

Receptor-interacting kinase 3 (RIP3) is a critical regulator of programed necrosis and is regulated by post-translation modifications. In this review, Moriwaki and Chan discuss the underlying mechanisms that promote RIP3-dependent necrosis and its role in inflammatory disease.
Keywords: RIP1, MLKL, PGAM5, caspase 8, FADD, inflammation
Abstract
The receptor-interacting protein kinase 3 (RIP3/RIPK3) has emerged as a critical regulator of programmed necrosis/necroptosis, an inflammatory form of cell death with important functions in pathogen-induced and sterile inflammation. RIP3 activation is tightly regulated by phosphorylation, ubiquitination, and caspase-mediated cleavage. These post-translational modifications coordinately regulate the assembly of a macromolecular signaling complex termed the necrosome. Recently, several reports indicate that RIP3 can promote inflammation independent of its pronecrotic activity. Here, we review our current understanding of the mechanisms that drive RIP3-dependent necrosis and its role in different inflammatory diseases.
Cell number is tightly regulated by cell division and cell death to maintain tissue homeostasis in multicellular organisms. A classic example is found in Caenorhabditis elegans, in which unneeded or unwanted cells are eliminated by cell death at specific times during development (Sulston and Horvitz 1977). Because of its specific timing and pattern, the term programmed cell death was coined to describe cell death during normal animal development (Lockshin and Williams 1964). The morphology of the dying cells under electron microscopy revealed cell shrinkage, chromatin condensation, and cellular fragmentation. The name apoptosis was used to describe this type of cell death because programmed death resembles that of leaves falling off a tree (Kerr et al. 1972). Apoptosis during development is an inherently programmed phenomenon, as the discovery of ced-3, ced-4, and ced-9 in C. elegans demonstrates a genetic network that controls the process (Ellis and Horvitz 1986). Although programmed cell death and apoptosis are often used as synonyms, it is noteworthy that apoptosis can be induced by a myriad of external cues that do not regulate developmental processes.
The term necrosis describes cell death with swelling of organelles and plasma membrane rupture. Necrosis was once considered accidental cell death caused by overwhelming physical or chemical trauma. However, we now know that specific genes can induce necrosis in a regulated manner. The terms programmed necrosis, necroptosis, or regulated necrosis have been used to distinguish these types of cell death from accidental necrosis. Programmed necrosis is induced by many stimuli, such as intracellular ATP depletion, disturbance of Ca2+ homeostasis, mitochondrial depolarization, poly-(ADP-ribose) polymerase (PARP) activation, proteolysis by nonapoptotic proteases, increased reactive oxygen species (ROS), and cell surface receptor activation. There is evidence that these signals use distinct and yet overlapping mechanisms to induce necrosis (Vanlangenakker et al. 2012).
Death cytokines in the tumor necrosis factor (TNF) family are classic inducers of programmed necrosis. Receptor-interacting protein kinase 1 (RIP1) and RIP3, the two critical kinases that mediate TNF-dependent necrosis, have been in the spotlight lately because of their unique signaling mechanisms and pathological functions (Chan 2012). Carswell et al. (1975) originally discovered TNF as the factor that induces rapid hemorrhagic necrosis in cancer cells. However, most of the early work had focused on the molecular mechanisms and pathophysiological roles of TNF in inflammation and apoptosis. In 1988, TNF was shown to induce necrosis in a cell line derived from the L929 cells that Carswell et al. (1975) used to examine TNF activity (Laster et al. 1988). It was not until 2000 when Holler et al. (2000) demonstrated that RIP1 is a key regulatory molecule of necrosis induced by the death receptor ligands TNF, Fas ligand, and TNF-related apoptosis-inducing ligand (TRAIL). Unfortunately, progress in the field was stalled in part because RIP1−/− mice suffer from perinatal lethality (Kelliher et al. 1998). Research on necrosis got a significant boost when necrostatins, a class of chemical inhibitors of RIP1, became available (Degterev et al. 2005, 2008). In 2009, another RIP family member, RIP3, was identified as a crucial regulator of death receptor-induced necrosis (Cho et al. 2009; He et al. 2009; Zhang et al. 2009). The fact that RIP3−/− mice have no remarkable developmental defects dramatically facilitated the study of necrosis in pathophysiology.
RIP3—the key player in the necrosis signaling pathway
Structural organization of RIP3
RIP3 was first identified by two different groups through yeast two-hybrid screening and database searches as a RIP1-binding protein with homology with RIP1 and RIP2 (Sun et al. 1999; Yu et al. 1999). The human rip3 gene is located on chromosome 14 (Kasof et al. 2000), and its mRNA encodes a polypeptide of 518 amino acids (Fig. 1). RIP3 has an active kinase domain in its N terminus that is conserved in other RIP kinases and is essential for necrosis. Unlike the N termini, each RIP family member encodes a unique C terminus: death domain in RIP1, caspase recruitment domain (CARD) in RIP2, and an ankyrin repeat in RIP4. The unique C termini dictate their recruitment to different signaling scaffolds. For RIP3, a rather degenerate C-terminal motif termed the RIP homotypic interaction motif (RHIM), which is also present in the intermediate domain of RIP1, mediates its interaction with RIP1 and necrosis (Fig. 1; Sun et al. 2002).
Figure 1.

