

Abstract
Kainate receptors (KARs) play fundamentally important roles in controlling synaptic function and regulating neuronal excitability. Postsynaptic KARs contribute to excitatory neurotransmission but the molecular mechanisms underlying their activity-dependent surface expression are not well understood. Strong activation of KARs in cultured hippocampal neurons leads to the downregulation of postsynaptic KARs via endocytosis and degradation. In contrast, low-level activation augments postsynaptic KAR surface expression. Here, we show that this increase in KARs is due to enhanced recycling via the recruitment of Rab11-dependent, transferrin-positive endosomes into spines. Dominant-negative Rab11 or the recycling inhibitor primaquine prevents the kainate-evoked increase in surface KARs. Moreover, we show that the increase in surface expression is mediated via a metabotropic KAR signalling pathway, which is blocked by the protein kinase C inhibitor chelerythrine, the calcium chelator BAPTA and the G-protein inhibitor pertussis toxin. Thus, we report a previously uncharacterized positive feedback system that increases postsynaptic KARs in response to low- or moderate-level agonist activation and can provide additional flexibility to synaptic regulation.
Keywords: G-protein-coupled receptors, glutamate receptor, kainate receptor, neuron, neuronal transmission, receptor recycling, synapse
Kainate receptors (KARs) are a subtype of ionotropic glutamate receptor, which are tetrameric assemblies of combinations of five possible subunits (GluK1–5) 1. They are highly expressed and widely distributed throughout the central nervous system (CNS) where they play key roles in the regulation of neurotransmission and neuronal network activity 2. Presynaptic KARs modulate neurotransmitter release, postsynaptic KARs mediate excitatory neurotransmission and extrasynaptic KARs are involved in controlling neuronal excitability 1.
In addition to direct ligand-gated ion channel activity, KARs also signal through a pertussis toxin-sensitive G-protein pathway leading to intracellular Ca2+ release and protein kinase C (PKC) activation. This metabotropic KAR pathway is involved in the presynaptic facilitation of glutamate release and in the downregulation of GABA release 3,4. Postsynaptic metabotropic KARs increase neuronal excitability by inhibiting the hyperpolarization caused by the postspike potassium current I(sAHP) 5.
Interestingly, the functional consequences of KAR activation depend on the strength of stimulation and the developmental stage of the neurons. For example, low to moderate activation of presynaptic KARs at rat hippocampal mossy fibres enhances synaptic transmission, whereas strong KAR stimulation depresses transmission 6. In immature rat hippocampal slices KAR activation enhances the motility of axonal filopodial, whereas in mature slices KAR activation inhibits motility 7.
Dendritic spines are small, often mushroom-shaped protrusions that contain specialized postsynaptic machinery and compartmentalize the signalling molecules required for efficient excitatory synaptic transmission and plasticity 8. We have shown previously that diffuse surface-expressed, non-spine-associated KARs are rapidly internalized following sustained KAR or transient NMDAR stimulation. Surprisingly, however, a less intense, transient kainate application causes an initial decrease followed by a slow and long-lasting increase in surface-expressed KARs to levels significantly greater than those prior to the agonist challenge. We interpreted those data to suggest that transient agonist activation of KARs evokes increased levels of exocytosis after the initial endocytosis 9.
We have also reported that internalized GluK2-containing KARs undergo differential activity-dependent sorting into recycling or degradative pathways depending on the endocytotic stimulus 10. Recycling endosomes are highly dynamic microtubule-associated vesiculo-tubular membrane structures that mediate recycling in dendritic spines 11,12 and their membrane is defined by the presence of the small GTPase Rab11 13,14.
In this study, we provide a mechanistic explanation for kainate-evoked increases in surface-expressed KARs at the postsynaptic membrane. We show that the increase is independent of de novo exocytosis or lateral diffusion. Rather, it is mediated by enhanced KAR recycling, which is accompanied by the accumulation of transferrin-positive vesicles in spines and blocked by dominant-negative Rab11 or primaquine. Furthermore, we show that these effects of KAR activation depend on G-protein signalling pathway. Taken together, our results reveal a novel metabotropic autoregulatory feedback pathway in which low or moderate levels of activation can increase KAR surface expression in dendritic spines by regulating receptor recycling.
Results
Transient kainate activation decreases KAR mobility and increases colocalization with PSD95
We investigated the dynamics of SEP-GluK2 in the plasma membrane of 20–25 DIV hippocampal neurons by fluorescence recovery after photobleach (FRAP) (Figure 1A). L689560 (5 mM), GYKI 53655 (40 mM) and tetrodotoxin citrate (TTX, 0.5 μM) were included to block NMDARs, AMPARs and suppress activity-dependent release of glutamate, respectively. Application of kainate (10 μM for 3 min) immediately before photobleaching the spine dramatically decreased the time course and extent of fluorescence recovery compared with the prebleached level of fluorescence (normalized to 100% in Figure 1B). The difference between the prebleach level of fluorescence and the recovered steady state after bleaching, achieved at ∼200 seconds, reflects the immobile fraction of SEP-GluK2. This is the proportion of photobleached SEP-GluK2 that is either anchored and does not diffuse away or diffuses only at very slow rates. This may be due to direct or indirect tethering of KARs to the cytoskeleton and/or their confinement in membrane microdomains 15. In control conditions, the steady state after FRAP was 96.2 ± 3.7% of the prebleached fluorescence. Following kainate application, however, the steady state after FRAP was 63.89 ± 2.4%. These results demonstrate that 10 μM kainate for 3 min increases the anchoring of surface-expressed SEP-GluK2 in spines.
Figure 1. Kainate activation retains KARs in spines.

A) Representative glow-scale images of FRAP timeline for SEP-GluK2 in a dendritic spine. The spine indicated by the circle in the second image was photobleached. To confirm that the FRAP was from surface-expressed SEP-GluK2 we briefly washed cells in pH 6.0 buffer to eclipse the surface of fluorescence. Scale bar 2 µm. B) Normalized traces of SEP-GluK2 FRAP in spines in control (black circles) and kainate (10 μM, 3 min, white circles)-treated neurons. Cells were bleached immediately after kainate treatment (t = 0 min). Mean ± SEM. Left, histograms showing SEP-GluK2 recovery in control and after kainate application. Recovery was defined as the mean value of the steady-state level (400–800 seconds). C) Fitted curves and histograms showing the diffusion coefficients in spines under control and kainate conditions. For (B) and (C) the data are the mean ± SEM, n = 25–36 spines, p < 0.001. D) Left panel, colocalization of endogenous GluK2 (red) and PSD95 (green). Right panel, colocalization of the NR1 (red) and PSD95 (green). Scale bar 2 µm, p < 0.001. Histograms show Pearson's coefficient for the colocalization ± SEM.
As detailed in the Materials and Methods, we fitted these FRAP data to a Brownian diffusion model that incorporates the possibility of an immobilized fraction 15 (Figure 1C). The SEP-GluK2 diffusion coefficient was sixfold slower after kainate application (control = 0.06 ± 0.012 µm2/second and kainate = 0.011 ± 0.004 µm2/second). Thus, there is a kainate-dependent increase in the proportion of KARs that are immobilized and/or a decrease in the rate of movement of KARs that remain mobile. Furthermore, these results indicate a marked increase in the proportion of SEP-GluK2 retained at spines after kainate application and that, compared with non-stimulated controls, KARs accumulate in spines over the time. Consistent with this, kainate application increased colocalization of endogenous GluK2/3 with the postsynaptic marker PSD95 after 3 min of kainate stimulation and 20 min of recovery (Figure 1D, left), with no change in the extent of NR1/PSD95 colocalization (Figure 1D, right; see also Figure S1A,B, Supporting Information).
Kainate activation increases GluK2 in spines but not in the dendritic shaft
Time-lapse experiments demonstrated that transient kainate stimulation increased surface SEP-GluK2 in spines, whereas levels were stable, or slightly reduced, in dendritic shaft regions. These results indicate that the kainate-invoked immobilization of SEP-GluK2 occurs specifically in spines (Figure 2A,B).
