number transcriptional lacZ fusion vector pHRP309 that harbors a gentamicin resistance gene and a promoterless lacZ reporter gene [19]. Correct cloning was verified by DNA sequencing. An empty pHRP309 plasmid was also introduced into each strain tested as the negative control. V. parahaemolyticus strains transformed with recombinant or empty pHRP309 plasmids were grown as above to measure the β-galactosidase activity in cellular extracts using a β-Galactosidase Enzyme Assay System (Promega) [4,5].
Preparation of 6× His-tagged OpaR (His-OpaR) and AphA (His-AphA) proteins
The preparation of purified His-OpaR or His-AphA protein was done as described previously [4,5]. The entire coding region of opaR or aphA of strain RIMD 2210633 was cloned between BamHI and HindIII sites of plasmid pET28a (Novagen). The recombinant plasmid encoding His-OpaR or His-AphA was transformed into E. coli BL21λDE3 cells, and grown in the Luria-Bertani (LB) broth at 37 °C with shaking at 200 rpm for 4 to 5 h. The resulting culture was diluted 1/100 into 200 to 300 ml of fresh LB broth, and grown under the above conditions to an OD600 of about 0.5. The culture was shifted to 18 °C for 1 h, and then induced with 1 mM IPTG for 16 to 18 h with shaking at 100 rpm. Cells were collected by centrifugation and frozen at -60 °C. The pellet was resuspended in 10 ml of 50 mM sodium phosphate buffer, pH 7.4, 500 mM NaCl, and 5 mM imidazole. Cells were disrupted using a cell cracker, and the insoluble material was pelleted by centrifugation at 12,000 rpm. The clarified supernatant was applied to a 3 ml Ni-NTA Agarose Column (Qiagen), and the overproduced protein was purified under native conditions. Fractions from a homogenous peak were pooled, and the final preparation was dialyzed against 10 mM Tris HCl, pH 7.4, 10 mM NaCl, 1 mM EDTA, 0.1 mM DTT, and 20% glycerol. The purified protein was stored at -60 °C, and the protein purity was verified by SDS-PAGE.
Electrophoresis mobility shift assay (EMSA)
The promoter-proximal DNA region of each indicated gene was amplified by PCR. For EMSA [4,5], the 5′ ends of DNA were labeled using [γ-32P] ATP and T4 polynucleotide kinase. DNA binding was performed in a 10 µl reaction volume containing binding buffer [1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5) and 0.05 mg/ml poly-(dI-dC)], labeled DNA (1000 to 2000 c.p.m/µl), and increasing amounts of His-AphA. Three controls were included in each EMSA experiment: 1) cold probe as specific DNA competitor (the same promoter-proximal DNA region unlabeled), 2) negative probe as non-specific DNA competitor (the unlabeled coding region of the 16S rRNA gene), and 3) non-specific protein competitor [rabbit anti-F1-protein polyclonal antibodies]. The F1 protein is the protective antigen from Yersinia pestis [23]. After incubation at room temperature for 30 min, the products were loaded onto a native 4% (w/v) polyacrylamide gel, and electrophoresed in 0.5× TBE buffer for about 50 min at 220 V. Radioactive species were detected by autoradiography after exposure to Kodak film at -70 °C.
DNase I footprinting
For DNase I footprinting [4,5], the promoter-proximal DNA regions with a single 32P-labeled end were PCR amplified with either sense or antisense primer being end-labeled. The PCR products were purified using the QiaQuick columns (Qiagen). Increasing amounts of His-AphA or His-OpaR were incubated with the purified, labeled DNA fragment (2 to 5 pmol) for 30 min at room temperature, in a final 10 µl reaction volume containing the binding buffer used in EMSA. Before DNA digestion, 10 µl of Ca2+/Mg2+ solution (5 mM CaCl2 and 10 mM MgCl2) was added, followed by incubation for 1 min at room temperature. The optimized RQ1 RNase-Free DNase I (Promega) was then added to the reaction mixture, and the mixture was incubated at room temperature for 40 to 90 s. The reaction was quenched by adding 9 µl of stop solution (200 mM NaCl, 30 mM EDTA, and 1% SDS), followed by incubation for 1 min at room temperature. The partially digested DNA samples were extracted with phenol/chloroform, precipitated with ethanol, and analyzed in 6% polyacrylamide/8 M urea gel. Protected regions were identified by comparison with sequencing ladders. The templates for sequencing were the same as DNA fragments for DNase I footprinting. Radioactive species were detected as above.
