

Abstract
Molecular signaling of messages emanating from cellular membranes through receptor tyrosine kinases (RTKs) is a major mechanism for intercellular communication and transduction during development and metabolism, as well as in disease-associated processes. The phosphorylation status and signaling activity of RTKs are determined by a dynamic equilibrium of the activity of both RTKs and protein tyrosine phosphatases (PTPs). RTKs are essentially a class of cell-surface receptors for growth factors and other extracellular ligands, the most conspicuous perhaps are members of the vascular endothelial growth factor (VEGF) gene family, which plays a fundamental role in the growth and differentiation of vascular, as well as lymphatic endothelial cells. In particular, VEGF is a major regulator of normal (physiologic) and abnormal (cancerous) angiogenesis, including that associated with tumors and cancer. Blockers/inhibitors and regulators of RTKs are indeed promising cancer interventions, their specific mechanisms are yet to be unraveled. In this cutting-edge synopsis, I elaborate on breakthroughs/advances and current concepts of RTK regulation, further shedding light on exploring the role of potential regulators, particularly the RTK inhibitor Semaxanib, and the mechanisms associated with tumorigenesis in an effort to understand a potentially alleviating pharmacologic therapeutic intervention. This survey also tackles the loopholes and shortcomings of the aforementioned inhibitory role of Semaxanib, especially its inefficacy and ultimate discontinuation of relevant clinical trials.
Keywords: Bevacizumab, Cell signaling, Development, Disease, Gene regulation, Immunopharmacology, Kinase, Phosphorylation, RTK, Semaxanib, Transcription factors
1. Introduction and background
The ramifications of anti-angiogenesis approach, via the regulation of specific kinases and their receptors, as a targeted therapy in many types of cancer are indeed perplexing. In my search for small molecules that have been in clinical trials and/or in markets targeting receptor kinases whose biochemistry is tailored for the support of angiogenesis, a hallmark process in cancer evolution and propagation, I witnessed a myriad of developing drugs, including Semaxanib and bioengineered monoclonal antibodies, commonly dubbed as MABs. It is not my intention though to hijack authoritative references involved with anti-angiogenesis control in just one or two molecules that are or have been potentially promising drugs for cancer therapeutics. Furthermore, I do not intend under any circumstance to fringe upon any patented rights or legal issues pertaining to discussed drugs, rather I endeavor to present what is common in the literature around us as thematically and objectively as I am possibly capable of and to the best of my ability.
I will present in this synopsis an overview of receptor tyrosine kinase regulation and involvement in reinforcing the mechanics of the process of angiogenesis, commonly involved with the evolution of many types of cancer as I have and will be indicating throughout. I then touch base with the chronological design and development of the so called SU-5416, or Semazanib, walking the reader through its pharmacokinetics and undertaken clinical trials, emphasizing the ups and downs, the efficacy and loopholes, and finally its discontinuation. From that I build on the recent advances in therapeutic approaches replacing the first generation of molecules targeting receptor tyrosine kinases involved in vascular endothelial growth factor-mediated angiogenesis and the ensuing development of cancer. One such example I will stress on is the second generation of MABs, particularly pointing finger to a very promising anti-angiogenesis drug, I emphatically meant bevacizumab (Avastin®). Lessons learned throughout are a take-home message: “Today’s drug market is inundated with specific and rather non-specific, or more accurately non-efficacious molecules, yet what is seemingly promising may not be as such in the upcoming years.” Burgeoning research that is continuing apace is pioneering and challenging at the same time in and of itself to try and identify not only potential targets but also effective drugs for cancer treatment and prevention.
2. Receptor tyrosine kinase regulation – an overview
Tyrosine kinases (TKs) are a subclass of specific and selective protein kinases that can transfer a phosphate group from ATP to another protein; conspicuously, they function as an “on” or “off” switch in many cellular/molecular functions (Hanks et al., 1988). The phosphate group is usually attached to the amino acid tyrosine on the protein. Moreover, TKs are considered a subgroup of the larger class of protein kinases that attach phosphate groups to other amino acids (particularly, serine and threonine) (Radha et al., 1996).
Phosphorylation of proteins via the action of kinases is understandably an important mechanism in intracellular signal transduction and in regulating cellular activity, such as the ensuing cell cycle and cell division. Protein kinases can become mutated thus lock in the “on” position, for instance, and therefore cause unregulated cellular growth, one of the hallmarks of imposing and developing cancer (Ruetten and Thiemermann, 1997; Schaller et al., 1992; Dengjel et al., 2009). Therefore, it is reasonable to deduce that kinase inhibitors and regulators are often effective in cancer treatment. On the other hand, most tyrosine kinases have an associated protein tyrosine phosphatase, which removes the phosphate group, and hence acts as an internal regulator (Hanks et al., 1988).
Receptor tyrosine kinases (RTKs) are the high-affinity cell-surface receptors for many polypeptide growth factors, cytokines, and hormones. Effectively, of the 90 unique tyrosine kinase genes identified in the human genome, approximately 58 encode receptor tyrosine kinase proteins. Receptor tyrosine kinases have been shown not only to be key regulators of normal cellular processes, but also to have a critical role in the development and progression of many types of cancer, as has been earlier alluded to (Fig. 1) (Hanks et al., 1988; Radha et al., 1996; Ruetten and Thiemermann, 1997).
Figure 1.
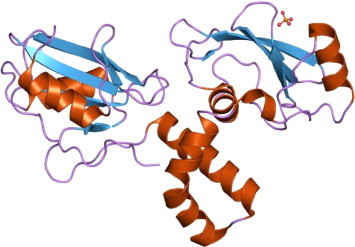
The molecular structure of tyrosine kinase (TK).
3. Receptor tyrosine kinase families – a synopsis
The tyrosine kinases are divided into two main families: the transmembrane receptor-linked kinases and those that are cytoplasmic proteins. Approximately 2000 kinases are known, and more than 90 protein tyrosine kinases (PTKs) have been found separately and in clusters within the human genome (Dengjel et al., 2009). They are divided into two classes: receptor and non-receptor PTKs. Currently, 58 RTKs are well known, grouped into 20 subfamilies. They play pivotal roles in diverse cellular activities including growth, differentiation, metabolism, adhesion, motility, and death (apoptosis) (Hanks et al., 1988; Radha et al., 1996; Ruetten and Thiemermann, 1997; Schaller et al., 1992; Dengjel et al., 2009).
Structurally, RTKs are composed of an extracellular domain, which is able to bind a specific ligand, a transmembrane domain, and an intracellular catalytic domain, which is able to bind and phosphorylate the selected substrates. Binding of a ligand to the extracellular region causes a series of structural (morphologic) rearrangements in the RTK that lead to its enzymatic activation. In particular, movement of some parts of the kinase domain gives free access to ATP and the substrate to the active site. This triggers a cascade of events through phosphorylation of intracellular proteins that ultimately transduce the extracellular signal, causing changes in the profile of gene expression (Dengjel et al., 2009; Blanke et al., 2008; le Coutre et al., 2008).
Many RTKs are involved in oncogenesis, either by gene mutation, or chromosome translocation, or simply by over-expression. In every case, the result is a hyperactive kinase, which confers an aberrant, ligand-independent, non-regulated growth stimulus to the cancer cells. In humans, particularly, there are 32 cytoplasmic PTKs. The first non-receptor tyrosine kinase identified was the v-src oncogenic protein (Fig. 2) (Blanke et al., 2008; le Coutre et al., 2008; Kuwai et al., 2008).
Figure 2.
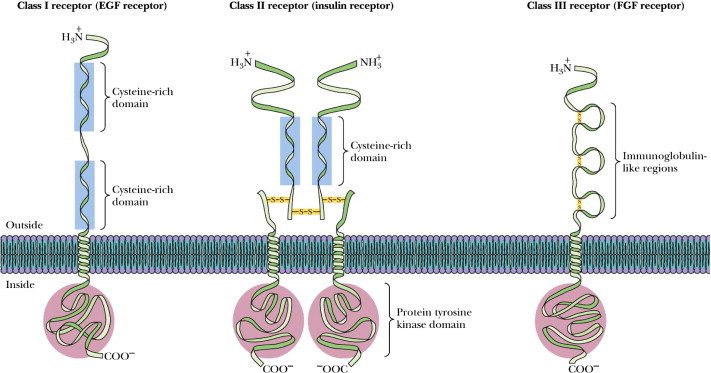
The major families of RTKs. Receptor tyrosine kinases span the membrane of the cell and have a hormone receptor on the outside and a tyrosine kinase portion on the inside. There are several subclasses of these receptors as shown here.
Most animal cells contain one or more members of the Src family of tyrosine kinases. For instance, a chicken sarcoma virus was found to carry mutated versions of the normal cellular Src gene. The mutated v-src gene has lost the normal built-in inhibition of enzyme activity that is characteristic of cellular Src (c-src) genes. Src family members have been found to regulate many cellular processes (Wiley and Burke, 2001; Rinker et al., 2008; Silvennoinen et al., 1997; Bhise et al., 2004). For example, the T-cell antigen receptor leads to intracellular signaling by the activation of Lck and Fyn, two proteins that are structurally similar to Src (Gunby et al., 2007; Kris et al., 2003; Sordella et al., 2004).
In upcoming paragraphs, the author is inclined to point the attention of the reader to RTK-related families and subgroups, tailored particularly for VEGF. Although related families such as fibroblast growth factor receptors (FGFRs) and RET, a receptor for members of the glial cell line-derived neurotrophic factor (GDNF) family of extracellular signaling molecules or ligands (GFLs) have been identified, I will specifically highlight and emphasize the VEGF/RTKs connection relevant to Semaxanib. There are excellent references the readers may want to consult with pertaining to the aforementioned families of FGFR and RET (Okamoto, 2010; Druker et al., 2001; Joensuu et al., 2001; Robinson et al., 2001; Zwick et al., 2001; Pawson, 1995; Ornitz and Itoh, 2001; Duchesne and Tissot, 2006; Coutts and Gallagher, 1995; Sleeman and Fraser, 2001; Robinson and Stringer, 2001; Myers and Eng, 1995; Baloh and Enomoto, 2000; Airaksinen et al., 1999; Arighi et al., 2005).
3.1. Vascular endothelial growth factor receptor (VEGFR) family
Vascular endothelial growth factor (VEGF) is one of the main activators and inducers of endothelial cell proliferation and the permeability of blood vessels (Robinson and Stringer, 2001). At least two RTKs bind to VEGF at the cell surface: VEGFR-1 (termed Flt-1) and VEGFR-2 (termed KDR/Flk-1). The VEGF receptors have an extracellular portion consisting of seven immunoglobulin (Ig)-like domains so, like FGFRs, they belong to the immunoglobulin superfamily. They also possess a single transmembrane spanning region and an intracellular portion containing a split tyrosine kinase domain. Moreover, VEGF-A specifically binds to VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1) receptors.
On the functional side, while VEGFR-2 appears to mediate almost all of the pervasive, well known cellular responses to VEGF, the function of VEGFR-1 is less well defined, although it is thought to modulate VEGFR-2 signaling. Another function of VEGFR-1 may be to act as a dummy/decoy receptor, sequestering VEGF from VEGFR-2 binding (this appears to be particularly important during vasculogenesis in the developing embryo). Relatively recently, another receptor has been discovered (termed VEGFR-3); however, what is actually known is that VEGF-A is not a ligand for this receptor. Furthermore, VEGFR-3 seems to mediate lymph angiogenesis in response to VEGF-C and VEGF-D (Robinson and Stringer, 2001).
3.2. Receptor tyrosine kinases (RTKs)
These receptors are transmembrane proteins that span the plasma membrane just once. Almost 58 different RTKs are known in humans. Some of the ligands that can trigger RTKs are: (i) insulin; (ii) insulin-like growth factor-1 (IGF-1); (iii) vascular endothelial growth factor (VEGF); (iv) platelet-derived growth factor (PDGF); (v) epidermal growth factor (EGF); and (vi) fibroblast growth factor (FGF) (Fig. 2). (Notably, a mutation in one of these receptors causes typical achondroplasia – the most common type of dwarfism (Hanks et al., 1988; Radha et al., 1996; Ruetten and Thiemermann, 1997; Schaller et al., 1992; Dengjel et al., 2009).)
Technically, binding of the ligand to two adjacent receptors forms an active homodimer. This activated dimer is essentially a tyrosine kinase, an enzyme that attaches phosphate groups to certain tyrosine (Tyr) residues – first on itself, then on other proteins converting them into an active state. Many of these other proteins are also tyrosine kinases (the human genome encodes almost 90 different tyrosine kinases) and in this way a cascade of expanding phosphorylation occurs within the cytosol. Furthermore, some of these cytosolic tyrosine kinases act directly on gene transcription by barging onto the nucleus and transferring their phosphate to transcription factors thus activating them (Hanks et al., 1988). To cite an example, the cytosol of B cells contains the Btk (Bruton’s tyrosine kinase), which is essential to turning on appropriate gene expression when a B cell encounters a specific antigen. Inherited mutations in the gene encoding Btk have been reported to cause X-linked agammaglobulinemia in males. This mutation can lead to downregulation of the manufacturing of antibodies and, therefore, may induce recurrent bacterial infections unless the afflicted individuals are given periodic injections of immune globulin (IG). Moreover, others act indirectly through the production of second messengers (Hanks et al., 1988).
At the regulatory level, it is apparent that a cell must also be able to stop responding to a signal. For growth factor receptors, failure to do so could lead to uncontrolled mitosis, or probably cancer. For the RTKs, this is done by quickly engulfing and destroying the ligand-receptor complex by receptor-mediated endocytosis (Dengjel et al., 2009; Wiley and Burke, 2001). One might expect that anything that leads to the inappropriate expression of receptors that trigger cell division could lead to uncontrolled cell division and cancer. In fact, the gene encoding PDGF, which is termed SIS, is a proto-oncogene, and mutated versions purportedly participate in making the cell cancerous. Furthermore, the genes encoding receptors for EGF are allegedly considered proto-oncogenes and are expressed at abnormally high levels in several human cancers. Two monoclonal antibodies that target these receptors recently identified are: Trastuzumab (Herceptin®) that inactivates HER2 (human epidermal growth factor receptor 2) and Cetuximab (Erbitux®) that inactivates HER1; both regulators show astounding promise against breast cancer (Kuwai et al., 2008; Wiley and Burke, 2001; Rinker et al., 2008; Silvennoinen et al., 1997; Bhise et al., 2004).
