

Kitade et al. sought to reveal the relative contribution of HGF/MET and EGF/Egfr to fate decisions of adult hepatic progenitor cells (HPCs). They found that both receptors collaborate to increase the self-renewal of HPCs through ERK activation. MET induced hepatocyte differentiation by activating AKT and STAT3, while Egfr induced Notch1 to promote cholangiocyte specification and suppress hepatocyte commitment. Liver-specific deletion of Egfr facilitated HPC-mediated liver regeneration by switching HPC differentiation from cholangiocytes toward hepatocytes. These results provide new mechanistic insights into the properties of adults HPCs.
Keywords: liver progenitor cells, MET, EGFR, lineage commitment
Abstract
The relative contribution of hepatocyte growth factor (HGF)/MET and epidermal growth factor (EGF)/EGF receptor (EGFR), two key signal transduction systems in the normal and diseased liver, to fate decisions of adult hepatic progenitor cells (HPCs) has not been resolved. Here, we developed a robust culture system that permitted expansion and genetic manipulation of cells capable of multilineage differentiation in vitro and in vivo to examine the individual roles of HGF/MET and EGF/EGFR in HPC self-renewal and binary cell fate decision. By employing loss-of-function and rescue experiments in vitro, we showed that both receptors collaborate to increase the self-renewal of HPCs through activation of the extracellular signal-regulated kinase (ERK) pathway. MET was a strong inducer of hepatocyte differentiation by activating AKT and signal transducer and activator of transcription (STAT3). Conversely, EGFR selectively induced NOTCH1 to promote cholangiocyte specification and branching morphogenesis while concomitantly suppressing hepatocyte commitment. Furthermore, unlike the deleterious effects of MET deletion, the liver-specific conditional loss of Egfr facilitated rather than suppressed progenitor-mediated liver regeneration by switching progenitor cell differentiation toward hepatocyte lineage. These data provide new insight into the mechanisms regulating the stemness properties of adult HPCs and reveal a previously unrecognized link between EGFR and NOTCH1 in directing cholangiocyte differentiation.
Healthy liver possesses a strong regenerative potential due to the unlimited proliferative potential of fully differentiated hepatocytes and biliary epithelial cells, also referred to as cholangiocytes (Bohm et al. 2010; Michalopoulos 2010; Riehle et al. 2011). However, regeneration of a diseased liver relies on activation of resident stem cells to replace the irreversibly damaged hepatocytes and biliary cells, which lose the capacity to proliferate (Thorgeirsson et al. 1993; Thorgeirsson and Grisham 2003; Duncan et al. 2009; Boulter et al. 2013). Hepatic stem cells are thought to reside within the terminal bile ductules, also referred to as Hering canals (Alison et al. 1996; Theise et al. 1999). Upon severe liver injury, they expand and give rise to a heterogeneous multipotent transient amplifying cell population known as “oval cells” or hepatic progenitor cells (HPCs) that can differentiate to hepatocytes and cholangiocytes and replenish their loss, as reported both in rodents and humans (Roskams et al. 2010; Tanaka et al. 2011).
There is strong evidence that HPC expansion and direction of differentiation depend on the signals provided by the tissue-specific stem cell niche (Thorgeirsson and Grisham 2003; Duncan et al. 2009; Greenbaum and Wells 2011). Among these, there is hepatocyte growth factor (HGF), a mesenchyme-derived multifunctional molecule that elicits its activities by binding to the cognate receptor tyrosine kinase (RTK) MET (Bertotti and Comoglio 2003; Birchmeier et al. 2003). Since it was first described as a potent mitogen for hepatocytes, HGF/MET signaling has been subsequently linked to pleiotropic morphogenic activities critical for embryonic development, tissue regeneration, and maintenance of HPCs (Trusolino et al. 2010; Nakamura et al. 2011). Gene knockout studies in mice confirmed that up-regulation of MET expression occurs early during development (embryonic day 13.5 [E13.5]) and is required at multiple steps of liver organogenesis from stimulating progenitor cell proliferation and resistance to apoptosis to hepatocyte differentiation (Schmidt et al. 1995; Suzuki et al. 2002). Furthermore, using conditional liver-specific Met inactivation, we showed recently that progenitor-mediated regeneration is dependent on MET signaling. Adult HPCs defective in MET signaling failed to expand and could not commit to hepatocyte lineage in a model of chronic toxic liver injury (Ishikawa et al. 2012).
Epidermal growth factor receptor (EGFR) is another key mediator of hepatic homeostasis (Michalopoulos and DeFrances 1997). Like MET, deregulated EGFR signaling has been implicated in liver regeneration, and genetic loss of Egfr decreased survival after partial hepatectomy in mice (Natarajan et al. 2007). Although extensive studies implicate EGFR in regulating stemness properties in many types of stem cells (Brill et al. 2009; Aguirre et al. 2010), much less is known about a role of EGFR in HPC biology. Prior work from our and other laboratories demonstrated that both receptors were activated and functional during HPC expansion and promoted progenitor cell hyperplasia induced by severe liver injury in the rat 2-acetylaminofluorene–70% hepatectomy (AAF/PHx) model (Evarts et al. 1993; Nagy et al. 1996; Hasuike et al. 2005).
Unlike MET, EGFR is activated by numerous paracrine and endocrine signals, including EGF, and, upon ligand binding, can form homodimers or heterodimers with either EGFR or other members of the ERBB or EGFR family of related RTKs (Burgess 2008). However, despite the diversity of ligands and binding partners complicating analysis of this pathway in the liver, Egfr, similar to Met, is capable of triggering the same primary downstream signaling cascades in cells, including the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK), phosphatidylinositol 3-kinase (PI3K)/AKT, and signal transducer and activator of transcription (STAT3) pathways, involved in control of cell proliferation, motility regulation, apoptosis protection, and differentiation (Trusolino et al. 2010). Moreover, depending on the signal strength and cellular context, the cross-communication between MET and EGFR has also been reported (Presnell et al. 1997; Jo et al. 2000), although the direct contribution of each signal transducer to HPC proliferation and fate specification has not been fully addressed.
In this study, we established HPC lines from Metfl/fl and Egfrfl/fl mice and confirmed that they were capable of effective differentiation toward both hepatocytic and biliary epithelial cell lineages. Using conditional genetic ablation and defined medium, we then examined how a specific loss of either Met or Egfr affects the binary cell fate decisions of HPCs. Our loss-of-function and gain-of-function experiments in vitro showed that MET is a strong inducer of hepatocyte differentiation via activation of AKT and STAT3, whereas EGFR is required for NOTCH1-controlled expression of cholangiocyte-specific genes and ductular morphogenesis. Both stimulation of NOTCH1 expression and biliary epithelial differentiation were reduced during progenitor cell-mediated liver regeneration in EGFR conditional knockout mice, supporting a role of the EGFR/NOTCH1 positive feedback loop for commitment of adult HPCs toward biliary epithelial cell lineage.
Results
Generation of HPC lines
To evaluate the individual role of EGFR and MET in the control of growth and bipotential properties of HPCs, we generated several clonal cell lines from mice harboring Egfr and Met floxed alleles. To activate HPCs, mice were maintained for 2 wk on a diet containing the porphyrinogenic agent 3,5-diethoxycarbonyl-1, 4-dihydro-collidine (DDC) (Preisegger et al. 1999). The primary HPCs (also known as oval cells) were isolated at high purity by fluorescence-activated cell sorting (FACS) after costaining with a cell-specific anti-EpCAM and lineage cocktail antibodies against CD3c, CD11b, CD45R, TER-119, LY-6C, and LY-6G to exclude cells of hematopoietic origin (Supplemental Fig. S1A,B; Okabe et al. 2009; Ishikawa et al. 2012). The EpCAM+/Lineage− cells were then cultured in nonadherent conditions to select for cells possessing self-renewal capacity (Cicalese et al. 2009) and established as parental Egfrfl/fl and Metfl/fl HPC lines.
First, we compared the transcriptome of Egfrfl/fl and Metfl/fl cells to define key molecular similarities/differences between independently derived clonal cell lines (n = 3 of each genotype). Transcriptome profiling confirmed a high degree of molecular similarity between both parental cell lines. The Pearson correlation coefficient across the averaged signal intensities in >25,000 probes showed a linearity with R2 = 0.9 (Fig. 1A). The subsequent bioequivalence test also demonstrated near 100% identity between the significant genes, with a cut off at fold change = 1.5 and P < 0.001 (Fig. 1B). Therefore, for simplicity, in the following, only one of the representative parental cell lines is shown unless otherwise indicated.