Computational promoter analysis
The 400 bp upstream regions of the genes tested (Table 2) were retrieved from the genome sequence of RIMD 2210633 with the ‘retrieve-seq’ program [18]. The position-specific scoring matrix (PSSM) representing the conserved signals for AphA [5] or OpaR [4] recognition was used for pattern matching within target DNA regions, by using the matrices-paster tool [18].
Table 2. Prediction of AphA/OpaR box-like sequences within upstream DNA regions.
| First gene |
AphA box-like sequence |
OpaR box-like sequence |
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Operon |
ID |
Name |
Position&
|
Sequence |
Score | Position&
|
Sequence |
Score | |||||||||||||||
| VPA1027-1024 | VPA1027 | hcp2 | R-119. . -100 | ATACGCTCCTTTATATCTTT | 3.98 | D-389…- 370 | TAATGACATTGTAGACAATA | 9.01 | |||||||||||||||
| D-87… 68 | TTTTGATACATCAATCATTA | 8.29 | |||||||||||||||||||||
| D-57. . -38 | TTCAGATAATTTAATTAATA | 9.45 | |||||||||||||||||||||
| VPA1043-1028 | VPA1043 | NA | R-196. . -177 | ATATCCAACCAGGTTCAAAT | 2.51 | D-356…- 337 | TATTTATAGATTTGTCTTTA | 9.33 | |||||||||||||||
| VPA1044-1046 | VPA1044 | NA | D-143. . -124 | ATATCCAACCAGGTTCAAAT | 2.51 | D-250. . -231 | TATTAACATTAAGATTAATA | 9.9 | |||||||||||||||
&, ‘D’ indicates the direct sequence while ‘R’ the reverse one; minus numbers denote the nucleotide positions upstream of indicated genes. NA, Not Applicable
Experimental replicates and statistical methods
For LacZ fusion assays, experiments were performed with at least three independent bacterial cultures, and values were expressed as mean ± standard deviation (SD). Statistical testing of difference was made by Student’s paired t test, and a P value of <0.01 was taken as significant. For primer extension, EMSA, and footprinting, representative data from at least two independent biological replicates were shown.
Results
Predicted AphA/OpaR box-like sequences within T6SS2 gene cluster
The first genes of the three T6SS2 operons VPA1027-1024, VPA1043-1028, VPA1044-1046 were subjective to computational promoter analysis and further gene regulation experiments. The previously characterized PSSMs of AphA [5] and OpaR [4] were used to statistically predict the presence of AphA/OpaR box-like elements within the promoter-proximal regions of the above three ‘first genes’. This analysis generated weight scores for each target promoter, and the higher score values indicated the higher probability of AphA/OpaR-promoter association [17]. When a frequently used score of seven was taken as the cutoff value, OpaR box-like sequences were found for all the three genes, while none of AphA box-like elements could be identified for them (Table 2).
Negative regulation of T6SS2 genes by AphA
For the following primer extension and LacZ fusion assays, bacterial cells were harvested at an OD600 value of about 0.2 to simulate the LCD conditions at which AphA was predominantly expressed. The primer extension assay (Figure 2a) indicated that the mRNA levels of all the three genes VPA1027, VPA1043, and VPA1044 evidently enhanced in ΔahpA relative to WT. The transcriptional lacZ fusion vector that contained the target promoter-proximal DNA region and the promoterless lacZ gene was transformed into WT and ΔaphA to compare the promoter activities of each of the above three genes in these two strains (Figure 2b). The LacZ fusion experiments disclosed that the promoter activity of each of the above three genes significantly enhanced in ΔaphA relative to WT. The promoter-proximal DNA regions of the above three genes were amplified, radioactively labeled, and subjected to EMSA with a purified His-AphA protein (Figure 2c). Negative EMSA results were observed for all the above three genes, but positive results were observed for the positive control gene opaR as described previously [5] (data not shown). Further DNase I footprinting experiments (Figure 2d) could not detected footprint for all the above three genes; these were consistent with the EMSA results. Taken together, V. parahaemolyticus AphA appears to negatively regulate the transcription of VPA1027-1024, VPA1043-1028, and VPA1044-1046 in an indirect manner.