In parallel, two tyrosine kinase inhibitors, termed Gefitinib (Iressa®) and Erlotinib (Tarceva®), have recently been shown to block the action of the EGF receptors on the cells of certain lung cancers and have shown some promise against these cancers (Hanks et al., 1988; Radha et al., 1996). Mutant versions of some of the “second-order” kinases are also associated with cancer: The oncogene SRC encodes a mutated version of a normal tyrosine kinase associated with the inner face of the plasma membrane. In addition, the fusion protein BCR/ABL produced by the Philadelphia chromosome activates constitutively the cytosolic tyrosine kinase ABL that normally would be activated only when the cell is stimulated by a growth factor (e.g., PDGF). The result is the debilitating chronic myelogenous leukemia (CML) (Rinker et al., 2008; Silvennoinen et al., 1997; Bhise et al., 2004; Gunby et al., 2007; Kris et al., 2003; Sordella et al., 2004). Another promising treatment is Imatinib mesylate (Gleevec®, also known STI571). This molecule fits into the active site of the ABL protein thus preventing ATP from binding there. Without ATP as a phosphate donor, the ABL protein cannot phosphorylate its substrate(s) (Blanke et al., 2008).
Furthermore, the RAF kinase has been reported to participate in a signaling pathway that links RTKs to gene activation. Binding of a ligand to the RTK activates an intracellular molecule called RAS, which then activates RAF. In mammals, this pathway promotes cellular mitosis. Excessive activities of the RAS gene or mutations in RAS and/or RAF are associated with many types of cancer, so RAS and RAF are considered proto-oncogenes. Almost 15% of all human tumors contain a mutated RAF, and 66% of melanomas – a highly malignant skin cancer of melanocytes – contain a mutated RAF (called BRAF) (Rinker et al., 2008; Silvennoinen et al., 1997; Bhise et al., 2004; Gunby et al., 2007; Kris et al., 2003; Sordella et al., 2004).
Given the wealth of the aforementioned, it is not surprising to indicate that in order to reduce enzyme activity, inhibitor molecules should tightly bind to the enzymes. Reducing enzyme activity can disable a pathogen or correct an incorrectly function system; as such, many enzyme inhibitors are developed to be used as drugs for the general public. Gastrointestinal stromal tumors (GIST), for example, are mesenchymal tumors that affect the gastrointestinal tract (Hanks et al., 1988; Radha et al., 1996; Ruetten and Thiemermann, 1997; Schaller et al., 1992; Dengjel et al., 2009). Treatment options have been limited. However Imatinib, as an inhibitor to the malfunctioning enzyme, can be effective. If Imatinib does not work, patients with advanced chronic myelogenous leukemia can use Nilotinib, another inhibitor to the malfunction enzyme that allegedly causes the leukemia. This inhibitor is a highly selective Bcr–Abl tyrosine kinase inhibitor (Blanke et al., 2008). Sunitinib, on the other hand, is an oral tyrosine kinase inhibitor that acts upon VEGFR, platelet-derived growth factor receptor (PDGFR), stem cell factor receptor (SCFR), and colony-stimulating factor (CSF)-1 receptor. Furthermore, Gefitinib inhibits the tyrosine kinase domain of epidermal growth factor receptor (EGFR). Kinase inhibitors can also mediate; paracrine signaling, for instance, mediates the response to EGFR kinase inhibitors. Paracrine activates epidermal growth factor receptor in endothelial cells of the tumor to do this (Hanks et al., 1988; Dengjel et al., 2009).
4. Chronological assessment of receptor tyrosine kinase inhibition – the case of Semaxanib (SU-5416)
Semaxanib (also known as semaxinib or SU-5416) is a drug initially intended for the treatment of cancer. It was still at an experimental stage and as such has not yet received a license for use on human patients (except, however unsuccessfully, in the setting of a clinical trial). Semaxanib was considered a potent and selective synthetic inhibitor of the Flk-1/KDR VEGF receptor tyrosine kinase; the inhibitor targets the VEGF pathway, and both in vivo and in vitro studies have demonstrated anti-angiogenic potential (O’Donnell et al., 2005; Lockhart et al., 2006; Hoff, 2006).
SU-5416 {3-[(2,4-dimethylpyrrol-5-yl)methylidenyl]-indolin-2-one} (Fig. 3), the Sugen (Pharmacia) Semaxanib compound identified from a large screen of potential inhibitors of phosphotyrosine kinases, is considered a potent and selective inhibitor of the kinase-insert domain-containing receptor (KDR)/Flk-1 RTK (Haluska and Adjei, 2001; Mendel et al., 2000a), a high-affinity receptor for the VEGF family of growth factors. SU-5416 has been developed as an anti-angiogenic compound for the potential therapeutic treatment of solid tumors mediated by suppression of metastasis and angiogenesis. Subsequently, Sugen Inc. (USA) and Taiho Pharmaceutical Co., Ltd. have agreed to pursue a joint development program for Sugen’s angiogenesis inhibitors.
Figure 3.
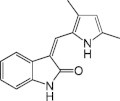
The molecular structure of Semaxanib, SU-5416.
As of July 1998, Taiho was providing a proportion of the funding for the development of Sugen’s angiogenesis inhibitors and was to receive marketing rights in Japan. In August 1998, a patent, which covered the composition of inhibitory compounds for the treatment of a variety of diseases (including cancer), was issued to Sugen covering a family of compounds, including SU-5416 (Haluska and Adjei, 2001; Mendel et al., 2000a). Phase I trials with SU-5416 began at UCLA School of Medicine in September 1997 to assess safety and dosage range in approximately 30 patients with advanced malignancies (Hannah, 1997; Rosen et al., 1998). In May 1998, interim results of the phase I study were presented at the 34th ASCO meeting in Los Angeles, CA, indicating tolerance at a dose range of 4.4–65 mg/m2.
In June 1998, plans for two additional trials were reported: The first was a phase I/II investigation conducted at the Cancer Research Campaign Center for Cancer Therapeutics at the Institute for Cancer Research and the Royal Marsden Hospital in London, UK, to assess leakage of tumor blood vessels as a biological marker for the angiogenic process, in addition to monitoring safety and pharmacokinetic parameters for SU-5416; the second study which was initiated at the Arizona Cancer Center, assessed alternative dose regimen for the compound in patients with advanced malignancies, including those with multiple tumors (Hannah, 1997; Rosen et al., 1998). Complete analysis of the results was reported at the Biologic Principles for the Therapy of Human Colon Cancer meeting in November 1998. Final experimental results of the phase I trial for SU-5614 were presented in May 1999 at the 35th ASCO meeting in Atlanta, GA, which showed that this drug was well tolerated for chronic administration at biologically active dose levels with the demonstration of clinical activity in certain tumor types (Mendel et al., 2000a; Hannah, 1997; Rosen et al., 1998).
By January 1999, phase III lung cancer studies were planned for the first half of 1999. In June 1999, the FDA agreed to the proposed design for phase III trials of the compound in colorectal and NSCLC. The trials were to compare Semaxanib against standard chemotherapy regimens in patients who had not yet received any chemotherapy. The primary endpoint of the trial was survival, with secondary endpoints of time-to-disease progression and objective response rates (Hannah, 1997). Furthermore, in April 2000, data from a phase I/II open-label, dose-escalating study of SU-5416 were reported at the 91st AACR meeting in San Francisco, CA. As a follow-up, pharmacokinetic data from four phase I and three phase II investigations were presented at the NCI-EORTC-AACR 11th symposium on New Drugs in Cancer in Amsterdam, The Netherlands. In May 2001, phase II data for SU-5416 in patients with metastatic melanoma were reported at the 37th ASCO meeting in San Francisco, CA, when the occurrence of one stable disease and one mixed response, coupled with preliminary data on tumor vascular perfusion has supported further investigation of this drug in this patient population (Haluska and Adjei, 2001; Mendel et al., 2000a; Hannah, 1997; Rosen et al., 1998).
5. Molecular mechanisms of receptor tyrosine kinase regulation and the Semaxanib connection
As indicated, molecular signaling of messages emanating from cell membranes through RTKs is a major mechanism for intercellular communication during development and metabolism, as well as in disease-associated processes (Fig. 4) (Bilder and Rojas, 1996; Shawver et al., 1997; Strawn and Shawver, 1998; King et al., 1993; McMahon et al., 1998; Traxler, 1998; Sun et al., 2000; Sun and McMahon, 2000; Teicher, 2000). The phosphorylation status and signaling activity of RTKs are determined by a dynamic equilibrium of the activity of both RTKs and protein tyrosine phosphatases (PTPs) (Spivak et al., 1992; Petrone and Sap, 2000; Yao et al., 1998). I have reported earlier on that RTKs are a class of cell-surface receptors for growth factors and other extracellular ligands, the most conspicuous perhaps are members of the VEGF gene family, which plays a fundamental role in the growth and differentiation of vascular as well as lymphatic endothelial cells (major requirement for angiogenesis). This family includes several members including VEGF, placenta growth factor (PlGF), VEGF-B, VEGF-C, VEGF-D and two VEGF-like proteins encoded by parapoxvirus orf virus. In particular, VEGF, referred to also as VEGF-A, is a major regulator of normal and abnormal angiogenesis, including those associated with tumors (Oliver et al., 1995).
Figure 4.
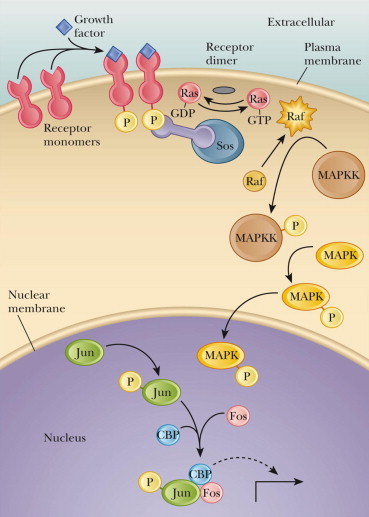
The growth factor receptor interactions involving RTKs and MAPK signaling pathways.
Angiogenesis is a mechanistic developmental process, which involves the generation and growth of new blood vessels (neo-vascularization) from pre-existing vasculature (Vlodavsky et al., 1990; Klagbrun and D’Amore, 1991; Pili et al., 2001). The angiogenic process is defined by the activation of quiescent endothelial cells in pre-existing blood vessels followed by the growth and migration of the aforementioned cells leading to dissolution of the vessel basement membrane. The migration and proliferation of endothelial cells then form new capillary lumina and loops that lead to the creation of a new basement membrane and maturation of microvessels. In physiological conditions, angiogenesis has been regarded as tightly controlled process that requires the balance of the anti-angiogenic and pro-angiogenic factors. However, angiogenesis may well play a detrimental role in the growth of solid tumors and their invasion and metastasis in pathophysiological conditions (see the diagrammatic representation; Fig. 1) (Oikawa, 1995; Le et al., 1993; Plate, 1996; Ono et al., 1997).
The growth of tumor cells is dependent on growth factors derived from tumor (autocrine stimulation) or microenvironment (paracrine stimulation). PDGF and FGF, for instance, are secreted from tumor cells and neighboring stromal and normal tissues (brain, liver, lung, bone, etc.) thereby leading to enhanced growth and survival. VEGF, FGF and PDGF stimulate the branching, extension and survival of endothelial cells resulting in the formation of new blood vessels during the progression of tumor angiogenesis (Neufeld et al., 1999). Moreover, VEGF-A and its related family members VEGF-B and VEGF-C, which elicit a pronounced angiogenic response, are mitogens for vascular endothelial cells derived from arteries, veins and lymphatics, and are involved in a variety of physiological and pathological neo-vascularization process, including wound healing, coronary and peripheral arterial collateral development (Ware and Simon, 1997), primary and secondary tumors (Folkman, 1995), retinopathy (Aiello et al., 1997, 1994), rheumatoid arthritis, psoriasis and other bullous skin diseases (Brown et al., 1995).
VEGFs are secreted and expressed by tumor cells and surrounding stromal tissue in response to hypoxia. These growth factors transduce their biological activities following binding to RTKs (Dumas, 2001; McMahon, 2000). VEGF-A, perhaps the most intensively studied growth factor family member, exerts its biological effects via interaction with three high-affinity receptors: VEGF-R1 (fms-like tyrosine kinase; Flt-1) (Hanks et al., 1988; Dengjel et al., 2009; Fong et al., 1995; Seetharam et al., 1995; Sun and McMahon, 2000), VEGF-R2 (fetal liver tyrosine kinase 1/kinase insert domain-containing receptor; Flk-1/KDR) (Quinn et al., 1993; Gerber et al., 1998), and neurophilin-1 (Soker et al., 1998). Both Flt-1 and Flk-1/KDR have seven immunoglobulin (Ig)-like domains in the extracellular domain, a single transmembrane region, and a tyrosine kinase domain, which is interrupted by a kinase-insert domain.
There is compelling evidence that Flk-1/KDR receptor is the key signaling receptor involved in mediating the initiation and propagation of embryonic vasculogenesis as well as angiogenesis during adult life (Strawn et al., 1996; Shawver, 1999), since knockout investigations revealed embryonic lethality associated with gross abnormality of vascularization (Shalaby et al., 1995). Furthermore, Flk-1/KDR is indispensable for tumor angiogenesis and, hence, plays a pivotal role in tumor growth, invasion and metastasis, since inhibition of Flk-1/KDR signaling by a dominant negative construct effectively suppressed glioblastoma growth in vivo (Millauer et al., 1994). Flt-1, on the other hand, functions, at least in some circumstances, as a decoy receptor with high-affinity to recombinant human VEGF, whereas neurophilin-1 acts by potentiating VEGF binding to Flk-1/KDR (Keyt et al., 1996) (see the diagrammatic representation; Fig. 5).
Figure 5.