Domain organization of human RIP family proteins. The phospho-acceptor sites are highlighted with the letter P. The crucial lysine residues in the ATP-binding pocket are shown (K45 in RIP1, K47 in RIP2, K50 in RIP3, and K51 in RIP4). M92 is the “gatekeeper residue” in RIP1 that is important for kinase activity (Lu et al. 2011).
The role of RIP3 in apoptosis, NF-κB activation, and necrosis
Early experiments using overexpression systems implicate RIP3 in apoptosis and NF-κB signaling (Sun et al. 1999; Yu et al. 1999; Meylan et al. 2004). However, RIP3−/− thymocytes responded normally to different apoptosis stimuli, and RIP3−/− fibroblasts or macrophages were normal for TNF- and toll-like receptor (TLR) ligand-induced NF-κB activation (Newton et al. 2004). While we cannot rule out that RIP3 might regulate apoptosis or NF-κB signaling in unique circumstances, these results indicate that RIP3 does not have a central role in apoptosis or NF-κB activation.
The first hint that RIP3 might regulate necrosis also came from overexpression studies. Feng et al. (2007) showed that human RIP3 was cleaved at Asp328 (D328). When a noncleavable mutant of human RIP3 was expressed in cells, it led to caspase-independent cell death (Feng et al. 2007). Similar cleavage of mouse RIP3 has since been reported (Zhang et al. 2009). However, Feng et al. (2007) concluded that the cell death caused by the mutant RIP3 was apoptosis. Subsequently, three independent studies unequivocally demonstrated that RIP3-driven cell death was necrotic rather than apoptotic (Cho et al. 2009; He et al. 2009; Zhang et al. 2009). Since then, RIP3 has been shown to be a critical switch that drives necrosis induced by TNF-like death receptors TLR3 and TLR4 and the T-cell receptor (Cho et al. 2009; Ch'en et al. 2011; He et al. 2011). The most extensively characterized pathway leading to RIP3 activation during necrosis is initiated by TNF. The ligation of TNF to TNF receptor 1 (TNF-R1) causes the formation of the membrane-associated TNF-R1 signaling complex termed complex I, which consists of multiple protein adaptors, including TNFR-associated death domain (TRADD), RIP1, cellular inhibitor of apoptosis 1 (cIAP1), cIAP2, TNFR-associated factor 2 (TRAF2), and the linear ubiquitin chain assembly complex (LUBAC) (Fig. 2). RIP1 ubiquitination mediated by cIAPs and LUBAC in complex I is a critical event to trigger the NF-κB pathway (Gerlach et al. 2011) and block apoptosis and necrosis (O'Donnell et al. 2012). Deubiquitination of RIP1 by CYLD promotes apoptosis and necrosis through the formation of the cytoplasmic death-inducing signaling complex termed complex II (Hitomi et al. 2008, Wang et al. 2008). In addition to RIP1, complex II also contains caspase-8, Fas-associated protein with death domain (FADD), and cellular FLICE-like inhibitory protein (cFLIP). FADD and caspase-8 promote apoptosis by activating downstream caspases and inhibit necrosis through cleavage of RIP1, RIP3, and CYLD (Lin et al. 1999; Chan et al. 2003; Feng et al. 2007; O'Donnell et al. 2011). When caspase-8 activity is blocked by genetic ablation, chemical inhibitors, or viral caspase inhibitors, RIP1 and RIP3 turn complex II into a necrosis-inducing signaling complex that is often referred to as the necrosome (Declercq et al. 2009).
Figure 2.
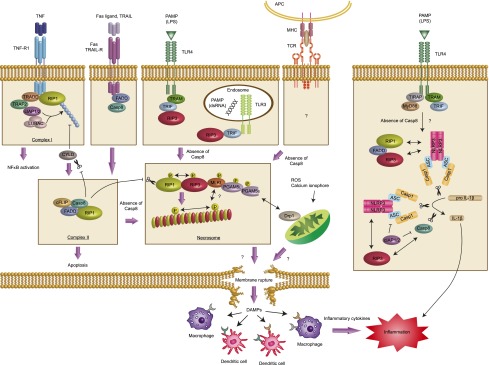
Overview of RIP3-dependent necrosis and inflammation. Ligation of TNF to TNF-R1 leads to the formation of complex I, where RIP1 is ubiquitinated by cIAP1/2 and LUBAC. Deubiquitination of RIP1 by CYLD converts the signal from cell survival to cell death by facilitating the formation of complex II. Fas ligand and TRAIL also induce complex II formation. Active caspase-8 in this complex induces apoptosis and inhibits necrosis by cleaving RIP1, RIP3, and CYLD. When caspase-8 is inhibited, the amyloid RIP1/RIP3 necrosome is formed. Necrosome formation causes necrosis through the mitochondrial pathway mediated by MLKL, recruitment of PGAM5L and PGAM5s, and activation of Drp1. PGAM5 and Drp1 also mediate necrosis induced by ROS and calcium ionophore. TLR3 and TLR4 activation by pathogen-associated molecular patterns (PAMPs) such as dsRNA and LPS lead to RIP3 binding to TRIF and necrosis. For the T-cell receptor (TCR), the mechanism that activates RIP3-dependent necrosis is still unknown. Membrane rupture results in the release of DAMPs to an extracellular space where innate immune cells such as macrophages and DCs are located. These innate immune cells recognize DAMPs during infection or tissue damage to elicit an inflammatory cytokine response. Besides necrosis, RIP3 can also drive IL-1β production in caspase-8-deficient DCs or when IAP proteins are eliminated by SMs. (APC) Antigen-presenting cells; (MHC) major histocompatibility complex.