Figure 2. Regulation of T6SS2 genes by AphA.

Lanes C, T, A, and G represent the Sanger sequencing reactions. The minus and positive numbers indicated the nucleotide positions upstream and downstream of indicated genes. a) Primer extension. An oligonucleotide primer was designed to be complementary to the RNA transcript of each gene tested. The primer extension products were analyzed with an 8 M urea-6% acrylamide sequencing gel. The transcriptional start sites were indicated by arrows with nucleotides and positions. b) LacZ fusion. The target promoter-proximal DNA region was cloned into the lacZ transcriptional fusion vector pHRP309 and then transformed into WT or ΔAphA to determine the promoter activity, i.e., the β-galactosidase activity (miller units) in the cellular extracts. c) EMSA. The radioactively labeled promoter-proximal DNA fragments were incubated with increasing amounts of purified His-AphA protein and then subjected to 4% (w/v) polyacrylamide gel electrophoresis. If there was the association of His-AphA and target DNA, the band of free DNA disappeared with increasing amounts of His-AphA, resulting in a retarded DNA band with decreased mobility, which presumably represented the DNA-AphA complex. Shown also was the schematic representation of the EMSA design. d) DNase I footprinting. Labeled coding or non-coding DNA probes were incubated with increasing amounts of purified His-AphA (Lanes 1, 2, 3, and 4 containing 0, 35.1, 46.8, and 58.5 pmol, respectively) and then subjected to DNase I footprinting assay. The footprint regions were indicated by vertical bars with positions.
Positive regulation of T6SS2 genes by OpaR
Bacterial cells were harvested at an OD600 value of about 1.2 to simulate the HCD conditions at which OpaR was predominantly expressed. Both primer extension (Figure 3a) and LacZ fusion (Figure 3b) assays disclosed that the transcription of the three genes VPA1027, VPA1043, and VPA1044 decreased in ΔopaR relative to WT. As determined by EMSA (Figure 3c), a purified His-OpaR protein was able to bind to the upstream DNA fragments of all these three genes in a dose-dependent manner. By using DNase I footprinting (Figure 3d), His-OpaR protected a single DNA region upstream of each of VPA1027, VPA1043, and VPA1044 in a dose-dependent manner, which was consistent with the EMSA results. Each of the footprints detected for VPA1027, VPA1043, and VPA1044 contained one or more OpaR box-like sequences as predicted in Table 2, and was considered as the OpaR site for each target gene. Taken together, V. parahaemolyticus OpaR can bind to the promoter regions of VPA1027-1024, VPA1043-1028, and VPA1044-1046 to stimulate their transcription.
Figure 3. Regulation of T6SS2 genes by OpaR.

The primer extension (a), LacZ fusion (b), EMSA (c), DNase I footprinting (d) assays were performed to characterize the regulation of VPA1027-1024, VPA1043-1028, and VPA1044-1046 operons by OpaR. See Figure 2 for detail annotations.
Cell density-dependent transcription of aphA, opaR, and VPA1027
The mRNA levels of aphA, opaR, and VPA1027 (as a representative of T6SS2 genes) were measured in WT grown at various OD600 values (i.e. at different cell densities) by the primer extension assay (Figure 4). The aphA mRNA levels decreased considerably with the increasing of cell density, whereas the mRNA levels of opaR and VPA1027 increased dramatically with increasing of cell density. These results not only confirmed the rationality of cell harvest at an OD600 value of about 0.2 or 1.2 for characterizing AphA- or OpaR-mediated gene regulation, respectively, but also indicated the gradual elevation of mRNA levels of T6SS2 genes with transition from LCD to HCD.