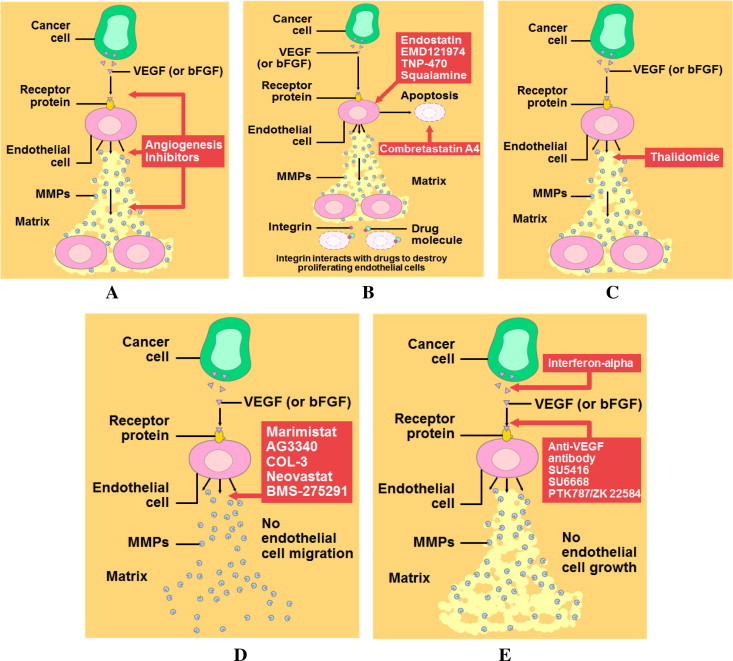
A series of regulatory pathways involving the role of growth factors in promoting the process of angiogenesis, a hallmark activity of developing tumor. (A) Angiogenesis inhibitors in the treatment of human cancer; these inhibitors fall into several different categories, depending on their mechanism of action. Some inhibit endothelial cells directly, while others inhibit the angiogenesis signaling cascade or block the ability of endothelial cells to break down the extracellular matrix. (B) Drugs that inhibit angiogenesis directly; one class of angiogenesis inhibitors being tested in cancer patients are molecules that directly inhibit the growth of endothelial cells. Included in this category is endostatin, the naturally occurring protein known to inhibit tumor growth in animals. Another drug, combretastatin A4, causes growing endothelial cells to commit suicide (apoptosis). Other drugs, which interact with a molecule called integrin, also can promote the destruction of proliferating endothelial cells. (C) Old drug with a new use; another interesting drug is thalidomide, a sedative used in the 1950s that was subsequently taken off the market because it caused birth defects when taken by pregnant women. Although this drug clearly would not be suitable for pregnant women, its ability to prevent endothelial cells from forming new blood vessels might make it useful in treating non-pregnant cancer patients. (D) Drugs that block extracellular matrix breakdown; another group of angiogenesis inhibitors are directed against one of the initial products made by growing endothelial cells, namely, the MMPs (matrix metalloproteinases), enzymes that catalyze the breakdown of the extracellular matrix. Because breakdown of the matrix is required before endothelial cells can migrate into surrounding tissues and proliferate into new blood vessels, drugs that target MMPs also can inhibit angiogenesis. Several synthetic and naturally occurring molecules that inhibit the activity of MMPs are currently being tested to see if interfering with this stage of angiogenesis will prolong the survival of cancer patients. (E) Drugs that block the angiogenesis signaling cascade and the mechanism of action of Semazanib (SU-5416); an active group of angiogenesis inhibitors being tested in human clinical trials are molecules that interfere with steps in the angiogenesis signaling cascade. Included in this category are anti-VEGF antibodies, including Semazanib, that block the VEGF receptor from binding growth factor. Another agent, interferon-alpha, is a naturally occurring protein that inhibits the production of bFGF and VEGF, preventing these growth factors from starting the signaling cascade. Also, several synthetic drugs capable of interfering with endothelial cell receptors are being tested in cancer patients.
During the past decade, chemically diverse small-molecule protein kinase inhibitors have been discovered to be of potential therapeutic value for the treatment of various human diseases, such as retinopathy, atherosclerosis and solid tumor growth (Harris, 1998; Raymond, 1998; Oo and Baumgartner, 1998; Stover et al., 1999; Traxler and Furet, 1999; Arasteh and Hannah, 2000; Vajkoczy et al., 2000; Ellis et al., 2000; Puduvalli and Sawaya, 2000; Liekens et al., 2001; Brostjan et al., 2000; Mitsuyasu, 2000; Ryan and Wilding, 2000; Deplanque and Harris, 2000). SU-5416 (Semaxanib) was found to be a potent and selective inhibitor of Flk-1/KDR receptor when tested using both kinase and cellular assays. In particular, it selectively inhibits VEGF-dependent mitogenesis and migration of human endothelial cells without inhibiting the growth of a variety of tumor cells in tissue culture systems. However, SU-5416 significantly inhibited the growth of many tumor cell lines when grown as subcutaneous xenografts in mice, suggesting the involvement of a paracrine pathway consistent with the angiogenesis mechanism.
Furthermore, SU-5416 altered tumor vascular density and vascular leakage after tumor implantation, and induced the regression of established tumors and apoptosis of endothelial cells in a model of colon metastasis. SU-5416 exhibited a novel biologic activity in patients with AIDS-related Kaposi’s sarcoma, a disease characterized by small tumors on the skin that are fueled by angiogenesis. This makes SU-6416 a strategically well positioned drug with promises for future applications in cancer therapy, provided its safety and efficacy persists in clinical trials (Vacca et al., 2000; Philip, 2000; Liekens et al., 2001). It is worth mentioning that albeit the initial trials for Semaxanib were relatively successful, further trials especially phase III trials, were not – this prompted the withdrawal of any further investigation, especially when a new generation of RTK inhibitors have been injected into the market.
6. Semaxanib synthesis and SAR
Recent advances in the understanding of the signaling mechanisms involved in neo-vascularization, which suggested that intervening against certain growth factor receptors and their associated signaling pathways may provide a promising approach to the development of angiogenesis inhibitors, have paved the way for Sugen Inc. and Allergan Inc. to enter into an exclusive collaboration to identify, develop and commercialize novel pharmaceutical compounds utilizing Sugen’s proprietary small-molecule signal transduction inhibition technology (Hanks et al., 1988). In May 1997, Sugen announced the solution of the three-dimensional co-crystal structure of FGF RTK with two of Sugen’s TK inhibitors. With this breakthrough in crystallography, Sugen and its collaborators have elucidated the structure of inhibitor compounds bound to the active site of FGF receptor, enabling the research team to design very specific and selective drug candidates, including Flk-1, PDGF-R, as well as other types of protein kinases. Sugen’s TK inhibitor compounds were initially identified by random screening for selective inhibitors, thereby providing the basis for adding computational chemistry and rational drug design into Sugen’s technology platform (Hanks et al., 1988; Dengjel et al., 2009).
Chronologically, in 1998, synthesis and biological evaluations of a novel class of TK inhibitors were reported, where 3-substituted indolin-2-ones exhibited selectivity toward particular RTKs. Subsequently, SU-5416 was screened and proposed as a potent and selective Flk-1/KDR kinase inhibitor that blockaded receptor autophosphorylation, endothelial cell mitogenesis and tumor cell growth (Fong et al., 1998, 1999). Co-crystallization analysis revealed that SU-5416 binds at the ATP domain of Flk-1/KDR, in essentially the same manner as its counterpart SU-4984. Therefore, on the basis of crystallography and the IC50 value on kinase activity of this and related compounds, it can be inferred that the indolin-2-one core interferes with the binding of the adenine core of ATP to RTK (Sun et al., 1998; Toledo et al., 1999), thus preventing phosphate transfer to the tyrosine residue. The fact that this particular substituted indolin-2-one preferentially exists as the Z-isomer is important for its specificity with regard to the Flk-1 tyrosine kinase. Synthesis of 3-indolin-2-ones in 4 steps, 63 analogous prepared isomer analysis in vitro tyrosine kinase activity (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001). Synthesis of indolin-2-ones as potent and selective inhibitors of Flk-1/KDR for the inhibition of tumor angiogenesis was subsequently documented. The 3-substituted indolin-2-ones 1–3 were initially identified as potent inhibitors, although compound 2 was non-selective with respect to other related receptor tyrosine kinases. A number of structural modifications of these compounds led to the conclusions that: (i) N1-methylation reduces activity; (ii) placing electron-donating groups in the phenyl para position provides high potency and selectivity (in contrast, installing electron-withdrawing substituents in this same position results in inactive compounds); and (iii) methylation of the vinyl group gave inactive analogs (Hanks et al., 1988).
Co-crystallization of an inhibitor with Flk-1 also provided insight into the importance of the double bond stereochemistry. In the bound structure of SU-4984, an analog of compound 2 where the isopropyl substituent was replaced by 4-methylpiperidine, the oxindole moiety occupied the ATP binding site. Interestingly, the compound 2 analog was bound as the Z-isomer, even though it was the E isomer in solution. Thus the E form must be converted to the Z form before binding. Apparently, this conversion was shown to be mediated by acid, base or light. The pyrrole moiety of compound 3 was replaced with other heterocycles (thiophene, furan, pyrazole), but the pyrrole ring was the superior compound. In this series, all the E isomers were inactive, and the vinyl proton was again determined to be essential for activity (Hanks et al., 1988; Dengjel et al., 2009).
7. Semaxanib pharmacology and pharmacokinetics
Since its foundation in 1991, Sugen has focused on the development of small-molecule drugs specifically targeting tyrosine kinase, tyrosine phosphatase and serine-threonine kinase families of signal transduction molecules, and their signaling pathways (Antonian et al., 1999). Data on the pharmacology of SU-5416, a novel synthetic compound, deals with its selectivity and in vitro and in vivo efficacy (Mendel et al., 2000b). SU-5416, a potent and selective inhibitor of the Flk-1/KDR receptor tyrosine kinase, was shown to inhibit vascular endothelial growth factor-dependent mitogenesis of human endothelial cells without inhibiting the growth of a variety of tumor cells in vitro. Being part of the screening procedure to identify this drug from a large pool of related compounds, SU-5416 is highly effective as an inhibitor of Flk-1/KDR tyrosine kinase. In cultured human endothelial cells (HUVECs), SU-5416 (0.1 μM) inhibited VEGF-stimulated thymidine uptake, whereas FGF-1-stimulated thymidine uptake was inhibited only at a concentration of 10 μM, further supporting selectivity of SU-5416 for the flk-1 tyrosine kinase (Antonian et al., 1999; Mendel et al., 2000b).
Although these in vitro data alone are insufficient to demonstrate the anti-angiogenic activity of SU-5416, further in vivo data are available on various tumors in nude mice. Systemic administration of SU-5416 at non-toxic doses in mice resulted in the inhibition of subcutaneous tumor growth of cells derived from various tissue origins. Specifically, SU-5416 in a dose of 25 mg/kg/day, reduced growth of a variety of tumors implanted in nu/nu mice, with a follow-up varying from 13 to 38 days. The anti-tumor effect of SU-5416 was accompanied by the appearance of pale white tumors that were resected from drug-treated animals, supporting the anti-angiogenic property of this agent. No direct data were given on the vascularity of these tumors, but a reduced vascularity was assumed on the basis of their pale appearance. These findings support that pharmacological inhibition of the enzymatic activity of the vascular endothelial growth factor receptor represents a novel strategy for limiting the growth of a wide variety of tumor types (Liekens et al., 2001; Fong et al., 1998, 1999; Sun et al., 1998, 1999, 2000; Toledo et al., 1999; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b).
The phosphorylation of the tyrosine residues in Flk-1/KDR receptor leads to homo-dimerization of receptors (Bold et al., 2000; Mendel et al., 2000c) and a sequence of events that is generally believed to mediate the angiogenic effect of VEGF. SU-5416 selectivity profile is based on various phosphorylation studies in appropriate cell lines and includes lack of inhibitory effect on phosphorylation of the tyrosine residues of PDGF-R, MAPK, EGFR, the insulin receptor (IR) and insulin receptor substrate (IRS)-1. Only IRS-1 phosphorylation was inhibited at a 100-fold higher concentration of SU-5416 than was needed to inhibit Flk-1 phosphorylation. Thus, as far as its specific effect on receptor tyrosine phosphorylation is concerned, SU-5416 can be considered selective for the Flk-1 receptor.
8. Semaxanib metabolism
SU-5416 has a short plasma half-life but prolonged in vivo effects in mouse tumor xenograft models and humans. In HUVECs, for example, SU-5416 did not inhibit VEGF-dependent KDR phosphorylation by reducing the surface expression of KDR, or the affinity or amount of VEGF binding to KDR, but probably by subcellular localization in cells. Furthermore, it was shown that, in addition to inhibiting KDR/Flk-1 receptor tyrosine kinase, SU-5416 also inhibits the Flt-1 receptor tyrosine kinase and MAP kinase activity, and prevents VEGF-dependent cell migration of vascular endothelial cells (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b,c; Bold et al., 2000). A phase II trial of SU-5416 in combination with standard-dose paclitaxel (175 mg/m2, q3w) shows the regimen is well tolerated, with no effect on paclitaxel pharmacokinetics. Su-5416 was also studied at doses of 85 or 145 mg/m2 twice weekly in combination with fluorouracil and leucovorin (given either weekly or daily for 5 days every 4 weeks) in patients with untreated metastatic colon cancer. The response rate was 36% with a time to progression of 9 months, a time to failure of 8.5 months and a median survival time of 22.6 months (Antonian et al., 1999; Mendel et al., 2000b). These results were compared with historical data from a phase III study of fluorouracil and leucovorin with or without irinotecan (CPT-11; Yakult Honsha KK/Daiichi Seiyaku Co. Ltd./Pharmacia Corp.). The time to progression and survival time appeared to be better than in the arm comprising fluorouracil and leucovorin alone and similar to the arm that included irinotecan (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b).
Another extensive study was designed to investigate the in vitro metabolism of SU-5416 by mouse, rat, dog, monkey, and human liver microsomes and to identify the major metabolites of SU-5416. A high-pressure liquid chromatography (HPLC) procedure analysis was developed and validated to resolve and quantify SU-5416 and its metabolites. To evaluate the in vitro metabolism of SU-5416, pooled liver microsomes from mice, rats, dogs, monkeys, and humans were incubated with SU-5416 (25 μM) in the presence of an NADPH-generating system. In the presence of NADPH, mouse, rat, dog, monkey, and human liver microsomes converted SU-5416 to at least 12, 9, 9, 7, and 6 polar metabolites, respectively (Sun et al., 1998, 1999, 2000; Toledo et al., 1999; Sielecki et al., 2000).
Microsomal metabolism of SU-5416 showed marked species differences in the levels of different metabolites formed. The overall rate of SU-5416 metabolism by liver microsomes from the species examined followed the rank order: monkey ⩾ mouse ≈ rat > dog > human. Two major metabolites of SU-5416 were identified, a hydroxymethyl derivative of SU-5416 (M12) and a carboxylic acid derivative of SU-5416 (M6), by spectroscopic methods and comparison with authentic compounds. Both of these oxidative metabolites were further metabolized in vivo through glucuronidation. The metabolic fate of SU-5416 in microsomes from various species as well as data from in vivo biotransformation in the rat was assessed and discussed (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b; Bold et al., 2000).
9. Semaxanib toxicity and cytotoxicity – contraindications
SU-5416 was investigated in phase I dose escalating trials in patients with advanced malignancies. The drug exhibited only mild toxicity, and the elimination t1/2 was approximately 50 min. A dose level of 145 mg/m2 was subsequently recommended for phase II trials, based on AUC. Of note, some comparative differences were reported in the context toxicity profile in rats following intravenous (i.v.) administration of SU-5416 and SU-6668, another tyrosine kinase inhibitor developed by Sugen (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b). Furthermore, in a phase I/II study of SU-5416 in combination with 5-FU in patients with metastatic colorectal cancer administered with SU-5416 (85 and 145 mg/m2) twice weekly, combined with a standard dose of 5-FU, it was described that 5-FU dose-limiting toxicities (DLTs) predominated; no SU-5416-related DLTs have been observed. The metabolic pharmacokinetics of the combination, in addition, was not significantly different from SU-5416 administered alone (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b,c; Bold et al., 2000).