Figure 1.

Metfl/fl and Egfrfl/fl cell lines possess similar molecular and functional properties of HPCs. (A,B) Bioequivalence tests of transcriptomic similarities between Metfl/fl and Egfrfl/fl cell lines (n = 3 for each genotype). (A) Pearson correlation of gene expression changes. Red lines represent plus/minus twofold changes of all detected genes (detection P-value < 0.01). (FC) Fold change. (B) Hierarchical cluster representation of significant genes (±1.5-fold change at P < 0.001). (C) Metfl/fl and Egfrfl/fl cells exhibit markers of both hepatocytic and biliary lineages. Relative mRNA expression levels of indicated genes were measured by quantitative RT–PCR (qRT–PCR). Gapdh was used as internal control for qRT–PCR. Data are mean ± SEM (n = 4). (D) Metfl/fl and Egfrfl/fl cells differentiate into hepatocytic lineage in vitro when stimulated with HGF and EGF. Double immunofluorescence staining for hepatocyte markers HNF4α (red) and albumin (green). (GFs) Growth factors. Nuclei were counterstained with DAPI. Bar, 50 μm. (E) Metfl/fl and Egfrfl/fl cells differentiate into duct-like cells when stimulated by type 1A collagen and HGF and EGF. Bright-field images. Bar, 200 μm. (F) Liver repopulation in MUP-uPA/SCID mice by Metfl/fl cells. Representative confocal images of livers 32 wk after intrasplenic injection of Metfl/fl cells transduced with mCherry reporter gene (1 × 106 cells per mouse, n = 3). Bars, 100 μm. (G) The repopulating mCherry-positive cells display properties characteristic for mature hepatocytes. Confocal images show coexpression of mCherry with albumin and glutamine synthetase (GS), a marker of pericentral hepatocytes (arrow). Bar, 100 μm. (PV) Portal vein; (CV) central vein.
As previously reported (Okabe et al. 2009), both parental progenitor cell lines expressed hepatocytic (AFP) and cholangiocytic (CK7 and CK19) markers (Fig. 1C). Consistent with their stemness properties, Metfl/fl and Egfrfl/fl cells formed spheroids in ultralow attachment culture (data not shown) and were capable of differentiating along hepatocytic and cholangiocytic lineages when subjected to lineage-specific differentiation assays in vitro, as shown by increased expression of the hepatocyte-specific proteins albumin and HNF4α (Fig. 1D) and acquisition of tube-like branching morphology characteristic for cholangiocyte differentiation (Fig. 1E). Furthermore, when Metfl/fl HPCs transfected with mCherry reporter gene were transplanted into chimeric major urinary promoter–urokinase plasminogen activator (MUP-uPA)/severe combined immunodeficiency disease (SCID) mice (Giannini et al. 2003), they gave rise to functional hepatocytes that integrated into and replaced diseased parenchyma. Quantitative analysis of mCherry-positive areas using confocal images (Fig. 1F) showed that liver repopulation reached, on average, 12.0% ± 18.2% by 8 mo after intrasplenic transplantation of 1 × 106 Metfl/fl HPCs. The repopulating mCherry-positive cells displayed properties characteristic for mature hepatocytes. They expressed the same levels of albumin as the neighboring mCherry− hepatocytes and displayed a zonal pattern of staining with glutamine synthetase, a key enzyme in ammonia metabolism and known marker of pericentral cells (Fig. 1G), indicating functional integration into parenchyma. Together, these results indicate that Metfl/fl and Egfrfl/fl parental cell lines possessed properties ascribed to HPCs.
Deletion of Met or Egfr was performed at passage 4 by Adeno-CMV-Cre (Vector Biolabs) transfection as described (del Castillo et al. 2008) to generate early passage (passages 8–10) single-cell clonal Egfr−/− and Met−/− HPC lines (Supplemental Fig. S1A). A complete deletion of each gene was demonstrated by a genotyping PCR (Supplemental Fig. S1D) and Western blotting (Supplemental Fig. S1E). The loss of Egfr or Met did not affect either expression of genes coding for HPC markers (Afp, Ck7, and Ck19) or EGFR and MET cognate ligands (Egf, HB-Egf, Tgfα, and Hgf, respectively) (Fig. 1C). Also, it did not cause a compensatory up-regulation of EGFR family members ERBB2 and ERBB3 (Supplemental Fig. S1E). In total, we established four independent single-cell clonal cell lines for each genotype, including parental (Metfl/fl and Egfrfl/fl) and mutant (Met−/− and Egfr−/−) lines.
Effect of Egfr and Met deletion on downstream signaling
To assess the function of EGFR and MET signaling in HPCs, we analyzed the activity/phosphorylation status of each receptor and primary downstream effector implicated in the control of proliferation and differentiation (Burgess 2008; Trusolino et al. 2010) in response to a short-term treatment with HGF, EGF, or both growth factors (Fig. 2). To ensure the comparable signal intensity, titration experiments were performed to select the optimal concentrations for each ligand based on the degree of receptor phosphorylation (Supplemental Fig. S2A,B).
Figure 2.

Altered activation of MET and EGFR downstream signaling in Met−/− and Egfr−/− cells. HPCs of indicated genotypes were plated in two-dimensional (2D) cultures and treated with HGF (H; 20 ng/mL), EGF (E; 10 ng/mL), or HGF and EGF (H+E) for 15 min after overnight serum starvation. pMET (Y1234/1235), MET, pEGFR (Y1068), EGFR, pERK1/2 (T202/Y204), ERK1/2, pAKT (S473), AKT, pSTAT3 (Y705), and STAT3 were detected by Western blotting with actin used as a loading control. (NT) No treatment.
The deletion of Met did not change the protein levels of EGFR and vice versa (Fig. 2). As expected, Met−/− and Egfr−/− HPCs were nonresponsive to stimulation with a corresponding growth factor. Consequently, loss of Met completely blocked HGF-mediated STAT3 phosphorylation and strongly reduced ERK1/2 and AKT phosphorylation. In contrast, deletion of Egfr caused a preferential abrogation of pERK1/2 and pAKT without affecting pSTAT3 levels (Fig. 2). These results suggest that MET and EGFR may have a common and selective impact on the downstream signaling in HPCs.
Effect of Egfr and Met deletion on sphere-forming ability
Next, we conducted a sphere-forming assay using ultralow attachment plates and a serum-free defined medium containing HGF and/or EGF. In HPCs with competent MET and EGFR receptors, HGF and EGF were considerably more effective in increasing both sphere frequency and sphere size as compared with either treatment alone (Supplemental Fig. S3A–C). The deletion of Met or Egfr in HPC lines caused a significant decrease in sphere-forming capacity, suggesting that the two pathways cooperate to increase the sphere-forming potential of progenitor cells (Supplemental Fig. S3A–C).
To further understand the role of MET and EGFR in self-renewal growth, we blocked the activity of ERK1/2, AKT, and STAT3 in the parental HPCs by small-molecule inhibitors, including PD98059, LY294002, and WP1066, respectively. The optimal dose for each drug was selected based on cell viability (>60%) and degree of target gene inhibition (>80%) (Supplemental Fig. S4A,B). Inhibiting ERK1/2 activation with PD98059 was considerably more effective in reducing both the sphere frequency and sphere size as compared with blocking STAT3 using WP1066 or blocking AKT using LY294002 (Supplemental Fig. 3D,E). Thus, persistent MET and EGFR signaling was sufficient to establish and maintain the self-renewing growth of HPCs through a mechanism involving ERK1/2 and, to a lesser degree, AKT and STAT3 activation.