Figure 2. Kainate activation increases GluK2 in spines but not in dendrite shaft.
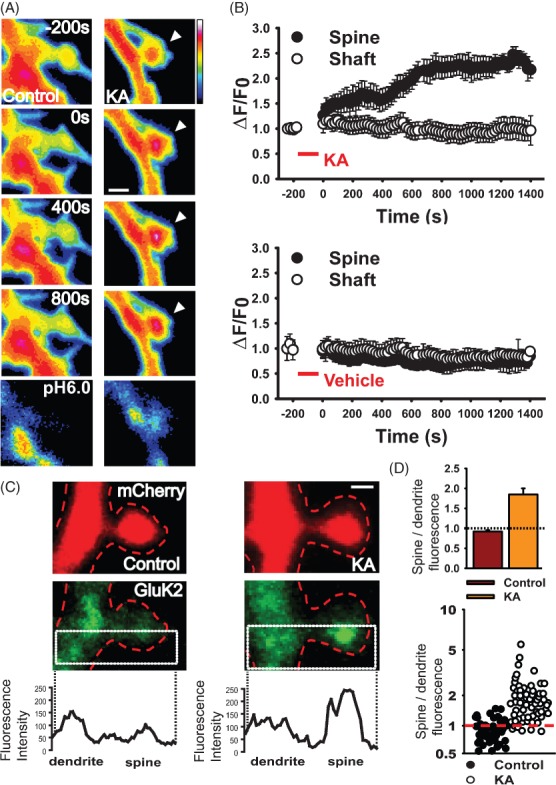
A) Representative images showing kainate-induced (10 μM, 3 min) increase of surface SEP-GluK2 in the spine head but not in the shaft. Scale bar 2 µm. B) Normalized fluorescence intensity values of spine/shaft pairs in time-lapse experiments after control (bottom panel) and kainate (upper panel). Mean ± SEM, n = 17–22 pairs spine/shaft per neuron from 8 to 10 neurons. C) Representative images showing the distribution of GluK2 (green) in the spine and the adjacent dendritic shaft in control and kainate-treated neurons expressing mCherry. Scale bar 1 µm. Line graphs show representative examples of the profiles of peak GluK2 fluorescence in the spine and adjacent shaft. For details of the analysis, see Materials and Methods and Figure S1C. D) Top, histogram of mean fluorescence ratio of spines and shafts in control and kainate-treated cells. Values were taken from the fluorescence intensity peaks of the GluK2 fluorescence profile across the spine/shaft. Values above 1 indicate higher GluK2 fluorescence in the spine compared with the shaft. p < 0.001. Bottom, scatter graph showing the distribution of all individual GluK2 spine/shaft ratios in control and kainate conditions. Values above 1 indicate higher GluK2 fluorescence in the spine compared with the shaft. n = 59–61 spine/shaft pairs.
We next analysed the distribution of native KARs in spines and adjacent dendritic shaft. We fixed cultured hippocampal neurons and used a gentle 0.05% digitonin plasma membrane permeabilization protocol to allow immunostaining with a specific C-terminal targeted anti-GluK2/3 antibody 16–18. We measured the localization of KARs in spines and adjacent shaft in two ways. First, we delineated a region of interest (ROI) with a box that longitudinally divided the spine and adjacent shaft in half and both halves were analysed as described previously 19. We then captured the profile of maximum GluK2 antibody fluorescence within these areas by line scans and normalized the peak value to mCherry fluorescence (see Materials and Methods and Figure S1C). This provided a profile of peak GluK2 distribution along the axis of spine and through the dendrite (Figure 2C,D). Second, we analysed the same raw image data by defining the area of the spine and an adjacent region of shaft, averaging the fluorescence in both regions and normalized this to the mCherry fluorescence in the same area (see Materials and Methods and Figure S1D). Both methods of analysis yielded similar results and demonstrate increased levels of KARs in spines following kainate activation.
Increased KAR surface expression in spines requires receptor recycling
The increase in GluK2/3 immunofluorescence in spines could be caused by increases in intracellular GluK2 or surface-expressed GluK2, or both. To determine if the increase was due to insertion directly into the spine membrane, we visualized newly inserted SEP-GluK2 in live neurons. We used an anti-GFP-Alexa 594 antibody, which recognizes SEP. In non-permeabilized cells the antibody has access only to the epitope in surface-expressed SEP-GluK2 [Figure 3A, see also Figure S120,21]. The rates of insertion in the spine and shaft were calculated from the increase in antibody labelling after 10 min (see Materials and Methods). Using this approach, we quantified the fraction of surface-expressed SEP-GluK2 that had not previously been present at the cell surface. This analysis excludes lateral diffusion and increases in fluorescence because of reinsertion/recycling of SEP-GluK2 labelled during the first anti-GFP-Alexa 594 incubation (Figure S1 20). The sensitivity of the experiment to detect de novo SEP-GluK2 in the plasma membrane was confirmed by preincubation of the cells with cycloheximide (CHM, Figure 3B,C, bottom panel). Importantly, kainate did not increase the exocytosis of GluK2 into the spines compared with the dendrites (Figure 3B,C), indicating that the kainate-induced increase in spines (Figure 2A–D) is not mediated by the exocytosis of naïve or newly synthesized receptors.
Figure 3. Acute increases in synaptic GluK2 do not involve insertion of de novo KARs.
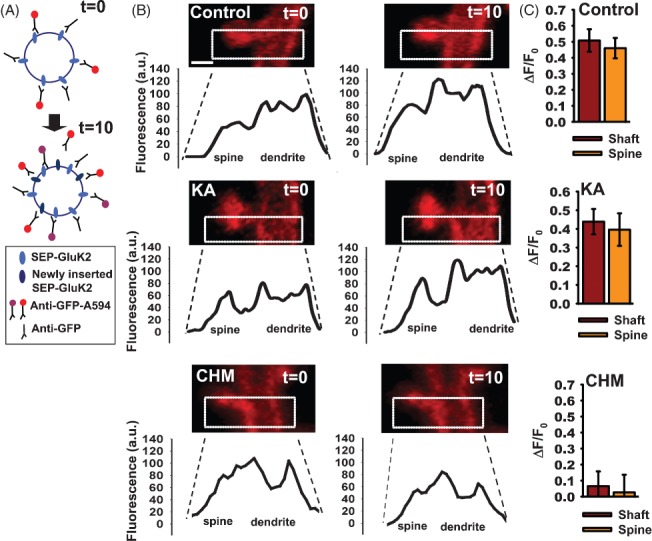
A) Schematic of protocol (see Materials and Methods for details). SEP-GluK2 inserted during the 10-min incubation interval illustrated by difference between the labelling at t = 0 and t = 10 min. Receptors previously surface expressed bound to unlabelled antibody and recycled back to the surface are not detected in this assay. Newly labelled receptors in each incubation are shown in red. Thus, receptors labelled at t = 0 are indicated as purple at t = 10, and the newly labelled receptors in red. B) Profiles of SEP-GluK2 insertion into spine and shaft. Images show SEP-GluK2 labelled with anti-GFP-Alexa 594 antibody at t = 0 and t = 10 in control cells, after kainate application or following 2-h incubation with cycloheximide. Note that in these experiments SEP is used as an extracellular epitope tag and only Alexa 594 fluorescence is shown and quantified. Scale bar 1 µm, n = 22–26 pairs spine/shaft per neuron from 10 to 12 neurons. C) Histograms showing the rate of insertion (calculated as ΔF/F0) in the spine and shaft of control cells (upper panel), kainate-treated cells (middle panel) or incubated with cycloheximide (bottom panel).