During the 36th annual meeting of the American Society of Clinical Oncology in New Orleans, LA, USA, a detailed dose escalating study of SU-5416 in AIDS-related Kaposi’s sarcoma was reported. SU-5416, a selective antagonist of Flk-1 located in stromal vessels around lesions, caused decreased VEGF phosphorylation, dimerization, and signaling. Men with Kapos’s sarcoma received doses of 65–145 mg/m2 i.v. biw for six cycles (of 29 days). There was no effect on CD4 count or viral load, but 10% had grade 3 migraine headaches that responded to sumatriptan (Glaxo Wellcome plc.) and 11 had phlebitis, requiring the use of central catheters. Reduced clearance occurred over time, most likely due to concomitantly administered protease inhibitors. Five responses and clinical improvement (which consisted of decreased edema and improved performance status) were observed in nine patients (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b,c; Bold et al., 2000).
Furthermore, it was reported that in a phase I study of SU-5416 in patients with a variety of tumor types, tumor vascular permeability was measured with dynamic contrast MRI to demonstrate that no dose limiting toxicity was seen at doses 48–190 mg/m2 bi-weekly × 4 weeks. Intrapatient dose escalation was permitted because the diluent for this agent is cremaphor, and so steroids and histamine blockers were administered (Vacca et al., 2000; Philip, 2000; Liekens et al., 2001; Fong et al., 1998, 1999; Sun et al., 1998, 1999; Toledo et al., 1999). There were no clinical responses, but six patients had tumor stabilization and in four of these, decreased tumor vascular permeability was seen. The investigators controlled the effect of the steroids on permeability, by performing scans before treatment with steroids also given. Another study reported a detailed phase I study of SU-5416 given as a 5-day load followed by 5-weekly maintenance infusions with the final doses achieved being 65 mg/m2 load and 190 mg/m2/week. Grade 3 headaches were the most significant toxicity. One patient with squamous cell carcinoma of the lung had a PR and three patients had SD. In addition, data were presented for a phase I/II study of SU-5416 given at escalating doses of 85–145 mg/m2 i.v. biw along with 5-fluorouracil and leucovorin to patients with metastatic colon cancer. No dose limiting toxicity attributable to the SU-5416 was observed. The chemotherapy did not alter SU-5416 pharmacokinetics. Patient responses included a complete response (CR), five PRs and nine patients with SD. In short, toxicities reported to date in patients treated with SU-5416 included headaches and gastrointestinal events, such as nausea, vomiting and diarrhea.
More data for SU-5416 were presented, including a phase I/II study in combination with paclitaxel and carboplatin, and a phase I study of an oral formulation. In the combined chemotherapy/Semaxanib study, full doses of carboplatin and paclitaxel and doses of SU-5416 (less than or equal to 145 mg/m2 i.v. twice weekly) were well tolerated, with no increases in thrombocytopenia, neutropenia or peripheral neuropathy, compared with toxicities reported for carboplatin and paclitaxel alone. Although paclitaxel and carboplatin PKs were not affected by SU-5416, the clearance of Semaxanib was reduced when given with paclitaxel, resulting in an increased AUC and systemic exposure. Among the 25 patients, there were four CRs and one PR (Liekens et al., 2001; Fong et al., 1998, 1999; Sun et al., 1998, 1999, 2000; Toledo et al., 1999; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000). In the study of the oral formulation, oral bioavailability was 45% when dosed in orange juice. Clearance was generally higher for oral vs. i.v. administration. Toxicities were mild but the taste of the drug was considered to be poor (Liekens et al., 2001; Fong et al., 1998, 1999; Sun et al., 1998, 1999, 2000; Toledo et al., 1999; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b,c; Bold et al., 2000; Smolich et al., 2001). Further work to improve the oral formulation is under way in order to use the drug on a more chronic basis for less advanced malignancies.
10. Semaxanib clinical development
10.1. Pre-clinical studies
An independent report on in vitro and in vivo efficacy of SU-5416 was published in 1997. Increased vascular permeability and excessive neo-vascularization are the hallmarks of endothelial dysfunction, which can lead to diabetic macular edema and proliferative diabetic retinopathy. VEGF was shown to be an important mediator of ocular neo-vascularization and a known vaso-permeability factor in non-ocular tissues (Mendel et al., 2000b,c; Bold et al., 2000; Smolich et al., 2001). In these studies, it was demonstrated that intra-vitreal injection of VEGF rapidly activated protein kinase C (PKC) in the retina at concentrations observed clinically, inducing membrane translocation of PKC isoforms α, βII, and δ and >threefold increases in retinal vaso-permeability in vivo. The effect of VEGF on retinal vascular permeability appeared to be mediated predominantly by the β-isoform of PKC with >95% inhibition of VEGF-induced permeability by intra-vitreal or oral administration of a PKC β-isoform-selective inhibitor that did not inhibit histamine-mediated effects.
In the HUVEC assay, a 3,5-dimethyl pyrrole-analog (SU-5416) was the superior compound, with an IC50 of 40 nM. The compound was also highly selective for Flk-1, being essentially inactive at other related tyrosine kinase receptors, such as PDGF, EGF and IGF-1. SU-5416 was chosen for further evaluation. Co-crystallization revealed that this compound binds at the ATP domain of Flk-1 in essentially the same manner as SU-4984. SU-5416 displayed dose-dependent inhibition of subcutaneous growth of A375 human melanoma in mice, and in preclinical studies, no toxic effects were noted at efficacious doses (Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b,c; Bold et al., 2000). This compound dramatically inhibited tumor size and tumor neo-vascularization, suggesting that tumor growth is not affected directly. SU-5416 displayed a broad anti-tumor spectrum: i.p. administration inhibited eight of 10 different cell lines grown subcutaneously in mice. Although the plasma half-life was short (30 min), the half-life for activity against Flk-1 was about 20 h, perhaps due to the lipophilicity of the compound.
SU-5416, among other candidate inhibitors, was screened as potent anti-angiogenic small-molecule inhibitor of receptor tyrosine kinases, including those of the VEGF and PDGF receptor families. The stem cell factor (SCF) receptor, c-kit, is structurally related to these receptors and, although not expressed on mature peripheral blood cells, is expressed in leukemic blasts derived from 60% to 80% of acute myeloid leukemia (AML) patients. The c-kit kinase inhibitory activity of SU-5416 was evaluated in MO7E cells, a human myeloid leukemia cell line. Tyrosine autophosphorylation of the receptor, induced by SCF, was inhibited in these cells by SU-5416 in a dose-dependent manner (inhibitory concentration of 50% [IC50] = 0.1–1 μM) (Smolich et al., 2001).
Inhibition of extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation, a signaling event downstream of c-kit activation, was also inhibited in a dose-dependent manner (Vajkoczy et al., 2000; Ellis et al., 2000; Puduvalli and Sawaya, 2000; Liekens et al., 2001; Brostjan et al., 2000; Mitsuyasu, 2000; Ryan and Wilding, 2000; Deplanque and Harris, 2000; Vacca et al., 2000). SU-5416 also inhibited SCF-induced proliferation of MO7E cells (IC50 = 0.1 μM). Furthermore, SU-5416 induced apoptosis in a dose- and time-dependent manner as measured by the increase in activated caspase-3 and the enhanced cleavage of its substrate poly(ADP-ribose) polymerase (Smolich et al., 2001). These findings with MO7E cells were extended to leukemic blasts from c-kit (+) patients. In patient blasts, SU-5416 inhibited SCF-induced phosphorylation of c-kit and ERK1/2 and induced apoptosis. These studies indicated that SU-5416 inhibited biologic functions of c-kit in addition to exhibiting anti-angiogenic properties and suggested that the combination of these activities may provide a novel therapeutic approach for the treatment of AML (Smolich et al., 2001).
10.2. Clinical investigations
In January 1997, Sugen announced that the company’s Flk-1 TK angiogenesis inhibitor program will move into human clinical studies in mid-1997. The objective of the angiogenesis clinical program was to establish the safety and efficacy of the company’s lead drug candidate as an anti-metastasis and angiogenesis inhibitory treatment for solid tumor cancers. The company has identified several small-molecule inhibitors of the Flk-1 receptor that meet its pre-clinical profile for an IND candidate in this program, and has proceeded into pharmacology and pharmaceutical development. It was subsequently planned to assess comprehensive human safety studies by late 1997, in order to permit the commencement of the first efficacy studies with the final drug candidate around the end of the year (Smolich et al., 2001).
Sugen’s animal studies have shown that Flk-1 TK inhibitors, including SU-5416, blocked the growth of most solid tumors by angiogenesis inhibition, and as a cause of death in many tumor types (Fig. 5) (Le et al., 1993; Plate, 1996; Ono et al., 1997; Neufeld et al., 1999; Ware and Simon, 1997; Folkman, 1995; Aiello et al., 1997). This was an important discovery because it demonstrated the potential utility of these compounds as part of a front-line therapy regimen, in addition to their applications in chronic therapy. In July 1997, Sugen announced that the company has filed an IND application with the FDA for the clinical testing of a synthetic small-molecule signal transduction inhibitor of Flk-1/KDR that has therapeutic potential as an anti-angiogenesis and anti-metastasis agent in solid tumor cancer patients.
In September 1997, Sugen announced that it has initiated the first phase I clinical trial of its angiogenesis inhibitor, SU-5416, for the treatment of solid tumors and tumor metastases. The target for this angiogenesis inhibitor is Flk-1/KDR, a molecular driver of blood vessel formation, which has been validated by Sugen scientists in collaboration with the Max Planck Institute. The phase I clinical study was being conducted under the guidance of Lee Rosen, MD, Director of the Jonson Comprehensive Cancer Center and the Cancer Therapy Development Program at the UCLA School of Medicine in Los Angeles, California, where the first patient went on study. The objective of this phase I trial was to assess the safety and dosage range of SU-5416 (Hanks et al., 1988). This study recruited patients with advanced malignancies who have failed previous drug therapy. Dosing was administered intravenously on a twice-weekly schedule and continued until unacceptable toxicity or progression of disease was seen. The study recruited approximately 30 patients and took up to six months to complete. In addition to the i.v. formulation used in phase I study, scientists have succeeded in developing an oral formulation for SU-5416 that has demonstrated bioavailability in animals (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b,c; Bold et al., 2000; Smolich et al., 2001).
This advance appeared to have cleared the way to introduce an oral dosage form, which was expected to enter into clinical trials in 1998 (Neufeld et al., 1999; Ware and Simon, 1997; Folkman, 1995; Aiello et al., 1997; Aiello et al., 1994; Brown et al., 1995; Dumas, 2001; McMahon, 2000). In May 1998, Sugen announced the initiation of a phase I/II study to investigate the safety and efficacy of SU-5416 in patients with Kaposi’s sarcoma who have failed currently available therapy. The study, expected to enroll 30 patients, was conducted at five US medical centers specializing in AIDS-related malignancies. The study endpoints included measurement of objective response and time-to-disease progression in addition to safety and pharmacokinetic parameters.
Interim phase I data of SU-5416 was presented at ASCO in 1998. To that date the study had enrolled at least 30 patients with advanced solid tumor malignancies, and indicated that SU-5416 was well tolerated at a dose range of 4.4–65 mg/m2. Patients with advanced malignancies, including those with colorectal and lung cancer, have been treated in the phase I dose-escalation study, which began in September 1997. SU-5416 was administered twice weekly in an i.v. formulation, with escalating doses up to a level of 145 mg/m2 (from an original starting dose of 4.4 mg/m2). SU-5416 was shown to be well tolerated with only mild side effects reported by the majority of patients (i.e., headache, pain at injection site). In patients treated for longer than six months, there had been no evidence of cumulative or chronic toxicities, indicating SU-5416’s potential use for chronic administration (Seetharam et al., 1995; Sun and McMahon, 2000; Quinn et al., 1993; Gerber et al., 1998; Soker et al., 1998; Strawn et al., 1996; Shawver, 1999; Shalaby et al., 1995; Millauer et al., 1994).
Pharmacokinetics data showed that the levels of SU-5416 detected in patient serum exceeded those predicted to be necessary to achieve an anti-angiogenesis effect based on preclinical animal studies. While phase I trials were not designed to measure efficacy, there have been observations of lesion shrinkage in Kaposi’s sarcoma, colorectal and basal cell cancer patients. Eight of these advanced disease patients (12%) remained on study for over six months, and another 14 patients (20%) remained on study for more than three months without any disease progression. Five out of 11 non-small cell lung cancer patients remained on study for over 150 days, including one patient who has remained on study for over 390 days. The phase I clinical trial, designed to assess the safety of SU-5416 in the escalating dose study, had demonstrated that, SU-5416 could be safely administered at the doses given. These investigations prepared the ground for the launch of multiple phase II studies later in 1998 (Mendel et al., 2000b,c; Bold et al., 2000; Smolich et al., 2001).
The clinical strategy for SU-5416 continued to focus primarily on the potential utility of this drug as a tumor growth inhibitor and anti-metastatic agent in solid tumor patient populations. In addition to Kaposi’s sarcoma study, Sugen planned to initiate multiple phase II trials in the course of 1998 to investigate the potential efficacy of SU-5416 in different solid tumor cancer indications. The phase I/II trial was conducted at the Cancer Research Campaign (CRC) Center for Cancer Therapeutics at the Institute for Cancer Research (ICR) and the Royal Marsden Hospital (RMH) in London. The study included 24 patients and assessed leakiness of tumor blood vessels as a biological marker for the angiogenic process using imaging methods, in addition to monitoring safety and pharmacokinetic parameters. By measuring the change in leakiness of the tumor blood vessels, a very early indication of how effective SU-5416 was at blocking the unwanted signals in human cancers was determined (Toledo et al., 1999; Sun et al., 1999, 2000; Sielecki et al., 2000).