MET induces HPC differentiation toward hepatocytes via AKT and STAT3
To determine the direct effects of MET and EGFR on the differentiation potency of HPCs, we used a modified hepatocyte differentiation assay and a defined medium containing HGF and/or EGF (Okabe et al. 2009). The parental HPCs with a competent MET/EGFR signaling were subjected to a single (HGF or EGF) or combined (EGF+HGF) treatment to evaluate the complimentary effects of each growth factor, whereas Met−/− and Egfr−/− cells, which are completely refractory to the corresponding ligand stimulation (Fig. 2), were treated with a combination of HGF+EGF for simplicity and uniformity. Addition of either a single or both growth factors to the parental HPC lines plated on Matrigel-coated culture dishes caused acquisition of hepatocyte-like morphology and a strong up-regulation of mature hepatocyte proteins albumin, HNF4α, and CYP2E1, as measured by Western blotting and double immunofluorescence confocal microscopy (Fig. 3A,B). Notably, the selective deletion of Met and Egfr had a differential impact on the induction of proteins associated with the hepatocyte phenotype. In Met-null cells, the directed differentiation toward hepatocytes was significantly impaired despite the presence of the functional Egfr. In contrast, the directed differentiation of Egfr−/− HPCs did not show the obvious defects in the morphological appearance or hepatocyte marker induction, indicating that Met was able to substitute at least in part for the lack of Egfr (Fig. 3A,B).
Figure 3.

Loss of Met blocks differentiation of progenitor cells toward hepatocytes in vitro. (A) Western blots for HNF4α, albumin (ALB), CYP2E1, and actin used as a loading control. [GF(−)] No growth factors; (H) HGF; (E) EGF; (H+E) HGF and EGF. Whole-cell lysate from primary hepatocyte culture was used as a positive control (PC). (B) Confocal microscopy images (bar, 50 μm) of immunofluorescence staining for albumin (green) and HNF4α (red). Nuclei were counterstained with DAPI. (C,D) Pharmacological inhibition of AKT (LY294002, 20 μM) and STAT3 (WP1066, 10 μM) but not ERK1/2 (PD98059, 20 μM) blocks hepatocyte differentiation in Metfl/fl cells. (C) Western blots for HNF4α, ALB, and actin. (LY) LY294002; (WP) WP1066; (PD) PD98059. (D) Confocal microscopy images (bar, 50 μm) of double immunofluorescence staining for albumin (green) and HNF4α (red). (E) Met knock-in (MetresWT) restores MET signaling. Western blots for pMET, MET, pAKT, AKT, pSTAT3, STAT3, and actin. Cells were treated with HGF (20 ng/mL) for 15 min after overnight serum starvation. (Mock) Empty vector. (F) Met knock-in (MetresWT) repairs hepatocyte differentiation defects in Met−/− progenitor cells. Confocal microscopy images (bar, 50 μm) of immunofluorescence staining for albumin (green) and HNF4α (red). Nuclei were counterstained with DAPI.
To check which of the MET-driven downstream effectors contribute to hepatocyte differentiation, we treated MET-competent parental HPC lines with the pharmacological inhibitors of MEK/ERK1/2, PI3K/AKT, and STAT3 using PD98059, LY294002, and WP1066, respectively (Fig. 3C,D). Inhibiting AKT and STAT3 but not MEK/ERK strongly reduced HNF4α and albumin expression, corresponding with the observation that genetic ablation of Met preferentially decreased AKT and STAT3 activation in response to both HGF and EGF (Fig. 2).
To further verify MET-specific effects, we rescued the Met−/− phenotype by reintroducing the Met gene into Met-null HPCs by a lentiviral-delivered expression vector and subjected cells to a hepatocyte differentiation protocol. Murine Met knock-in (referred as to MetresWT) restored MET signaling and up-regulated hepatocyte differentiation genes (Fig. 3E,F). These data establish that the MET pathway has a dominant role in facilitating hepatocytic differentiation via AKT/STAT3 activation.
EGFR is an essential mediator of the NOTCH1 pathway in cholangiocyte differentiation and branching morphogenesis
To induce cholangiocyte differentiation, we employed a three-dimensional (3D) morphogenesis assay using HGF and EGF as stimulants (Tanimizu et al. 2007; Okabe et al. 2009). HPCs were mixed with Matrigel/collagen1 and grown on Matrigel/collagen1 gel. Treating the parental HPCs with HGF and EGF alone or in combination resulted in the development of a characteristic interconnected tubular network, which was concordant with the up-regulation of biliary markers (Fig. 4A,B). Met-null HPCs grown in 3D cultures and subjected to biliary epithelial cell differentiation assay also acquired biliary cell marker expression (Fig. 4A) and were able to form branches, albeit with a strongly reduced tubule length (Fig. 4B–D). In striking contrast, the lack of Egfr−/− in HPCs suppressed both the marker expression and branching morphogenesis (Fig. 4A–D). In Egfr−/− cells that are not responsive to EGF stimulation (Fig. 2), HGF supplementation failed to support up-regulation of proteins associated with the biliary epithelial cell phenotype, including CK7, CK19, EpCAM, SOX9, NOTCH1, and NOTCH effector HES1 (Fig. 4A). The real-time RT–PCR analysis confirmed a strong down-regulation of Notch1 and Hes1, whereas the mRNA levels of other members of the HES family, including Hes3 and Hes5, were not changed (Supplemental Fig. 5). Furthermore, the number of branch points formed by Egfr−/− HPCs was decreased >10.3-fold and 7.6-fold as compared with the growth receptor-competent or Met mutant cultures (Fig. 4D) without affecting the primitive tube length (Fig. 4B,C).
Figure 4.

Loss of EGFR blocks differentiation of progenitor cells toward biliary lineage in vitro. (A) Western blots for CK7, CK19, EpCAM, SOX9, Notch1, Notch2, and HES1. Actin was used as a loading control. (GF−) No growth factors; (H) HGF; (E), EGF; (H+E) HGF and EGF. (B, top and middle) Bright-field images (bars, 50 μm) and confocal microscopy images (bars, 50 μm) of double immunofluorescence staining for CK19 (green) and SOX9 (red). Nuclei were counterstained with DAPI. Tube length (C) and branch point (D) numbers. Data are mean ± SEM. Fifty randomly selected cell clusters were analyzed from three independently derived samples. (*) P < 0.01 versus GF− cells of the same genotype; (#) P < 0.01 versus similarly treated Metfl/fl cells. (E, top images) Egfr knock-in (EgfrresWT) restores the ability of Egfr−/− cells to form tube-like branched structures characteristic for biliary differentiation. Bright-field images. Bar, 100 μm. (F) Egfr knock-in (EgfrresWT) restores Egfr signaling and NOTCH1 expression in Egfr−/− cells. Western blots for pEGFR, EGFR, and NOTCH1. Actin was used as a loading control. Monolayer cultures of indicated genotypes were treated with EGF (10 ng/mL) for 15 min after overnight serum starvation. (Mock) Empty vector. (G) Egfr knock-in (EgfrresWT) restores expression of Notch downstream targets Hes1 and Sox9 in Egfr−/− cells. mRNA expression levels measured by qRT–PCR. GAPDH was used as internal control for qRT–PCR. Data are mean ± SEM of two independent duplicate experiments. Expression levels are shown relative to untreated Egfrfl/fl cells. (*) P < 0.01 versus corresponding Egfr−/− cells.
To confirm the specificity of the described effects, we carried out rescue experiments by stably overexpressing wild-type human EGFR in the Egfr-deleted HPCs (referred to as EgfrrecWT). The reintroduction of EGFR effectively restored the branching phenotype as well as expression of NOTCH1 (Fig. 4E) and its downstream targets, Hes1 and Sox9 (Fig. 4G; Meier-Stiegen et al. 2010). The mRNA levels of other NOTCH target genes such as Hes3, Hes5, Hey1, and Hey2 were unaffected by EGFR restoration (data not shown). Based on these data, we hypothesized that EGFR controls the biliary gene expression and ability to form branches via stimulation of the NOTCH pathway. Consistent with this, the protein levels of NOTCH1 and SOX9 were reduced in the untreated Egfr-null HPCs as compared with both parental and Met−/− HPCs, whereas the NOTCH2 levels were unaffected (Supplemental Fig. S6A). The down-regulation of Notch1 but not Notch2 expression in the absence of Egfr signaling was confirmed in four additional Egfr-null HPC clonal lines (Supplemental Fig. S6B). The deletion of Egfr did not affect the expression of Jagged1 ligand for the Notch1 receptor or Numb1, a negative regulator of the NOTCH signaling pathway (Supplemental Fig. S6A).