To further explore the possible contributions from newly synthesized KARs, we preincubated SEP-GluK2-expressing neurons with cycloheximide for 2 h and then challenged with kainate. As expected, kainate elicited a transient increase in SEP-GluK2 surface expression in spines, but not in the dendritic shaft (Figure 4A,B). Further, this increase in spines corresponded to the initial phase of the biphasic increase in SEP-GluK2 shown in Figure 2. After ∼10 min, however, the kainate-invoked increase in spine SEP-GluK2 fluorescence started to decline in cycloheximide-treated neurons. These results suggest that the initial increase in GluK2 does not require new protein but that GluK2 synthesis is necessary to maintain raised GluK2 levels in spines.
Figure 4. Endosomal recycling mediates increased synaptic GluK2.
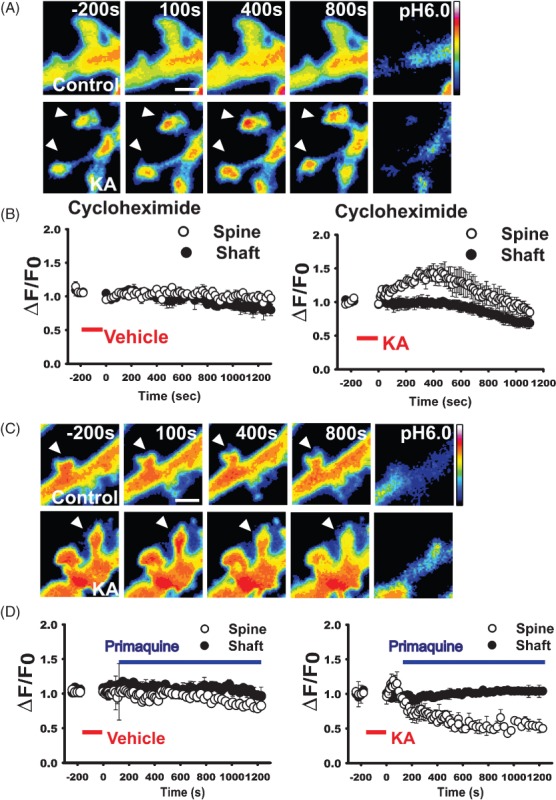
A) Transient increase in SEP-GluK2 in the spine (arrowheads at 400 seconds in lower panels), but not in the adjacent shaft, of kainate-treated neurons preincubated with cycloheximide for 2 h. Scale bar 1 µm. B) Quantification of normalized fluorescence intensity values of spine/shaft pairs showing that kainate increases synaptic GluK2 in the absence of new protein synthesis. Data = mean ± SEM, n = 20–28 pairs spine/shaft per neuron from 10 to 15 neurons. C) Representative images showing that the recycling blocker primaquine prevents kainate-evoked increases in surface SEP-GluK2. Scale bar 3 µm. D) Normalized fluorescence intensity values of spine/shaft pairs in control (left panel) and kainate condition (right panel). Mean ± SEM, n = 23–29 pairs spine/shaft from 12 to 15 neurons. Kainate application is indicated by the red bar and primaquine by the blue bar.
Next, we tested if recruitment of already surface-expressed receptors from outside the spine could account for increased levels of postsynaptic SEP-GluK2. We used a protocol combining FRAP and fluorescence loss in photobleaching (FLIP) to monitor SEP-GluK2 in neurons preincubated with cycloheximide to remove any contribution from newly synthesized receptors. This FRAP/FLIP protocol specifically measures SEP-GluK2 exocytosis because it prevents any component of FRAP from already surface-expressed SEP-GluK2 fluorescence by repetitively photobleaching a section of dendrite proximal to the ROI (Figure S2A,C, green line) 22–24. Because lateral membrane diffusion is excluded, the time taken for fluorescence recovery is dramatically slowed in FRAP-FLIP experiments (minutes) compared with FRAP experiments (seconds). Kainate stimulation can potentiate the mobility of dendrites and protrusions 7. To minimize these confounding influences, we selected a relatively large ROI and performed partial bleaching to 60–80% of the prebleach fluorescence. The kainate-induced increase in SEP-GluK2 surface expression in spines was still observed in these FRAP/FLIP experiments, indicating that increased synaptic SEP-GluK2 is unlikely because of redistribution of surface-expressed SEP-GluK2 from proximal to more distal areas (Figure S2).
Primaquine selectively inhibits membrane protein recycling but not endocytosis 2000 and we have shown previously that it blocks GluK2 recycling in neurons 21. In control cells, primaquine caused a gradual decline in SEP-GluK2 fluorescence in spines compared with the shaft (Figure 4D,E). Spine fluorescence decreased by 24.65 ± 0.25%, whereas the shaft decreased by 9.45 ± 0.29%. These values are the mean fluorescence between t = 1000 and 1200 seconds as a percentage of the mean baseline fluorescence determined prior to KA or vehicle addition (−240 to 0 seconds). This suggests that the contribution of constitutive recycling is higher in spines than in shaft. In kainate-treated neurons, there was no primaquine-induced decline in SEP-GluK2 fluorescence in the shaft (103.94 ± 0.328%). Furthermore, primaquine applied 2 min after transient kainate stimulation not only blocked but also reversed the SEP-GluK2 increase in spines (44.54 ± 0.18%, Figure 4D,E). Taken together, these results confirm that regulation of KAR recycling underpins the kainate-invoked increase in synaptic KARs.
To investigate the endosomal mechanisms involved in synaptic KAR recycling, we used wild-type and dominant-negative Rab proteins. Specifically, we investigated Rab4, Rab8 and Rab11. Rab4 mediates recycling from early endosomes back into the plasma membrane, whereas Rab11 promotes the activity-dependent translocation of endosomes from the shaft to the head of the spine 26–28. Overexpression of wild-type Rab11 dramatically enhanced, and dominant-negative Rab11 completely prevented, the kainate-induced increase in spine GluK2/3 immunoreactivity (Figure 5A,B). In contrast, overexpression of dominant-negative Rab4 or Rab8, which is associated with delivery from trans-Golgi membranes, had no effect (Figure S3). These results indicate that the agonist-evoked increase in GluK2 in the spine is dependent on Rab11-positive recycling endosomes.
Figure 5. Increased synaptic GluK2 requires Rab11-dependent recruitment of recycling endosomes.
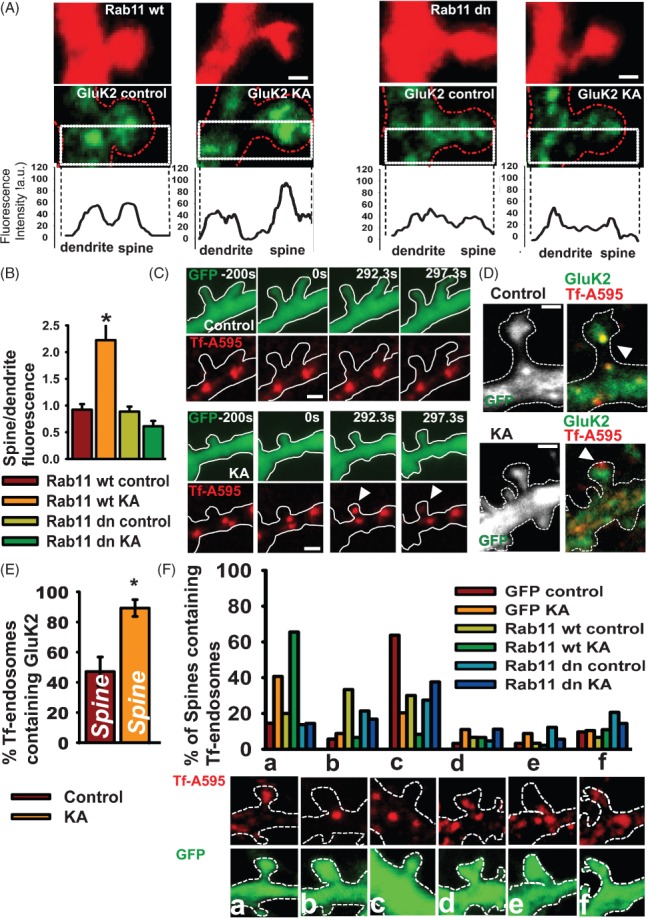
A) Representative images of GluK2 immunoreactivity (green) and the fluorescence profile along transects through the spine and shaft of neurons expressing RFP-Rab11 or RFP-Rab11-dominant-negative (Rab11dn). Scale bar 1 µm. B) Histogram of mean GluK2 fluorescence ratio of spines and shafts in RFP-Rab11 or RFP-Rab11dn-expressing neurons. p < 0.001, n = 19–32 pairs spine/shaft from 12 to 15 neurons. C) Time-lapse experiments showing the translocation of Alexa-transferrin-labelled recycling endosomes from the shaft to the head of the spine after kainate application. Cells were imaged before (t = −200 seconds) and after kainate treatment (t = 0). Arrows indicate the movement of endosomes into the spine from the shaft. Scale bar 2 µm. D) Representative images showing GluK2 colocalization with Tf-A595 endosomes in the spine following kainate application. Scale bar 1 µm. E) Quantification of the proportion of Tf-A595 endosomes containing GluK2 in the spine head, n = 12–19 cells. F) Classification of Tf-A595 endosomes according to their position in the head (a), neck (b), shaft (c), head and neck (d), neck and shaft (e) and head–neck–shaft (f) in cells overexpressing GFP, Rab11 or Rab11dn in control and 20 min after kainate application (3 min). n = 20–40 spines per neuron from 11 to 14 neurons. Scale bar 1 µm.