In May 1999, it was announced that Sugen intended to collaborate with the National Cancer Institute (NCI) on multiple phase II and I/II studies of SU-5416, to be conducted at approximately 20 leading cancer centers around the country. Under the Clinical Trials Agreement, the NCI had selected more than 20 studies out of 51 proposals from investigators at cancer centers and clinical research hospitals throughout the US to test SU-5416 as either a single agent or in combination with the other drugs in a variety of cancer indications. Between 500 and 700 patients were expected to be enrolled in these NCI-sponsored trials. SU-5416 was used as single agent therapy in several phase II studies, including renal, head and neck, sarcoma, and prostate cancers. SU-5416 was also tested in several phase I/II studies in combination with standard and investigational chemotherapy in patients with advanced malignancies, including colorectal, renal, ovarian, and breast cancers, and certain leukemias (Traxler and Furet, 1999; Arasteh and Hannah, 2000; Vajkoczy et al., 2000; Ellis et al., 2000; Puduvalli and Sawaya, 2000; Liekens et al., 2001; Brostjan et al., 2000; Mitsuyasu, 2000; Ryan and Wilding, 2000; Deplanque and Harris, 2000; Vacca et al., 2000; Philip, 2000; Liekens et al., 2001; Fong et al., 1998, 1999; Sun et al., 1998, 1999; Toledo et al., 1999).
These trials helped set the stage for potentially broad use of SU-5416 in a variety of therapeutic settings. Furthermore, a phase I/II study of SU-5416 in combination with 5-Flurouracil (FU)/Leucovorin (LV) in patients with metastatic colorectal cancer was presented in May 2000. The phase I/II study evaluated 28 patients to determine the safety and tolerability of SU-5416 when used in combination with 5-FU/LV Patients with untreated advanced colorectal cancer were treated with increasing doses of SU-5416 (85 and 145 mg/m2) administered intravenously twice each week, in combination with standard doses of 5-FU/LV therapy. In a preliminary analysis of this data, it was shown that patients receiving SU-5416 were found to be free of tumor growth and spread for a median time in excess of 9.2 months. Of the 28 patients who participated in the study, data to date show 41% had an objective response (either complete or partial response with 50% or greater shrinkage of tumor size). Safety data showed no dose-limiting toxicity attributable to SU-5416, with three patients remaining on therapy for more than one year. Toxicities were predominantly those known to occur with 5-FU/LV, including severe nausea, vomiting, diarrhea, mucositis and neutropenia (Sun et al., 1999, 2000; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b,c; Bold et al., 2000; Smolich et al., 2001).
In January 1999, Sugen announced today that it planned to accelerate the development of its lead angiogenesis inhibitor, SU-5416, moving directly from phase I and phase I/II studies into phase III colorectal and lung studies, and a phase II/III study in AIDS-related Kaposi’s sarcoma. Pending discussions with the FDA, both the phase III colorectal and lung studies were intended to compare the efficacy of standard chemotherapy regimens to those same regimens in combination with SU-5416. The equivalent of phase II data was gathered and analyzed at scheduled interim reviews. In March 1999, SU-5416 entered phase II/III clinical trials for AIDS related Kaposi’s sarcoma. In June 1999, Sugen announced that FDA had agreed with the company’s proposed clinical trial design for phase III trials of SU-5416, its lead angiogenesis inhibitor, in colorectal and non-small cell lung cancer (Liekens et al., 2001; Fong et al., 1998, 1999; Sun et al., 1998, 1999, 2000; Toledo et al., 1999; Sielecki et al., 2000; Krystal et al., 2001; Antonian et al., 1999; Mendel et al., 2000b).
The trials, which began in the US and Europe, compared SU-5416 against standard chemotherapy regimens in patients who have not yet received any chemotherapy. Both trials provided for scheduled interim analyses. The primary endpoint was survival, with secondary endpoints of time-to-disease progression and objective response rate (Stover et al., 1999; Traxler and Furet, 1999; Arasteh and Hannah, 2000; Vajkoczy et al., 2000; Ellis et al., 2000; Puduvalli and Sawaya, 2000; Liekens et al., 2001; Brostjan et al., 2000; Mitsuyasu, 2000; Ryan and Wilding, 2000; Deplanque and Harris, 2000; Vacca et al., 2000). The studies analyzed tumor samples, using Sugen’s proprietary gene expression array technology.
11. Semaxanib side effects and contraindications
Side effects and contraindications associated with SU-5416 reported to date (that the time of the actual performance of the myriad clinical trials) included headaches, minor gastrointestinal complications, such as nausea, vomiting and diarrhea, mucositis and neutropenia. Apart from that, no detailed data are currently available on the side effects and contraindications associated with SU-5416 administration. What the author has concluded from the aforementioned is that the stage at which Semaxanib was withdrawn from further experimentation and clinical trials was crucial at initiating the emerging of a new generation of RTK/VEGF regulators that are showing solid grounds in the therapeutic treatment of cancer via the regulation of angiogenesis (see Fig. 5).
12. Current therapy and opinion – the case of bevacizumab (Avastin®) and the anti-angiogenesis connection
Since first proposed few decades earlier, angiogenesis has been coined as an important and crucial factor in the etiology of many human diseases. In cancer, an assessment of tumor-associated angiogenesis has been particularly viewed as a potential indicator of tumor aggressiveness and patient prognosis. Of note, anti-angiogenesis therapy have been already investigated in pre-clinical models in combination with more traditional cytotoxic therapies, and many clinical studies are currently addressing the efficacy and safety of a combination of angiogenesis inhibitors and the conventional anti-cancer approaches, such as radiation and chemotherapy. These aforementioned approaches, which have incorporated a strategy to develop and commercialize selective inhibitors of RTKs, might certainly provide a novel method for the development of new agents for the treatment of human cancers and other associated diseases (see Figs. 2 and 5).
In recent years, the network of signal transduction pathways has been largely deciphered, thereby making more clearly the factor that any aberration in signaling elements is the root of many diseases. For instance, many of the known oncogenes and proto-oncogenes code for RTKs. This realization has identified these proteins as potential targets for disease therapy. Thus the concept of “signal transduction therapy” takes on a broader insight and can apply to proliferative diseases, such as cancers, psoriasis and retinosis, as well as inflammatory conditions, such as sepsis and multiple sclerosis. Apparently, agents that directly or indirectly modulate any of the converging transduction pathways targeting specific components would be introduced as a means to combating the pathologic condition (Haddad, 2001, 2004).
One such novel approach is the development of SU-5416, as a selective inhibitor of Flk-1/KDR kinase with an effective anti-angiogenesis and anti-tumorigenesis therapy. The mechanism of action and the selectivity profile makes SU-5416 an interesting and promising drug among a growing number of anti-angiogenic agents that aim to reduce or halt tumor growth. Prospects for continued development and evaluation in pre-clinical as well as clinical investigations are certainly outstanding and have proven to be efficacious. Despite the fact that competition in this field is fierce, the selectivity profile of the SU-5416 gives it a unique place in anti-angiogenic strategies. It is unknown, however, whether in tumor treatment such selectivity is warranted or beneficial, but even if multi-target approaches are being devised, it is likely that a selective anti-VEGF arm is part of such a strategy.
In the light of the aforementioned, I would want to point the reader’s attention to current developments in anti-VEGF therapy, particularly shedding light on the FDA-approved monoclonal chimera dubbed bevacizumab (Avastin®). Given this understanding, I can now ask from where the next generation of targeted therapies will arise. Clearly, VEGFR and EGFR signaling are validated target pathways for new treatments for colorectal cancer, the initial point of focus of Semaxanib. The following excerpts are highlighted from the acclaimed reference of Colorectal Cancer: Evidence-Based Chemotherapy Strategies, edited by Leonard B. Saltz, 2007 (Humana Press, an imprint of Springer-Verlag) (Saltz, 2007).
“Bevacizumab (Avastin®)”, a humanized antibody directed against VEGF, has demonstrated significant survival benefits when added to traditional chemotherapy in both the first- and second-line setting in metastatic colorectal cancer. The toxicities of bevacizumab appear modest relative to those of most cytotoxic chemotherapy. In addition to bevacizumab, several other promising agents are in late stages of clinical development. Important questions, particularly regarding duration of use, remain to be addressed in well-designed clinical trials.
As indicated, bevacizumab is a humanized antibody directed against the VEGFR-1 and -2 ligands. Preclinical studies demonstrated that bevacizumab binds to and neutralizes all human VEGF-A isoforms. In addition to its direct anti-angiogenic effects, bevacizumab may also improve the delivery of chemotherapy by reducing interstitial pressure in tumors. Phase I clinical trials initiated in 1997 showed that the antibody was relatively nontoxic as a single agent, and that adding it to standard chemotherapy did not significantly increase chemotherapy-associated toxicities. More encouraging efficacy results were seen when bevacizumab was combined with standard chemotherapeutic agents in CRC.
In an early randomized phase II trial, 104 previously untreated patients with metastatic CRC were randomly assigned to 5-fluorouracil (5-FU)/leucovorin (LV) (5-FU 500 mg/m2 and LV 500 mg/m2 once weekly for the first 6 weeks of an 8-wk cycle) alone or with bevacizumab at one of two different doses (either 5 or 10 mg/kg every 2 weeks). Comparing the 5 mg/kg dose with 5-FU alone demonstrated differences in response rate (40% vs. 17%), time to tumor progression (9 vs. 5.2 months), and median OS (21.5 vs. 13.8 months), and this dose was recommended for further use/testing. Higher-dose bevacizumab therapy plus chemotherapy did not offer further potential gains and indeed might have been inferior to lower dose drug. Thrombosis was the most significant adverse event (10.4% grade 3/4) and was fatal in one person, and grade 3/4 hypertension was seen in 16.4% of patients. In addition, 25.3% of patients receiving bevacizumab developed proteinuria or experienced worsening proteinuria. It is not clear why the 5 mg/kg dose of bevacizumab seemed to be more effective than the 10 mg/kg dose, though this was not a phase III trial designed to specifically compare the three arms in terms of efficacy, and it was pointed out that any imbalance in randomization in this relatively small study could have led to a higher number of poor-prognosis patients in the high-dose arm. The Eastern Cooperative Oncology Group (ECOG) has evaluated the combination of irinotecan, 5-FU/LV (IFL), and bevacizumab in patients with previously untreated advanced colorectal cancer in a phase II study (E2200). The first 20 patients received irinotecan (125 mg/m2), 5-FU (500 mg/m2), and LV (20 mg/m2) weekly for 4 of 6 weeks and higher-dose bevacizumab (10 mg/kg) every other week. Following a toxicity review of other trials using IFL, subsequent patients were enrolled at reduced starting doses of irinotecan (100 mg/m2) and 5-FU (400 mg/m2). Preliminary results are available for the first 87 patients. Although 36% of patients experienced grade 3/4 neutropenia, febrile neutropenia was uncommon (5%). There was one grade 4 epistaxis (requiring tamponade but no transfusion) and two grade 3 melena. There were six grade 3 thrombotic events and three grade 4 events (pulmonary embolism). Proteinuria and hypertension were infrequent. Overall response rate was 49%, and median progression-free survival (PFS) was 10 months. Median overall survival (OS) has not been reported, although the 1-yr OS rate was 85%.
In a subsequent pivotal phase III trial, 813 patients with previously untreated metastatic colorectal cancer were randomized to receive IFL plus either bevacizumab (5 mg/kg every 2 weeks) or placebo. A third treatment arm consisting of 5-FU/LV plus bevacizumab enrolled 110 patients before a planned interim safety analysis established an acceptable safety profile for the IFL plus bevacizumab treatment group; at that time, 100 patients had also been randomized to the IFL plus placebo treatment arm. The addition of bevacizumab improved median OS (20.3 vs. 15.6 months), PFS (10.6 vs. 6.2 months), and response rate (44.8% vs. 34.8%) compared with bolus IFL alone. Grade 3 hypertension was more common during treatment with IFL plus bevacizumab than with IFL plus placebo (11% vs. 2.3%), but was easily managed with standard antihypertensives. Unlike in the previously discussed phase II trial, no appreciable increases in thrombosis, bleeding, or proteinuria occurred with the addition of bevacizumab. However, bowel perforation occurred in 1.5% of patients receiving IFL plus bevacizumab, and one patient died as a direct result of this event. Based on the convincing proof of efficacy when added to IFL and 5-FU/LV as first-line treatment, bevacizumab was approved by the FDA in 2004 for use in the first-line setting in combination with intravenous 5-FU-based therapy.
An interesting randomized, placebo-controlled phase II study was conducted concurrently with the pivotal trial in patients deemed nonoptimal candidates for first-line irinotecan-containing regimens. Patients were required to have at least one of the following adverse characteristics: age 65 years or older, ECOG performing status (PS) of 1 or 2, serum albumin no more than 3.5 g/dL, or prior radiotherapy to abdomen or pelvis. A total of 209 patients were randomized to weekly 5-FU/LV plus either lower dose bevacizumab or placebo. Despite this higher risk study population, the regimen of 5-FU/LV plus bevacizumab appeared to be well tolerated. Patients receiving bevacizumab had a higher incidence of grade 3 hypertension (16% vs. 3%) and arterial thrombotic events (10% vs. 5%), but no differences were seen in venous thrombosis, grade 3/4 bleeding, or clinically significant proteinuria. The addition of bevacizumab to 5-FU/LV was associated with numerically superior median PFS (9.2 vs. 5.5 months) and response rates (26% vs. 15.2%) compared to 5-FU/LV alone. OS, the primary end point of the study, was longer in the group receiving bevacizumab (16.6 vs. 12.9 months), but this difference did not reach statistical significance. Despite the fact that these patients were deemed unfit for first-line irinotecan, the authors suggested that one possible explanation for the lack of a survival benefit is that more than 40% of patients received potentially effective post-progression therapy with irinotecan and/or oxaliplatin, conceivably diluting any survival advantage from first line therapy containing bevacizumab.
A combined analysis of raw primary data from three clinical trials further evaluated the addition of bevacizumab to 5-FU/LV. These three studies consisted of the two phase II studies discussed previously and the third treatment group in the pivotal randomized phase III trial (5-FU/LV plus bevacizumab). In the combined analysis of the three studies, 249 patients had received 5-FU/LV plus bevacizumab, and 241 patients had received either 5-FU/LV (n = 141 patients) or bolus IFL (n = 100 patients) without the addition of the anti-angiogenic agent. Stratified analysis showed that the patients receiving bevacizumab realized a significant increase in median survival (17.9 vs. 14.6 months), PFS (8.8 vs. 5.6 months), and response rate (34.1% vs. 24.5%) relative to patients receiving cytotoxic chemotherapy alone.
Oxaliplatin-containing regimens are also highly active against colorectal cancer, and investigators have combined FOLFOX with bevacizumab in the front-line setting. The TREE1 study randomized 147 previously untreated patients to one of three oxaliplatin/fluoropyrimidine regimens (mFOLFOX6, bolus 5-FU/oxaliplatin [bFOL], or capecitabine + oxaliplatin [CapOx]). Objective tumor responses were seen in 24–39% of patients in the three arms. The TREE2 study had identical eligibility criteria and then randomized 210 patients to one of the same three regimens (although the capecitabine dose was reduced from 1000 mg/m2 twice daily [TREE1] to 850 mg/m2 twice daily [TREE2] after excess toxicity was seen at the higher dose level), plus bevacizumab at a dose of either 5 mg/kg every 2 weeks (FOLFOX, bFOL) or 7.5 mg/kg every 3 weeks (CapOx). The addition of bevacizumab resulted in higher response rates in all three arms of the TREE2 study compared to the TREE1 study, as overall response rates were seen in 34–49% of patients. There appeared to be no significant additive toxicity with bevacizumab.