To support our hypothesis, we then carried out the Notch1 loss-of function and Egfr gain-of-function experiments in Egfr-competent and Egfr-null HPCs, respectively. First, we generated a stable lentivirus-transduced Egfrfl/fl cell line expressing shRNAs against Notch1 and clonally isolated cells with effective (>75%) Notch1 knockdown (Fig. 5A,B). Reduced expression levels of Notch1 in the Egfr-competent HPCs significantly decreased CK19 and SOX9 expression (Fig. 5C) and interfered with the ability to form branches (Fig. 5D). On the other hand, Notch1 knockdown increased the expression of genes coding for hepatocyte-specific functions during differentiation of shNotch1 cells toward hepatocytic lineage (Fig. 5E).
Figure 5.

EGFR-regulated NOTCH1 is required for differentiation of HPCs toward biliary lineage. (A) Depletion of Notch1 mRNA after a stable lentiviral transduction of Notch1 shRNA into the Egfrfl/fl cell line. Relative mRNA expression levels of indicated genes were measured by qRT–PCR. Gapdh was used as internal control for qRT–PCR. Data are mean ± SEM (n = 4). (*) P < 0.01 versus Egfrfl/fl. (B) Depletion of NOTCH1 protein after a stable lentiviral transduction of Notch1 shRNA into Egfrfl/fl cell line. (Mock) Empty vector. (C) Silencing of Notch1 by shRNA in Egfrfl/fl cells inhibits biliary epithelial marker expression. Western blots for CK19, SOX9, and actin. Monolayer cultures of indicated genotypes were treated with HGF (20 ng/mL) and EGF (10 ng/mL) for 15 min after overnight serum starvation. [GF(−)] No growth factors. (D) Silencing of Notch1 by shRNA in Egfrfl/fl cells reduces branching in 3D cultures (bar, 100 μm). (E) Silencing of Notch1 by shRNA in Egfrfl/fl cells promotes differentiation toward hepatocytic lineage. Western blots for HNF4α, albumin (ALB), and actin. (F) Transduction of NICD into Egfr−/− cells (EgfrresNICD) restores Notch1 expression. Western blots for NOTCH1, EGFR, and actin. (G) Transduction of NICD into Egfr−/− cells (EgfrresNICD) restores biliary epithelial cell marker expression. Monolayer cultures of indicated genotypes were treated with HGF (20 ng/mL) for 15 min after overnight serum starvation. (H) Transduction of NICD into Egfr−/− cells (EgfrresNICD) restores branching morphogenesis in Egfr−/− progenitor cells subjected to biliary epithelial cell differentiation assay. Bar, 250 μm.
Finally, to strengthen our conclusion that the impaired differentiation toward the biliary epithelial cell lineage seen in Egfr−/− HPCs was due to the loss of NOTCH1 signaling but not the intrinsic defect in differentiation potential in mutant cells, we introduced Notch1 intracellular domain (NICD) into Egfr-deficient HPCs. Lentiviral transduction of NICD into Egfr−/− HPCs (referred to as EGFRresNICD) restored NOTCH1 expression (Fig. 5F) and repaired biliary differentiation defects, as judged by cholangiocyte marker expression (Fig. 5G) and ability to develop tubular structures with extensive branching morphology (Fig. 5H). Together, these data indicate that EGFR-mediated up-regulation of NOTCH1 is a necessary step for initiating cholangiocytic differentiation in vitro.
Loss of Egfr leads to impaired differentiation of HPCs toward biliary lineage in vivo
To extend these findings in vivo, we used Egfrfl/fl;Mx1-Cre+/− conditional knockout mice. Western blotting confirmed the successful deletion of Egfr in Egfrfl/fl;Mx1-Cre+/− livers (Supplemental Fig. S7A). However, the phosphorylation of ERK1/2, AKT, and STAT3 was not affected, indicating that functional redundancy and overlapping functions of RTKs may substitute for Egfr loss. In striking contrast to Metfl/fl;Mx1-Cre+/− mice, which failed to mobilize HPCs and died from liver failure (Ishikawa et al. 2012), genetic loss of Egfr was beneficial rather than detrimental for liver regeneration induced by DDC liver injury. Accordingly, Egfrfl/fl;Mx1-Cre+/− mice exhibited higher liver to body mass ratios as compared with DDC-treated control animals and had decreased serum levels of alanine aminotransferase, alkaline phosphatase, and total bilirubin, reflecting a better liver function (Supplemental Fig. S7B).
Furthermore, differently from the Met conditional knockout model (Ishikawa et al. 2012), EGFR mutant mice developed a more extensive progenitor cell population as compared with Egfrfl/fl livers by week 4 on a DDC diet (Supplemental Fig. S8A,B). Notably, the expanding population of HPCs in Egfrfl/fl;Mx1-Cre+/− livers expressed the known common markers of biliary/progenitor cells A6, EpCAM, and CK19 (Supplemental Fig. S8C; Factor et al. 1994; Rountree et al. 2007; Okabe et al. 2009) but, consistent with impaired differentiation toward biliary lineage in vitro (Figs. 4, 5), did not remodel into characteristic bile duct structures with well-defined lumina (Supplemental Fig. S8A).
Given the impotence of EGFR-mediated NOTCH1 expression for biliary differentiation in vitro, we then compared the pattern of NOTCH1 expression in normal and regenerating livers from Egfrfl/fl and Egfr mutant mice. Double immunofluorescence analysis revealed that while the mature bile ducts of untreated livers were NOTCH1-negative (Fig. 6A), the DDC-treated EGFRfl/fl livers displayed a prominent accumulation of newly formed bile duct-like structures that coexpressed the A6 biliary/progenitor cell marker and Notch1 (Fig. 6A). Morphologically, they appeared as more mature enlarged ducts with well-recognizable lumina (Fig. 6A) that differed from irregular A6-positive ductules without obvious lumens. Quantitative estimates showed that the frequency of NOTCH1+/A6+ bile duct-like structures was drastically reduced in the EGFR mutant livers (Fig. 6B). Of note, the expression of NOTCH2 was not a selective marker of newly formed bile ducts, since NOTCH2 was up-regulated in both the hepatocytes and HPCs (Supplemental Fig. S9A). Supporting the differences in NOTCH staining and consistent with our in vitro data (Supplemental Fig. S6), untreated Egfrfl/fl;Mx1-Cre+/− livers showed reduced basal levels of Notch1 mRNA and, differently from control Egfrfl/fl livers, failed to up-regulate Notch1 during a progenitor-mediated liver regeneration (Supplemental Fig. S9B), whereas Notch2 expression was comparable in mice of both genotypes regardless of treatment (Supplemental Fig. S9C). The reduced frequency of the NOTCH1+/A6+ bile duct-like structures was paralleled by a more rapid accumulation of newly formed hepatocytes identified by coexpression of A6 and the mature hepatocyte marker HNF4α (Fig. 6C,D). The latter finding could explain a better functional performance and a more efficient liver regeneration in Egfrfl/fl;Mx1-Cre+/− mice after DDC damage (Supplemental Fig. S7B). Last, we stained the DDC-treated livers with αSMA, a marker of activated stellate cells (Supplemental Fig. S10A), an important component of the HPC niche affecting progenitor cell fate decision (Espanol-Suner et al. 2012). Because αSMA-positive stellate cells typically accompany progenitor ductular cell reaction, we performed a double immunofluorescence staining for A6 and αSMA (Supplemental Fig. S10A). Quantitative estimates of areas occupied by A6- and αSMA-positive cells revealed that the extent of stellate cell activation was proportional to the size of progenitor cell reaction in mice of both genotypes, supporting our conclusion that NOTCH1 down-regulation caused by Egfr loss rather than altered HPC niche composition switched the balance of HPC differentiation from cholangiocytes toward hepatocytes.
Figure 6.