Figure 4. Gene transcription pattern during growth.
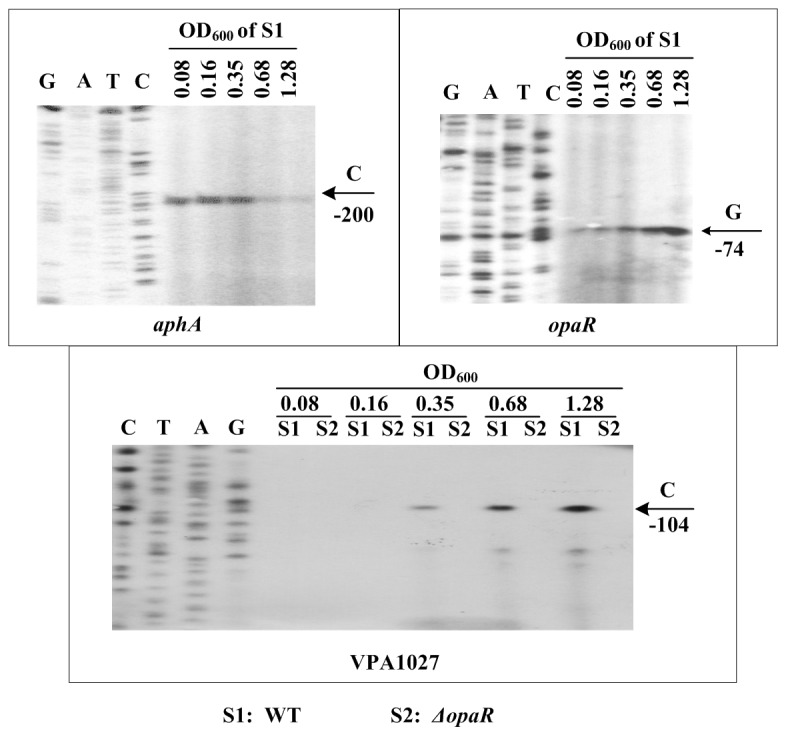
The bacterial cells were harvested at various OD600 values. An oligonucleotide primer was designed to be complementary to the RNA transcript of each gene tested. The primer extension products were analyzed with an 8 M urea-6% acrylamide sequencing gel. Lanes C, T, A, and G represented Sanger sequencing reactions. The transcriptional start sites were indicated by arrows with nucleotides and positions. The minus numbers under the arrows indicated the nucleotide positions upstream of the indicated genes.
Discussion
OpaR inhibits V. parahaemolyticus-induced cytotoxicity to host cells, mostly though acting on type three secretion system 1 (T3SS1) that is the major determinant of cytotoxicity in V. parahaemolyticus [24–26]. AphA enhances lethality in mice and cytotoxic activity in V. parahaemolyticus [27]. The detailed roles of OpaR and AphA in regulating virulence genes in V. parahaemolyticus need to be further elucidated.
As characterized in this study, OpaR and AphA acted as the positive and negative regulators of T6SS2, respectively, leading to a gradient elevation of transcriptional levels of T6SS2 with transition from LCD to HCD (Figure 1). The positive regulation of the three T6SS2 operons VPA1027-1024, VPA1043-1028, and VPA1044-1046 by OpaR achieved through direct association between OpaR and its target promoters. In contrast, AphA negatively regulated T6SS2 genes most likely through acting on OpaR, given the facts that none of the above promoter-proximal regions could be bound by AphA (this study) and that AphA and OpaR acted as transcriptional repressors to interact with each other [4,5]. These observations strongly supported the notion that T6SS2 might play important roles at the middle/late stages of growth/infection.