It should be noted that patients were randomized within TREE1 and TREE2, but that patients were not randomized between these two studies. Therefore, conclusions regarding the benefit of bevacizumab should be made cautiously. Numerous additional studies are either ongoing or planned to further examine the role of anti-angiogenesis strategies in the upfront treatment of advanced CRC. An industry-sponsored trial (No. 16966) randomized approximately 1500 previously untreated patients to either CapOx or FOLFOX4, with or without bevacizumab (dosed at 5 mg/kg every 2 weeks with FOLFOX4 or at 7.5 mg/kg every 3 weeks with CapOx), with its primary end point being PFS. This predominantly European study easily completed accrual prior to the commercial availability of bevacizumab in Europe, despite the fact that a similar National Cancer Institute (NCI) Intergroup study opened in the United States and closed prematurely when it became apparent that American patients, who had access to bevacizumab commercially, would not accept potential treatment on a study with nonbiological containing arms. A large, phase III Intergroup trial coordinated by the Southwest Oncology Group (SWOG) and the Cancer and Leukemia Group B (CALGB) activated in September 2005. This trial permits investigators to select upfront chemotherapy for individual patients (either mFOLFOX6 or FOLFIRI), and then randomizes patients among concurrent biological therapy with bevacizumab, cetuximab, or both.
The success of bevacizumab and VEGF targeting has stimulated other strategies to block new blood vessel growth in tumors. The precise mechanism by which bevacizumab improves cancer chemotherapy in colorectal is not precisely known. Originally, such strategies were designed to “starve” the tumor by blocking its blood supply; however, bevacizumab is most effective in combination with blood-borne chemotherapy and it has relatively modest activity as single agents.
One alternative hypothesis is that VEGF targeting actually normalizes tumor vasculature and enhances drug delivery to the growing tumor cells. Nonetheless, this lack of a complete mechanistic understanding of VEGF-directed therapy has not deterred other anti-angiogenic strategies from entering clinical development. One novel strategy has targeted a large and diverse family of cell adhesion proteins called integrins. These are a complex superfamily of transmembrane glycoproteins that exist as heterodimers composed of α and β chains. Different heterodimers have different specificities of ligand binding and are associated with a variety of functions in a broad range of tissues. There are 18 known α integrin subunits and 8 β subunits, which associate to form at least 24 heterodimeric α/β pairs. Integrins have dual functions, serving as cell-surface adhesion structural proteins and as signaling receptors, although unlike receptor tyrosine kinases, they have no intrinsic enzyme activity. Integrins mediate cell–extracellular matrix and cell–cell interactions. Certain integrins can stimulate endothelial and tumor cell survival, whereas others promote cell aggregation and can regulate cell trafficking functions. They are postulated to have major but complex roles in tumor angiogenesis, invasion, and metastases. Some integrin subfamilies are functionally interrelated to signaling via the PI3 K/Akt pathway in some systems such as hormone resistant prostate cancer cells. The various integrin heterodimers have different ligand-binding specificities. For example, the fibronectin protein binds to α4β1 and α5β1 integrins, whereas lamin and collagen bind to α3β1, α2β1, and α1β1 integrins.
A number of integrin-targeting agents have entered clinical trials. One such agent is M200 (volociximab), a chimeric IgG4 anti-α5β1 antibody being developed by Protein Design Laboratories, Inc. The α5β1 integrin is involved in cell adhesion, migration, and enhanced cell survival. It is present on tumor endothelial cells and it helps to regulate growth factor-activated endothelial cells. The α5β1 integrin is also present on some tumor cells and on monocytes; however, it is not seen in endothelial cells from normal blood vessels. Inhibition of α5β1 fibronectin binding leads to apoptosis of activated endothelial cells, inhibition of cell migration, and prevention of tumor-associated neoangiogenesis. The M200 antibody blocks binding of α5β1 to fibronectin and it can inhibit tumor endothelial cell α5β1-mediated angiogenesis. In laboratory studies it can inhibit human endothelial cell tube formation and it blocks choroidal neo-vascularization in primate models. It also actively blocks blood vessel growth in xenografts in chicken chorioallantoic membrane (CAM) assays. M200 cross reacts with chicken and primate α5β1 but not with rodent integrins, making it inactive in standard rodent-based models. In a Phase I study of intravenously administered M200 infused over 1 h weekly, it was well tolerated with no serious dose-limiting adverse events. Pharmacokinetics were non-dose proportional with decreased clearance seen at higher dose levels consistent with possible saturation tissue kinetics. The overall plasma half-life was approx. 16 days. In the first six evaluated patients, a partial response was observed in a patient with renal cell cancer and prolonged stable disease lasting longer than 4 months was seen in patients with non-small cell lung cancer, melanoma, and parotid cancers. No disease stabilization was seen in the four patients with colon cancer enrolled in this study. Currently, M200 is in phase II studies in renal cell cancer.
Eisai Pharmaceuticals has adopted a strategy of using small-molecule inhibitors of integrins in the development of E7820, a novel oral sulfonamide agent that reduces the expression of α2, α3, α5, and β1 mRNA in tumor tissues. E7820 blocks VEGF- and bFGF-induced human umbilical vascular endothelial cell tube formation in laboratory experiments and it effectively inhibits the growth of human breast and pancreas cancer xenografts. In a phase I study of daily oral E7820, the agent was well tolerated with dose-proportional pharmacokinetics. Major dose-limiting toxicities consisted of grade 3 thrombocytopenia and elevation of hepatic transaminases. Prolonged disease stabilization lasting longer than 6 months was noted in four total patients, two with colorectal cancer and one with bladder cancer and malignant melanoma. Current studies are examining E7820 in combination with irinotecan, cetuximab, and other targeted therapies. In preclinical in vitro and in vivo data, marked synergy was observed when E7820 was combined with the other chemotherapeutic and targeted agents.
“Other integrin-targeting agents in clinical development include MEDI522 (Vitaxin®) a humanized IgG1 anti-αvβ3 antibody developed by MedImmune. It is in phase II trials against melanoma, prostate, leiomyosarcoma, and colorectal cancer. Cilengitide (EMD121974) is a cycle peptide inhibitor of αvβ3 and αvβ5 developed by Merck KGaA. This agent is in phase II studies in gliomas, melanoma, and prostate cancers. Finally, CNTO 95 is a fully human IgG1 anti-αvβ3 and anti-αvβ5 antibody that is being developed by Medarex/Centocor. Thus, this remains an active area of clinical development.”
13. Conclusion and prospects
Currently, much of the pharmaceutical industry’s resources are geared toward developing drugs that mimic the effects of cetuximab or bevacizumab in hitting these same targets to try to develop better and more effective therapies. Although advances and refinements in our current treatments will undoubtedly arise from these efforts, a larger question is what new molecular targets will be important for the development of future targeted therapies. For the purposes of the aforementioned discussion, I wish to speculate as to what types of agents and targets will yield the next generation of therapies for cancer. There is no absolute way to be precise in these predictions, any more than a venture capitalist can reliably pick a winner in the stock market. However, it is possible to make some highly educated guesses (Saltz, 2007). The pathways and the inherent targets discussed here reflect our personal biases and are influenced by our own direct experience in cancer drug development. Thus, it is likely that when this manuscript is reviewed in 5 or 10 years, its likelihood of being highly accurate is small. Nonetheless, it is both exciting and exhilarating to consider select areas of applied scientific research with the highest potential for yielding the next generation of clinical advances in cancer therapeutics. I certainly have great confidence that the current ongoing basic research in cancer biology ultimately will lead to tomorrow’s great therapeutic advances (Tables 1–3).
Table 1.
SU-5416 at a glimpse.
| Originator: Sugen Inc., USA |
| Status: Phases I, II and III clinical trials; discontinuation and withdrawal |
| Indication: Angiogenesis disorder; colorectal tumor; Kaposi’s sarcoma; lung tumor; melanoma; myeloid leukemia; myeloproliferative disorder; neoplasm; solid tumor |
| Action: Angiogenesis inhibitor; protein tyrosine kinase (PTK) inhibitor; MAP kinase (MAPK) inhibitor; VEGF antagonist |
| Synonyms: Flk-1/KDR RTK antagonists; SU-5416; angiogenesis inhibitors; Semaxanib |
Table 2.
Developmental history.
| Developer | Country | Status | Indication | Date |
|---|---|---|---|---|
| Sugen Inc. | USA | DR | Neoplasm | 09-Nov-95 |
| Sugen Inc. | USA | DR | Angiogenesis disorder | 09-Nov-95 |
| Taiho Pharmaceutical Co. Ltd. | Japan | DR | Neoplasm | 09-Nov-95 |
| Sugen Inc. | USA | C1 | Solid tumor | 01-Oct-97 |
| Sugen Inc. | USA | C2 | Kaposi’s sarcoma | 21-Sep-98 |
| Taiho Pharmaceutical Co. Ltd. | Japan | C1 | Solid tumor | 13-Nov-98 |
| Taiho Pharmaceutical Co. Ltd. | Japan | C2 | Kaposi’s sarcoma | 21-Sep-98 |
| Sugen Inc. | USA | C1 | Myeloproliferative disorder | 17-Nov-98 |
| Sugen Inc. | USA | C3 | Colorectal tumor | 01-May-99 |
| Sugen Inc. | USA | C3 | Lung tumor | 01-May-99 |
| Sugen Inc. | Western Europe | C2 | Kaposi’s sarcoma | 29-Sep-99 |
| Sugen Inc. | USA | C2 | Myeloid leukemia | 01-Dec-00 |
| Sugen Inc. | USA | DR | Melanoma | 29-May-01 |
Table 3.
Literature classifications.
| Study type | Effect studied | Experimental model | Result |
|---|---|---|---|
| Biology | |||
| In vitro/in vivo | Development of a strategy to combat ophthalmic diseases with angiogenesis | Retinal cultures, explants and in vivo eye model | Analysis of angiogenesis inhibitors for the treatment of ophthalmic neo-vascular diseases, such as age-related macular degeneration and diabetic retinopathy |
| Computational chemistry | Screening for a novel class of broadly applicable drug candidates for the inhibition of RTKs | Computational chemistry | The solution of the three-dimensional co-crystal structure of FGF RTK with two of Sugen’s TK inhibitors |
| In vitro/in vivo | Tyrosine kinase inhibition, proliferation indexes endothelium tumor growth | Receptor phosphorylation in3T3 cells 3H-thymidine uptake by EC, tumor growth of A375, A431, Calu-6 C6, LNCAP, PH4-VEGF, 3T3HER2, 488G2m2, SF763T, and SF67T | IC50 = 1.04 μM, 100-fold less than other TRKs. Reduced 3H uptake. Reduced growth of tumors except for SF763T and SF67T |
| Synthesis chemistry | Synthesis and biological evaluation of RTK inhibitors | Synthesis chemistry | 3-Subsituted indolin-2-ones exhibited selectivity toward particular RTKs |
| In vitro | Assessment of SU-5416 selectivity against Flk-1/KDR kinase | Human endothelial cells | SU-5416 was reported as a potent and selective Flk-1/KDR kinase inhibitor, which blockaded receptor auto-phosphorylation, endothelial cell mitogenesis, tumor growth, angiogenesis and micro-hemodynamics |
| In vitro/in vivo | Assessment of SU-5416 potency against a broader range of RTK kinases | HUVECs and mice transplant | The derivative 3,5-dimethyl pyrrole-analog was found to be the superior compound, with an IC50 = 40 nM. SU-5416 was also highly selective for Flk-1/KDR, being essentially inactive at other RTKs. This compound dramatically inhibited tumor size and neo-vascularization with a broader anti-tumor spectrum: i.p. administration blockaded eight out of ten different cell lines grown subcutaneously. Although the plasma half-life was short (30 min), the half-life for activity against Flk-1 was about 20 h, ostensibly due to the lipophilicity of the compound |
| In vivo | Assessment of subcutaneous tumor growth of cells derived from various tissues | Mice | Systemic administration of SU-5416 at non-toxic doses (25 mg/kg/day) resulted in the inhibition of subcutaneous tumor growth of cells derived from various tissue origins. The anti-tumor effect of SU-5416 was accompanied by the appearance of pale white tumors that were resected from drug-treated animals |
| In vitro/in vivo | Evaluation of the biochemical and biological functions of SU-5416 and other RTK inhibitors | C-KIT | SU-5416 and SU-6668 inhibited the biochemical and biological functions of C-KIT (IC50 = 0.1–1 μM) |
| In vivo | Evaluation of SU-5416 possible inhibition of Flt-1 receptor | Mice and rats | SU-5416 inhibited the TK VEGF receptor, Flt-1. SU-5416 and SU-6668 exhibited comparative differences in toxicity profile following intravenous administration |
| Metabolism | |||
| In vitro | Analysis of VEGF-stimulated thymidine uptake | Cultured human endothelial cells (HUVECs) | SU-5416 inhibited VEGF-stimulated thymidine uptake at 0.1 μM, whereas FGF-1-mediated thymidine uptake was inhibited only at a concentration of 10 μM |
| In vivo | Determination of toxicity metabolism and elimination | Patients with advanced malignances | SU-5416 in a dose-escalating trial exhibited mild toxicity, and the elimination t½ was approximately 50 min |
| In vivo | Assessment of SU-5416 half-life and prolonged effects | Mice and human tumor xenograft models | SU-5416 exhibited a short plasma half-life, but prolonged in vivo effects. This compound inhibited VEGF-dependent KDR phosphorylation by blockading subcellular localization |
| In vitro/in vivo | To investigate the metabolism of SU-5416 and its major metabolites by HPLC | Mouse, rat, dog, monkey and human liver microsomes | Microsomes, incubated with SU-5416 (25 μM) in the presence of NADPH-generating system, showed the compound’s conversion to at least 6 polar metabolites, where the overall rate of metabolism followed the rank monkey ⩾ mouse≈ rat > dog > human |
| In vivo | The effect of a combination of SU-5416 and fluorouracil/leucovorin on the progression of colon cancer | Patients with untreated metastatic colon cancer | SU-5416 (85 or 145 mg/m2) exhibited as response rate of 36% with as time to progression of 9 months, a time to failure of 8.5 months and a median survival time of 22.5 months |
| Clinical | ||
| Initiation of clinical trials with Flk-1 TK angiogenesis inhibitors | Patients with solid tumor cancers | The safety and efficacy of the company’s lead compound, SU-5416, were established |
| Initiation of phase I angiogenesis inhibitor study | Patients with solid tumors and tumor metastasis | The initiation of this study represented the first time a small-molecule inhibitor targeting the angiogenic mechanism has been administered in humans. This was carried out to assess the safety and dosage range of SU-5416, which was administered intravenously on a twice-weekly schedule. SU-5416 has been well tolerated at a dose of 4.4 to 65 mg/m2. The drug exhibited only mild toxicity, and the elimination t½ was approximately 50 min. In four out of five patients experiencing tumor stabilization, a decrease in tumor permeability and leakiness was demonstrated |
| Initiation of phase I/II clinical trial in Kaposi’s sarcoma | Patients (n = 30) with AIDS-related Kaposi’s sarcoma | The study endpoints, carried out at five US medical centers specializing in AIDS-related malignancies, included measurement of objective response and time-to-disease progression in addition to safety and pharmacokinetic parameters |
| Initiation of registrational studies with SU-5416 | Patients with Kaposi’s sarcoma, colorectal and basal cell cancer | Of the patients who remained in the study evaluating various doses, the majority exhibited well tolerance with mild negative repercussion. Expected side effects included nausea, vomiting, headache and transient increase in liver transaminases. Most patients showed stable disease for a prolonged period of time |
| Initiation of multiple phase II and I/II studies | Patients with solid tumors (including renal, head and neck, sarcoma, and prostate cancers) (n = 500–700) in approximately 20 leading cancer centers in the USA | The trials planned to test SU-5416 as either a single agent or in combination with other drugs in a variety of cancer indications |
| Entrance of SU-5416 into phase II/III trials | Patients with AIDS-related Kaposi’s sarcoma, colorectal and non-small cell lung cancer | The study offered multiple information of the drug’s potency, safety and efficacy |
| Phase II trial in combination with paclitaxel | Patients with angiosarcoma and cervical cancer | SU-5416 in combination with standard-dose paclitaxel (175 mg/m2, q3w) showed the regimen is tolerated, with no effect on paclitaxel pharmacokinetics |
| Phase I/II study in combination with 5-fluorouracil and leucovorin | Patients with metastatic colorectal cancer | Patients were found to be free of tumor growth and spread for a median time of 9.2 months |
Acknowledgments
The author thanks the IDDB center for providing invaluable information about the RTK inhibitor, SU-5416 (London, UK). Dr. John Haddad held the Georges John Livanos Trust fellowship (London) and the NIH postdoctoral fellowship award at UCSF (California, USA).