Phosphorylation regulates necrosome assembly and activity
Necrostatins, which inhibit RIP1 kinase activity by binding to the kinase domain and locking RIP1 in its inactive conformation (Degterev et al. 2008; Xie et al. 2013), blocked necrosome assembly (Cho et al. 2009). Hence, the kinase activity of RIP1 is indispensable for necrosome assembly. Mass spectroscopy revealed that RIP1 is autophosphorylated at multiple serine residues in the kinase domain (Degterev et al. 2008), suggesting that phosphorylation might regulate its kinase activity. Surprisingly, alanine substitution of individual serine residue had minimal effects on RIP1 kinase activity (T McQuade and F Chan, unpubl.). Therefore, one can speculate that the cumulative effect of the negative charge on multiple phospho-serine residues might be important for activity. In addition to autophosphorylation, RIP1 kinase activity might also be regulated by RIP3. For instance, RIP3 could directly phosphorylate RIP1 in vitro, and necrosis-induced RIP1 phosphorylation was abrogated in RIP3−/− cells (Cho et al. 2009). In contrast, a kinase-inactive RIP3 mutant that could not mediate necrosis was nonetheless able to complex with RIP1 (He et al. 2009; Wang et al. 2012).
Like RIP1, RIP3 is highly phosphorylated upon necrosis induction. Because phosphorylation of RIP3 was inhibited by Nec-1, RIP1 is likely the upstream kinase that initiates necrosis signaling (Cho et al. 2009). One phosphorylation site on RIP3, Ser227 (Thr231/Ser232 for mouse RIP3), is particularly important for recruitment and activation of mixed-lineage kinase domain-like (MLKL), a crucial downstream substrate of RIP3 in the necrosis pathway (Sun et al. 2012; Chen et al. 2013). Although there are other phosphorylated serine/threonine residues on RIP3 (Chen et al. 2013), their roles in RIP3 activation are less defined. Collectively, these results are most consistent with a model in which RIP3 binding provides a scaffold on which RIP1 becomes activated. Once activated by autophosphorylation, RIP1 can proceed to phosphorylate downstream targets, including RIP3.
Necrosome formation is also tightly regulated by ubiquitination (see above) and acetylation. A recent study found that RIP1 was acetylated at Lys530 (Narayan et al. 2012). Deacetylation by sirtuin2 (SIRT2), which constitutively binds to RIP3, facilitated TNF-induced necrosis. These results suggest that one of the early events that stabilize the RIP1–RIP3 necrosome could be SIRT2-mediated deacetylation of RIP1. More work is required to confirm the role of SIRT2 and RIP1 acetylation in physiological necrosis.
The amyloidal nature of the necrosome
The interaction between RIP1 and RIP3 is mediated by the RHIM, a hydrophobic patch of β sheet (IQIGXXN for RIP1 [amino acids 539–542] and VQVGXXN for RIP3 [amino acids 458–461]) flanked by unstructured coiled-like residues. Detailed biochemical and biophysical study showed that the RHIMs of RIP1 and RIP3 mediate formation of a filamentous structure with classical characteristics of β-amyloids (Li et al. 2012). Mutations of the core RHIM residues of RIP1 (IQIG) or RIP3 (VQVG) disrupted the assembly of this β-amyloid structure, RIP kinase activation, and necrotic cell death. These results suggest that the RIP kinase activities and necrosome assembly might function in a feed-forward manner to amplify the pronecrotic signal (Fig. 2).
The amyloid scaffold of the necrosome raises several important questions. For example, since RIP1 and RIP3 can bind to pronecrotic (e.g., MLKL) as well as anti-necrotic factors (FADD and caspase-8), does amyloid assembly dictate whether apoptosis or necrosis will ensue upon TNF stimulation? Do RIP1 and RIP3 become resistant to caspase-8 cleavage and inhibition when they assemble into the β-amyloid complex? Does the structural similarity of the necrosome to neurotoxic amyloidal fibrils suggest a role for necrosis in neurodegeneration? These are some of the interesting and important questions to be addressed in the future.
RHIM in other innate immune signaling adaptors
The RHIM is also found in other adaptors with important functions in innate immunity and cell death. These include the TIR domain-containing adaptor-inducing interferon-β (IFNβ) (TRIF) (Meylan et al. 2004; Kaiser and Offermann 2005), the DNA-dependent activator of IFN regulatory transcription factors (DAI/ZBP1/DLM-1) (Kaiser et al. 2008; Rebsamen et al. 2009), and the viral inhibitor of RIP activation (vIRA/M45) from murine cytomegalovirus (MCMV) (Upton et al. 2008). In overexpression studies, RIP1 and RIP3 could bind to many of these RHIM-containing proteins. Interestingly, some of these “noncanonical” RHIM–RHIM interactions also appear to drive necrosis. For example, bacterial lipopolysaccharide (LPS), a ligand for TLR4, induced RIP3 and TRIF binding and necrosis in macrophages (He et al. 2011). In contrast, during MCMV infection, vIRA/M45 inhibits RIP3 binding to DAI to prevent premature necrosis of the infected cells, a process that is salient for productive viral replication (Upton et al. 2010, 2012). Thus, not all RHIM–RHIM interactions result in cell death. To complicate matters further, RHIM-mediated interaction between RIP1 and TRIF or DAI also drives downstream NF-κB activation (Kaiser et al. 2008; Rebsamen et al. 2009), an event that is usually associated with cell survival. It will be interesting to determine whether these non-death-inducing RHIM-dependent interactions also use β-amyloid assembly as a signaling platform.
