

Abstract
RIG-I is a cytosolic helicase that senses 5’-ppp-RNA contained in negative strand RNA viruses and triggers innate antiviral immune responses. Calorimetric binding studies establish that the RIG-I C-terminal regulatory domain (CTD) binds to blunt-end double-stranded 5’-ppp-RNA a factor of 17 more tightly than to its single-stranded counterpart. Here we report on the crystal structure of RIG-I CTD domain bound to both blunt-ends of a self-complementary 5’-ppp-dsRNA 12-mer, with interactions involving 5’-pp clearly visible in the complex. The structure, supported by mutation studies, defines how a lysine-rich basic cleft within the RIG-I CTD domain sequesters the observable 5’-pp of the bound RNA, with a stacked Phe capping the terminal base pair. Key intermolecular interactions observed in the crystalline state are retained in the complex of 5’-ppp-dsRNA 24-mer and full-length RIG-I under in vivo conditions, as evaluated from the impact of binding pocket RIG-I mutations and 2’-OCH3 RNA modifications on the interferon response.
Host defense against infection by invading viral and bacterial pathogens is critically dependent on the initiation and maintenance of the finely tuned primary innate immune response1–4, a rapid protective response that is coupled to subsequent adaptive immunity, thereby providing long-term protection based on immunological memory. The innate immune system is equipped with a number of pattern recognition receptors that detect characteristic microbial components, including foreign nucleic acids and their characteristic structures, ranging from long double-stranded RNA (dsRNA) and 5’-triphosphorylated (ppp) RNA, to unmethylated CpG DNA5. Such nucleic acid molecules are either less frequent or absent in the host organism, or they are localized in other cellular compartments than self nucleic acids. In contrast to acquired antibody-mediated immunity, nucleic acids of viral origin are detected by host cell constitutively expressed pathogen-associated molecular pattern (PAMP) recognition receptors (PRRs). These receptors are localized to either endosomal compartments commonly occupied by pathogens, or to the cytosol, resulting in the stimulation of innate antiviral immune responses including the induction of interferon regulatory factors (IRFs) and NFκB, that in turn regulate the production of type I interferons (IFNs) and proinflammatory cytokines and chemokines, respectively, as well as increased expression of costimulatory molecules essential for the activation of T cells. Immunorecognition of viral nucleic acids leads to the suppression of viral replication in the infected cell, and cytokine expression attracts and activates innate effector cells, such as NK (natural killer) cells, that eliminate virus-infected cells and guide the development of a subsequent adaptive immune response. Consequently, there is a great interest in developing molecularly defined immunostimulatory nucleic acids for the therapy of viral infection and cancer6,7.
Nucleic acid-sensing PRRs come in several flavors, including ‘toll-like receptors (TLRs), ‘retinoic-acid-inducible-gene-I (RIG-I)-like receptors (RLRs) and nucleotide oligomerization domain (NOD)-like receptors (NLRs). The RLR class is localized to the cytoplasm and specialized in RNA-sensing. Its members include RIG-I, ‘melanoma differentiation-associated gene 5’ (MDA5), and ‘laboratory of genetics and physiology-2’ (LGP2) receptors8–10.
RIG-I was initially identified as the RLR responsible for activation of the IFN-β promoter in response to sensing of viral RNAs11 from hepatitis C, Sendai, vesicular stomatitis, rabies, influenza and encephalitis viruses. RIG-I responds to RNAs containing a 5’-triphosphate by type I IFN production, apoptosis induction and inflammasome activation12,13. The RIG-I protein contains two N-terminal caspase recruitment domains (CARDs), a central DExH box helicase/adenosine triphosphatase (ATPase), and a C-terminal regulatory domain (CTD). A comparative sequence alignment of the CTD’s of RIG-I, MDA5 and LGP2 are listed in Fig. 1a. It has been proposed that the CARD domains of RIG-I are auto-inhibited by other domains of the protein, with RNA-ligand-binding inducing a conformational change, resulting in RIG-I dimerization6.
Figure 1.

Sequence alignment of RIG-I family of pattern recognition receptors and ITC studies of the binding of RIG-I CTD domain to RNA as a function of 5’-end phosphorylation and duplex/strand status. (a) Sequence alignment of C-terminal regulatory domains (CTDs) of RIG-I, MDA5 and LGP2. The secondary structure of RIG-I is shown over the alignments. Key amino acids contacting the α and β phosphates of the 5’-pp-dsRNA are shown by filled blue circles, while those proposed to contact the modeled γ-phosphate are shown by open blue circles. ITC binding curves for RIG-I CTD domain binding to (b) blunt-end 5’-ppp-dsRNA 12-mer and (c) single-stranded 5’-ppp-ssRNA in 100 mM NaCl and 2 mM MgCl2 buffer. ITC binding curves for RIG-I CTD domain binding to (d) blunt-end 5’-ppp-dsRNA 12-mer and (e) blunt-end 5’-OH dsRNA 12-mer in 250 mM NaCl and 2 mM MgCl2 buffer.
There is a considerable literature on RNA targets of RIG-I, with claims ranging from ssRNA to dsRNA, with and without phosphate-end modifications and overhang residues. Two initial studies independently established that RIG-I specifically targets 5’-triphosphate-containing viral RNAs14,15, thereby distinguishing viral from host transcripts, since the 5’-end of the latter are either capped by 7-meG or removed during the maturation process. More recently, two groups showed that RIG-I required base-paired structures in conjunction with attached 5’-triphosphate to trigger antiviral signaling16,17. In addition, it has been shown that besides viral RNA, RNA helicase RIG-I is also activated by RNA-polymerase III-mediated conversion of microbial DNA into 5’-triphosphorylated dsRNA18,19.
Single-molecule protein-induced fluorescent enhancement studies have established that RIG-I is a 5’-triphosphate RNA-dependent ATP-powered translocase along the entire length of dsRNA20. In addition, the 5’ to 3’ directed movement of RIG-I along the duplex axis appears to occur without unwinding the RNA duplex. These studies also demonstrated that the CARD regulatory domains suppressed translocation in the absence of 5’-triphosphorylated RNA.