Synaptic stimulation elicits the redistribution of transferrin-positive recycling endosomes from the dendritic shaft to the spine head 29,30; therefore, we tested if this process could underlie the increase in spine KARs following transient kainate activation. Neurons overexpressing GFP were incubated with fluorescent transferrin (Tf-A595) and the distribution of Tf-labelled endosomes monitored in control and kainate-treated cells (Figure 5C). Kainate application triggered a rapid translocation of Tf-A595 endosomes from the shaft into the spine head of live cells. We next analysed Tf-A595 colocalization with GluK2 in fixed neurons to determine the proportion of GluK2-containing endosomes in the spine head (Figure 5D,E). In control neurons, 47.1 ± 9.6% of the Tf-endosomes in the spine head contained GluK2, whereas in kainate-stimulated neurons 89.2 ± 5.5% of the Tf-endosomes in the spine head contained GluK2.
We also determined the effects of overexpressing wild-type or dominant-negative Rab11 on Trf-A595 endosomes. In control neurons, 63% of spines had Trf-A595-labelled recycling endosomes at their base and 15% at their head (Figure 5F). In contrast, 20 min after kainate stimulation, only 20% of spines had recycling endosomes in their base and 41% at their head. Thus, recycling endosomes translocate from the base of the spine into the spine after kainate stimulation. Overexpression of dominant-negative Rab11 completely blocked this kainate-induced translocation of recycling endosomes into the spine. Taken together, these results demonstrate that transient kainate stimulation increases KAR surface expression in spines via a mechanism that involves recruitment of recycling endosomes into the spine through a Rab11-dependent pathway.
The increase in GluK2 surface expression requires PKC
KAR surface expression and recycling are regulated by PKC phosphorylation of GluK2 9,18,31. Therefore, we investigated the role of PKC in kainate-induced increases in GluK2 surface expression by surface biotinylation. As expected, kainate stimulation caused an increase in surface GluK2 with no change in the total amount of GluK2 protein. Consistent with our previous data, this redistribution of GluK2 to the plasma membrane was prevented by incubation with primaquine. In addition, we show that it is also prevented by preincubation with the PKC inhibitor chelerythrine (Figure 6A,B), which we have shown previously block PKC-dependent recycling of GluK2-containing KARs 21. Note, however, that addition of chelerythrine immediately after stimulation did not prevent the kainate-induced increase of surface GluK2, indicating that PKC activity is required to trigger the increase but not for the maintenance of surface GluK2.
Figure 6. Increased surface expression of GluK2 requires PKC activation.
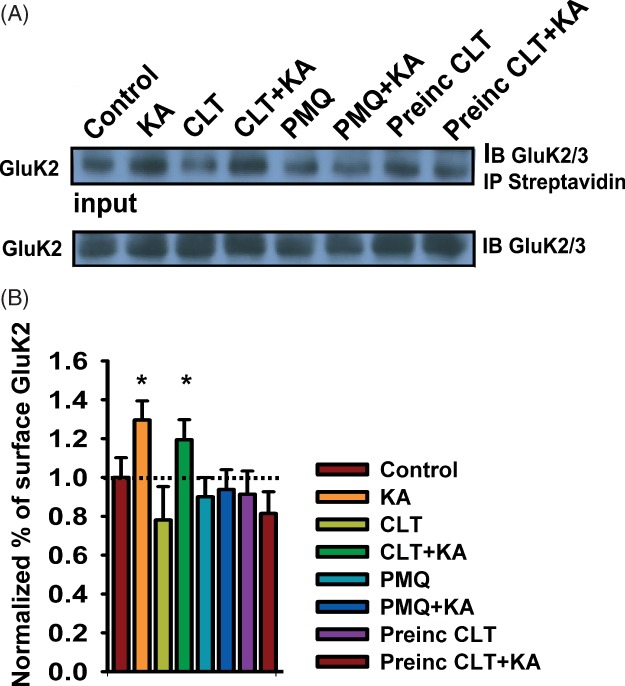
A) Representative biotinylation and immunoblots showing surface expression of GluK2 after kainate treatment. Application of chelerythrine (CLT) after kainate stimulation did prevent the kainate-induced increase of KARs in the plasma membrane (lanes 3 and 4), but preincubation with chelerythrine (lanes 7 and 8) or primaquine (PMQ, lanes 5 and 6) blocked the kainate-induced increase of KARs in the surface. Lower panel shows the KAR immunoreactivity in the lysates (input). B) Quantification of data shown, mean ± SEM, n = 4.
Recycling, endosomal trafficking and metabotropic actions of KARs
As KARs can signal via PKC-sensitive metabotropic pathways 4,32 we investigated if this regulates postsynaptic KAR recycling. Inhibition of PLC, G-protein activation and blocking intracellular Ca2+ release by incubating cells with BAPTA-AM [but not preventing Ca2+ influx by chelating extracellular calcium with ethylenediaminetetraacetic acid (EDTA)] all prevented the kainate-induced increase of surface GluK2 (Figure 7A,B). As reported previously 33, pretreatment with pertussis toxin increased the levels of both surface-expressed and total GluK2. Kainate, however, did not further increase the surface/total ratio of GluK2 in pertussis toxin-treated cells. These results indicate that G-protein activation and intracellular calcium signalling are involved in the kainate-induced increase in synaptic KARs.
Figure 7. Increased GluK2 recycling requires metabotropic actions of KARs.

A) Representative immunoblots showing GluK2 surface expression after kainate treatment. BAPTA-AM (lanes 5 and 6) but not EDTA 3,4 blocked the kainate-induced increase of KARs in the plasma membrane. Preincubation with the phospholipase C inhibitor (U73122, lanes 7 and 8) or the G-protein inhibitor pertussis toxin PTS (lanes 9 and 10) blocked the kainate-induced increase of KARs in the surface. n = 3 (note that PTS increased GluK2 in the total fraction and in the plasma membrane). n = 4. B) Histogram showing quantification of normalized data represented in A. C) Immunoblots showing that replacing extracellular Na+ with N-methyl-d-glucamine does not block the kainate-induced increase of KARs in the plasma membrane. n = 3. D) Histogram of quantified data represented in C. E) Immunolocalization of GluK2 (red) and PSD95 (green) in control neurons and neurons 20 min after kainate application (KA). Also shown are neurons treated with EDTA ± KA, BAPTA-AM ± KA, U73122 ± KA, chelerythrine (CLT) ± KA and pertussis toxin (PTS) ± KA. F) Histogram indicating Pearson's coefficient for the degree of colocalization of GluK2 and PSD95 under the conditions shown in A–B. Mean ± SEM. p < 0.001, n = 11–15 neurons. G) Percentage of spines with TfR-A495-positive endosomes in the spine head or in the dendritic shaft at the spine base. Mean ± SEM, p < 0.001 for control versus kainate and EDTA versus EDTA kainate and p > 0.1 for the rest of the treatments. n = 18–20 spines per neuron from 10 to 12 neurons.