Collection of data of translation/transcription start sites, core promoter -10 and -35 elements, OpaR sites, OpaR box-like sequences, Shine-Dalgarno (SD) sequences (ribosomal binding sites) enabled us to depict the organization of AphA/OpaR-dependent promoters characterized herein (Figure 5). The OpaR sites for VPA1043-1028 and VPA1044-1046 were upstream of promoter -35 elements and, thus, both of these OpaR-dependent promoters might have a class I transcriptional stimulation that depends on the RNAP α subunit C-terminal domain for function [28]. Binding of OpaR to the upstream region of VPA1027-1024 was highly unusual, because two different OpaR sites, upstream and downstream of the -35 and -10 core promoter regions, respectively, were identified.
Figure 5. Organization of promoter-proximal DNA regions.

The promoter-proximal DNA regions of indicated genes were derived from RIMD 221063.
Three additional gene loci have been shown to be positively regulated by HMRs in a direct manner in different Vibrio species. V. harveyi LuxR binds to the upstream DNA region of the luminescence operon luxCDABEGH to stimulate its transcription [29]. Vibrio cholerae HapR [30] and V. vulnificus SmcR [31] stimulate the metalloprotease genes hapA and vvpE, respectively. Given the fact that the reciprocally gradient production of AphA and HMR is a conserved mechanism employed by multiple Vibrio species [3–6], the above three HMR-stimulated gene loci would show an elevated production in a gradient manner with transition from LCD to HCD.
Supporting Information
Primer extension assay for validation of non-polar deletion of opaR or aphA. For complementation of ΔopaR or ΔaphA, a PCR-generated DNA fragment composed of the entire coding region of opaR or aphA, respectively, together with a upstream synthetic ribosome binding site (Table 1), was cloned into between the XbaI and HinDIII sites of pBAD33 vector [42] harboring an arabinose PBAD promoter and a chloramphenicol resistance gene. The resulting recombinant plasmid pBAD33-opaR or pBAD33-aphA, respectively, was then introduced into ΔopaR or ΔaphA through electrotransformation, yielding the complemented mutant strain ΔopaR/pBAD33-opaR or ΔaphA/pBAD33-aphA, respectively. In addition, the empty vector pBAD33 was introduced into WT or ΔopaR or ΔaphA to generate the strain named WT/pBAD33 or ΔopaR/pBAD33 or ΔaphA/pBAD33, respectively. Bacteria were cultivated as described in the main text, with the modification that 5 µg/ml chloramphenicol and 0.1% arabinose were added in cell cultures. The primer extension experiments were subsequently done to determine the relative mRNA levels of VPA1027 in WT/pBDA33, ΔopaR/pBDA33, ΔopaR/pBDA33-opaR, ΔaphA/pBDA33, and ΔaphA/pBDA33-aphA. The mRNA level was significantly repressed in ΔopaR/pBDA33 relative to either WT/pBDA33 or ΔopaR/pBDA33-opaR, and, yet, it was significantly enhanced in ΔaphA/pBDA33 compared to either WT/pBDA33 or ΔaphA/pBDA33-aphA. These results confirmed that the opaR or aphA deletion was nonpolar.
(TIF)
Acknowledgments
We thank Professor Mitsuaki Nishibuchi from Kyoto University for kindly providing strain RIMD 2210633. All the experiments of this study were done in Dongsheng Zhou’s laboratory.