Previous studies determined the crystal structure of the C-terminal domain of RIG-I in the free state, and based on NMR titration studies identified a basic RNA-binding surface within the CTD that appeared to be critical for 5’-ppp-RNA recognition21,22. Of note, these studies were performed by using 5′-triphosphate RNA generated by in vitro transcription, known to result in RNA molecules that are not well-defined and that contain unexpected contamination of single-strand RNA with duplex RNA. Recently, we used a chemical synthetic approach to generate defined 5’-ppp-RNA, and found that RIG-I required base-paired structures in conjunction with attached 5’-triphosphate to trigger antiviral signaling17. These results suggested that the minimal complex should contain the CTD domain of RIG-I and a blunt-end RNA duplex containing 5’-ppp termini. Here we provide new biophysical and crystallographic studies, complemented by functional studies further characterizing this interaction.
RESULTS
Binding affinity of 5’-ppp-dsRNA versus 5’-ppp-ssRNA
We used isothermal titration calorimetry (ITC) to measure and compare binding affinities of RIG-I CTD for 5’-ppp-modified dsRNA and ssRNA. We measured a binding affinity of 29.2 nM for complex formation between RIG-I CTD and self-complementary blunt-end 5’-ppp-dsRNA 12-mer in 100 mM NaCl solution (Fig. 1b; the complete ITC data are plotted in Supplementary Fig. 1). The stoichiometry of binding was approx. two CTD’s bound per duplex, reflecting CTD molecules bound to both 5’-ppp ends of the self-complementary RNA duplex. The binding affinity decreased by a factor of 16.7 to 488 nM for complex formation between RIG-I CTD and single-stranded 5’-ppp-ssRNA (Fig. 1c), consistent with earlier results that established a requirement for a blunt-end 5’-ppp-dsRNA for maximal binding affinity16,17.
Binding affinity of 5’-ppp-dsRNA versus 5’-OH-dsRNA
We used isothermal titration calorimetry (ITC) to measure binding affinities of RIG-I CTD for dsRNA as a function of 5’-triphosphorylation. The binding affinity of RIG-I CTD for self-complementary blunt-end 5 ’-ppp-dsRNA 12-mer was dependent on NaCl concentration, with the binding affinity decreasing by a factor of 7.3 from 29.2 nM in 100 mM NaCl solution (Fig. 1b to 215 nM in 250 mM NaCl solution (Fig. 1d).
The 5’-phosphorylation status of the dsRNA is also important since the binding affinity decreased by a factor of 5.1 from 215 nM for blunt-end 5’-ppp-dsRNA 12-mer (Fig. 1d) to 1.10 μM for its 5’-OH-dsRNA counterpart (Fig. 1e), both in 250 mM NaCl solution.
Crystal structure of 5’-ppp-dsRNA bound to RIG-I CTD
We previously demonstrated that structural characterization of protein-RNA interactions involving either 5’- or 3’-end RNA recognition by protein modules in a duplex context was facilitated by use of self-complementary duplexes, whereby the protein modules bound to both ends of the duplex. Using this approach, we structurally characterized PAZ (Piwi/Argonaute/Zwille) domain recognition of 2-nt overhangs at 3’-ends of dsRNA23, Piwi (P-element induced wimpy testis) protein recognition of 5’-monophosphate ends of dsRNA24, and La protein recognition of UUU-3’ RNA ends25. More recently, the same approach has been adopted by other groups to structurally characterize recognition of 5’-OH ends of dsRNA by the CTD domains of LPG26 and Ebola viral protein VP35 protein27.
So far, there has been no structural information on protein-RNA complexes of 5’-ppp-RNA bound to either RIG-I or MDA5. By contrast, the structure of TLR3 had been solved with bound dsRNA28. We initiated crystallization of the RIG-I CTD with self-complementary 5’-ppp-dsRNA of 10-mer, 12-mer and 14-mer lengths, with the goal of generating complexes involving RIG-I CTD recognition of 5’-triphosphate termini of blunt-end dsRNA (details of protein expression and chemical synthesis of 5’-ppp-RNA are described in Online Methods). We obtained crystals of RIG-I CTD (positions 792–925, Fig. 1a) bound to both ends of a chemically synthesized self-complementary 5’-pppGACGCUAGCGUC 12-mer dsRNA and solved the structure to 2.55 Å resolution (details of crystallization and structure determination are described in Online Methods, while x-ray statistics are listed in Table 1).
Table 1.
Data collection and refinement statistics
| RIG-I CTD bound to 5’-ppp-dsRNA | |
| Data Collection | |
| Space group | P65 |
| Cell dimensions | |
| a, b, c (Å) | 83.45, 83.45, 110.34 |
| α, β, γ(º) | 90.0, 90.0, 120.0 |
| Resolution (Å) | 50–2.55 (2.59–2.55)* |
| Rsym | 8.7 (40.1) |
| I/σI | 23.9 (4.0) |
| Completeness (%) | 99.9 (100.0) |
| Redundancy | 9.6 (8.2) |
| Refinement | |
| Resolution (Å) | 30–2.55 |
| No. reflections | 14,219 |
| Rwork/Rfree | 19.5/23.0 |
| No. atoms | |
| Protein | 1,978 |
| Ligand/ion | 524 |
| Water | 103 |
| B-factors | |
| Protein | 39.3 |
| Ligand/ion | 71.8 |
| Water | 35.5 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (º) | 1.3 |
Highest resolution shell is shown in parenthesis.
The crystal structure of the complex is shown in Fig. 2a. The self-complementary 5’-ppp-dsRNA 12-mer forms a fully-paired duplex (omit electron density map shown in Supplementary Fig. 2), with RIG-I CTD domains (in salmon) bound at either end of the duplex (in green) in the complex (intermolecular contacts shown schematically in Fig. 2b). To our surprise, we could only trace two phosphates at the 5’-end of the RNA (Fig. 2c), indicative of hydrolytic cleavage29 and loss of the terminal phosphate of bound 5’-ppp-dsRNA during generation and/or crystallization of the complex.
Figure 2.