We next replaced Na+ with an equimolar concentration of the non-permeant cation N-methyl-d-glucamine 3 to block the ionotropic function of KARs. Lowering the extracellular concentration of Na+ did not block the kainate-induced increase of surface GluK2 (Figure 7C,D), demonstrating that this process is largely independent of KAR channel activity.
Finally, we measured the degree of colocalization of GluK2 and PSD95 in randomly selected fields of view (see Materials and Methods) in cultured neurons. Consistent with the biotinylation data, blocking intracellular Ca2+, PKC, PLC or G-protein activation prevented the kainate-induced increase of GluK2 colocalization with PSD95 (Figure 7E,F). Furthermore, the kainate-dependent translocation of recycling endosomes from the shaft to the spine head was prevented by blockade of the metabotropic signalling pathway (Figure 7G).
Discussion
KARs are unusual in that the same receptor complex signals via both ionotropic and metabotropic pathways. This was first shown by electrophysiological studies using concentrations of kainate that do not activate inward currents (for reviews, see 1, 32, and 34). Here, we demonstrate that this non-canonical metabotropic pathway mediates a positive feedback system that results in increased levels of surface-expressed postsynaptic GluK2-containing KARs.
Relatively low-level kainate stimulation (in our culture 10 μM, 3 min) elicits KAR recycling, whereas longer kainate stimulation (10 μM, 30 min) causes endocytosis and degradation 9,10. We interpret our results to suggest that metabotropic KAR signalling could represent a priming system to recruit KARs to synapses under conditions of low activity to increase synaptic gain. At active synapses, where KARs are subjected to higher levels of the endogenous neurotransmitter glutamate, ionotropic signalling leads to KAR internalization. Thus, this bidirectional feedback system constitutes an elegant scaling mechanism to increase KARs at weakly active synapses decreases KARs at strongly active synapses.
PKC dynamically regulates KARs trafficking and is required to maintain KAR synaptic responses in different brain regions 35–37. Both sustained and transient agonist effects on KARs are dependent on PKC activation 10. The GluK2 C-terminal residues Ser-846 and Ser-868 are phosphorylated by PKC and this regulates KAR biosynthesis and maturation, and also endocytosis from the plasma membrane 31. Further, PKC phosphorylation of Ser-868 regulates SUMOylation of Lys-886, which leads to KAR endocytosis and degradation 18. PKC phosphorylation of Ser-868 in the absence of SUMOylation, however, enhances GluK2 recycling between endosomal compartments and the plasma membrane. Thus, SUMOylation may act as the switch between PKC-dependent enhancement or reductions in the surface expression of KARs 21.
Our data indicate that the initial increase in surface GluK2 involves redistribution of existing GluK2 but that maintenance requires new protein synthesis. The FLIP/FRAP experiments demonstrate that the increase in synaptic GluK2 is not via lateral diffusion of already surface-expressed GluK2 from the soma or the proximal segment of the dendrite. Rather, we show that the increase requires the translocation of recycling endosomes into the spine head. It should be noted, however, that our data do not completely exclude a contribution of KARs inserted into the membrane at extrasynaptic sites diffusing to adjacent synapses 30,38. Transient kainate stimulation recruits recycling endosomes into spines. Under resting conditions, ∼10% of Tf-containing endosomes are present in the spine head compared with ∼67% after kainate stimulation. Consistent with the synaptic activity-dependent translocation of Tf vesicles 39, this redistribution of recycling endosomes is Rab11 dependent. Thus, low or moderate kainate activation appears to increase the endosomal recycling in the spine via a metabotropic pathway that requires PKC, G-protein activation and Rab11. These findings also support a role for kainate stimulation in raised levels of protein transport and membrane insertion, consistent with overexpression of KAR and/or KAR activation causing pronounced effects on axonal and dendritic outgrowth, development of philopodia and increased spine density 7,9,40–42.
In summary, we demonstrate that kainate activation elicits an increase in surface expression of GluK2-containing KARs in dendritic spines. This occurs via KAR metabotropic signalling that promotes KAR recycling in spines by the Rab11-dependent recruitment of recycling endosomes into the spine head. This represents a previously unsuspected KAR autoregulatory pathway that provides additional flexibility to synaptic regulation and will likely have important physiological and pathophysiological consequences in the control of neuronal excitability and synaptic transmission. Furthermore, KARs are expressed in a range of polarized cells in addition to neurons 43 and the processes we define here likely represent generalized KAR trafficking mechanisms for other tissues outsidethe CNS.
Materials and Methods
Dissociated neuronal cultures
Rat embryonic hippocampal (for imaging) or cortical (for biochemistry) neuronal cultures were prepared as previously described 18. Briefly, hippocampi from E18 Wistar rats of either sex were dissected and the neurons dissociated by enzymatic digestion with trypsin for 15 min and mechanical dissociation. Cells were then plated at a density of 500 000 per 35-mm dish or 50 000 onto 22-mm glass coverslips coated with poly-l-lysine (Sigma). The culture medium was composed of neurobasal medium (Gibco) supplemented with horse serum (10%), B27 (Gibco) and 2 mM glutamine. On the second day, the media were changed for neurobasal medium supplemented with B27 and neurons were then fed each week with this glutamine-free medium until use (20–25 days in vitro).
Image acquisition
Images were captured using Zeiss LSM 510 META confocal system or an Ultraview Spinning Disk system (Perkin Elmer). Fluorescence was excited using 63× oil objective (numerical aperture, 1.4) by 488-nm laser light, and emission was detected through a 505-nm long-pass filter (or 505- to 550-nm bandpass filter when imaged simultaneously with Alexa 594; Zeiss LSM). Imaging of Alexa 594-labelled anti-GFP antibody fluorescence used 543-nm excitation and a long-pass 600-nm emission filter.
Time-lapse assays
Neurons were infected with Sindbis virus-expressing SEP-GluK2 and imaged the next day or transfected using Lipofectamine 2000 and used for experiments 5–6 days later. Cells were imaged in modified aCSF (in mM: 119 NaCl, 10 glucose, 25 HEPES pH 7.4, 2 NaHCO3, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4 saturated with 95% O2 and 5% CO2) at 37°C containing L689560 (5 μM), GYKI 53655 (40 μM) and TTX (0.5 μM) to block NMDARs, AMPARs and suppress activity-dependent release of glutamate, respectively. A total of 1–10 μM kainate was applied for 3 min and the neurons were washed twice and incubated in aCSF without kainate for 20 min at 37°C. Time series were collected as image stacks that were compressed into two dimensions using a maximum projection algorithm and filtered using a 3 × 3 low-pass kernel. Any images that exhibited movements in the x, y or z planes during the experiments were discarded.
The dynamics of surface-expressed SEP-GluK2 were measuring using surface fluorescence at the indicated time points and after an acid wash at the end of the experiment. The residual fluorescence after the acid wash was subtracted from each time point and then normalized to the fluorescence before kainate treatment (time = 0). For measurements of the spine/shaft fluorescence ratio in the time-lapse experiments shown in Figure 2, fluorescence measurements at each time point were performed by manually drawing around the area of interest and normalizing to its membrane area. The change in fluorescence (ΔF) was normalized to the averaged fluorescence intensity (F0) before kainate treatment. ΔF was calculated for each frame by subtraction of F0 from the measured fluorescence intensity at each time point.