Loss of Egfr suppresses progenitor cell differentiation toward biliary lineage while promoting differentiation toward hepatocytes in Egfrfl/fl;Mx1-Cre+/− mice fed a DDC diet. (A) Confocal microscopy images of double immunofluorescence staining for A6 (green) and NOTCH1 (red). Bar, 50 μm. (Top images) No NOTCH1 positivity was found in mature bile ducts from untreated livers of both genotypes. At 4 wk after DDC diet, Egfrfl/fl livers displayed numerous A6+/NOTCH1+ duct-like structures with clearly defined round lumens (arrows), whereas only few were present in Egfrfl/fl;Mx1-Cre+/− livers. Bar in bottom images, 25 μm. Nuclei counterstained were with DAPI. (B) Time-course changes in the frequency of A6+/Notch1+ bile duct-like structures. Quantification was performed in 10 independent images taken with a confocal microscope at 200×. Data are mean ± SEM, n = 4 per each group of mice. (*) P < 0.05 versus Egfrfl/fl mice. (C) Confocal microscopy images (bar, 50 μm) of double immunofluorescence staining for A6 (green) and HNF4α (red; white arrows) 2 wk after DDC diet. Nuclei were counterstained with DAPI. (D) Time-course changes in the frequency of A6+/HNF4α+ cells. Quantification was performed in 10 independent images taken with confocal microscope at 200×. Data are mean ± SEM, n = 4 per each group of mice. (*) P < 0.05 versus EGFRfl/fl mice.
Discussion
Extensive studies link various biological processes controlled by EGFR and MET to specific signaling pathways. Here we used a growth factor-defined in vitro system of single-cell-derived HPC lines to delineate novel mechanisms by which the two key hepatic RTKs govern the binary fate decisions of adult HPCs. Using sphere-forming assay in combination with genetic and pharmacological approaches, we found that the two pathways cooperate to increase the self-renewal proliferation of HPCs through ERK1/2 activation. However, MET-initiated up-regulation of STAT3 and AKT was a specific driving force of HPC differentiation toward hepatocytes. Notably, signaling through EGFR failed to compensate for the lack of MET receptor in HPCs, while HGF stimulation of Egfr-null progenitor cells or MET restoration in MET mutant HPCs was sufficient to promote hepatocyte differentiation. These observations corroborate prior findings regarding the role of HGF/MET signaling in facilitating progenitor, embryonic stem cell, and induced pluripotent stem cell differentiation along hepatocyte lineage (Si-Tayeb et al. 2010; Ishikawa et al. 2012; Lee et al. 2012).
We further identify EGFR as a key player in cholangiocyte differentiation through a mechanism requiring NOTCH1. Genetic loss of Egfr in HPCs strongly suppressed NOTCH1 expression. Reintroduction of Egfr in Egfr-null HPCs restored NOTCH levels, indicating that Egfr can directly up-regulate NOTCH1. This is consistent with studies reporting interactions between EGFR and NOTCH1. Thus, during asymmetric division of mouse neural stem cells, NICD expression was higher in a daughter cell expressing high levels of EGFR (Andreu-Agullo et al. 2009). Similarly, up-regulation of Egfr mRNA was observed in a Drosophila cell line upon Notch activation, and loss of either Notch or Egfr resulted in related defects in muscle development (Krejci et al. 2009). The EGFR–RAS–ERK pathway has also been implicated in a positive control of the Notch ligand Delta (Tsuda et al. 2002; Chen and Greenwald 2004), while NOTCH has been shown to negatively regulate EGF expression (Ligoxygakis et al. 1998; Karp and Greenwald 2003). More recent data demonstrated the involvement of p53 in the reciprocal regulation of EGFR and NOTCH1 expression through the EGFR effector C-JUN (Kolev et al. 2008; Purow et al. 2008). Future work is required to determine the detail mechanisms of the Egfr–Notch1 relationship.
Our data confirm and extend prior findings regarding a key role of NOTCH1 signaling in the biliary fate decision of HPCs in human and experimental animal models (Li et al. 1997; Oda et al. 1997; McCright et al. 2002; Geisler et al. 2008; Tchorz et al. 2009; Zong et al. 2009; Sparks et al. 2010; Boulter et al. 2012). The EGFR/NOTCH signaling was required at multiple steps of biliary lineage commitment. The HPCs with a targeted deletion of Egfr or Notch1 failed to up-regulate marker proteins CK7, CK19, SOX9, and EpCAM. Furthermore, in the branching assay, HGF stimulation of HPC cultures lacking Egfr or Notch1 caused only rudimentary ductal branching. However, after reinstallation of NICD in Egfr−/− HPCs, HGF treatment successfully augmented branching morphogenesis and caused up-regulation of biliary marker proteins, providing evidence that EGFR is primarily responsible for NOTCH1-mediated induction of branching morphogenesis while limiting MET activity to the control of primitive tube formation. This is in agreement with earlier findings that NOTCH signaling is essential for biliary duct architecture (Sparks et al. 2010).
There are four homologs of NOTCH receptor (NOTCH1, NOTCH2, NOTCH3, and NOTCH4) in mammals (Harper et al. 2003). NOTCH2 was identified as the main contributor to embryonic and early postnatal bile duct formation (McCright et al. 2002; Geisler et al. 2008; Jeliazkova et al. 2013), and Notch2 silencing was reported to promote hepatoblast differentiation into hepatocytes (Tanimizu and Miyajima 2004). In our study, loss of Egfr caused down-regulation of NOTCH1 but not NOTCH2, critically attenuating HPC differentiation toward biliary epithelial cells both in vitro and in vivo. Supporting a role for NOTCH1 in the adult intrahepatic bile duct maintenance, a selective expression of NOTCH1, but not NOTCH2 or NOTCH3, was found in the expanding population of progenitor cells in the rat AAP-PH model (Jensen et al. 2004). Furthermore, overexpression of NICD increased the intrahepatic bile duct density in the adult liver, whereas knock-in Notch2 intracellular domain just temporally promoted intrahepatic bile duct development during embryonic and early postnatal life (Tchorz et al. 2009; Sparks et al. 2010). More work will elucidate the significance of each Notch receptor for liver development and regeneration.
Our results also support the notion that EGFR-mediated NOTCH1 expression may serve as a negative regulator of HPC commitment to hepatocyte lineage. Thus, silencing of Notch1 by shRNA in wild-type HPCs increased the efficiency of hepatocyte differentiation in vitro, suggesting that EGFR-controlled NOTCH1 activity may switch the balance toward the alternate differentiation options. Accordingly, HPCs activated in Egfr conditional knockout mice showed a preferential differentiation into hepatocytes, likely contributing to a faster and more efficient recovery from DDC toxicity. Conversely, these mice had noticeably fewer newly formed bile duct-like structures as compared with the Egfrfl/fl livers. Consistent with these data, the impaired bile duct maturation during DDC treatment was recently reported in two other genetic mouse models in which Cre-mediated excision of Notch2 and RBP-Jk was under the control of the albumin enhancer promoter and thus was limited only to epithelial compartment (Fiorotto et al. 2013). We realize the obvious limitations of our conventional conditional knockout mouse model, which uses a ubiquitous interferon-responsive promoter to drive Cre-recombinase expression in numerous hepatic cell types. This model does not allow the definite conclusion about the cell fate decisions of HPCs, which depend on both the intrinsic and extrinsic signaling cascades. Nevertheless, it is worth noting that the phenotype of progenitor cell response in Egfrfl/fl; Mx1-Cre+/− livers recapitulated the differentiation defects of Egfr−/− HPCs described in the in vitro setting, supporting our overall conclusion that the EGFR/NOTCH1 signaling axis contributes to lineage decisions of adult HPCs. Further work using compartment-specific lineage tracing studies is required for a definitive demonstration of differentiation options and evolution of HPCs.
In conclusion, using an in vitro model of adult HPCs, we demonstrate that HGF and EGF, two primary liver growth stimuli, elicit differential responses in adult HPCs via selective activation of nonoverlapping downstream networks. EGFR-mediated NOTCH1 signaling was crucial in controlling commitment of HPCs to the biliary lineage, while MET-driven AKT/STAT3 activity promoted differentiation toward hepatocytes (Fig. 7).
Figure 7.