Funding Statement
National Natural Science Foundation of China (31170127, 31071093, and 31170129) and the National Key Program for Infectious Disease of China (2013ZX10004216). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cámara M, Hardman A, Williams P, Milton D (2002) Quorum sensing in Vibrio cholerae. Nat Genet 32: 217-218. doi:10.1038/ng1002-217. PubMed: 12355076. [DOI] [PubMed] [Google Scholar]
- 2. Defoirdt T, Boon N, Sorgeloos P, Verstraete W, Bossier P (2008) Quorum sensing and quorum quenching in Vibrio harveyi: lessons learned from in vivo work. Isme J 2: 19-26. doi:10.1038/ismej.2007.92. PubMed: 18180744. [DOI] [PubMed] [Google Scholar]
- 3. Rutherford ST, van Kessel JC, Shao Y, Bassler BL (2011) AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev 25: 397-408. doi:10.1101/gad.2015011. PubMed: 21325136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lesic B, Starkey M, He J, Hazan R, Rahme LG (2009) Quorum sensing differentially regulates Pseudomonas aeruginosa type VI secretion locus I and homologous loci II and III, which are required for pathogenesis. Microbiology 155: 2845-2855. doi:10.1099/mic.0.029082-0. PubMed: 19497948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishikawa T, Rompikuntal PK, Lindmark B, Milton DL, Wai SN (2009) Quorum sensing regulation of the two hcp alleles in Vibrio cholerae O1 strains. PLOS ONE 4: e6734. doi:10.1371/journal.pone.0006734. PubMed: 19701456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Kessel JC, Rutherford ST, Shao Y, Utria AF, Bassler BL (2013) Individual and Combined Roles of the Master Regulators AphA and LuxR in Control of the Vibrio harveyi Quorum-Sensing Regulon. J Bacteriol 195: 436-443. doi:10.1128/JB.01998-12. PubMed: 23204455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCarter LL (1998) OpaR, a homolog of Vibrio harveyi LuxR, controls opacity of Vibrio parahaemolyticus. J Bacteriol 180: 3166-3173. PubMed: 9620967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin B, Wang Z, Malanoski AP, O’Grady EA, Wimpee CF et al. (2010) Comparative genomic analyses identify the Vibrio harveyi genome sequenced strains BAA-1116 and HY01 as Vibrio campbellii. Environ Microbiol Rep 2: 81-89. PubMed: 20686623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML et al. (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406: 477-483. doi:10.1038/35020000. PubMed: 10952301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen CY, Wu KM, Chang YC, Chang CH, Tsai HC et al. (2003) Comparative genome analysis of Vibrio vulnificus, a marine pathogen. Genome Res 13: 2577-2587. doi:10.1101/gr.1295503. PubMed: 14656965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng J, Shin OS, Cameron DE, Mekalanos JJ (2010) Quorum sensing and a global regulator TsrA control expression of type VI secretion and virulence in Vibrio cholerae. Proc Natl Acad Sci U S A 107: 21128-21133. doi:10.1073/pnas.1014998107. PubMed: 21084635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Broberg CA, Calder TJ, Orth K (2011) Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect 13: 992-1001. doi:10.1016/j.micinf.2011.06.013. PubMed: 21782964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yeung PS, Boor KJ (2004) Epidemiology, pathogenesis, and prevention of foodborne Vibrio parahaemolyticus infections. Foodborne Pathog Dis 1: 74-88. doi:10.1089/153531404323143594. PubMed: 15992266. [DOI] [PubMed] [Google Scholar]
- 14. Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T et al. (2003) Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet 361: 743-749. doi:10.1016/S0140-6736(03)12659-1. PubMed: 12620739. [DOI] [PubMed] [Google Scholar]
- 15. Yu Y, Yang H, Li J, Zhang P, Wu B et al. (2012) Putative type VI secretion systems of Vibrio parahaemolyticus contribute to adhesion to cultured cell monolayers. Arch Microbiol, 194: 827–35. PubMed: 22535222. [DOI] [PubMed] [Google Scholar]