FRAP and FRAP-FLIP
FRAP and FLIP protocols were performed as described previously 9,20,22. Briefly, photobleaching was achieved by on-demand activation (controlled by acousto-optical tunable filter) of maximal laser power targeted to predefined circular ROIs. Depending on the precise experiment photobleaching lasted ∼1–4 seconds. Images were acquired within 5 seconds of the end of photobleaching. After acquisition the fluorescence data were normalized to correct for any photobleaching during the sampling. This was done by analysing a region in a different cell remote from the specifically photobleached ROI. Background bleaching was never more than 10% of the total fluorescence. Each FRAP dataset was then expressed as a percentage of prebleach fluorescence (average of five images immediately before photobleaching) and then fitted to the equation that models Brownian two-dimensional diffusion in a membrane after photobleaching and incorporates the possibility of an immobile population 15. The quality of FRAP plot fitting was estimated by R2 values. From this fit, the time constant and R values were extracted for each experiment. Averaged diffusion coefficients were calculated by applying the equation D = A2/C t1/2 44, where D is the diffusion coefficient, A is the area of the membrane in the bleached region for each time point and C is a constant value for two-dimensional diffusion. More detailed protocols are published in video format 23,24.
Transferrin assay
Neurons expressing GFP were preincubated with serum/B27-free neurobasal for 1 h and then incubated with 10 µg/mL of Alexa 594-conjugated transferrin (Molecular Probes) for 10–15 min. Cells were then washed twice and then placed in modified aCSF (see above) at 37°C containing L689560 (5 μM), GYKI 53655 (40 μM) and TTX (0.5 μM) to block NMDARs, AMPARs and suppress activity-dependent release of glutamate, respectively. For live-cell experiments, neurons were recorded for 7–10 min, then 10 μM kainate was applied for 3 min, the neurons washed twice and placed in aCSF without kainate. Sequential sets of z-stacks (0.15-µm spacing between single slices) were acquired in both channels using a spinning disk confocal microscope.
For fixed cells, treatments were performed as described above. In EDTA (0.5 mM) and BAPTA-AM (1 mM) experiments, CaCl2 was replaced with NaCl in the aCSF modified media. For PLC and G-protein inhibition, neurons were preincubated with PLC inhibitor U73122 (1 μM) or pertussis toxin (PTX, 0.5 μM) for 10–15 min before the kainate challenge. Following 20-min recovery at 37°C, neurons were washed twice, fixed in 2.0% paraformaldehyde, mounted and imaged. Series of z-stacks (0.15-µm spacing between single confocal slices) were collected by sequential acquisition of the same focal plane in both channels.
GluK2 distribution and colocalization assays
Neurons were treated as described above, fixed in 2.0% paraformaldehyde, permeabilized with 0.05% digitonin and blocked with 5% of horse serum for half an hour and then stained with specific primary antibodies against GluK2 (rabbit polyclonal anti-GluK2 antibody, Millipore) or NR1 (NB100-41105, Novus Biologicals) and Cy3-conjugated secondary antibody (Jackson Laboratory). For PSD95 (clone 7E3-1B8, Millipore) staining cells were treated with methanol for 5 min at −20°C, incubated with the primary antibody and then with Cy5-conjugated secondary antibody (Jackson Laboratory). To assess the distribution of GluK2 in spines/dendrite, neurons were transfected with mCherry or GFP to fill the neuron and define the spines. After 48–72 h neurons were fixed, permeabilized and stained with specific primary antibodies against GluK2 followed by Cy2-conjugated secondary antibodies (The Jackson Laboratory).
Receptor exocytosis experiments
To measure the insertion of new receptors directly into the spine membrane, we incubated neurons expressing SEP-GluK2 with excess anti-GFP antibody (rAb; 1:200, Invitrogen) at room temperature for 5 min to cross-link SEP-GluK2 and prevent lateral diffusion between the shaft and spine. Neurons were then placed on a microscope stage at 37°C and incubated with diluted anti-GFP rAb directly coupled to Alexa 594 (anti-GFP-Alexa 594, 1:500) for 2 min to visualize the plasma membrane. The neurons were quickly washed twice and the Alexa 594 fluorescence acquired, this was designated as time t = 0. Confocal sensitivity was adjusted to produce the maximum dynamic range of detection while avoiding saturation. Cells were incubated either a further 10 min to allow exocytosis of new receptors to occur and then reprobed with anti-GFP-Alexa 594 (1:200), rapidly washed twice and imaged to determine the SEP-GluK2 inserted during the 10-min incubation interval. Newly inserted receptors were visualized with anti-GFP-Alexa 594 antibody and the rates of insertion in the spine head and in the shaft were calculated as the increase of Alexa-594 fluorescence after 10 min. Note that only the insertion of receptors not labelled during the first anti-GFP-Alexa 594 incubation, but labelled during the second incubation contribute to the increase of fluorescence after 10 min. Thus, this approach allows the quantification of SEP-GluK2 from the intracellular pool (i.e. which had not previously been present at the cell surface) or newly synthesized de novo SEP-GluK2. Any reinsertion/recycling of SEP-GluK2 labelled during the first anti-GFP-Alexa 594 incubation did not contribute to the increase of fluorescence after 10 min in this assay. We validated that the GFP antibody preincubation effectively cross-linked surface SEP-GluK2 by demonstrating that there was no FRAP in areas in the shaft and the spine of cells incubated with cycloheximide (see Figure S1A–C). The concentration of antibody needed to saturate membrane SEP-GluK2 at t = 0 was established by performing an additional incubation with anti-GFP-Alexa595 immediately after the first, without time interval. Saturation was considered when less than 5% of increase in the fluorescence was detected (see Figure S1D).
For each pair of spine/dendrites spine and dendrite fluorescence were quantified independently as the increase of fluorescence at t = 10 normalized to the fluorescence at t = 0. The ratio of insertion was also calculated as surface-expressed SEP-GluK2 at t = 10 minus that t = 0 in the spine divided by surface-expressed SEP-GluK2 in the dendrite at t = 10 minus that at t = 0. For control this ratio was 0.907 ± 0.04 and for kainate-stimulated it was 0.905 ± 0.06.
Image analysis and quantification
Analysis of endosomes localization was performed as previously described 30 and blind with respect to the treatment. Briefly, cell-filled images (GFP) and recycling endosome-labelled images (Alexa595) were thresholded at 1.20- to 1.80-fold (for cell-filled images) and at 1.30- to 2.10-fold (for recycling endosome-labelled images) and the image stacks were reassembled into a set of red-green three-dimensional (3D) stereo images (ImageJ, NIH). Endosome localization relative to spines was classified according to the presence of an endosomal structure at the head (a), in the neck (b), in the base (c), head and neck (d) or head, neck and base of the spine. When a portion of the endosome region extended into the dendritic shaft, defined by a perpendicular line drawn at the base of the spine, it was also counted as being present at the base.
Three-dimensional volumes of z-stacks (0.25-µm spacing between single confocal slices) were analysed using ImageJ (NIH). The degree of colocalization between immunolabels was assessed in whole-cell volumes and subvolumes by calculating the Pearson's correlation coefficient in the ROI using the JaCoP plugin 45. The Pearson's correlation coefficient was calculated for the original data and for a large set (500) of images randomized with a grain size determined by the point spread function of the microscope objective. If the Pearson's correlation coefficient of the original image was not greater than 95% of the randomized images, then the samples were not used. Cells displaying saturated or low, near-threshold signals were also discarded. Automated thresholding was used to avoid user bias in setting analysis parameters. Histograms presenting the mean correlation coefficient (derived from 19 to 31 cells assessed per treatment condition) are shown ± standard deviation (SD).
For GluK2 localization in endosomal structures, 3D images were segmented using ImageJ Fiji software and then the analysis of centroids and intensity centres was performed as described 45 using the object-based colocalization analysis plugin in ImageJ. Data are shown as percentage of colocalizing structures in both channels ± SD. These percentages were compared to a random distribution by shifting 180° one of the channels and analysing them in the same way.