Details of the crystal structure of the RIG-I CTD bound to blunt-end 5’-pp-dsRNA 12-mer. (a) Crystal structure of the RIG-I CTD bound to 5’-pp-dsRNA 12-mer (in green). CTD (in salmon) are bound to both ends of the 5’-pp-dsRNA 12-mer. (b) Schematic representation highlighting intermolecular hydrogen-bonding and stacking contacts in the complex. (c) Details of the intermolecular contacts in the structure of the complex. The CTD is in a ribbon representation (salmon color) and the 5’-pp-dsRNA 12-mer is in a stick representation (green color with backbone phosphorus atoms in yellow). Intermolecular hydrogen bonds between the α and β 5’-phosphates to amino acid side chains lining the CTD recognition pocket are shown as red dotted lines. The shaded aromatic ring of F853 is stacked on both bases of the terminal base pair. (d) A view similar to panel c except for an electrostatic representation of the 5’-phosphorylated RNA-binding surface of the CTD. The RNA is in a ribbon representation and the CTD-binding pocket in an electrostatic surface representation. Blue and red patches define basic and acidic regions, respectively.
Intermolecular contacts in the complex
Intermolecular contacts towards one end of the complex are highlighted in Fig. 2c, with the observable 5’-pp end of the bound dsRNA sequestered within a basic lysine-rich patch on the RIG-I CTD in the complex (Fig. 2d). The side chain of F853 stacks over both bases of the terminal base pair of the dsRNA in the complex, thereby readily explaining the requirement for a blunt-end duplex (Fig. 2c). Specifically, the aromatic ring of F853 partially stacks on 5’-G1, while the Cα-Cβ bond of F853 stacks on C12 (Supplementary Fig. 3). Individual phosphates of the 5’-triphosphated end are labeled α, β and γ, with the α phosphate closest to the terminal base. The non-bridging phosphate oxygens of the α phosphate are hydrogen-bonded to the side chains of K861 and K888, while the phosphate oxygens of the β phosphate are hydrogen bonded to the side chains of K861, H847 and K858 (Fig. 2c). In addition, the non-bridging phosphate oxygen between nucleosides 2 and 3 hydrogen bonds to the side chain K907 in the complex. Complex formation results in a buried surface area of 328 Å2 at either end of the complex.
We modeled the missing γ phosphate within the basic 5’-phosphorylated end-binding pocket in our structure of RIG-I bound to blunt-end 5’-pp-dsRNA (Fig 3a), so as to generate a complex with bound 5’-ppp-dsRNA. In this model of the complex, the γ phosphate oxygens could form potential hydrogen bonds with K849 and K851, though this should be considered to be speculative at this time.
Figure 3.

Role of 5’-phosphorylated ends in the crystal structure of the RIG-I CTD bound to blunt-end 5’-pp-dsRNA 12-mer and comparison of the electrostatics of the binding surfaces of RIG-I, MDA5 and LGP2 CTD’s for 5’-ppp-dsRNA. (a) Modeling of the γ 5’-phosphate onto the bound 5’-pp-dsRNA 12-mer in the structure of the complex. The RNA is in a ribbon representation and the CTD-binding pocket in is an electrostatic surface representation. (b) Comparison in stereo of the superposed crystal structures of the RIG-I CTD bound to blunt-end 5’-pp-dsRNA 12-mer (salmon color; only two terminal base pairs are shown in the interest of clarity) with RIG-I CTD in the free state (blue color; PDB ID: 2QFB). Conformational changes are seen for the loop spanning positions 847–856, and also in the alignment of F853, between the superposed structures and these are highlighted in darker colors. (c) The electrostatics of the binding surface of RIG-I CTD recognized by blunt-end 5’-ppp-dsRNA 12-mer. This region is shown by dashed red circle. (d) The electrostatics of the corresponding binding surface of MDA5 CTD. (e) The electrostatics of the corresponding binding surface of LGP2 CTD.
Conformational change in RIG-I CTD on complex formation
Superposition of the previously reported crystal structure of the free RIG-I CTD (in blue) with the current structure of the complex with bound blunt-end 5’-pp-dsRNA 12-mer (in salmon) is shown in stereo in Fig. 3b. The CTD domains superposed well (r.m.s.d. = 0.49 Å), except for differences in the loop spanning positions 847–856 (in bold colors) and the orientation of F853, whose aromatic ring and Cα-Cβ bond are involved in stacking interactions with the terminal base pair in the complex. It is important to stress that the conformation of the CTD loop in the structure of the present complex with bound 5’-pp-dsRNA could differ from that of the as yet undetermined complex with bound 5’-ppp-dsRNA.
5’-ppp-dsRNA binding pockets of RIG-I, MDA5 and LGP2
A comparison of the sequences of the CTD domains (Fig. 1a), show that RIG-I residues Lys888 is conserved between RIG-I, MDA5 and LGP2, that RIG-I residue Lys851 is conserved between RIG-I and MDA5, while there is no conservation between RIG-I residues His847, Lys849, Lys858 and Lys861 and their counterparts in MDA5 and LGP2 (Fig. 1a). As a result, the highly basic nature of the 5’-ppp binding pocket (dashed red-circled region) within the CTD of RIG-I (Fig. 3c), becomes less basic in the corresponding surface segment of MDA5 (Fig. 3d), and becomes hydrophobic for LGP2 (Fig. 3e). In addition, while cavities exist on this surface segment (dashed red-circled region) for RIG-I (Fig. 3c) and, to some extent, LGP2 (Fig. 3e), there is an outwards protrusion for MDA5 (Fig. 3d). These results suggest that the complex of RIG-I CTD bound to a blunt-end 5’-phosphorylated RNA duplex reported here is distinct from corresponding complexes with MDA5 and LGP2. There are adjacent basic patches in the CTD domains of MDA5 and LGP2 (lower left hand corner of Figs. 3d,e) that could provide alternate recognition sites for 5’-ppp-RNAs in these complexes, but the involvement of these sites in the recognition of MDA5 or LGP2 CTDs is unclear at present.