Downstream execution of RIP3-dependent necrosis
As discussed above, MLKL is a recently identified RIP3 substrate. MLKL binds RIP3 through its C-terminal kinase-like domain, which is phosphorylated by RIP3 at Thr357 and Ser358. Mutations of MLKL at these residues blocked necrosis but not binding to RIP3 (Sun et al. 2012). Although the name implies that it may not be an authentic kinase, MLKL expressed in HEK293T cells could weakly phosphorylate myelin basic protein (MBP) in vitro (Zhao et al. 2012). Moreover, expression of an MLKL mutant in which the lysine residues in the ATP-binding pocket were substituted with alanines failed to restore TNF-induced necrosis in fibroblasts lacking MLKL expression (Zhao et al. 2012). Whether this weak kinase activity of MLKL is important for necrosome activity remains to be tested.
A fundamental feature of necrosis is the loss of cellular membrane potentials, which may be caused by depletion of intracellular ATP, damaged membrane lipids, and/or loss of function of ATP-dependent ion pumps. Disrupted membrane potential can lead to organelle swelling, mitochondrial damage, and plasma membrane rupture that are often associated with necrosis. How might RIP3 regulate these cellular changes? One recent study shows that phosphoglycerate mutase 5 (PGAM5), a resident mitochondrial protein, may play a role in the execution of cellular necrosis (Wang et al. 2012). PGAM5 exists in two isoforms: PGAM5 long and short (PGAM5L and PGAM5s). When PGAM5s is recruited to the RIP1/RIP3 necrosome by MLKL and becomes phosphorylated, it dephosphorylates and activates the mitochondrial fission protein Drp1. Drp1 activation causes mitochondrial fission and ROS production. Hence, PGAM5 may promote ROS production that causes the cellular damage observed in necrosis. Although this is a fascinating model, ROS scavengers could not inhibit all forms of necrosis (Temkin et al. 2006). Does that mean that PGAM5 and ROS are only required for necrosis in certain cell types? Further work is needed to clarify the role of PGAM5 in different cell types and identify whether additional effectors beyond Drp1 are involved.
RIP3 function in embryonic development
Unlike apoptosis, necrosis is not normally detected during embryogenesis. However, mice deficient in the essential apoptosis regulators Bax and Bak developed normally (Wei et al. 2001), indicating that nonapoptotic cell death can contribute to embryonic development when apoptosis is impaired. In contrast, caspase-8−/− and FADD−/− mice die on embryonic day 10.5 (E10.5) due to a defect in hematopoietic precursors in endothelium and failure in yolk sac vascularization (Varfolomeev et al. 1998; Yeh et al. 1998; Kang et al. 2004). The lethality might be due to excessive RIP kinase-dependent necrosis. Indeed, the lethality of caspase-8−/− and FADD−/− mice was rescued by germline inactivation of RIP1 or RIP3 (Kaiser et al. 2011; Oberst et al. 2011; Zhang et al. 2011). Mice deficient in the anti-apoptotic protein cFLIP also suffer from similar embryonic lethality (Yeh et al. 2000). Surprisingly, RIP3 deletion did not rescue this phenotype because the caspase-8 homodimer could still form to promote apoptosis (Dillon et al. 2012). This surprising finding is consistent with the fact that the caspase-8/cFLIP heterodimer has restricted cleavage activity compared with the caspase-8 homodimer, such that it cannot induce apoptosis but is still sufficient to cleave and inhibit RIP1 and/or RIP3 (Oberst et al. 2011). Consistent with this proposition, RIP3/FADD double deficiency rescued the lethality of cFLIP−/− mice (Dillon et al. 2012).
Besides rescuing the lethality of FADD−/− and caspase-8−/− mice, RIP3 deficiency also partially inhibited the lethality of mice lacking multiple cellular IAPs. The IAPs are RING-containing E3 ligases that promote cell survival through canonical NF-κB activation (cIAP1 and cIAP2) or by direct binding to caspases (XIAP) (Silke and Meier 2013). Germline deletion of individual IAP had no overt effects on development (Harlin et al. 2001; Conze et al. 2005; Conte et al. 2006). In contrast, cIAP1−/−cIAP2−/− or cIAP1−/−XIAP−/− mice died around E10.5 due to hemorrhages and cardiovascular defect (Moulin et al. 2012), suggesting certain functional redundancy among the IAPs. Interestingly, TNFR-1 deficiency allowed cIAP1−/−cIAP2−/− mice to survive until birth (Moulin et al. 2012). However, inactivation of RIP3 only prolonged survival for a few days during embryogenesis. Surprisingly, cIAP1−/−XIAP−/− mice that were hemizygous for RIP1 were born alive, while cIAP1−/−XIAP−/−RIP1−/− mice did not survive past E14.5 (Moulin et al. 2012). The molecular mechanisms that contribute to these confounding observations are unclear at present. Nonetheless, they do indicate that gene dosage can greatly impact the outcome of RIP kinase signaling during embryonic development.