- 16. Salomon D, Gonzalez H, Updegraff BL, Orth K (2013) Vibrio parahaemolyticus Type VI Secretion System 1 Is Activated in Marine Conditions to Target Bacteria, and Is Differentially Regulated from System 2. PLOS ONE 8: e61086. doi:10.1371/journal.pone.0061086. PubMed: 23613791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sheng L, Gu D, Wang Q, Liu Q, Zhang Y (2012) Quorum sensing and alternative sigma factor RpoN regulate type VI secretion system I (T6SSVA1) in fish pathogen Vibrio alginolyticus. Arch Microbiol 194: 379-390. doi:10.1007/s00203-011-0780-z. PubMed: 22173829. [DOI] [PubMed] [Google Scholar]
- 18. Sana TG, Hachani A, Bucior I, Soscia C, Garvis S et al. (2012) The second type VI secretion system of Pseudomonas aeruginosa strain PAO1 is regulated by quorum sensing and Fur and modulates internalization in epithelial cells. J Biol Chem 287: 27095-27105. doi:10.1074/jbc.M112.376368. PubMed: 22665491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang W, Xu S, Li J, Shen X, Wang Y et al. (2011) Modulation of a thermoregulated type VI secretion system by AHL-dependent quorum sensing in Yersinia pseudotuberculosis. Arch Microbiol 193: 351-363. PubMed: 21298257. [DOI] [PubMed] [Google Scholar]
- 20. Khajanchi BK, Sha J, Kozlova EV, Erova TE, Suarez G et al. (2009) N-acylhomoserine lactones involved in quorum sensing control the type VI secretion system, biofilm formation, protease production, and in vivo virulence in a clinical isolate of Aeromonas hydrophila. Microbiology 155: 3518-3531. doi:10.1099/mic.0.031575-0. PubMed: 19729404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gode-Potratz CJ, McCarter LL (2011) Quorum sensing and silencing in Vibrio parahaemolyticus. J Bacteriol 193: 4224-4237. doi:10.1128/JB.00432-11. PubMed: 21705592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma L, Zhang Y, Yan X, Guo L, Wang L et al. (2012) Expression of the Type VI Secretion System 1 Component Hcp1 Is Indirectly Repressed by OpaR in Vibrio parahaemolyticus. Scientific World J: 7: 982140 PubMed: 22924031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andrews GP, Heath DG, Anderson GW Jr., Welkos SL, Friedlander AM (1996) Fraction 1 capsular antigen (F1) purification from Yersinia pestis CO92 and from an Escherichia coli recombinant strain and efficacy against lethal plague challenge. Infect Immun 64: 2180-2187. PubMed: 8675324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gode-Potratz CJ, McCarter LL (2011) Quorum sensing and silencing in Vibrio parahaemolyticus. J Bacteriol, 193: 4224–37. PubMed: 21705592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hiyoshi H, Kodama T, Iida T, Honda T (2010) Contribution of Vibrio parahaemolyticus virulence factors to cytotoxicity, enterotoxicity, and lethality in mice. Infect Immun 78: 1772-1780. doi:10.1128/IAI.01051-09. PubMed: 20086084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Henke JM, Bassler BL (2004) Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus. J Bacteriol 186: 3794-3805. doi:10.1128/JB.186.12.3794-3805.2004. PubMed: 15175293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang L, Ling Y, Jiang H, Qiu Y, Qiu J et al. (2013) AphA is required for biofilm formation, motility, and virulence in pandemic Vibrio parahaemolyticus. Int J Food Microbiol 160: 245-251. doi:10.1016/j.ijfoodmicro.2012.11.004. PubMed: 23290231. [DOI] [PubMed] [Google Scholar]
- 28. Ishihama A (2000) Functional modulation of Escherichia coli RNA polymerase. Annu Rev Microbiol 54: 499-518. doi:10.1146/annurev.micro.54.1.499. PubMed: 11018136. [DOI] [PubMed] [Google Scholar]
- 29. Swartzman E, Silverman M, Meighen EA (1992) The luxR gene product of Vibrio harveyi is a transcriptional activator of the lux promoter. J Bacteriol 174: 7490-7493. PubMed: 1385389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsou AM, Zhu J (2010) Quorum sensing negatively regulates hemolysin transcriptionally and posttranslationally in Vibrio cholerae. Infect Immun 78: 461-467. doi:10.1128/IAI.00590-09. PubMed: 19858311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jeong HS, Lee MH, Lee KH, Park SJ, Choi SH (2003) SmcR and cyclic AMP receptor protein coactivate Vibrio vulnificus vvpE encoding elastase through the RpoS-dependent promoter in a synergistic manner. J Biol Chem 278: 45072-45081. doi:10.1074/jbc.M308184200. PubMed: 12947096. [DOI] [PubMed] [Google Scholar]