In vitro analysis of RIG-I CTD mutants on binding affinity
To assess the importance of amino acids lining the basic 5’-ppp recognition patch, single, double and triple RIG-I CTD domain mutants were recombinantly expressed. We monitored the binding of blunt-end 5’-ppp-dsRNA to wild-type and mutant RIG-I CTD domains by isothermal titration calorimetry (ITC). The 5’-ppp-dsRNA bound to wild-type RIG-I CTD in 100 mM NaCl, 2 mM MgCl2, buffer with a binding affinity of 29.2 nM, with approx. two CTD domains bound per duplex (Fig. 4a). This affinity dropped to 162 nM for the single H847A CTD mutant (Fig. 4b), to 649 nM for the double H847A/K858A mutant (Fig. 4c) and to >15 μM for the triple H847A/K858A/K851A mutant (Fig. 4d).
Figure 4.

ITC and electrophoretic mobility shift studies of the binding of blunt-end 5’-ppp-dsRNA 12-mer to wild-type and mutants of RIG-I CTD. ITC binding curves for blunt-end 5’-ppp-dsRNA 12-mer binding to (a) wild-type, (b) H847A single mutant, (c) H847A/K858A double mutant and (d) H847A/K858A/K851A triple mutant of RIG-I CTD domain in 100 mM NaCl and 2 mM MgCl2 buffer. (e) Palindromic radiolabeled 5’-ppp-dsRNA 12-mer was incubated at increasing concentrations of recombinant protein under the same salt and buffer conditions as the ITC experiment. The complexes were resolved on a native polyacrylamide gel. Increasing numbers of mutation in the triphosphate-binding pocket of the protein weaken RNA binding and reduce the distinct gel shift seen for the wild-type protein to a smear. To ensure that the palindromic 5’-ppp-RNA was exclusively present as dsRNA rather than a hairpin, we pre-annealed the RNA at 5 μM strand concentration and only diluted it to 20 nM prior to the incubation with protein.
We also assessed RIG-I CTD ppp-dsRNA-binding by electrophoretic mobility shift analysis. A triphosphorylated 11-mer was labeled at the 3'-end using RNA ligase and radioactive pCp, yielding the 12-mer palindromic sequence used in crystallographic or calorimetric studies, although with an additional 3'-phosphate. Consistent with the binding constants determined by calorimetry, 100 nM protein completely shifted the 20 nM input ppp-dsRNA, and H847A mutation reduced binding by a factor of approx. 10 (Fig. 4e). Double or triple mutations weakened the interaction to a point where the complex was no longer stable under electrophoresis conditions, even in the presence of 10 μM protein.
In vivo analysis of RIG-I mutants lining the binding pocket
To confirm the biological significance of the observed intermolecular contacts involving the 5’-phosphorylated end of the RNA in the crystal structure of the 5’-pp-dsRNA bound to RIG-I CTD, all lysines (K), histidine (H) or phenylalanine (F) that line the observed binding pocket were mutated to alanine (A) in full-length RIG-I (see Supplementary Table 1 for a complete list of RIG-I mutants). Wild-type and mutants of full-length human RIG-I were overexpressed in human HEK293 cells. Cells were stimulated with synthetic 5’-ppp-ssRNA, 5’-ppp-dsRNA or 5’-OH-dsRNA. RIG-I activation in cells was analyzed based on the formation of the interferon-inducible chemokine IP-10 as measured in the supernatants. Mutation of K858 (β phosphate contact), K861 (α/β phosphate contact) and K888 (α phosphate contact) strongly or completely abrogated activation of RIG-I by 5’-ppp-dsRNA (Fig. 5a; mutated positions shown in Fig. 5b). By contrast, the H847 (β phosphate contact) had little impact on the RIG-I activity in cells. In the same setting the 5’-OH-dsRNA was inactive. Mutation of F853, which is important for terminal base pair stacking, strongly reduced activation, while mutation of K907 that hydrogen bonds internucleotide phosphate between nucleosides 2 and 3, completely abolished 5’-ppp-dsRNA-induced IP-10 (Fig. 5a; mutated positions shown in Fig. 5b). Although the K849 and K851 single mutants showed little impact on activity, their functional contribution is confirmed by dose response analysis of the K849/K851 double mutant (Fig. 5a and Supplementary Fig. 4).
Figure 5.

In vivo analysis of the impact of point mutants on the biological activity of RIG-I. (a) Human RIG-I (wt) or RIG-I mutants as indicated were overexpressed in human HEK293 cells and stimulated with 5 nM single-stranded (5’-ppp-ssRNA, 5’-ppp-GFP2) or double-stranded (5’-ppp-dsRNA, 5’-ppp-GFP2+AS GFP2) synthetic triphosphorylated RNA or non-modified double-stranded RNA (5’-OH-dsRNA, 5’-OH-GFP2+As GFP2). 24 hours after stimulation, IP-10 was analyzed in the supernatants of cells. Data from three independent experiments are depicted as mean values ± SEM. (b) Positions of amino acids that were mutated in the crystal structure of the 5’-pp-dsRNA 12-mer bound to RIG-I CTD.
In vivo analysis of RNA 2′-OCH3 substituent effects
In efforts to evaluate the in vivo relevance of the binding orientation observed in the crystal structure of RIG-I CTD bound to 5’-ppRNA duplex, a series of 5’-ppp-RNA analogs were synthesized containing 2′-OCH3 residues at positions 1 to 6 within a blunt-end 5’-ppp-dsRNA 24-mer context and the interferon stimulating activity of these duplexes was analyzed in human peripheral blood mononuclear cells (PBMC). To avoid recognition of short RNA by immune receptors other than RIG-I, such as TLR7 and TLR8, PBMC was stimulated in the presence of chloroquine. In this setting, immune recognition of short dsRNA in monocytes is exclusively mediated by RIG-I. The sequences of the first 6 nucleotides of the test 5’-ppp-dsRNA 24-mer and of the self-complementary 5’-ppp-dsRNA 12-mer used for crystallization were identical.
We found that 2’-OCH3 incorporation at nucleosides 1 and 2 results in complete and 30% loss of in vivo activity respectively, while 2’-OCH3 incorporation at nucleoside 3 to 6 had no impact on the in vivo activity (Fig. 6a). The orientations of the 2’-OH groups of nucleosides 1, 2 and 3 are highlighted in red in the structure of the complex in Fig. 6b.
Figure 6.