RIP3 in viral and bacterial infections
Apoptosis is a major host defense mechanism against viral infection. To circumvent apoptosis of infected cells and establish successful infection, many viruses encode caspase inhibitors. When the apoptosis pathway is blocked, RIP3-dependent necrosis can function as a backup host defense mechanism to limit viral replication. Consistent with this notion, RIP3−/− mice failed to eliminate vaccinia virus and succumbed to the infection (Cho et al. 2009). In contrast, MCMV has evolved and developed inhibitors to prevent both apoptosis and necrosis (see above). Failure to block necrosis compromised the fitness of the virus, leading to abortive infection in the host (Upton et al. 2010, 2012).
Severe bacterial infection can lead to sepsis that is characterized by overwhelming cytokine production and life-threatening systemic organ failure. TNF is a prime mediator of inflammation in septic shock, implying that RIP3 might also participate in bacterial-induced sepsis (Bhatia et al. 2009). Indeed, RIP3−/− mice were protected from TNF or cecal ligation and puncture (CLP)-induced systemic inflammation, two experimental models for clinical sepsis (Duprez et al. 2011; Linkermann et al. 2012). In addition, RIP3 also plays a crucial role in the pathogenesis of Salmonella enterica serovar Typhimurium. S. Typhimurium is a facultative intracellular pathogen that replicates in intestinal epithelia to cause gastroenteritis. Early reports show that S. Typhimurium causes caspase-1-dependent pyroptosis in macrophages (Fink et al. 2008; Miao et al. 2010). Similar to necrosis, pyroptosis is marked by plasma membrane rupture and release of danger-associated molecular patterns (DAMPs), which induce inflammation by activating cognate pattern recognition receptors. However, necrosis and pyroptosis are regulated by distinct molecular machineries. Recently, S. Typhimurium was shown to kill macrophages in a type I IFN- and RIP3-dependent manner (Robinson et al. 2012). IFNα/β receptor-deficient or RIP3−/− mice exhibited reduced macrophage necrosis and better control of S. Typhimurium. Although the investigators conclude that this is directly due to RIP3-dependent necrosis, one should also consider the possibility that the dampened cytokine response due to reduced necrosis and DAMP release could have also contributed to the protection in RIP3−/− mice.
RIP3 in lymphocyte homeostasis
Lymphocyte homeostasis is finely balanced between cell proliferation and cell death. This concept is best illustrated by mutations in the death receptor Fas/CD95/APO-1, which lead to autoimmune lymphoproliferative diseases in humans and mice that resemble lupus. Because Fas is a major inducer of caspase-dependent apoptosis, apoptosis was thought to be the dominant mechanism by which autoreactive lymphocytes are eliminated. Paradoxically, T cells that lack FADD or caspase-8 or those that express a dominant-negative version of FADD developed immunodeficiency instead of lymphoproliferation (Newton et al. 1998; Walsh et al. 1998; Zhang et al. 1998, 2005; Salmena et al. 2003). In response to antigen receptor stimulation, these T cells failed to clonally expand due to premature cell death by necrosis. Normal proliferation was restored by genetic ablation of RIP1 or RIP3 (Osborn et al. 2010; Ch'en et al. 2011; Lu et al. 2011; Zhang et al. 2011). Moreover, caspase-8−/−RIP3−/− mice developed lpr/gld-like lymphoproliferation and lupus-like disease (Oberst et al. 2011). Therefore, while caspase-dependent apoptosis is the dominant cell death mechanism for lymphocytes, immune homeostasis also requires the coordinate effort of RIP kinase-dependent necrosis.
RIP3 in sterile inflammation
Hepatocytes are highly susceptible to the cytotoxic effects of TNF. Normally, hepatocytes are protected from the death-inducing effect of TNF because of NF-κB-induced expression of survival factors. As such, hepatocytes that lacked NF-κB essential modulator (NEMO) underwent spontaneous apoptosis, leading to inflammation, steatosis, liver fibrosis, and hepatocellular carcinoma (Luedde et al. 2007). Caspase-8ΔHepa mice also developed spontaneous liver inflammation and enhanced nonapoptotic liver injury (Liedtke et al. 2011; Hatting et al. 2013). Combined hepatocyte-specific caspase-8 and NEMO deletion further exacerbated liver necrosis and cholestasis (Liedtke et al. 2011). Since RIP1/RIP3/FADD complexes were detected in these cases, it is tempting to speculate that RIP3-dependent necrosis was responsible for the liver pathologies. In support of this hypothesis, RIP3−/− mice were protected from ethanol-induced expression of proinflammatory cytokines, hepatocyte injury, and steatosis (Roychowdhury et al. 2013).
Deficiency of caspase-8 or FADD in intestinal or skin epithelium spontaneously caused massive inflammation, which was rescued by additional deletion of RIP3 (Kovalenko et al. 2009; Bonnet et al. 2011; Gunther et al. 2011; Welz et al. 2011). Interestingly, TNF-R1 deficiency did not fully rescue excessive necrosis and inflammation in the intestine and skin, indicating that physiological necrosis can often be induced by ligands other than TNF (Bonnet et al. 2011; Welz et al. 2011).