- 32. Henke JM, Bassler BL (2004) Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J Bacteriol 186: 6902-6914. doi:10.1128/JB.186.20.6902-6914.2004. PubMed: 15466044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Waters CM, Bassler BL (2006) The Vibrio harveyi quorum-sensing system uses shared regulatory components to discriminate between multiple autoinducers. Genes Dev 20: 2754-2767. doi:10.1101/gad.1466506. PubMed: 17015436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS et al. (2004) The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118: 69-82. doi:10.1016/j.cell.2004.06.009. PubMed: 15242645. [DOI] [PubMed] [Google Scholar]
- 35. Tu KC, Bassler BL (2007) Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev 21: 221-233. doi:10.1101/gad.1502407. PubMed: 17234887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shao Y, Bassler BL (2012) Quorum-sensing non-coding small RNAs use unique pairing regions to differentially control mRNA targets. Mol Microbiol 83: 599-611. doi:10.1111/j.1365-2958.2011.07959.x. PubMed: 22229925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Long T, Tu KC, Wang Y, Mehta P, Ong NP et al. (2009) Quantifying the integration of quorum-sensing signals with single-cell resolution. PLOS Biol 7: e68. doi:10.1371/journal.pbio.1000068. PubMed: 19320539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tu KC, Long T, Svenningsen SL, Wingreen NS, Bassler BL (2010) Negative feedback loops involving small regulatory RNAs precisely control the Vibrio harveyi quorum-sensing response. Mol Cell 37: 567-579. doi:10.1016/j.molcel.2010.01.022. PubMed: 20188674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Teng SW, Schaffer JN, Tu KC, Mehta P, Lu W et al. (2011) Active regulation of receptor ratios controls integration of quorum-sensing signals in Vibrio harveyi. Mol Syst Biol 7: 491 PubMed: 21613980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pompeani AJ, Irgon JJ, Berger MF, Bulyk ML, Wingreen NS et al. (2008) The Vibrio harveyi master quorum-sensing regulator, LuxR, a TetR-type protein is both an activator and a repressor: DNA recognition and binding specificity at target promoters. Mol Microbiol 70: 76-88. doi:10.1111/j.1365-2958.2008.06389.x. PubMed: 18681939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tu KC, Waters CM, Svenningsen SL, Bassler BL (2008) A small-RNA-mediated negative feedback loop controls quorum-sensing dynamics in Vibrio harveyi. Mol Microbiol 70: 896-907. PubMed: 18808382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177: 4121-4130. PubMed: 7608087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer extension assay for validation of non-polar deletion of opaR or aphA. For complementation of ΔopaR or ΔaphA, a PCR-generated DNA fragment composed of the entire coding region of opaR or aphA, respectively, together with a upstream synthetic ribosome binding site (Table 1), was cloned into between the XbaI and HinDIII sites of pBAD33 vector [42] harboring an arabinose PBAD promoter and a chloramphenicol resistance gene. The resulting recombinant plasmid pBAD33-opaR or pBAD33-aphA, respectively, was then introduced into ΔopaR or ΔaphA through electrotransformation, yielding the complemented mutant strain ΔopaR/pBAD33-opaR or ΔaphA/pBAD33-aphA, respectively. In addition, the empty vector pBAD33 was introduced into WT or ΔopaR or ΔaphA to generate the strain named WT/pBAD33 or ΔopaR/pBAD33 or ΔaphA/pBAD33, respectively. Bacteria were cultivated as described in the main text, with the modification that 5 µg/ml chloramphenicol and 0.1% arabinose were added in cell cultures. The primer extension experiments were subsequently done to determine the relative mRNA levels of VPA1027 in WT/pBDA33, ΔopaR/pBDA33, ΔopaR/pBDA33-opaR, ΔaphA/pBDA33, and ΔaphA/pBDA33-aphA. The mRNA level was significantly repressed in ΔopaR/pBDA33 relative to either WT/pBDA33 or ΔopaR/pBDA33-opaR, and, yet, it was significantly enhanced in ΔaphA/pBDA33 compared to either WT/pBDA33 or ΔaphA/pBDA33-aphA. These results confirmed that the opaR or aphA deletion was nonpolar.
(TIF)