

Abstract
Seemingly unrelated mutations that drive cancer share the ability to promote nutrient uptake and metabolism conducive to cell growth.
Human cancers occur as a consequence of genetic alterations that promote uncontrolled cell proliferation outside the context of normal tissues. Mutations in oncogenes and tumor suppressor genes influence various cellular processes, such as signal transduction and gene expression, but it remains unclear how different combinations of diverse genetic alterations synergize to cause unrestricted cell growth. Two recent reports1,2 in Science provide new evidence that otherwise unrelated oncogenic events elicit the common effect of facilitating glucose uptake and metabolism (Fig. 1). The studies provide further insights into the central role of nutrient uptake and altered metabolism in early tumorigenesis and suggest that treatment strategies that exploit the unique metabolic needs of cancer cells may be less susceptible to the development of drug resistance.
Figure 1.
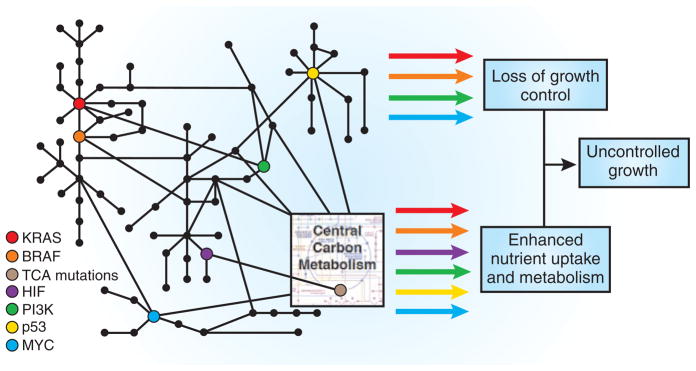
Genetic events shown to be important in human cancer influence signal transduction through a complex network that results in inappropriate cell proliferation. Arrow colors depict signal outputs that correspond to the various oncogenic mutations. These mutations lead to a dysregulation of cell growth control as well as alterations in central carbon metabolism. One consequence that is shared among the various mutations in oncogenes and tumor suppressor genes is promotion of nutrient uptake. Acquiring sufficient nutrients to allow accumulation of biomass is essential for cell proliferation and provides an attractive target for novel therapeutics. “TCA mutations” includes mutation of SDH5.
In one study, Hao et al.1 showed that the nuclear mitochondrial gene SDH5 is frequently mutated in a form of hereditary paraganglioma, a rare neuroendocrine tumor. SDH5 is not part of a signaling pathway that controls cell growth but an essential component of central carbon metabolism. The authors found that SDH5 is indispensable for the assembly of a functional succinate dehydrogenase complex, which acts both as the electron transport chain complex II (ETC II) and as an enzyme in the tricarboxylic acid cycle, where it catalyzes the oxidation of succinate to fumarate. As a consequence, the loss-of-function mutations identified in SDH5 will disrupt tricarboxylic acid cycle flux and likely cause succinate accumulation.
Interestingly, mutations in three subunits of the succinate dehydrogenase complex, all of which decrease the activity of ETC II, are associated with forms of hereditary paraganglioma1,3. The oncogenic effects of ETC II mutations are thought to occur through indirect stabilization of the hypoxia-inducible transcription factor (HIF)4. The α subunit of HIF is hydroxylated in an oxygen-dependent reaction and targeted for degradation. However, the catalytic mechanism of HIF hydroxylation requires decarboxylation of α-ketoglutarate to form succinate, such that succinate accumulation can also inhibit HIF hydroxylation through product inhibition5. Thus, either depletion of oxygen or build-up of succinate resulting from decreased ETCII activity despite normal oxygen levels stabilizes HIF and induces the expression of hundreds of genes, including the glucose transporter GLUT1 and enzymes involved in glycolysis6. All mutations known to cause hereditary paraganglioma therefore lead to altered gene expression that drives glucose uptake and metabolism and promotes cancer cell proliferation. Similarly, mutations in other components of the tricarboxylic acid cycle increase HIF expression and are believed to be oncogenic in other cancer types7.
In a separate study, Yun et al.2 studied pairs of constructed isogenic cell lines that differ only in the mutational status of KRAS and BRAF. The KRAS and BRAF genes encode signal transduction proteins and frequently acquire mutations in human cancer that render their products constitutively active8. However, mutations in these oncogenes are mutually exclusive, suggesting that the advantage they provide for tumor growth is redundant. The authors showed that GLUT1 was one of only three genes consistently upregulated in cell lines harboring one of these mutations. They then focused on a KRAS-mutated cell line and subjected a wild-type version of these cells to glucose deprivation. Although most of the cells perished in this environment, a significant fraction of the ones that survived spontaneously acquired new KRAS mutations, and all of them increased GLUT1 expression.