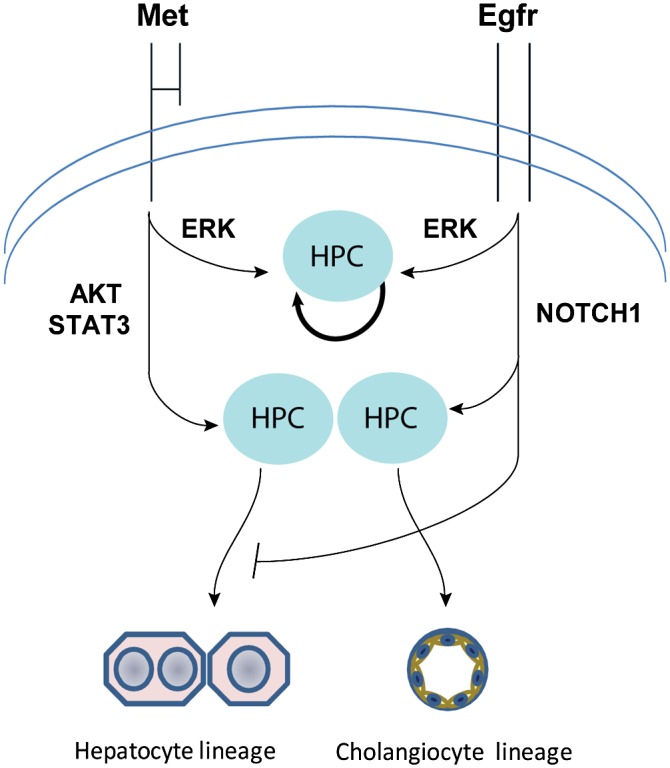
A model of EGFR and MET function in HPCs. EGFR is an essential mediator of NOTCH1, which is required for commitment of progenitor cells to biliary lineage and branching morphogenesis. MET-driven AKT/STAT3 activity promotes differentiation toward hepatocytic lineage and supports primitive tubulogenesis.
Materials and methods
Mouse genetics and establishment of HPC single-cell clonal lines
Generation of Metfl/fl and Egfrfl/fl mice was described elsewhere (Huh et al. 2004; Lee and Threadgill 2009). Eight-week-old mice were fed with a diet containing 0.1% DDC (Bioserv) for 2 wk to induce the HPC response. Progenitor cells were isolated as described by Okabe et al. (2009) with modifications (Supplemental Fig. S1A–C). First, the bulk of nonparenchymal cells was isolated by a two-step collagenase perfusion method and stained with PE-EpCAM (kind gift from Dr. Atsushi Miyajima, Tokyo University) and mouse lineage cocktail antibodies containing anti-CD3c, anti-CD11b, anti-CD45R, anti-TER-119, anti-LY-6C, and anti-LY-6G conjugated with APC (BD Biosciences) (Okabe et al. 2009; Ishikawa et al. 2012). EpCAM+/Lineage− cells were then FACS-sorted using BD FACSVantage (BD Biosciences), plated on a 3D gel formed by a growth factor-reduced Matrigel (BD Biosciences) at a concentration of 1 × 105 per square centimeter, and incubated with a basal growth medium described by Okabe et al. (2009), which was supplemented with 20 ng/mL HGF and 10 ng/mL EGF (Peprotech) for 7–10 d to select for cells possessing sphere-forming capacity. Here and thereafter, the HGF and EGF were replenished every other day. The spheres were dissociated into a single-cell suspension using 0.25% trypsin solution (GIBCO). Next, the cells were seeded on collagen1-coated plastic dishes (BD Biosciences) at a concentration of 1 × 105 per square centimeter and expanded in a basal growth medium containing HGF (10 ng/mL) and EGF (5 ng/mL) as passages 1–4. Starting from passage 4, cells were grown on plastic dishes in the basal growth medium. Deletion of Met or Egfr was performed at passage 4 by Adeno-CMV-Cre (Vector Biolabs) transfection as described (del Castillo et al. 2008). Adeno-Null vector (Vector Biolabs) is referred to as Mock. Parental and Adeno-CMV-Cre transfected cells were single-cell-cloned at passage 4, expanded, and subjected to PCR genotyping and Western blotting at passage 7. Primers used for genotyping are listed in Supplemental Table S1A (Huh et al. 2004). In total, we established four independent single-cell clonal cell lines for each genotype, including two parental (Metfl/fl and Egfrfl/fl) and two mutant (Met−/− and Egfr−/−) lines. The cells kept the capacity to differentiate along biliary epithelial lineage for at least 20 passages, but efficiency of differentiation toward hepatocytic lineage was declining after the 10th passage. Therefore, all experimental work was performed within eight to 10 passages.
Liver transplantation
The homozygous MUP-uPA/SCID mice were generated by cross-breeding MUP-uPA transgenic mice originally described by Weglarz et al. (2000) with SCID-BEIGE mice purchased from Taconic. mCherry-tagged Metfl/fl HPCs (passage 8, 106 per mouse) were injected into the inferior pole of the spleen of 3-wk-old mice of both sexes. Cellular engraftment in the liver was examined at 32 wk after transplantation (n = 5 mice).
Sphere-forming assay
HPCs (2 × 103 cells) were plated in ultralow attachment plates (Costar) in serum-free growth medium containing 1% methylcellulose solution (R&D Systems) and 1× B-27 (Invitrogen) in the presence or absence of 20 ng/mL HGF and/or 10 ng/mL EGF. The average number and sphere size were calculated using NIH ImageJ software in three independent duplicate experiments. The spheres with a diameter <50 μm were excluded from analysis.
Differentiation assays
Cells were subjected to the differentiation protocols toward hepatocyte and biliary epithelial cells as described with modifications (Tanimizu et al. 2007; Okabe et al. 2009). Briefly, for hepatocyte differentiation, HPCs were plated at 90% confluence on a plastic plate containing an underlay of 1% solidified Matrigel and treated with 20 ng/mL HGF and/or 10 ng/mL EGF in growth medium containing 1% fetal bovine serum (FBS) and 1 mM dexamethasone for 7 d. Next, the cultures were overlayed with the same medium containing 5% reduced growth factor Matrigel (BD Biosciences) for 5 d.
For biliary cell differentiation, 1 × 105 HPCs were grown in 3D cultures in 1 mg/mL collagen1 (BD Biosciences) mixed with Matrigel in a 1:1 ratio for 7 d in growth medium containing 1% FBS in the presence or absence of 20 ng/mL HGF and/or 10 ng/mL EGF. At least three independent experiments were performed for each assay using three HPC lines for each genotype.
Pharmacological inhibition of ERK, AKT, and STAT3
To observe the effect of individual downstream targets of MET and EGFR signaling on HPC properties, the sphere-forming and differentiation assays were performed with inhibitors for MEK/ERK, AKT, and STAT3 using 20 μM PD98059 (Cell Signaling), 20 μM LY294002, and 10 μM WP1066 (Calbiochem), respectively, in the presence of 20 ng/mL HGF and 10 ng/mL EGF. The optimal concentration of each inhibitor was defined based on at least 80% inhibition of target signals and no less than 60% viability (Supplemental Fig. S5).
Egfr, Met, and Notch1 restoration
Egfr and Met were reintroduced in Egfr- and Met-null HPCs, respectively, by retroviral-delivered expression constructs, and NICD was reintroduced in EGFR-null HPCs by lentiviral-delivered expression construct. WT-EGFR cDNA-inserted vector (Addgene plasmid 11011) was developed by Dr. Matthew Meyerson (Dana Farber Cancer Institute) (Greulich et al. 2005). A full-length murine Met cDNA-inserted vector (Addgene, plasmid 17493) was developed by Dr. Joan Brugge (Harvard Medical School) (Wrobel et al. 2004), and pBABE-puro (Addgene, plasmid no. 1764) expression vector without insert was developed by Dr. Robert Weinberg (Whitehead Institute for Biomedical Research) (Morgenstern and Land 1990). NICD cDNA-inserted vector (Addgene, plasmid 17623) was developed by Dr. Linzhao Cheng (The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins) (Greulich et al. 2005).
Notch1 knockdown by shRNA
Notch1 knockdown was performed using lenti-shRNA vector against mouse Notch1 (Sigma-Aldrich) in Egfrfl/fl HPC lines according to the manufacturer's protocol. The efficiency of transfection was verified by RT–PCR and Western blotting 14 d after puromycin selection.
RT–PCR, Western blotting, and immunofluorescence
Real-time PCR, Western blotting, and immunofluorescence were performed using standard assays described elsewhere (O'Brien et al. 2006; Factor et al. 2010; Marquardt et al. 2011; Ishikawa et al. 2012). Primer sequences are listed in Supplemental Table S1B, and antibodies are listed in Supplemental Table S2. Fluorescently stained cells and tissues were viewed by a Zeiss LSM 510 NLO and Zeiss LSM 710 NLO (Zeiss).