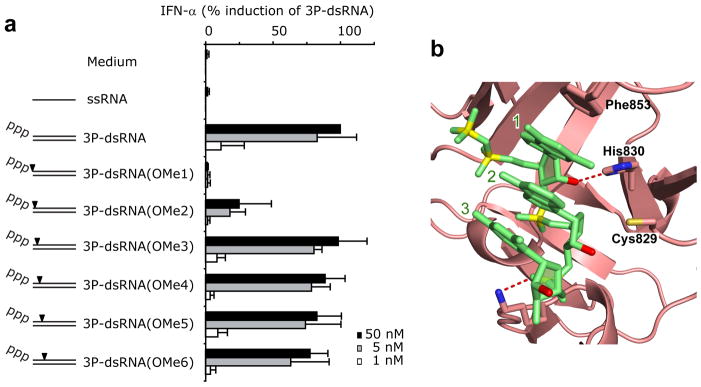
In vivo analysis of 5’-ppp-RNA strand 2′-OCH3 substituent effects. (a) Synthetic 5’-ppp-GFP2 and the corresponding 2’-O-methyl-derivatives (5’-ppp-GFP2 OMe1 to OMe6) were hybridized with the complementary antisense strand (GFP2 AS) and transfected into human chloroquine-treated PBMCs. GFP2 AS was used as a control. IFN-α production was analyzed 20 hr after stimulation. Data from four independent donors are depicted as normalized mean values ± SEM. (b) Position of 2’-OH groups (in red) of nucleosides 1, 2 and 3 adjacent to the 5’-phosphorylated end in the crystal structure of the 5’-pp-dsRNA 12-mer bound to RIG-I CTD. Amino acids that either hydrogen bond or are in close proximity of the 2’-OH groups are also labeled in the figure.
DISCUSSION
Unique features of α and β 5’-pp recognition
The RIG-I CTD targets both ends of the self-complementary 5’-pp-dsRNA 12-mer in the crystal structure of the complex. The α and β 5’-phosphates are anchored in a basic pocket through electrostatic and multiple hydrogen-bonding interactions (Fig. 2c), with their being sufficient room in the basic pocket to model in the γ 5’-phosphate (Fig. 3a). The side chains of K861 and K888 hydrogen bond to the α 5’-phosphate, the side chains of H847 and K858 hydrogen bond to the β 5’-phosphate (Fig. 2c), while the side chains of additional lysines, including K849 and K851, could provide potential electrostatic and hydrogen-binding sites for interaction with the γ 5’-phosphate (Fig. 3a). Functional data support the involvement of K849 and K851 for lower concentrations of RNA (Supplementary Fig. 4).
The RIG-I CTD recognizes blunt-end duplexes as a consequence of the aromatic ring and Cα-Cβ bond of F853 stacking on the terminal base pair (Fig. 2c). More specifically, the observed partial stacking of the F853 on the 5’-base G1 (Supplementary Fig. 3), could provide an explanation for better recognition of 5’-purine bases with which viruses initiate their RNA replication. Further, the loop that contains F853, undergoes a conformation transition on complex formation that facilitates this stacking interaction (Fig. 3b). In addition, alignment of nucleoside 1 at the 5’-ppp end is facilitated by a hydrogen bond between the purine N7 position and the side chain of K858 (Fig. 2c).
The RIG-I CTD also contacts the sugar-phosphate backbone spanning nucleosides 1 to 3 counting from the 5’-ppp end of the dsRNA duplex in the complex. Specifically, the sugar 2’-OH of nucleoside 1 is hydrogen-bonded to the side chain of H830, perhaps contributing to the preference of RIG-I for RNA over DNA, while the phosphate linking nucleosides 2 and 3 is hydrogen bonded to side chain of K907 (Fig. 6b). In addition, K849 forms hydrogen bonds with the backbone phosphate between residues 3’ and 4’ (Fig. 2b), though this could reflect crystal packing interactions.
Finally, only one CTD domain targets the 5’-ppp blunt-end of the RNA duplex, and we can eliminate models of the complex in which the CTD dimerizes on the same end of the RNA.
These results unambiguously define the elements associated with recognition of the 5’-pp end of the dsRNA by the CTD of RIG-I, and by extension the intact protein. This conclusion is validated by our supporting data discussed below, that analyzes the impact of binding pocket mutations and 2’-OCH3 RNA modifications on the interferon response, within the context of full-length RIG-I.
Correlation of structure with binding pocket mutation data
The results of binding pocket mutations identify key positions (K861, K888 and K907) that are all absolutely required for in vivo functional activation of RIG-I upon binding of 5’-ppp-dsRNA (Fig. 5a). According to the crystal structure, K861 is involved in hydrogen bonds to both the α and β phosphates at the 5′-end of the bound RNA (Fig. 5b). K888 contributes two hydrogen bonds to the α 5’-phosphate, while K907 contributes a hydrogen bond to the bridging phosphate between the second and the third nucleoside of the 5′-end of the bound dsRNA (Fig. 6b). Furthermore, considerable contribution to functional RIG-I activity comes from K858 (Fig. 5a) that binds to the β 5’-phosphate and to the N7 of the guanosine base of the first nucleoside of the 5’-end of dsRNA (Fig. 5b) and from F853 (Fig. 5a) that stacks on the first base pair of the blunt end in the structure of the complex (Fig. 5b and Supplementary Fig. 3).
Lysines K849 and K851 that form part of the basic patch, and are potential candidates for interaction with the γ 5’-phosphate (Fig. 3a), are not required but contribute to functional activation at low concentrations of RNA (Supplementary Fig. 4). Furthermore, H847, which forms one hydrogen bond to the β 5’-phosphate (Fig. 5b) makes a functional contribution, as confirmed by results obtained from the double mutant of H847 and K858 (Fig. 5a).
Further support for a functional role of H847 comes from the binding studies using recombinant CTD of RIG-I with a H847A mutation (Fig. 4b). ITC-based in vitro binding studies establish a decrease in binding affinity by a factor of 5.6 for the H847A mutant (Fig. 4b) and a further decrease in binding affinity of a factor of 4.0 for the H847A/K858 dual mutant (Fig. 4c). Both H847 and K858 are coordinated to the β phosphate in the crystal structure of RIG-I CTD bound to 5’-pp-dsRNA (Fig. 2c).