Martin Raff9 proposed a number of years ago that multicellular organisms control tissue size by requiring cells to compete for limited amounts of survival signals provided by growth factors and/or contact with the extra-cellular matrix. This is in contrast to unicellular organisms, which regulate cell proliferation by nutrient availability. It has been suggested that the same survival signals that regulate mammalian cell number also control nutrient uptake10. Thus, for cells in a tissue to proliferate auto nomously and survive outside of their normal environment, they must acquire mutations that further increase nutrient uptake. This idea is highlighted by a recent study demonstrating a requirement of altered metabolic activity for survival when cells are detached from the extracellular matrix 11.
Accumulating evidence therefore supports the theory that seemingly disparate genetic alterations known to promote cancer lead to a single converging metabolic phenotype characterized by enhanced nutrient uptake. The studies by Hao et al.1 and Yun et al.2 raise the intriguing possibility that specific environmental perturbations facilitate adaptation to hyperactive metabolic states by selecting for mutations that enhance nutrient uptake and reorganize metabolism. In Hao et al.1, a state of pseudo-hypoxia caused by defective mitochondrial respiration resulted in the activation of oncogenic transcription factors that control glucose metabolism. In Yun et al.2, glucose deprivation drove the emergence of cells harboring a constitutively active KRAS gene that allowed these cells to compensate by increasing GLUT1 expression. In several cancers, various major oncogenic events, such as PI3K pathway activation and loss of the tumor suppressor p53, have been associated with increased glucose uptake and metabolism12,13. In addition, uptake of the nutrient glutamine can be driven by the MYC oncogene14. This suggests that reorganization of metabolic pathways by genetic alterations in cancer cells supports cell-autonomous metabolism with biosynthetic capabilities necessary for the rapid production of a new cell.
Attempts at therapies that target one or two signaling proteins have met with limited success in the clinic, in part because cancer cells use redundant pathways to circumvent target inhibition. The finding that cells in which mutant KRAS has reverted to wild type are selected to acquire new KRAS mutations implies that KRAS mutation may impose dependencies that can be exploited for therapy, although a majority of the clones acquired the ability to proliferate by unidentified genetic means. However, in all cases upregulation of GLUT1 was observed. This suggests that many if not all mutations that permit continued cell proliferation enhance glucose metabolism. Targeting this common metabolic dependency may prove less susceptible to development of drug resistance.
Yun et al.2 found that targeting metabolic pathways could be successful in treating KRAS mutant tumors. The alkylating agent 3-bromo-pyruvate (3BrPA) selectively inhibited cancer cell proliferation both in culture and in vivo, (using tumor xenografts), presumably through effects on glucose metabolism, and might be effective against any tumor with a genetic alteration that increases glucose uptake and metabolism. 3BrPA has been proposed to act through inhibition of hexokinase, although some reports suggest that it is an effective antiproliferative agent in concentrations at which no hexokinase inhibition is observed15. Understanding the exact mechanism by which 3BrPA disrupts cellular metabolism may provide valuable insight into metabolic targeting as an antitumor therapy.
Our understanding of cancer cell metabolism is complicated by the enormous complexity of the interaction between metabolic pathways and the genetic aberrations that alter these pathways. Future progress will require new technologies and conceptual frameworks, such as high-throughput metabolomics and mathematical models that can parse the effects of many simultaneous interactions. Nevertheless, the insights of Hao et al.1 and Yun et al.2 provide validation that investing this effort could result in potent new treatments for people with cancer.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturebiotechnology/.
Contributor Information
Jason W Locasale, Department of Systems Biology, Harvard Medical School, and Beth Israel Deaconess Medical Center, Department of Medicine, Division of Signal Transduction, Boston, Massachusetts, USA.
Lewis C Cantley, Email: Lewis_Cantley@hms.harvard.edu, Department of Systems Biology, Harvard Medical School, and Beth Israel Deaconess Medical Center, Department of Medicine, Division of Signal Transduction, Boston, Massachusetts, USA.
Matthew G Vander Heiden, Email: Matthew_VanderHeiden@dfci.harvard.edu, Department of Systems Biology, Harvard Medical School, and Beth Israel Deaconess Medical Center, Department of Medicine, Division of Signal Transduction, Boston, Massachusetts, USA. Dana Farber Cancer Institute, Department of Medical Oncology, Boston, Massachusetts, USA.
References
- 1.Hao HX, et al. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yun J, et al. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baysal BE, et al. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 4.Selak MA, et al. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 5.Ivan M, et al. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 6.Denko NC. Nat Rev Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 7.Thompson CB. N Engl J Med. 2009;360:813–815. doi: 10.1056/NEJMe0810213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogelstein B, Kinzler KW. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 9.Raff MC. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 10.Vander Heiden MG, et al. Mol Cell Biol. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schafer ZT, et al. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bensaad K, Vousden KH. Trends Cell Biol. 2007;17:286–291. doi: 10.1016/j.tcb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Engelman JA, Luo J, Cantley LC. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 14.Gao P, et al. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira da Silva AP, et al. Biochem J. 2009;417:717–726. doi: 10.1042/BJ20080805. [DOI] [PubMed] [Google Scholar]