Induction of HPC response in liver-specific EGFR conditional knockout mice
For Mx1-Cre-mediated Egfr inactivation, both 6-wk-old Egfrfl/fl ;MxCre+/− and Egfrfl/fl;MxCre−/− mice (referred to as EGFRfl/fl) received three intraperitoneal (i.p.) injections of 300 μg of pIpC (Sigma-Aldrich) in saline at 2-d intervals, which resulted in Egfr gene deletion in 3–4 wk (data not shown). To induce HPC response, mice were given a diet containing 0.1% DDC (Bioserv) 4 wk after the first pIpC injection. Serum and liver samples were obtained after 1, 2, and 4 wk of DDC treatment. All experimental procedures were approved by the NCI-Bethesda Animal Care and Use Committee (ACUC) under LEC-049. The NCI animal program meets or exceeds requirements of the Public Health Service Policy on Humane Care and Use of Animals and is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International.
Microarray profiling
Metfl/fl and Egfrfl/fl single-cell clonal lines (n = 3 each, passage 10) were grown on plastic in the basal growth medium containing 10% FBS. When cells reached 70% confluency, they were serum-starved for 16 h. Total RNAs were isolated and precipitated by RNeasy minikit (Qiagen). Linear amplification of 200 ng of RNA was performed with Illumina TotalPrep RNA amplification kit (Life Technologies). A total of 750 ng of cRNA for each sample was hybridized on Sentrix Mouse Expression BeadChips version 2 (Illumina). Microarray data collection, preprocessing, and quantile normalization were performed with GenomeStudio2011.1 (Illumina), allowing for a detection of P-value < 0.01. The similarity between groups of arrays was analyzed using bioequivalence tests. The test allows for ±1.5-fold change at P < 0.001. Comparison of gene expression changes between Metfl/fl and Egfrfl/fl cells was performed using Pearson correlation. Microarray data were submitted to Gene Expression Omnibus (accession no. GSE46981) and are accessible at http://www.ncbi.nlm.nih.gov/geo.
Statistical analysis
The data were analyzed by Bootstrap t-test with 10,000 replications, Tukey's pairwise comparison test, Poisson GLM test, and Pearson's χ2 test as indicated.
Acknowledgments
We are grateful for a kind gift of EpCAM antibody from Dr. Atsushi Miyajima, Tokyo University. We thank B. Taylor and S. Banerjee for assistance with flow-cytometry, S. Garfield and P. Mannan for help with imaging, and T. Hoang and A. Ton for technical assistance. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.214601.113.
References
- Aguirre A, Rubio ME, Gallo V 2010. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature 467: 323–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alison MR, Golding MH, Sarraf CE 1996. Pluripotential liver stem cells: Facultative stem cells located in the biliary tree. Cell Prolif 29: 373–402 [DOI] [PubMed] [Google Scholar]
- Andreu-Agullo C, Morante-Redolat JM, Delgado AC, Farinas I 2009. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat Neurosci 12: 1514–1523 [DOI] [PubMed] [Google Scholar]
- Bertotti A, Comoglio PM 2003. Tyrosine kinase signal specificity: Lessons from the HGF receptor. Trends Biochem Sci 28: 527–533 [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF 2003. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 4: 915–925 [DOI] [PubMed] [Google Scholar]
- Bohm F, Kohler UA, Speicher T, Werner S 2010. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol Med 2: 294–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, Ridgway RA, Seo SS, Spee B, Van Rooijen N, et al. 2012. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med 18: 572–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter L, Lu WY, Forbes SJ 2013. Differentiation of progenitors in the liver: A matter of local choice. J Clin Invest 123: 1867–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill LM, Xiong W, Lee KB, Ficarro SB, Crain A, Xu Y, Terskikh A, Snyder EY, Ding S 2009. Phosphoproteomic analysis of human embryonic stem cells. Cell Stem Cell 5: 204–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AW 2008. EGFR family: Structure physiology signalling and therapeutic targets. Growth Factors 26: 263–274 [DOI] [PubMed] [Google Scholar]
- Chen N, Greenwald I 2004. The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev Cell 6: 183–192 [DOI] [PubMed] [Google Scholar]
- Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG 2009. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 138: 1083–1095 [DOI] [PubMed] [Google Scholar]
- del Castillo G, Factor VM, Fernandez M, Alvarez-Barrientos A, Fabregat I, Thorgeirsson SS, Sanchez A 2008. Deletion of the Met tyrosine kinase in liver progenitor oval cells increases sensitivity to apoptosis in vitro. Am J Pathol 172: 1238–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Dorrell C, Grompe M 2009. Stem cells and liver regeneration. Gastroenterology 137: 466–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espanol-Suner R, Carpentier R, Van Hul N, Legry V, Achouri Y, Cordi S, Jacquemin P, Lemaigre F, Leclercq IA 2012. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 143: 1564–1575.e7 [DOI] [PubMed] [Google Scholar]
- Evarts RP, Hu ZY, Fujio K, Marsden ER, Thorgeirsson SS 1993. Activation of hepatic stem-cell compartment in the rat: role of transforming growth-factor-α, hepatocyte growth-factor, and acidic fibroblast growth-factor in early proliferation. Cell Growth Differ 4: 555–561 [PubMed] [Google Scholar]
- Factor VM, Radaeva SA, Thorgeirsson SS 1994. Origin and fate of oval cells in dipin-induced hepatocarcinogenesis in the mouse. Am J Pathol 145: 409–422 [PMC free article] [PubMed] [Google Scholar]
- Factor VM, Seo D, Ishikawa T, Kaposi-Novak P, Marquardt JU, Andersen JB, Conner EA, Thorgeirsson SS 2010. Loss of c-Met disrupts gene expression program required for G2/M progression during liver regeneration in mice. PLoS ONE 5: e12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorotto R, Raizner A, Morell CM, Torsello B, Scirpo R, Fabris L, Spirli C, Strazzabosco M 2013. Notch signaling regulates tubular morphogenesis during repair from biliary damage in mice. J Hepatol 59: 124–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler F, Nagl F, Mazur PK, Lee M, Zimber-Strobl U, Strobl LJ, Radtke F, Schmid RM, Siveke JT 2008. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology 48: 607–616 [DOI] [PubMed] [Google Scholar]
- Giannini C, Morosan S, Tralhao JG, Guidotti JE, Battaglia S, Mollier K, Hannoun L, Kremsdorf D, Gilgenkrantz H, Charneau P 2003. A highly efficient, stable, and rapid approach for ex vivo human liver gene therapy via a FLAP lentiviral vector. Hepatology 38: 114–122 [DOI] [PubMed] [Google Scholar]
- Greenbaum LE, Wells RG 2011. The role of stem cells in liver repair and fibrosis. Int J Biochem Cell Biol 43: 222–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, et al. 2005. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2: e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JA, Yuan JS, Tan JB, Visan I, Guidos CJ 2003. Notch signaling in development and disease. Clin Genet 64: 461–472 [DOI] [PubMed] [Google Scholar]
- Hasuike S, Ido A, Uto H, Moriuchi A, Tahara Y, Numata M, Nagata K, Hori T, Hayashi K, Tsubouchi H 2005. Hepatocyte growth factor accelerates the proliferation of hepatic oval cells and possibly promotes the differentiation in a 2-acetylaminofluorene/partial hepatectomy model in rats. J Gastroenterol Hepatol 20: 1753–1761 [DOI] [PubMed] [Google Scholar]
- Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS 2004. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci 101: 4477–4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Factor VM, Marquardt JU, Raggi C, Seo D, Kitade M, Conner EA, Thorgeirsson SS 2012. Hepatocyte growth factor (HGF)/c-Met signaling is required for stem cell mediated liver regeneration. Hepatology 55: 1215–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeliazkova P, Jors S, Lee M, Zimber-Strobl U, Ferrer J, Schmid RM, Siveke JT, Geisler F 2013. Canonical Notch2 signaling determines biliary cell fates of embryonic hepatoblasts and adult hepatocytes independent of Hes1. Hepatology 57: 2469–2479 [DOI] [PubMed] [Google Scholar]