Together, a comparison of structural data with in vivo functional and in vitro binding data reveal that binding to the α and β 5’-phosphates, the first guanosine base, the bridging phosphate between the second and the third nucleoside and the first base pair of the blunt-end are the primary sites that determine the interaction between RIG-I and 5’-ppp-dsRNA. Our in vivo functional data shed no light currently on the potential role of K849 and K851 in targeting the γ phosphate. The in vivo functional contribution of H847, which forms a hydrogen bond to the β 5’-phosphate in the crystal structure of the complex (Fig. 5b), and its potential candidate role in targeting the γ 5’-phosphate, requires further investigation. Further clarification on this point will require detection of the γ 5’-phosphate and its intermolecular contacts in the structure of RIG-I CTD bound to 5’-ppp-dsRNA.
Correlation of structure with 2’-OCH3 modification data
The complete loss of activity observed with the 5’-ppp-RNA 24-mer duplex containing 2’-OCH3 at the 1 position (Fig. 6a) most likely reflects the expected steric conflict between the trans-C3′ oriented 2′-OCH3 group and the side chain of H830 (Fig. 6b). This nucleoside is additionally fixed through Watson-Crick base pairing with the 3′-terminal nucleoside from the opposite strand and through a stacking interaction with the aromatic side chain of F853, as well as through multiple hydrogen bonding interactions involving its α and β 5’-phosphates (Fig. 5b), thereby precluding formation of productive alternative configurations.
The observed 30% loss in activity following incorporation of the 2′-OCH3 group at nucleoside 2 (Fig. 6a) most likely reflects this positions proximity to side chain of C829 (Fig. 6b). Moreover, the internucleotide bond between the second and third nucleosides of the bound 5’-ppRNA duplex is also involved in a critical helix orienting interaction with the side chain of K907 (Figs. 6b).
The 2’-OH groups of resides 3 to 6 on the 5’-ppp-strand of the RNA duplex are solvent oriented and make no minor groove contacts with protein side chains in the crystal structure of the complex. This explains the experimental observation that nucleosides at positions 3 to 6 can tolerate 2’-OCH3 substitutions without loss of activity (Fig. 6a).
These 2’-OCH3 modification data provide evidence that the same binding orientation and intermolecular contacts are adopted for blunt-end 5’-ppp-RNA duplex recognition in its complex with bound RIG-I CTD in the crystal (bound as 5’-pp-RNA duplex) compared to its complex with full-length RIG-I under in vivo conditions in solution.
Future Studies
In the future, we plan to attempt crystallization and structure determination of complexes of longer 5’-ppp-RNA and RIG-I constructs that include the helicase domain, so as to define the fold of the helicase domain, as well as identify the nature of the intramolecular (associated with potential dimerization of RIG-I) and intermolecular (associated with potential helicase-RNA recognition) contacts in the bound state. These will be supplemented with in vivo functional assays to analyze the impact of intermolecular contacts associated with the helicase domain on the interferon response.
ONLINE METHODS
Protein expression and purification
An expression vector (PET-SUMO, Invitrogen) was constructed to produce the CTD of human RIG-I. E. coli cells were harvested 8 hours after incubation with 0.1 mM IPTG at 20°C. The cells were lysed in 20 mM Tris-HCl buffer, 0.2 M NaCl, 2 mM MgCl2, 1 mM DTT, pH 7.5, on ice. The protein was first purified using His-select nickel agarose resin (GE). The recovered protein was digested using ulp protease (Invitrogen), dialyzed against 20 mM Tris-HCl, 0.1 M NaCl, 1 mM DTT, pH 7.5, overnight at 4°C, and further purified by His-select nickel agarose and gel-filtration chromatography (GE Healthcare). Finally, it was concentrated to 15 mg ml−1 in 20 mM Tris-HCl, 50 mM NaCl and 1 mM DTT. The RIG-I CTD mutants were made with the Quick-change kit (Stratagene) and verified by sequencing. We expressed and purified the mutant proteins following the same protocol as that used for wide-type protein.
Chemical synthesis of 10-, 12- and 14-mer 5’-ppp-dsRNAs
Oligoribonucleotides were synthesized using DMT-2'-O-TBDMS-rC(ac), DMT-2'O-TBDMS-rA(bz), DMT-2'-O-TBMDS-rG(ib), DMT-2'-O-TBDMS-rU amidites on 1 micromole scale. The CPG bound oligomers containing free 5′-OH were triphosphorylated using the standard cyclotriphosphate protocol of triphosphate synthesis30, which was modified in order to allow product specific labeling with a lipophilic gamma amidate purification tag. (J.L. manuscript in preparation) Tagged and non-tagged products were cleaved from the column and base deprotected with 40% methylamine-ammonia 1:1 at 65° for 10 min followed by 2′-O-TBDMS deprotection with 1 M TBAF for 16 hr at room temperature and desalting on a NAP-25 column. Preparative RP HPLC purification of the tagged 5’-ppp-RNA was performed on a Hamilton PRP-1 column (4.1 × 250 mm, 10 μm) using linear gradient of methanol (Gradient: 1–100 % B in 40 min, A = 0.05 M TEAB ; B = 80% methanol 0.05 M TEAB). In contrary to untagged palindromic sequences, which give inseparable product peaks on RP HPLC31, this procedure results in clean separation of triphosphorylated tagged 10-, 11- and 12-mers from non-phosphorylated and shorter synthesis failure sequences. The tagged intermediate was converted in a second step into 5’-ppp-RNA using acid hydrolysis at pH = 3.8 at 60° for 60–90 min. Quantitative conversion into 5’-ppp-RNA was controlled by HPLC and MALDI analysis. 5’-ppp-RNA was ethanol precipitated as Na+ salt from the hydrolysis mixture.
Analytical data
MALDI TOF characterization of 5’-ppp-RNA products and lipophilic tagged intermediates. Spectra were measured on a Bruker Biflex III with linear detection mode using a proprietary Sequenom matrix by Metabion/Martinsried (Germany). The mass spectroscopic analysis is presented in Supplementary Table 2.