References
- Aiello L.P., Avery R.L., Arrigg P.G., Keyt B.A., Jampel H.D., Shah S.T., Pasquale L.R., Thieme H., Iwamoto M.A., Park J.E. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- Aiello L.P., Bursell S.E., Clermont A., Duh E., Ishii H., Takagi C., Mori F., Ciulla T.A., Ways K., Jirousek M., Smith L.E., King G.L. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective (-isoform-selective inhibitor. Diabetes. 1997;46:1473–1480. doi: 10.2337/diab.46.9.1473. [DOI] [PubMed] [Google Scholar]
- Airaksinen M.S., Titievsky A., Saarma M. GDNF family neurotrophic factor signaling: four masters, one servant? Mol. Cell. Neurosci. 1999;13(5):313–325. doi: 10.1006/mcne.1999.0754. [DOI] [PubMed] [Google Scholar]
- Antonian L., Yang C., Sukbuntherng J., Fong T.A.T., Shenoy N., Schreck R., Wagner G., Shawver L. The pharmacokinetic and pharmacodynamic properties of SU-5416, a novel VEGF receptor inhibitor. Proc. Am. Assoc. Cancer Res. 1999;40:388–394. [Google Scholar]
- Arasteh K., Hannah A. The role of vascular endothelial growth factor (VEGF) in AIDS-related Kaposi’s sarcoma. Oncologist. 2000;5:28–31. doi: 10.1634/theoncologist.5-suppl_1-28. [DOI] [PubMed] [Google Scholar]
- Arighi E., Borrello M.G., Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16(4-5):441–467. doi: 10.1016/j.cytogfr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Baloh R.H., Enomoto H. The GDNF family ligands and receptors – implications for neural development. Curr. Opin. Neurobiol. 2000;10(1):103–110. doi: 10.1016/s0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- Bhise, S.B., Abhijit, D. Nalawade, Hitesh Wadhawa, 2004. Role of protein tyrosine kinase inhibitors in cancer therapeutics. Ind. J. Biochem. Biophys. 41, 273–280. [PubMed]
- Bilder G.E., Rojas C.J. Inhibitors of the platelet-derived growth factor receptor tyrosine kinase. Cardiovasc. Drug Rev. 1996;14:380–399. [Google Scholar]
- Blanke C.D., Demetri G.D., von Mehren M. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 2008;26(4):620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- Bold G., Altmann K.H., Frei J., Lang M., Manley P.W., Traxler P., Wietfeld B., Bruggen J., Buchdunger E., Cozens R., Ferrari S., Furet P. New anilinophthalazines as potent and orally well-absorbed inhibitors of the VEGF receptor tyrosine kinases useful as antagonists of tumor-driven angiogenesis. J. Med. Chem. 2000;43:2310–2323. doi: 10.1021/jm9909443. [DOI] [PubMed] [Google Scholar]
- Brostjan C., Roka S., Gornikiewicz A., Friedl J. Anti-angiogenic approaches in tumor therapy. Acta Chir. Austriaca. 2000;32:251–254. [Google Scholar]
- Brown L.F., Harrist T.J., Yeo K.T., Stahle Backdahl M., Jackman R.W., Berse B., Tognazzi K., Dvorak H.F., Detmar M. Increased expression of vascular permeability factor (vascular endothelial growth factor) in bullous pemphigoid, dermatitis herpetiformis, and erythema multiforme. J. Invest. Dermatol. 1995;104:744–749. doi: 10.1111/1523-1747.ep12606974. [DOI] [PubMed] [Google Scholar]
- Coutts J.C., Gallagher J.T. Receptors for fibroblast growth factors. Immunol. Cell Biol. 1995;73(6):584–589. doi: 10.1038/icb.1995.92. [DOI] [PubMed] [Google Scholar]
- Dengjel J., Kratchmarova I., Blagoev B. Receptor tyrosine kinase signaling: a view from quantitative proteomics. Mol. BioSyst. 2009;5(10):1112–1121. doi: 10.1039/b909534a. [DOI] [PubMed] [Google Scholar]
- Deplanque G., Harris A.L. Anti-angiogenic agents: clinical trial design and therapies in development. Eur. J. Cancer A Gen. Top. 2000;36:1713–1724. doi: 10.1016/s0959-8049(00)00149-0. [DOI] [PubMed] [Google Scholar]
- Druker B.J., Talpaz M., Resta D.J., Peng B., Buchdunger E., Ford J.M., Lydon N.B., Kantarjian H., Capdeville R., Ohno-Jones S., Sawyers C.L. Efficacy and safety of a specific inhibitor of the BCR–ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001;344(14):1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Duchesne L., Tissot B. N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding. J. Biol. Chem. 2006;281(37):27178–27189. doi: 10.1074/jbc.M601248200. [DOI] [PubMed] [Google Scholar]
- Dumas J. Protein kinase inhibitors: emerging pharmacophores1997 – 2000. Expert. Opin. Ther. Pat. 2001;11:405–429. [Google Scholar]
- Ellis L.M., Takahashi Y., Liu W., Shaheen R.M. Vascular endothelial growth factor in human colon cancer: biology and therapeutic implications. Oncologist. 2000;5:11–15. doi: 10.1634/theoncologist.5-suppl_1-11. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other diseases. Nat. Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- Fong G.H., Rossant J., Gertsenstein M., Breitman M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- Fong T.A., Shawver L.K., Sun L., Tang C., App H., Powell T.J., Kim Y.H., Schreck R., Wang X., Risau W., Ullrich A. SU-5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59:99–106. [PubMed] [Google Scholar]
- Fong T.A.T., Shawver L.K., App H., Sun L., Tang C., Rice A., Kim Y.H., Schreck R., Chen J., Dowd B., Suto E., Vasile S., Wang X., Hirth K.P., McMahon G. SU-5416: a potent and selective Flk-1-KDR kinase inhibitor that blocks Flk-1 phosphorylation, endothelial cell mitogenesis, and tumor growth. Proc. Am. Assoc. Cancer Res. 1998;39:560–561. [Google Scholar]
- Gerber H.P., McMurtrey A., Kowalski J., Yan M., Keyt B.A., Dixit V., Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- Gunby R.H., Sala E., Tartari C.J., Puttini M., Gambacorti-Passerini C., Mologni L. Oncogenic fusion tyrosine kinases as molecular targets for anti-cancer therapy. Anticancer Agents Med. Chem. 2007;7(6):594–611. doi: 10.2174/187152007784111340. [DOI] [PubMed] [Google Scholar]
- Haddad J.J. Reduction-oxidation mediated regulation of MAPKp38-dependent pro-inflammatory cytokine biosynthesis: On the mechanism of glutathione as a novel immunoregulatory antioxidant thiol. Int. Arch. Biosci. 2001;1:1001–1013. [Google Scholar]
- Haddad J.J. Redox and oxidant-mediated regulation of apoptosis signaling pathways: immuno-pharmaco-redox conception of oxidative siege versus cell death commitment. Int. Immunopharmacol. 2004;4:475–493. doi: 10.1016/j.intimp.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Haluska P., Adjei A.A. Receptor tyrosine kinase inhibitors. Curr. Opin. Invest. Drugs. 2001;2:280–286. [PubMed] [Google Scholar]
- Hanks S.K., Quinn A.M., Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Hannah A. Trials underway, SU-5416, # 23680. Clin. Trials Monit. 1997;6:9–11. [Google Scholar]
- Harris A.L. Are angiostatin and endostatin cures for cancer? Lancet. 1998;351:1598–1599. doi: 10.1016/S0140-6736(98)22022-8. [DOI] [PubMed] [Google Scholar]
- Hoff P.M. A phase I study of escalating doses of the tyrosine kinase inhibitor Semaxanib (SU5416) in combination with irinotecan in patients with advanced colorectal carcinoma. Jpn. J. Clin. Oncol. 2006;36(2):100–103. doi: 10.1093/jjco/hyi229. [DOI] [PubMed] [Google Scholar]
- Joensuu H., Roberts P.J., Sarlomo-Rikala M., Andersson L.C., Tervahartiala P., Tuveson D., Silberman S., Capdeville R., Dimitrijevic S., Druker B., Demetri G.D. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N. Engl. J. Med. 2001;344(14):1052–1056. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- Keyt B.A., Nguyen H.V., Berleau L.T., Duarte C.M., Park J., Chen H., Ferrara N. Identification of vascular endothelial growth factor determinants for binding KDR and Flt-1 receptors. Generation of receptor-selective VEGF variants by site-directed mutagenesis. J. Biol. Chem. 1996;271:5638–5646. doi: 10.1074/jbc.271.10.5638. [DOI] [PubMed] [Google Scholar]
- King I.C., Feng M., Catino J.J. High-throughput assay for inhibitors of the epidermal growth factor receptor-associated tyrosine kinase. Life Sci. 1993;53:1465–1468. doi: 10.1016/0024-3205(93)90619-e. [DOI] [PubMed] [Google Scholar]
- Klagbrun M., D’Amore P.A. Regulators of angiogenesis. Annu. Rev. Physiol. 1991;53:217–239. doi: 10.1146/annurev.ph.53.030191.001245. [DOI] [PubMed] [Google Scholar]
- Kris M.G., Natale R.B., Herbst R.S., Lynch T.J., Prager D., Belani C.P., Schiller J.H., Kelly K., Spiridonidis H., Sandler A., Albain K.S., Cella D., Wolf M.K., Averbuch S.D., Ochs J.J., Kay A.C. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290(16):2149–2158. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- Krystal G.W., Honsawek S., Kiewlich D., Liang C., Vasile S., Sun Li, McMahon G., Lipson K.E. Indolinone tyrosine kinase inhibitors block kit activation and growth of small cell lung cancer cells. Cancer Res. 2001;61:3660–3668. [PubMed] [Google Scholar]
- Kuwai T., Nakamura T., Sasaki T., Kim S.J., Fan D., Villares G.J., Zigler M., Wang H., Bar-Eli M., Kerbel R.S., Fidler I.J. Phosphorylated epidermal growth factor receptor on tumor-associated endothelial cells is a primary target for therapy with tyrosine kinase inhibitors. Neoplasia. 2008;10(5):489–500. doi: 10.1593/neo.08200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Coutre P., Ottmann O.G., Giles F., Kim D.W., Cortes J., Gattermann N., Apperley J.F., Larson R.A., Abruzzese E., O’Brien S.G., Kuliczkowski K., Hochhaus A., Mahon F.X., Saglio G., Gobbi M., Kwong Y.L., Baccarani M., Hughes T., Martinelli G., Radich J.P., Zheng M., Shou Y., Kantarjian H. Nilotinib (formerly AMN107), a highly selective BCR–ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–1839. doi: 10.1182/blood-2007-04-083196. [DOI] [PubMed] [Google Scholar]
- Le Q., Duval D., Tobelem G. Tumor angiogenesis. Bailliere’s Clin. Hematol. 1993;6:711–730. doi: 10.1016/s0950-3536(05)80195-7. [DOI] [PubMed] [Google Scholar]
- Liekens S., De Clercqe E., Neyts J. Angiogenesis: regulators and clinical applications. Biochem. Pharmacol. 2001;61:253–270. doi: 10.1016/s0006-2952(00)00529-3. [DOI] [PubMed] [Google Scholar]
- Liekens S., De Clercqe, Neyts J. Angiogenesis: regulators and clinical applications. Biochem. Pharmacol. 2001;61:253–270. doi: 10.1016/s0006-2952(00)00529-3. [DOI] [PubMed] [Google Scholar]
- Lockhart A., Cropp G., Berlin J., Donnelly E., Schumaker R., Schaaf L., Hande K., Fleischer A., Hannah A., Rothenberg M. Phase I/pilot study of SU5416 (semaxinib) in combination with irinotecan/bolus 5-FU/LV (IFL) in patients with metastatic colorectal cancer. Am. J. Clin. Oncol. 2006;29(2):109–115. doi: 10.1097/01.coc.0000199882.53545.ac. [DOI] [PubMed] [Google Scholar]
- McMahon G. VEGF receptor signaling in tumor angiogenesis. Oncologist. 2000;5:3–10. doi: 10.1634/theoncologist.5-suppl_1-3. [DOI] [PubMed] [Google Scholar]
- McMahon G., Sun L., Liang C., Tang C. Protein kinase inhibitors: structural determinants for target specificity. Curr. Opin. Drug Discov. Dev. 1998;1:131–146. [PubMed] [Google Scholar]
- Mendel D.B., Laird A.D., Smolich B.D., Blake R.A., Liang C., Hannah A.L., Shaheen R.M., Ellis L.M., Weitman S., Shawver L.K., Cherrington J. Development of SU-5416, a selective small molecule inhibitor of VEGF receptor tyrosine kinase activity, as an anti-angiogenesis agent. J. Anti-Cancer Drug Des. 2000;15:29–41. [PubMed] [Google Scholar]
- Mendel D.B., Schreck R.E., West D.C., Li G., Strawn L.M., Tanciongco S.S., Vasile S., Shawver L.K., Cherrington J.M. The angiogenesis inhibitor SU-5416 has long-lasting effects on vascular endothelial growth factor receptor phosphorylation and function. Clin. Cancer Res. 2000;6:4848–4858. [PubMed] [Google Scholar]
- Mendel D.B., Schreck R., West D., Li G., Shawver L., Cherrington J. SU-5416, a small molecule VEGF receptor inhibitor, exhibits durable inhibition of VEGF-dependent phosphorylation of Flk-1/KDR in cells. Proc. Am. Assoc. Cancer Res. 2000;5:344–353. [Google Scholar]