Although necrosis is optimally induced when caspases are inhibited, it can also occur in the presence of intact FADD or caspase-8 function. For example, cerulein-induced acute necrotizing pancreatitis was reduced in RIP3−/− mice (He et al. 2009; Zhang et al. 2009). RIP3 deficiency inhibited photoreceptor cell death in a retinal detachment model and cone cell death in Rd10 mice that develop retinitis pigmentosa (Trichonas et al. 2010; Murakami et al. 2012). Necrotic macrophages have been observed in atherosclerosis lesions from human patients and animal models of atherosclerosis (Tabas 2010). Interestingly, RIP3 deficiency alleviated macrophage necrosis in advanced atherosclerosis lesions in atherosclerosis-prone LDL-R−/− or ApoE−/− mice (Lin et al. 2013), indicating that RIP3-dependent necrosis is a key driver for inflammation in atherosclerosis.
Necrosis-independent function of RIP3 in inflammation
The release of cellular DAMPs is believed to be the main driver of inflammation in physiological necrosis. Recently, several reports show that RIP3 can also facilitate inflammation independent of necrosis. Production of the inflammatory cytokine IL-1β requires two distinct signals: a first signal that induces de novo pro-IL-1β gene transcription through NF-κB and a second inflammasome-dependent signal that cleaves pro-IL-1β to produce the mature cytokine. The inflammasome is a macromolecular complex composed of caspase-1, the adaptor ASC1, and an upstream sensor such as NLRP3. Thus, stimulation of TLR4 with LPS alone, which provides the first signal, is not sufficient to induce mature IL-1β expression in bone marrow-derived macrophages. Surprisingly, SMAC mimetic (SM), which targets the IAPs for proteasomal degradation, promotes NLRP3 inflammasome activation and IL-1β maturation in LPS-primed macrophages and dendritic cells (DCs) (Vince et al. 2012). Similar results were obtained with cIAP1−/−cIAP2−/−XIAP−/− macrophages. Strikingly, inflammasome activation in these conditions requires RIP3 but not necrosis.
In addition to SM, caspase-8 deficiency also appears to turn on RIP3-dependent inflammasome activation in DCs. Mice with DC-specific deletion of caspase-8 developed an aggressive systemic inflammatory disease and were highly susceptible to the lethal effect of LPS (Kang et al. 2013). LPS stimulation of caspase-8−/− DCs led to enhanced activation of the NLRP3/ASC/caspase-1 inflammasome and IL-1β expression. Inhibition of RIP1 by necrostatins or RIP3 deficiency restored normal inflammasome activity. Surprisingly, using RNAi, the investigators showed that the downstream necrosis regulators MLKL and PGAM5 were also required for heightened inflammasome activation in caspase-8−/− DCs. Although the investigators concluded that enhanced in vitro IL-1β secretion in caspase-8−/− DCs was independent of the pronecrotic role of RIP3, low level of necrosis could still occur to promote inflammation in vivo because caspase-8−/− cells are highly sensitive to necrosis. Spontaneous inflammation was also observed in mice lacking FADD in DCs. However, in contrast to the results from the DC-specific caspase-8−/− mice, the phenotypes were caused by excessive exposure to commensal microbiota and DC necrosis (Young et al. 2013). More research into the role of RIP3 in systemic inflammation and DC biology is required to reconcile the discrepant interpretations from these studies.
Potential role of RIP3 in cancer
Although the concept that apoptosis can serve as a natural barrier to cancer development has been well established, the role of necrosis in cancers remains unknown. It is noteworthy that necrosis is a characteristic feature of many advanced solid tumors (Hanahan and Weinberg 2011). However, there is no clear evidence to indicate whether necrosis is beneficial or harmful in cancers. One of the challenges in clinical cancer therapy is the resistance of cancers to apoptosis. Since many anti-cancer drugs are inducers of apoptosis, inducing RIP3-dependent necrosis is an attractive strategy to circumvent apoptosis resistance of cancer cells. In addition to DNA alkylating agents, which induce necrosis in the PARP-dependent manner (Zong et al. 2004; Fu et al. 2013; Sosna et al. 2013), some drugs were reported to induce RIP3-dependent necrosis in certain conditions (Tenev et al. 2011; Bray et al. 2012; Basit et al. 2013). Because necrosis also facilitates inflammation, its effect on inflammation in the tumor microenvironment has to be carefully considered (Mantovani et al. 2008). Further work is required to answer these important questions and understand how we can harness the power of RIP3-dependent necrosis in cancer therapy.
Closing thoughts
Much has been learned about the role of RIP3 in different disease pathologies in recent years. With this newfound understanding, it is perhaps tempting to ponder how we might integrate this knowledge into clinical applications. However, several hurdles must first be overcome. For instance, we must learn whether RIP3 actually induces necrosis and inflammation in human diseases when FADD and caspase-8 functions are intact. Technologies that allow us to monitor the activation status of RIP3 and quantify RIP3-dependent necrosis will be vital in this endeavor. At present, electron microscopy remains the most reliable way to distinguish necrosis in diseased tissues. More quantitative methods to detect necrosis and differentiate it from apoptosis need to be developed. In addition, we need to create ways to differentiate active RIP3 during necrosis versus inflammasome activation. Finally, one must consider whether RIP3 is indeed a good target for therapeutic intervention. Given the diverse biological processes that RIP1 regulates, it is safe to assume that RIP3 is a better therapeutic target than RIP1. However, as in the case of many other cell death regulators, RIP3 can have other “day jobs,” such as that in inflammasome activation. Therefore, the success of RIP3-targeted therapies will depend on whether we can differentiate RIP3 functions in various biological responses.