Crystallization and data collection
All crystals were grown by the hanging-drop vapor diffusion method. The RIG-I CTD was mixed with self-complementary 5’-ppp-RNA 12-mer duplex at 1:1 molar ratio. Crystals were grown from 27% (v/v) polyethylene glycerol 3350 (PEG 3350), 50 mM Tris-HCl, pH 7.5, 0.1 M KCl, and 10 mM MgCl2 at 4°C. Crystals were flash-frozen with the reservoir solution as cryoprotectant.
Diffraction data were collected at 100 K at beamline X-29 at the Brookhaven National Laboratory. Data was processed with the HKL2000 suite32 and data processing statistics are summarized in Table 1. The complex of RIG-I CTD domain and 5’-phosphorylated 12-mer RNA duplex belonged to the P65 space group and diffracted to 2.55 Å.
Structure determination and refinement
The structure of RIG-I CTD domain bound to 5’-pp-RNA 12-mer duplex was solved by molecular replacement with the program Phaser33, using the published CTD domain structure in the free state22 (PDB ID 2QFB) as a search model. Model building and refinement were undertaken with Coot34 and Phenix35. The stereochemistry of the refined structure was analyzed by program Procheck36. The final model (Rwork = 19.5%; Rfree = 23.0%) complex contained two RIG-I CTD molecules and one RNA duplex in the asymmetric unit. Residues 803–923 of CTD domain and all nucleotides of duplex RNA could be traced in the complex. Surprisingly, we observed a 5’-pp rather than the expected 5’-ppp terminus at the duplex ends.
Isothermal titration calorimetry (ITC) measurements
Calorimetric experiments were conducted at 25.0°C with a MicroCal™ iTC200 instrument (GE Healthcare). The RIG-I CTD samples were dialyzed against the titration buffer containing 25 mM HEPES-NaOH, pH 7.5, 100 mM NaCl (or 250 mM NaCl for high salt condition), and 2 mM MgCl2. Protein concentration was determined by absorbance spectroscopy at 280 nm (Tyr ε280=1,420 M−1 cm−1; Trp ε280=5,600 M−1 cm−1; Cys ε280=125 M−1 cm−1). Lyophilized RNA samples were prepared in the titration buffer, annealed at high concentration (3.0 mM, 75°C for 1min, 25°C for 2 hr), and then diluted to required concentration for ITC titration. The RNA samples are quantified by their absorbance at 260 nm (1OD = 50 μg/ml for duplex and 1OD = 40 μg/ml for single-stranded RNA). All titrations were performed in a ‘ligand-in-cell’ strategy, in which concentrated RIG-I CTD samples (~400 μM × 40 μl) were injected in seventeen successive points into an RNA pool (~20 μM × 200 μl) inside the sample cell. Acquired calorimetric titration data were analyzed using software Origin 7.0 based on the ‘One Set of Sites’ fitting model.
Electrophoretic mobility shift assay
5’-Triphosphorylated 11-mer (ppp-GACGCUAGCGU) was chemically synthesized, and its sequence is identical to nucleotides 1–11 of the palindromic 12-mer used for crystallization. The 11-mer (10 pmol) was then radiolabeled at its 3' end with 10 pmol alpha-32P-pCp in a 10 μl reaction containing 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 10% DMSO using T4 RNA ligase 1. Prior to the addition of enzyme, the reaction mixture was denatured by heating to 90ºC for 1 min, then placed on ice for 1 min, and followed by the addition of 1 μl RNA ligase (20 U). The ligation reaction was then incubated on ice in the cold room for 10 h. Following the addition of 40 μl of water, the reaction solution was applied onto a G25 column (GE Life Science) for removal of unincorporated radioactive pCp. This was followed by the addition of 90 pmol unlabeled triphosphorylated 12-mer ppp-GACGCUAGCGUC, the same material as used for crystallization, 1/10 of the volume of 3 M NaCl, and 3 volumes of 100% ethanol. The RNA was precipitated, and the dried pellet was resuspended in 10 μl of 100 mM KOAc, 30 mM HEPES-KOH, pH 7.4, 2 mM Mg(OAc)2, denatured for 1 min at 90ºC and then incubated at 37ºC for 3 h, yielding a final duplex concentration of 5 μM.
20 nM duplex was incubated in 10 μl aliquots of 100 mM NaCl, 2 mM MgCl2, 50 mM HEPES-KOH, pH ,7.5, 5% glycerol with increasing amounts of recombinant protein at 25°C for 15 min. Binding was visualized by running samples on native 10% 19:1 acrylamide:bisacrylamide gels (0.5×TBE supplemented with 100 mM NaCl Gels were pre-run for 30 min at room temperature at 12 V/cm, and for another 45 min upon sample loading, thereafter transferred to 3MM Whatman paper and dried at 60ºC for 1 h. Radioactive signal was detected using phosphorimaging.
Cloning of full-length RIG-I mutants
Site-directed mutagenesis of full-length RIG-I was performed by PCR-amplification of the whole expression plasmid (pEF-Bos RIG-I-FLAG) with phosphorylated primers (Supplementary Table 1) and ligation after DpnI digestion and gel extraction. Positive clones were confirmed by sequencing. Protein expression of RIG-I mutants was confirmed to be at the same levels by Western blot using an antibody against FLAG (Sigma).
Cell isolation and culture
Human PBMCs were isolated from whole blood of healthy volunteers by Ficoll-Hypaque density-gradient centrifugation (Biochrom Berlin). Red blood cells were lysed with red blood cell lysis buffer (Sigma). Cells were kept in RPMI 1640 containing 10% FCS, 1.5 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin.
Cell stimulation
Human PBMCs (4×105 cells per 96 wells) were preincubated with chloroquine (2.5 μg/ml), followed by transfection with 200 ng, 20 ng or 4 ng of RNA using Lipofectamine (Invitrogen).
For analysis of biological activity of RIG-I mutants, HEK293 cells were seeded into 96 well plates and transfected with 20 ng expression plasmid using Genejuice (Merck). Then cells were stimulated with 5 nM RNA complexed with Lipofectamine 2000 (Invitrogen). Human IP-10 was assessed in the supernatants of cells 24 hr after stimulation using a commercial ELISA kit (BD Biosciences).
ELISA/ Detection of Cytokines
The amount of IFN-α production was determined with the IFN-α module set from Bender MedSystems. The ELISA assay was performed according to the manufacturer’s protocol.