- Millauer B., Shawver L.K., Plate K.H., Risau W., Ullrich A. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367:576–579. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- Mitsuyasu R.T. Update on the pathogenesis and treatment of Kaposi’s sarcoma. Curr. Opin. Oncol. 2000;12:174–180. doi: 10.1097/00001622-200003000-00013. [DOI] [PubMed] [Google Scholar]
- Myers S.M., Eng C. Characterization of RET proto-oncogene 3′ splicing variants and polyadenylation sites: a novel C-terminus for RET. Oncogene. 1995;11(10):2039–2045. [PubMed] [Google Scholar]
- Neufeld G., Cohen T., Gengrinovitch S., Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- O’Donnell A., Padhani A., Hayes C., Kakkar A., Leach M., Trigo J., Scurr M., Raynaud F., Phillips S., Aherne W., Hardcastle A., Workman P., Hannah A., Judson I. A Phase I study of the angiogenesis inhibitor SU5416 (Semaxanib) in solid tumours, incorporating dynamic contrast MR pharmacodynamic end points. Br. J. Cancer. 2005;93(8):876–883. doi: 10.1038/sj.bjc.6602797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T. Strategy for overcoming cancer through inhibition of angiogenesis. Biotherapy. 1995;9:494–500. [Google Scholar]
- Okamoto I. Epidermal growth factor receptor in relation to tumor development: EGFR-targeted anticancer therapy. FEBS J. 2010;277(2):309–315. doi: 10.1111/j.1742-4658.2009.07449.x. [DOI] [PubMed] [Google Scholar]
- Oliver S.J., Cheng T.P., Banquerigo M.L., Brahn E. Suppression of collagen-induced arthritis by an angiogenesis inhibitor, AGM-1470, in combination with cyclosporin: reduction of vascular endothelial growth factor (VEGF) Cell. Immunol. 1995;166:196–206. doi: 10.1006/cimm.1995.9978. [DOI] [PubMed] [Google Scholar]
- Ono M., Kuwano M., JAP J. Development of anti-tumor agents targeting angiogenesis. Cancer Chemother. 1997;24:178–186. [PubMed] [Google Scholar]
- Oo W.C., Baumgartner M. Angiogenesis inhibitors as wonder drug against cancer? Dtsch. Apoth. Ztg. 1998;138:48–49. [Google Scholar]
- Ornitz D.M., Itoh N. Fibroblast growth factors. Genome Biol. 2001;2(3) doi: 10.1186/gb-2001-2-3-reviews3005. Reviews 3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signaling networks. Nature. 1995;373(6515):573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- Petrone A., Sap J. Emerging issues in receptor protein tyrosine phosphatase function: lifting fog or simply shifting? J. Cell Sci. 2000;113:2345–2354. doi: 10.1242/jcs.113.13.2345. [DOI] [PubMed] [Google Scholar]
- Philip S. Angiogenesis inhibitors in oncology: the research continues. Cancer Pract. 2000;8:148–150. doi: 10.1046/j.1523-5394.2000.83010.x. [DOI] [PubMed] [Google Scholar]
- Pili R., Kruszewski M.P., Hager B.W., Lantz J., Carducci M.A. Combination of phenylbutyrate and 13-cis retinoic acid inhibits prostate tumor growth and angiogenesis. Cancer Res. 2001;61:1477–1485. [PubMed] [Google Scholar]
- Plate K.H. Gene therapy of malignant glioma via inhibition of tumor angiogenesis. Cancer Metastasis Rev. 1996;15:237–240. doi: 10.1007/BF00437477. [DOI] [PubMed] [Google Scholar]
- Puduvalli V.K., Sawaya R. Anti-angiogenesis – therapeutic strategies and clinical implications for brain tumors. J. Neuro Oncol. 2000;50:189–200. doi: 10.1023/a:1006469830739. [DOI] [PubMed] [Google Scholar]
- Quinn T.P., Peters K.G., De Vries C., Ferrara N., Williams L.T. Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc. Natl. Acad. Sci. USA. 1993;90:7533–7537. doi: 10.1073/pnas.90.16.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radha V., Nambirajan S., Swarup G. Association of Lyn tyrosine kinase with the nuclear matrix and cell-cycle-dependent changes in matrix-associated tyrosine kinase activity. Eur. J. Biochem. 1996;236(2):352–359. doi: 10.1111/j.1432-1033.1996.00352.x. [DOI] [PubMed] [Google Scholar]
- Raymond E. Tumor angiogenesis inhibitors, or the medias and science. Presse Med. 1998;27:1221–1224. [PubMed] [Google Scholar]
- Rinker S., Ke Y., Liu Y., Chhabra R., Yan H. Self-assembled DNA nanostructures for distance-dependent multivalent ligand-protein binding. Nat. Nanotechnol. 2008;3(7):418–422. doi: 10.1038/nnano.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson C.J., Stringer S.E. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 2001;114(5):853–865. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- Robinson D.R., Wu Y.M., Lin S.F. The protein tyrosine kinase family of the human genome. Oncogene. 2001;19(49):5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- Rosen, L., Kabbinavar, F., Rosen, P., Mulay, M., Quigley, S., Hannah, A. 1998. Phase I trial of SU-5416, a novel angiogenesis inhibitor, in patients with advanced malignancies. Proc. Am. Soc. Clin. Oncol. (Abs. 843).
- Ruetten H., Thiemermann C. Effects of tyrphostins and genistein on the circulatory failure and organ dysfunction caused by endotoxin in the rat: a possible role for protein tyrosine kinase. Br. J. Pharmacol. 1997;122(1):59–70. doi: 10.1038/sj.bjp.0701345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan C.J., Wilding G. Angiogenesis inhibitors: new agents in cancer therapy. Drugs Aging. 2000;17:249–255. doi: 10.2165/00002512-200017040-00001. [DOI] [PubMed] [Google Scholar]
- Saltz L.B., editor. Colorectal cancer: evidence-based chemotherapy strategies. Humana Press; Berlin: 2007. [Google Scholar]
- Schaller M.D., Borgman C.A., Cobb B.S., Vines R.R., Reynolds A.B., Parsons J.T. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl Acad. Sci. USA. 1992;89(11):5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetharam L., Gotoh N., Maru Y., Neufeld G., Yamaguchi S., Shibuya M. A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene. 1995;10:135–147. [PubMed] [Google Scholar]
- Shalaby F., Rossant J., Yamaguchi T.P., Gertsenstein M., Wu X.F., Breitman M.L., Schuh A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Shawver L.K. Tyrosine kinase inhibitors: from the emergence of targets to their clinical development. Asco Educ. Book. 1999;35:29–47. [Google Scholar]
- Shawver L.K., Lipson K.E., Fong T.A.T., McMahon G., Plowman G.D., Strawn L.M. Receptor tyrosine kinases as targets for inhibition of angiogenesis. Drug Discov. Today. 1997;2:50–63. [Google Scholar]
- Sielecki T.M., Boylan J.F., Benfield P.A., Trainor G.L. Cyclin-dependent kinase inhibitors: useful targets in cell cycle regulation. J. Med. Chem. 2000;43:1–18. [PubMed] [Google Scholar]
- Silvennoinen O., Saharinen P., Paukku K., Takaluoma K., Kovanen P. Cytokine receptor signal transduction through Jak tyrosine kinases and Stat transcription factors. APMIS. 1997;105(7):497–509. doi: 10.1111/j.1699-0463.1997.tb05047.x. [DOI] [PubMed] [Google Scholar]
- Sleeman M., Fraser J. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 2001;271(2):171–182. doi: 10.1016/s0378-1119(01)00518-2. [DOI] [PubMed] [Google Scholar]
- Smolich B.D., Yuen H.A., West K.A., Giles F.J., Albitar M., Cherrington J.M. The anti-angiogenic protein kinase inhibitors SU-5416 and SU-6668 inhibit the SCF receptor (C-KIT) in a human myeloid leukemia cell line and in acute myeloid leukemia blasts. Blood. 2001;97:1413–1421. doi: 10.1182/blood.v97.5.1413. [DOI] [PubMed] [Google Scholar]
- Soker S., Takashima S., Miao H.Q., Neufeld G., Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- Sordella R., Bell D.W., Haber D.A., Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Spivak J.L., Fisher J., Isaacs M.A., Hankins W.D. Protein kinases and phosphatases are involved in erythropoietin-mediated signal transduction. Exp. Hematol. 1992;20:500–504. [PubMed] [Google Scholar]
- Stover D.R., Lydon N.B., Nunes J.J. Recent advances in protein kinase inhibition: current molecular scaffolds used for inhibitor synthesis. Curr. Opin. Drug Discov. Dev. 1999;2:274–285. [PubMed] [Google Scholar]
- Strawn L.M., Shawver L.K. Tyrosine kinases in disease: overview of kinase inhibitors as therapeutic agents and current drugs in clinical trials. Exp. Opin. Invest. Drugs. 1998;7:553–573. doi: 10.1517/13543784.7.4.553. [DOI] [PubMed] [Google Scholar]
- Strawn L.M., mcmahon G., App H., Schreck R., Kuchler W.R., Longhi M.P., Hui T.H., Tang C., Levitzki A., Gazit A., Chen I., Keri G., Orfi L., Risau W., Flamme I., Ullrich A., Hirth K.P., Shawver L.K. Flk-1 as a target for tumor growth inhibition. Cancer Res. 1996;56:3540–3545. [PubMed] [Google Scholar]
- Sun L., McMahon G. Inhibition of tumor angiogenesis by synthetic receptor tyrosine kinase inhibitors. Drug Discov. Today. 2000;5:344–353. doi: 10.1016/s1359-6446(00)01534-8. [DOI] [PubMed] [Google Scholar]
- Sun L., Tran N., Tang F., App H., Hirth P., McMahon G., Tang C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J. Med. Chem. 1998;41:2588–2603. doi: 10.1021/jm980123i. [DOI] [PubMed] [Google Scholar]
- Sun L., Tran N., Liang C., Tang F., Rice A., Schreck R., Waltz K., Shawver L.K., mcmahon G., Tang C. Design, synthesis and evaluations of substituted 3-[(3- or 4-carboxyethylpyrrol-2-yl)methylidenyl]indolin-2-ones as inhibitors of VEGF, FGF and PDGF receptor tyrosine kinases. J. Med. Chem. 1999;42:5120–5130. doi: 10.1021/jm9904295. [DOI] [PubMed] [Google Scholar]
- Sun L., Tran N., Liang C., Hubbard S., Tang F., Lipson K., Schreck R., Zhou Y., McMahon G., Tang C. Identification of substituted 3-[(4,5,6,7-tetrahydro-1H-indol-2-yl)methylene]-1,3-dihydroindol-2-ones as growth factor receptor inhibitors for VEGF-R2 (Flk-1/KDR), FGF-R1, and PDGF-Rβ tyrosine kinases. J. Med. Chem. 2000;43:2655–2663. doi: 10.1021/jm9906116. [DOI] [PubMed] [Google Scholar]
- Teicher B.A. Molecular targets and cancer therapeutics: discovery, development and clinical validation. Drug Resist. Updat. 2000;3:67–73. doi: 10.1054/drup.2000.0123. [DOI] [PubMed] [Google Scholar]
- Toledo L.M., Lydon N.B., Elbaum D. The structure-based design of ATP-site directed protein kinase inhibitors. Curr. Med. Chem. 1999;6:775–805. [PubMed] [Google Scholar]
- Traxler P. Tyrosine kinase inhibitors in cancer treatment (Part II) Exp. Opin. Ther. Pat. 1998;8:1599–1625. [Google Scholar]
- Traxler P., Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999;82:195–206. doi: 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- Vacca A., Ribatti D., Pellegrino A., Dammacco F. Angiogenesis and anti-angiogenesis in human neoplasms: recent developments and therapeutic prospects. Ann. Ital. Med. Int. 2000;15:7–19. [PubMed] [Google Scholar]
- Vajkoczy P., Thurnher A., Hirth K.P., Schilling L., Schmiedek P., Ullrich A., Menger M.D. Measuring VEGF-Flk-1 activity and consequences of VEGF-Flk-1 targeting in vivo using intravital microscopy: clinical applications. Oncologist. 2000;5:16–19. doi: 10.1634/theoncologist.5-suppl_1-16. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I., Korner G., Ishai-Michaeli R., Bashkin P., Bar-Shavit R., Fuks Z. Extracellular matrix-resident growth factors and enzymes: possible involvement in tumor metastasis and angiogenesis. Cancer Metastasis Rev. 1990;9:203–226. doi: 10.1007/BF00046361. [DOI] [PubMed] [Google Scholar]
- Ware J.A., Simon M. Angiogenesis in ischemic heart disease. Nat. Med. 1997;3:158–164. doi: 10.1038/nm0297-158. [DOI] [PubMed] [Google Scholar]
- Wiley H.S., Burke P.M. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001;2(1):12–18. doi: 10.1034/j.1600-0854.2001.020103.x. [DOI] [PubMed] [Google Scholar]
- Yao Z.J., Ye B., Wu X.-W., Wang S., Wu L., Zhang Z.-Y., Burke T.R. Structure-based design and synthesis of small molecule protein tyrosine phosphatase inhibitors. Bioorg. Med. Chem. 1998;6:1799–1810. doi: 10.1016/s0968-0896(98)00140-0. [DOI] [PubMed] [Google Scholar]
- Zwick E., Bange J., Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr. Relat. Cancer. 2001;8(3):161–173. doi: 10.1677/erc.0.0080161. [DOI] [PubMed] [Google Scholar]