Acknowledgments
This work is supported by NIH grant AI083497 to F.C. F.C. is a member of the University of Massachusetts DERC (DK32520). K.M. is supported by a post-doctoral fellowship from the Japan Society for the Promotion of Science.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.223321.113.
References
- Basit F, Cristofanon S, Fulda S 2013. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ doi: 10.1038/cdd.2013.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, He M, Zhang H, Moochhala S 2009. Sepsis as a model of SIRS. Front Biosci 14: 4703–4711 [DOI] [PubMed] [Google Scholar]
- Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M 2011. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 35: 572–582 [DOI] [PubMed] [Google Scholar]
- Bray K, Mathew R, Lau A, Kamphorst JJ, Fan J, Chen J, Chen HY, Ghavami A, Stein M, DiPaola RS, et al. 2012. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS ONE 7: e41831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B 1975. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci 72: 3666–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK 2012. Fueling the flames: Mammalian programmed necrosis in inflammatory diseases. Cold Spring Harb Perspect Biol 4: a008805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ 2003. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278: 51613–51621 [DOI] [PubMed] [Google Scholar]
- Ch'en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM 2011. Mechanisms of necroptosis in T cells. J Exp Med 208: 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, Zhang Y, Ma H, Huang D, Li W, et al. 2013. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-Like (MLKL) interaction in necroptotic signaling. J Biol Chem 288: 16247–16261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK 2009. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte D, Holcik M, Lefebvre CA, Lacasse E, Picketts DJ, Wright KE, Korneluk RG 2006. Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival. Mol Cell Biol 26: 699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conze DB, Albert L, Ferrick DA, Goeddel DV, Yeh WC, Mak T, Ashwell JD 2005. Posttranscriptional downregulation of c-IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol Cell Biol 25: 3348–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declercq W, Vanden Berghe T, Vandenabeele P 2009. RIP kinases at the crossroads of cell death and survival. Cell 138: 229–232 [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J 2005. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119 [DOI] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al. 2008. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4: 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR 2012. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep 1: 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P 2011. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35: 908–918 [DOI] [PubMed] [Google Scholar]
- Ellis HM, Horvitz HR 1986. Genetic control of programmed cell death in the nematode C. elegans. Cell 44: 817–829 [DOI] [PubMed] [Google Scholar]
- Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M 2007. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 19: 2056–2067 [DOI] [PubMed] [Google Scholar]
- Fink SL, Bergsbaken T, Cookson BT 2008. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci 105: 4312–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D, Jordan JJ, Samson LD 2013. Human ALKBH7 is required for alkylation and oxidation-induced programmed necrosis. Genes Dev 27: 1089–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW, et al. 2011. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471: 591–596 [DOI] [PubMed] [Google Scholar]
- Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, et al. 2011. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477: 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2011. Hallmarks of cancer: The next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB 2001. Characterization of XIAP-deficient mice. Mol Cell Biol 21: 3604–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatting M, Zhao G, Schumacher F, Sellge G, Masaoudi MA, Gabetaler N, Boekschoten M, Muller M, Liedtke C, Cubero FJ, et al. 2013. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in mice. Hepatology 57: 2189–2201 [DOI] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X 2009. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137: 1100–1111 [DOI] [PubMed] [Google Scholar]
- He S, Liang Y, Shao F, Wang X 2011. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci 108: 20054–20059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J 2008. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135: 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J 2000. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489–495 [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Offermann MK 2005. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol 174: 4942–4952 [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Mocarski ES 2008. Receptor-interacting protein homotypic interaction motif-dependent control of NF-κB activation via the DNA-dependent activator of IFN regulatory factors. J Immunol 181: 6427–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES 2011. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471: 368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, et al. 2004. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol 173: 2976–2984 [DOI] [PubMed] [Google Scholar]
- Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D 2013. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38: 27–40 [DOI] [PubMed] [Google Scholar]
- Kasof GM, Prosser JC, Liu D, Lorenzi MV, Gomes BC 2000. The RIP-like kinase, RIP3, induces apoptosis and NF-κB nuclear translocation and localizes to mitochondria. FEBS Lett 473: 285–291 [DOI] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P 1998. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8: 297–303 [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR 1972. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko A, Kim JC, Kang TB, Rajput A, Bogdanov K, Dittrich-Breiholz O, Kracht M, Brenner O, Wallach D 2009. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med 206: 2161–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laster SM, Wood JG, Gooding LR 1988. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol 141: 2629–2634 [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. 2012. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke C, Bangen JM, Freimuth J, Beraza N, Lambertz D, Cubero FJ, Hatting M, Karlmark KR, Streetz KL, Krombach GA, et al. 2011. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology 141: 2176–2187 [DOI] [PubMed] [Google Scholar]
- Lin Y, Devin A, Rodriguez Y, Liu ZG 1999. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13: 2514–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Li H, Yang M, Ren J, Huang Z, Han F, Huang J, Ma J, Zhang D, Zhang Z, et al. 2013. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep 3: 200–210 [DOI] [PubMed] [Google Scholar]
- Linkermann A, Brasen JH, De Zen F, Weinlich R, Schwendener RA, Green DR, Kunzendorf U, Krautwald S 2012. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-α-induced shock. Mol Med 18: 577–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockshin RA, Williams CM 1964. Programmed cell death. II. Endocrine potentiation of the breakdown of the intersegmental mucles of silmoths. J Insect Physiol 10: 643–649 [Google Scholar]