Sequences
5’-ppp-dsRNA sequences containing 2’-OCH3 modifications are listed in Supplementary Table 3. Upper case: ribonucleotides; lower case: 2′-OCH3 ribonucleotides. Synthesis of these 5’-ppp-RNA sequences was performed as described for the self-complementary 5’-ppp RNA 12-mer sequence.
Supplementary Material
Acknowledgments
The authors are grateful to the staff of X-29 beamline at Brookhaven National Laboratory for their assistance during synchrotron data collection. We thank Dhirendra Shimanshu and Yuan Tian for their assistance in synchrotron data collection, Michaela Aigner (Micura lab) for chemical synthesis of 5’-ppp-RNAs undertaken in Innsbruck, and Greg Wardle (Tuschl lab) for MS analysis in the RU proteomics facility to quality control the 5’-ppp-RNAs. DJP was supported by funds from the Abby Rockefeller Mauze Trust and the Maloris Foundation. R.M. was supported by the Austrian Science fund FWF (I317). GH was supported by grants of the Bundesministerium für Bildung und Forschung Biofuture and GoBio and of the Deutsche Forschungsgemeinschaft (SFB704, SFB670, SFB832 and KFO177).
Footnotes
Accession codes: Protein Data Bank: The atomic coordinates and structure factors for the RIG-I CTD domain-bound to blunt-end 12-mer ppRNA have been deposited under accession code 3NCU.
AUTHOR CONTRIBUTIONS
Y.W. expressed the RIG-I CTD domain, with G.S. assisting with protein expression and purification. Y.W. crystallized and solved the structure of the complex. J.L. was responsible for chemical synthesis of palindromic and non-palindromic 5’-ppp-RNAs and 2’-O-methyl-modified 5’-ppp-RNAs. C.S., M.G. and M.S. were responsible for the in vivo functional assays, including expression of full-length RIG-I mutants. H.L. and Y.W. were responsible for ITC titration assays. S.J. was responsible for gel shift assays and prepared the plasmids for mutant RIG-I CTD expression. R.M. provided earlier batches of short 5’-ppp-RNAs for crystallization trials. The paper was written by D.J.P., G.H. and T.T. with the assistance of the other authors.
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests. T.T. is a co-founder and scientific advisor of Alnylam Pharmaceuticals, and also an advisor of Regulus Therapeutics.
References
- 1.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 3.Wilkins C, Gale M., Jr Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rehwinkel J, Reis e Sousa C. RIGorous detection: Exposing virus through RNA sensing. Science. 2010;327:284–286. doi: 10.1126/science.1185068. [DOI] [PubMed] [Google Scholar]
- 5.Coch C, et al. Higher activation of TLR9 in plasmacytoid dendritic cells by microbial DNA compared with self-DNA based on CpG-specific recognition of phosphodiester DNA. J Leukoc Biol. 2009;86:663–670. doi: 10.1189/jlb.0509314. [DOI] [PubMed] [Google Scholar]
- 6.Poeck H, et al. 5'-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14:1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 7.Barchet W, Wimmenauer V, Schlee M, Hartmann G. Accessing the therapeutic potential of immunostimulatory nucleic acids. Curr Opin Immunol. 2008;20:389–395. doi: 10.1016/j.coi.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Barral PM, et al. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: Key regulators of innate immunity. Pharmacol Ther. 2009;124:219–234. doi: 10.1016/j.pharmthera.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 11.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in dsRNA-induced innate antiviral response. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 12.Besch R, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest. 2009;119:2399–2411. doi: 10.1172/JCI37155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poeck H, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 14.Hornung V, et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 15.Pichlmair A, et al. RIG-I mediated antiviral responds to single-stranded RNA bearing 5’-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt A, et al. 5’-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Scis USA. 2009;106:12067–12072. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlee M, et al. Recognition of the 5’-phosphate by RIG-I helicase requires short blunt dsRNA as contained in the panhandle of negative-strand virus. Immunity. 2009;31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ablasser A, et al. RIG-I dependent sensing of poly(dA:dT) through the induction of an RNA pol III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu YH, et al. RNA pol III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myong S, et al. Cytosolic viral sensor RIG-I is a 5’-triphosphate-dependent translocase on dsRNA. Science. 2009;323:1070–1074. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahasi K, et al. Non-self RNA-sensing mechanism of RNA-I helicase and activation of antiviral immune responses. Mol Cell. 2008;29:428–440. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 22.Cui S, et al. The C-terminal regulatory domain is the RNA 5’-triphosphate sensor of RIG-I. Mol Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 23.Ma J-B, Ye K, Patel DJ. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature. 2004;429:318–322. doi: 10.1038/nature02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma JB, et al. Structural basis for 5’-end-specific recognition of the guide RNA strand by the A. fugidus PIWI protein. Nature. 2005;434:666–670. doi: 10.1038/nature03514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teplova M, et al. Structural basis for recognition and sequestration of UUUOH 3’-terminii of nascent mRNA polymerase III transcripts by La autoantigen. Mol Cell. 2006;21:75–85. doi: 10.1016/j.molcel.2005.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, et al. The RIG-I-like receptor LGP2 recognizes the termini of dsRNA. J Biol Chem. 2009;284:13881–13891. doi: 10.1074/jbc.M900818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leung DW, et al. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat Struct Mol Biol. 2010;17:165–172. doi: 10.1038/nsmb.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, et al. Structural basis of Toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Utyanskaya EZ, Lidskii BV, Neihaus MG, Shilov AE. Mathematical modeling of kinetics of adenosine 5’-triphosphate hydrolysis catalyzed by Zn2+ ion in the pH range 7.1 to 7.4. J Inorg Chem. 2000;81:239–258. doi: 10.1016/s0162-0134(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 30.Ludwig J, Eckstein F. Rapid and Efficient Synthesis of Nucleoside 5'-O-(1-Thiotriphosphates), 5'-Triphosphates and 2',3'-Cyclophosphorothioates Using 2-Chloro-4h-1,3,2-Benzodioxaphosphorin-4-One. Journal of Organic Chemistry. 1989;54:631–635. [Google Scholar]
- 31.Anderson AC, et al. HPLC purification of RNA for crystallography and NMR. RNA. 1996;2:110–117. [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.