- Jensen CH, Jauho EI, Santoni-Rugiu E, Holmskov U, Teisner B, Tygstrup N, Bisgaard HC 2004. Transit-amplifying ductular (oval) cells and their hepatocytic progeny are characterized by a novel and distinctive expression of delta-like protein/preadipocyte factor 1/fetal antigen 1. Am J Pathol 164: 1347–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC 2000. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem 275: 8806–8811 [DOI] [PubMed] [Google Scholar]
- Karp X, Greenwald I 2003. Post-transcriptional regulation of the E/Daughterless ortholog HLH-2, negative feedback, and birth order bias during the AC/VU decision in C. elegans. Genes Dev 17: 3100–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev V, Mandinova A, Guinea-Viniegra J, Hu B, Lefort K, Lambertini C, Neel V, Dummer R, Wagner EF, Dotto GP 2008. EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat Cell Biol 10: 902–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci A, Bernard F, Housden BE, Collins S, Bray SJ 2009. Direct response to Notch activation: Signaling crosstalk and incoherent logic. Sci Signal 2: ra1. [DOI] [PubMed] [Google Scholar]
- Lee TC, Threadgill DW 2009. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 47: 85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Seo D, Choi D, Park KY, Holczbauer A, Marquardt JU, Conner EA, Factor VM, Thorgeirsson SS 2012. Contribution of hepatic lineage stage-specific donor memory to the differential potential of induced mouse pluripotent stem cells. Stem Cells 30: 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, et al. 1997. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 16: 243–251 [DOI] [PubMed] [Google Scholar]
- Ligoxygakis P, Yu SY, Delidakis C, Baker NE 1998. A subset of notch functions during Drosophila eye development require Su(H) and the E(spl) gene complex. Development 125: 2893–2900 [DOI] [PubMed] [Google Scholar]
- Marquardt JU, Raggi C, Andersen JB, Seo D, Avital I, Geller D, Lee YH, Kitade M, Holczbauer A, Gillen MC, et al. 2011. Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology 54: 1031–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCright B, Lozier J, Gridley T 2002. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 129: 1075–1082 [DOI] [PubMed] [Google Scholar]
- Meier-Stiegen F, Schwanbeck R, Bernoth K, Martini S, Hieronymus T, Ruau D, Zenke M, Just U 2010. Activated Notch1 target genes during embryonic cell differentiation depend on the cellular context and include lineage determinants and inhibitors. PLoS ONE 5: e11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalopoulos GK 2010. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am J Pathol 176: 2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalopoulos GK, DeFrances MC 1997. Liver regeneration. Science 276: 60–66 [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H 1990. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18: 3587–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P, Bisgaard HC, Santoni-Rugiu E, Thorgeirsson SS 1996. In vivo infusion of growth factors enhances the mitogenic response of rat hepatic ductal (oval) cells after administration of 2-acetylaminofluorene. Hepatology 23: 71–79 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Sakai K, Matsumoto K 2011. Hepatocyte growth factor twenty years on: Much more than a growth factor. J Gastroenterol Hepatol 26: 188–202 [DOI] [PubMed] [Google Scholar]
- Natarajan A, Wagner B, Sibilia M 2007. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci 104: 17081–17086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien LE, Yu W, Tang K, Jou TS, Zegers MM, Mostov KE 2006. Morphological and biochemical analysis of Rac1 in three-dimensional epithelial cell cultures. Methods Enzymol 406: 676–691 [DOI] [PubMed] [Google Scholar]
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, et al. 1997. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 16: 235–242 [DOI] [PubMed] [Google Scholar]
- Okabe M, Tsukahara Y, Tanaka M, Suzuki K, Saito S, Kamiya Y, Tsujimura T, Nakamura K, Miyajima A 2009. Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development 136: 1951–1960 [DOI] [PubMed] [Google Scholar]
- Preisegger KH, Factor VM, Fuchsbichler A, Stumptner C, Denk H, Thorgeirsson SS 1999. Atypical ductular proliferation and its inhibition by transforming growth factor β1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Invest 79: 103–109 [PubMed] [Google Scholar]
- Presnell SC, Stolz DB, Mars WM, Jo M, Michalopoulos GK, Strom SC 1997. Modifications of the hepatocyte growth factor/c-met pathway by constitutive expression of transforming growth factor-α in rat liver epithelial cells. Mol Carcinog 18: 244–255 [PubMed] [Google Scholar]
- Purow BW, Sundaresan TK, Burdick MJ, Kefas BA, Comeau LD, Hawkinson MP, Su Q, Kotliarov Y, Lee J, Zhang W, et al. 2008. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis 29: 918–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle KJ, Dan YY, Campbell JS, Fausto N 2011. New concepts in liver regeneration. J Gastroenterol Hepatol 26: 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskams T, Katoonizadeh A, Komuta M 2010. Hepatic progenitor cells: An update. Clin Liver Dis 14: 705–718 [DOI] [PubMed] [Google Scholar]
- Rountree CB, Barsky L, Ge S, Zhu J, Senadheera S, Crooks GM 2007. A CD133-expressing murine liver oval cell population with bilineage potential. Stem Cells 25: 2419–2429 [DOI] [PubMed] [Google Scholar]
- Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C 1995. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 373: 699–702 [DOI] [PubMed] [Google Scholar]
- Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA 2010. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51: 297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks EE, Huppert KA, Brown MA, Washington MK, Huppert SS 2010. Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology 51: 1391–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Zheng YW, Kaneko S, Onodera M, Fukao K, Nakauchi H, Taniguchi H 2002. Clonal identification and characterization of self-renewing pluripotent stem cells in the developing liver. J Cell Biol 156: 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Itoh T, Tanimizu N, Miyajima A 2011. Liver stem/progenitor cells: Their characteristics and regulatory mechanisms. J Biochem 149: 231–239 [DOI] [PubMed] [Google Scholar]
- Tanimizu N, Miyajima A 2004. Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J Cell Sci 117: 3165–3174 [DOI] [PubMed] [Google Scholar]
- Tanimizu N, Miyajima A, Mostov KE 2007. Liver progenitor cells develop cholangiocyte-type epithelial polarity in three-dimensional culture. Mol Biol Cell 18: 1472–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchorz JS, Kinter J, Muller M, Tornillo L, Heim MH, Bettler B 2009. Notch2 signaling promotes biliary epithelial cell fate specification and tubulogenesis during bile duct development in mice. Hepatology 50: 871–879 [DOI] [PubMed] [Google Scholar]
- Theise ND, Saxena R, Portmann BC, Thung SN, Yee H, Chiriboga L, Kumar A, Crawford JM 1999. The canals of Hering and hepatic stem cells in humans. Hepatology 30: 1425–1433 [DOI] [PubMed] [Google Scholar]
- Thorgeirsson SS, Grisham JW 2003. Overview of recent experimental studies on liver stem cells. Semin Liver Dis 23: 303–312 [DOI] [PubMed] [Google Scholar]
- Thorgeirsson SS, Evarts RP, Bisgaard HC, Fujio K, Hu Z 1993. Hepatic stem cell compartment: Activation and lineage commitment. Proc Soc Exp Biol Med 204: 253–260 [DOI] [PubMed] [Google Scholar]
- Trusolino L, Bertotti A, Comoglio PM 2010. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 11: 834–848 [DOI] [PubMed] [Google Scholar]
- Tsuda L, Nagaraj R, Zipursky SL, Banerjee U 2002. An EGFR/Ebi/Sno pathway promotes delta expression by inactivating Su(H)/SMRTER repression during inductive notch signaling. Cell 110: 625–637 [DOI] [PubMed] [Google Scholar]
- Weglarz TC, Degen JL, Sandgren EP 2000. Hepatocyte transplantation into diseased mouse liver: Kinetics of parenchymal repopulation and identification of the proliferative capacity of tetraploid and octaploid hepatocytes. Am J Pathol 157: 1963–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel CN, Debnath J, Lin E, Beausoleil S, Roussel MF, Brugge JS 2004. Autocrine CSF-1R activation promotes Src-dependent disruption of mammary epithelial architecture. J Cell Biol 165: 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ 2009. Notch signaling controls liver development by regulating biliary differentiation. Development 136: 1727–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]