Analysis of the distribution of fluorescence in the spine and shaft was performed as described elsewhere 19 with the specified modifications. All analyses were carried out blind with respect to the treatment condition. Briefly, ROIs were delineated with a box that divided longitudinally the spine and adjacent shaft in half and both halves were quantified. The shaft extension was defined in each pair of spine/shaft by drawing a perpendicular line at the base of the spine equal to the radius of the spine head (Figure S1C). The fluorescence profiles across the spine/head pairs were obtained, the peaks of fluorescence intensity were normalized to mCherry fluorescence in each region and the values obtained used to quantify the receptor distribution along the spine and shaft. We repeated the analysis by using averaged fluorescence in the selected areas of the spine and shaft (Figure S1D) normalized to mCherry in the same area. Data are presented as histograms of the averaged ratios of the peak values in pairs of spine/shaft ± standard error of the mean (SEM).
Biotinylation
Treatments were performed at 37°C in modified aCSF (with L689560, GYKI 53655 and TTX) plus the indicated drugs. Neurons were incubated with membrane-impermeant Sulfo-NHS-SS-Biotin (0.15 mg/mL, Pierce) in PBS for 10 min on ice as described previously 9. Cells were lysed, solubilized and centrifuged and supernatants were incubated with streptavidin beads to isolate biotinylated proteins. Tubulin was used as an intracellular control to ensure that there was no biotinylation of intracellular proteins.
Statistical analyses
Statistical significance for differences between paired combinations of images was calculated using the two-tailed Student's t-test. Statistical analysis of differences between experimental groups was performed using one-way anova followed by post hoc Tukey test calculated using SigmaStat software. Data are presented as mean ± SEM.
Acknowledgments
We are grateful to the Wellcome Trust, ERC, MRC and BBSRC for financial support. I. M. G.-G. was funded by an EMBO Fellowship. We are grateful to Philip Rubin for technical assistance and Frédéric Jaskolski for invaluable assistance with the live-cell imaging protocols and analysis. We thank Jose Esteban for the Rab8, Rab11 and Rab4 plasmids. We also thank Drs Kevin Wilkinson, Stéphane Martin, Frédéric Jaskolski and Mike Ashby for critical reading of the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure 1. Methods for quantifying GluK2 in spines and controls demonstrating surface cross-linking blocks lateral diffusion of SEP-GluK2. A) Three-dimensional reconstruction of a dendritic segment from a 17 DIV neuron. B) Two-dimensional projection of a z-stack of a dendritic segment filled with GFP and orthogonal view (right panel). PSD95 (left panel, red) was superimposed onto GFP images showing that clusters over the dendrite correspond to spines and protrusion perpendicular to the dendrite in z (white arrow). Scale bar = 1 μm. C) Illustration demonstrating the method used for line scan analysis. The spine was bisected in two halves by a longitudinal line through the spine and shaft. A region of shaft equal to half of the diameter of the spine head was analysed (yellow arrows). Fluorescence profiles were constructed from the maximum intensity values for each column of pixels (one of which is illustrated by the yellow vertical box) within the selected area. Scale bar = 0.5 μm. D) Alternative quantification methods using averaged fluorescence intensities within the spine or the dendrite (encompassed with a white line). Bar graph shows the quantification using averaged fluorescence in control and kainate-treated neurons. E and F) Representative FRAP experiments in unstimulated neurons expressing S'EP-GluK2 and preincubated with cycloheximide and GFP antibodies (1:200) and then with GFP-Alexa595 (1:500). There is little or no FRAP (measured at 600–900 seconds) in spine (E) or dendrite (F) demonstrating that antibody cross-linking of surface GluK2 blocks lateral diffusion. Scale bar = 5 μm. G) Quantification of the SEP-GluK2 recovery after FRAP in the spine and dendrite of untreated cells and cells preincubated with an anti-GFP antibody (GFP) to cross-link surface SEP-GluK2. H) Images demonstrating that saturating concentrations of unlabelled anti-GFP antibody were used to cross-link surface SEP-GluK2. In exocytosis experiments neurons were incubated with unlabelled anti-GFP antibodies (1:100–1:1000), washed twice and then immediately incubated with anti-GFP-Alexa595 antibody (1:500). Fluorescence after the first incubation with anti-GFP-Alexa595 antibody (top) was compared to the fluorescence after the second incubation (bottom). At saturating concentrations of unlabelled anti-GFP antibody, there should be no change between the first and second incubation with anti-GFP-Alexa595 antibody. As expected, a very low signal was detected as denoted by the blue colour. The unlabelled anti-GFP antibody concentration was considered saturated when the increase in the fluorescence of GFP-Alexa595 between the first and the second incubation was lower than 5%. Scale bar = 8 μm.
Figure 2. The GluK2 increase in spines is not because of surface diffusion. A) Example of FRAP-FLIP experiment where the region of interest (ROI) was bleached (FRAP, middle panel, white box) and a segment of dendrite proximal to the FRAP ROI was continuously bleached (FLIP, left panel, green box) to prevent any fluorescence recovery from lateral diffusion. B) SEP-GluK2 in spines and shaft inside the ROI of the FRAP-FLIP experiments. Upper panels = control; lower panels = kainate-treated. There is no discernible difference in recovery rates. Scale bar = 2 μm. C) Normalized fluorescence intensity values of spine/shaft pairs of FRAP-FLIP experiments after control (left panel) and kainate (right panel). In both conditions, neurons were preincubated with cycloheximide for 2 h, transferred to the microscope stage and bleached immediately after the kainate treatment (10 μM, 3 min). Data show mean ± SEM, n = 16–22 pairs spine/shaft from eight to nine neurons.
Figure 3. GluK2 increase in spines is independent of Rab4 and Rab8. A) Representative confocal images of endogenous GluK2 (green) localization in the spine and shaft of neurons expressing RFP-Rab4, RFP-Rab4dn, RFP-Rab8 or RFP-Rab8dn. Scale bar = 1 μm. B) Mean GluK2 intensities through the spine head and shaft in cells expressing RFP-Rab8 or RFP-dominant-negative Rab8 after kainate treatment. p < 0.001, n = 12–22 pairs spine/shaft from 10 to 13 neurons. C) Averaged values of GluK2 intensities through the spine head and shaft in cells expressing RFP-Rab4 or RFP-dominant-negative Rab4 after kainate treatment. p < 0.001, n = 18–28 pairs spine/shaft from 13 to 15 neurons.