- Lu JV, Weist BM, van Raam BJ, Marro BS, Nguyen LV, Srinivas P, Bell BD, Luhrs KA, Lane TE, Salvesen GS, et al. 2011. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc Natl Acad Sci 108: 15312–15317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M 2007. Deletion of NEMO/IKKγ in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 11: 119–132 [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F 2008. Cancer-related inflammation. Nature 454: 436–444 [DOI] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J 2004. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κB activation. Nat Immunol 5: 503–507 [DOI] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A 2010. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11: 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulin M, Anderton H, Voss AK, Thomas T, Wong WW, Bankovacki A, Feltham R, Chau D, Cook WD, Silke J, et al. 2012. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J 31: 1679–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y, Miller JW, Vavvas DG 2012. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc Natl Acad Sci 109: 14598–14603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan N, Lee IH, Borenstein R, Sun J, Wong R, Tong G, Fergusson MM, Liu J, Rovira II, Cheng HL, et al. 2012. The NAD-dependent deacetylase SIRT2 is required for programmed necrosis. Nature 492: 199–204 [DOI] [PubMed] [Google Scholar]
- Newton K, Harris AW, Bath ML, Smith KG, Strasser A 1998. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J 17: 706–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Sun X, Dixit VM 2004. Kinase RIP3 is dispensable for normal NF-κBs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24: 1464–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR 2011. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT 2011. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol 13: 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell MA, Hase H, Legarda D, Ting AT 2012. NEMO inhibits programmed necrosis in an NFκB-independent manner by restraining RIP1. PLoS ONE 7: e41238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn SL, Diehl G, Han SJ, Xue L, Kurd N, Hsieh K, Cado D, Robey EA, Winoto A 2010. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci 107: 13034–13039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J 2009. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep 10: 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S 2012. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13: 954–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE 2013. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 57: 1773–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, et al. 2003. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev 17: 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, Meier P 2013. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect Biol 5: a008730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P, Herdegen T, Linkermann A, Rittger A, Chan FK, et al. 2013. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci doi: 10.1007/s00018-013-1381-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR 1977. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 56: 110–156 [DOI] [PubMed] [Google Scholar]
- Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM 1999. RIP3, a novel apoptosis-inducing kinase. J Biol Chem 274: 16871–16875 [DOI] [PubMed] [Google Scholar]
- Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM 2002. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem 277: 9505–9511 [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. 2012. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227 [DOI] [PubMed] [Google Scholar]
- Tabas I 2010. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 10: 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin V, Huang Q, Liu H, Osada H, Pope RM 2006. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol 26: 2215–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, et al. 2011. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43: 432–448 [DOI] [PubMed] [Google Scholar]
- Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG 2010. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci 107: 21695–21700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES 2008. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem 283: 16966–16970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES 2010. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7: 302–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES 2012. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11: 290–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlangenakker N, Vanden Berghe T, Vandenabeele P 2012. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ 19: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276 [DOI] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O'Reilly L, Mason K, Gross O, Ma S, Guarda G, et al. 2012. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36: 215–227 [DOI] [PubMed] [Google Scholar]
- Walsh CM, Wen BG, Chinnaiyan AM, O'Rourke K, Dixit VM, Hedrick SM 1998. A role for FADD in T cell activation and development. Immunity 8: 439–449 [DOI] [PubMed] [Google Scholar]
- Wang L, Du F, Wang X 2008. TNF-α induces two distinct caspase-8 activation pathways. Cell 133: 693–703 [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X 2012. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148: 228–243 [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ 2001. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M 2011. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477: 330–334 [DOI] [PubMed] [Google Scholar]
- Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A, Yuan J, Shi Y 2013. Structural basis of RIP1 inhibition by necrostatins. Structure 21: 493–499 [DOI] [PubMed] [Google Scholar]
- Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, et al. 1998. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279: 1954–1958 [DOI] [PubMed] [Google Scholar]
- Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, et al. 2000. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12: 633–642 [DOI] [PubMed] [Google Scholar]
- Young JA, He TH, Reizis B, Winoto A 2013. Commensal microbiota are required for systemic inflammation triggered by necrotic dendritic cells. Cell reports 3: 1932–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu PW, Huang BC, Shen M, Quast J, Chan E, Xu X, Nolan GP, Payan DG, Luo Y 1999. Identification of RIP3, a RIP-like kinase that activates apoptosis and NFκB. Curr Biol 9: 539–542 [DOI] [PubMed] [Google Scholar]
- Zhang J, Cado D, Chen A, Kabra NH, Winoto A 1998. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392: 296–300 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Rosenberg S, Wang H, Imtiyaz HZ, Hou YJ, Zhang J 2005. Conditional Fas-associated death domain protein (FADD): GFP knockout mice reveal FADD is dispensable in thymic development but essential in peripheral T cell homeostasis. J Immunol 175: 3033–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J 2009. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325: 332–336 [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J 2011. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471: 373–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG 2012. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci 109: 5322–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB 2004. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 18: 1272–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]