References
- 1.Contractor A, Mulle C, Swanson GT. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 2011;34:154–163. doi: 10.1016/j.tins.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaskolski F, Coussen F, Mulle C. Subcellular localization and trafficking of kainate receptors. Trends Pharmacol Sci. 2005;26:20–26. doi: 10.1016/j.tips.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 4.Rozas JL, Paternain AV, Lerma J. Noncanonical signaling by ionotropic kainate receptors. Neuron. 2003;39:543–553. doi: 10.1016/s0896-6273(03)00436-7. [DOI] [PubMed] [Google Scholar]
- 5.Melyan Z, Wheal HV, Lancaster B. Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron. 2002;34:107–114. doi: 10.1016/s0896-6273(02)00624-4. [DOI] [PubMed] [Google Scholar]
- 6.Schmitz D, Mellor J, Breustedt J, Nicoll RA. Presynaptic kainate receptors impart an associative property to hippocampal mossy fiber long-term potentiation. Nat Neurosci. 2003;6:1058–1063. doi: 10.1038/nn1116. [DOI] [PubMed] [Google Scholar]
- 7.Tashiro A, Dunaevsky A, Blazeski R, Mason CA, Yuste R. Bidirectional regulation of hippocampal mossy fiber filopodial motility by kainate receptors. A two-step model of synaptogenesis. Neuron. 2003;38:773–784. doi: 10.1016/s0896-6273(03)00299-x. [DOI] [PubMed] [Google Scholar]
- 8.Rochefort NL, Konnerth A. Dendritic spines: from structure to in vivo function. EMBO Rep. 2012;13:699–708. doi: 10.1038/embor.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin S, Bouschet T, Jenkins EL, Nishimune A, Henley JM. Bidirectional regulation of kainate receptor surface expression in hippocampal neurons. J Biol Chem. 2008;283:36435–36440. doi: 10.1074/jbc.M806447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin S, Henley JM. Activity-dependent endocytic sorting of kainate receptors to recycling or degradation pathways. EMBO J. 2004;23:4749–4759. doi: 10.1038/sj.emboj.7600483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prekeris R, Foletti DL, Scheller RH. Dynamics of tubulovesicular recycling endosomes in hippocampal neurons. J Neurosci. 1999;19:10324–10337. doi: 10.1523/JNEUROSCI.19-23-10324.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooney JR, Hurlburt JL, Selig DK, Harris KM, Fiala JC. Endosomal compartments serve multiple hippocampal dendritic spines from a widespread rather than a local store of recycling membrane. J Neurosci. 2002;22:2215–2224. doi: 10.1523/JNEUROSCI.22-06-02215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 14.Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91:119–149. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feder TJ, Brust-Mascher I, Slattery JP, Baird B, Webb WW. Constrained diffusion or immobile fraction on cell surfaces: a new interpretation. Biophys J. 1996;70:2767–2773. doi: 10.1016/S0006-3495(96)79846-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mulle C, Sailer A, Perez-Otano I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- 17.Coussen F, Perrais D, Jaskolski F, Sachidhanandam S, Normand E, Bockaert J, Marin P, Mulle C. Co-assembly of two GluR6 kainate receptor splice variants within a functional protein complex. Neuron. 2005;47:555–566. doi: 10.1016/j.neuron.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 18.Konopacki FA, Jaafari N, Rocca DL, Wilkinson KA, Chamberlain S, Rubin P, Kantamneni S, Mellor JR, Henley JM. Agonist-induced PKC phosphorylation regulates GluK2 SUMOylation and kainate receptor endocytosis. Proc Natl Acad Sci U S A. 2011;108:19772–19777. doi: 10.1073/pnas.1111575108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerges NZ, Brown TC, Correia SS, Esteban JA. Analysis of Rab protein function in neurotransmitter receptor trafficking at hippocampal synapses. Methods Enzymol. 2005;403:153–166. doi: 10.1016/S0076-6879(05)03013-2. [DOI] [PubMed] [Google Scholar]
- 20.Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chamberlain SE, Gonzalez-Gonzalez IM, Wilkinson KA, Konopacki FA, Kantamneni S, Henley JM, Mellor JR. SUMOylation and phosphorylation of GluK2 regulate kainate receptor trafficking and synaptic plasticity. Nat Neurosci. 2012;15:845–852. doi: 10.1038/nn.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaskolski F, Mayo-Martin B, Jane D, Henley JM. Dynamin-dependent membrane drift recruits AMPA receptors to dendritic spines. J Biol Chem. 2009;284:12491–12503. doi: 10.1074/jbc.M808401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hildick KL, Gonzalez-Gonzalez IM, Jaskolski F, Henley JM. Lateral diffusion and exocytosis of membrane proteins in cultured neurons assessed using fluorescence recovery and fluorescence-loss photobleaching. J Vis Exp. 2012 doi: 10.3791/3747. (60). pii: 3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez-Gonzalez IM, Jaskolski F, Goldberg Y, Ashby MC, Henley JM. Measuring membrane protein dynamics in neurons using fluorescence recovery after photobleach. Methods Enzymol. 2012;504:127–146. doi: 10.1016/B978-0-12-391857-4.00006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Weert AW, Geuze HJ, Groothuis B, Stoorvogel W. Primaquine interferes with membrane recycling from endosomes to the plasma membrane through a direct interaction with endosomes which does not involve neutralisation of endosomal pH nor osmotic swelling of endosomes. Eur J Cell Biol. 2000;79:394–399. doi: 10.1078/0171-9335-00062. [DOI] [PubMed] [Google Scholar]
- 26.Kapitein LC, Schlager MA, Kuijpers M, Wulf PS, van Spronsen M, MacKintosh FC, Hoogenraad CC. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol. 2010;20:290–299. doi: 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
- 27.Ng EL, Tang BL. Rab GTPases and their roles in brain neurons and glia. Brain Res Rev. 2008;58:236–246. doi: 10.1016/j.brainresrev.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 28.Brown TC, Correia SS, Petrok CN, Esteban JA. Functional compartmentalization of endosomal trafficking for the synaptic delivery of AMPA receptors during long-term potentiation. J Neurosci. 2007;27:13311–13315. doi: 10.1523/JNEUROSCI.4258-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 30.Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nasu-Nishimura Y, Jaffe H, Isaac JT, Roche KW. Differential regulation of kainate receptor trafficking by phosphorylation of distinct sites on GluR6. J Biol Chem. 2010;285:2847–2856. doi: 10.1074/jbc.M109.081141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez-Moreno A, Sihra TS. Metabotropic actions of kainate receptors in the CNS. J Neurochem. 2007;103:2121–2135. doi: 10.1111/j.1471-4159.2007.04924.x. [DOI] [PubMed] [Google Scholar]
- 33.Rivera R, Rozas JL, Lerma J. PKC-dependent autoregulation of membrane kainate receptors. EMBO J. 2007;26:4359–4367. doi: 10.1038/sj.emboj.7601865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lerma J. Kainate receptor physiology. Curr Opin Pharmacol. 2006;6:89–97. doi: 10.1016/j.coph.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Coussen F, Mulle C, Dev KK, Coutinho V, Meyer G, Isaac JT, Collingridge GL, Henley JM. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron. 2003;37:625–638. doi: 10.1016/s0896-6273(02)01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho K, Francis JC, Hirbec H, Dev K, Brown MW, Henley JM, Bashir ZI. Regulation of kainate receptors by protein kinase C and metabotropic glutamate receptors. J Physiol. 2003;548(Pt 3):723–730. doi: 10.1113/jphysiol.2003.040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park Y, Jo J, Isaac JT, Cho K. Long-term depression of kainate receptor-mediated synaptic transmission. Neuron. 2006;49:95–106. doi: 10.1016/j.neuron.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 38.Maletic-Savatic M, Malinow R. Calcium-evoked dendritic exocytosis in cultured hippocampal neurons. Part I: trans-Golgi network-derived organelles undergo regulated exocytosis. J Neurosci. 1998;18:6803–6813. doi: 10.1523/JNEUROSCI.18-17-06803.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerges NZ, Backos DS, Rupasinghe CN, Spaller MR, Esteban JA. Dual role of the exocyst in AMPA receptor targeting and insertion into the postsynaptic membrane. EMBO J. 2006;25:1623–1634. doi: 10.1038/sj.emboj.7601065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki F, Makiura Y, Guilhem D, Sorensen JC, Onteniente B. Correlated axonal sprouting and dendritic spine formation during kainate-induced neuronal morphogenesis in the dentate gyrus of adult mice. Exp Neurol. 1997;145:203–213. doi: 10.1006/exnr.1997.6469. [DOI] [PubMed] [Google Scholar]
- 41.Hirbec H, Martin S, Henley JM. Syntenin is involved in the developmental regulation of neuronal membrane architecture. Mol Cell Neurosci. 2005;28:737–746. doi: 10.1016/j.mcn.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 42.Ibarretxe G, Perrais D, Jaskolski F, Vimeney A, Mulle C. Fast regulation of axonal growth cone motility by electrical activity. J Neurosci. 2007;27:7684–7695. doi: 10.1523/JNEUROSCI.1070-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skerry TM, Genever PG. Glutamate signalling in non-neuronal tissues. Trends Pharmacol Sci. 2001;22:174–181. doi: 10.1016/s0165-6147(00)01642-4. [DOI] [PubMed] [Google Scholar]
- 44.Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224(Pt 3):213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.