

RAB10 and RAB14 function at sequential steps of insulin-stimulated GLUT4 translocation to the plasma membrane. RAB14 functions upstream of RAB10 in GLUT4 sorting to the specialized transport vesicles, and RAB10 and its GAP protein comprise the main signaling module that regulates the accumulation of GLUT4 transport vesicles at the plasma membrane.
Abstract
Adipocyte glucose uptake in response to insulin is essential for physiological glucose homeostasis: stimulation of adipocytes with insulin results in insertion of the glucose transporter GLUT4 into the plasma membrane and subsequent glucose uptake. Here we establish that RAB10 and RAB14 are key regulators of GLUT4 trafficking that function at independent, sequential steps of GLUT4 translocation. RAB14 functions upstream of RAB10 in the sorting of GLUT4 to the specialized transport vesicles that ferry GLUT4 to the plasma membrane. RAB10 and its GTPase-activating protein (GAP) AS160 comprise the principal signaling module downstream of insulin receptor activation that regulates the accumulation of GLUT4 transport vesicles at the plasma membrane. Although both RAB10 and RAB14 are regulated by the GAP activity of AS160 in vitro, only RAB10 is under the control of AS160 in vivo. Insulin regulation of the pool of RAB10 required for GLUT4 translocation occurs through regulation of AS160, since activation of RAB10 by DENND4C, its GTP exchange factor, does not require insulin stimulation.
INTRODUCTION
Regulated trafficking of the glucose transporter 4 (GLUT4) in adipose and muscle tissue in response to insulin is necessary for physiological regulation of glucose homeostasis (Abel et al., 2001; Dugani and Klip, 2005; Graham and Kahn, 2007; Huang et al., 2007), and disruption of this process can lead to the insulin resistance that underlies type 2 diabetes (Abel et al., 2001; Chen et al., 2011a). Work from our lab and others led to the development of a dual-cycle model of insulin-regulated GLUT4 trafficking in adipocytes (Blot and McGraw, 2008a; Dugani et al., 2008; Martin et al., 2000, 2006). In the basal, non–insulin-stimulated condition, GLUT4 is sorted from the endosome to either a perinuclear storage compartment or specialized GLUT4 storage vesicles (GSVs; Blot and McGraw, 2008a). Cycling between the endosome and these two compartments leads to retention of GLUT4 inside the cell under basal conditions. A slow rate of cycling between the GSV compartment and the plasma membrane (PM) exists, although GSVs do not efficiently engage with the PM in the basal state (Dugani and Klip, 2005; Xiong et al., 2010). In addition, some GLUT4 traffics to the PM in the basal state via a constitutive, transferrin receptor (TR)-containing, general trafficking pathway between the endosome and the PM (Blot and McGraw, 2008a; Xiong et al., 2010; Chen et al., 2012).
Insulin signaling stimulates GSV recruitment to and fusion with the PM. Briefly, insulin binding to its receptor on the cell surface stimulates a signaling cascade, one result of which is phosphorylation of the GTPase-activating protein (GAP) AS160 by its upstream kinase, AKT (Zeigerer et al., 2004; Gonzalez and McGraw, 2009). In basal conditions, the activity of the GAP domain of AS160 maintains its targets, small-GTPase Rab proteins, in an inactive, GDP-bound state. On insulin stimulation, the phosphorylation of AS160 by AKT renders the GAP domain of AS160 inactive, thereby relieving the repression on target Rab proteins and leading to an increase in their active, GTP-bound forms (Sano et al., 2003, 2007, 2011; Zeigerer et al., 2004; Dugani and Klip, 2005; Eguez et al., 2005; Larance et al., 2005). Active Rabs, located on GLUT4 vesicles, in turn promote the recruitment to and subsequent functional engagement or docking of GSVs with the PM, thus preparing vesicles for fusion (Gonzalez and McGraw, 2006; Xiong et al., 2010). Of interest, the relevant Rab proteins appear to differ between myocytes and adipocytes, leading to tissue specificity of the downstream GLUT4 trafficking pathway (Ishikura et al., 2007; Ishikura and Klip, 2008; Sun et al., 2010). In addition to AKT-AS160–regulated GSV accumulation at the PM, insulin also stimulates the redistribution of GLUT4 from the perinuclear storage compartment to the endosome (Zeigerer et al., 2004) and a modest increase in translocation of endosomal TR-positive vesicles to the PM (Blot and McGraw, 2008a; Chen et al., 2012). Fusion of GSVs in response to insulin stimulation results in an increase in the amount of GLUT4 on the cell surface, thereby increasing transport of glucose into the cell (Huang et al., 2007). Insulin signaling also inhibits GLUT4 endocytosis (Jhun et al., 1992; Czech and Buxton, 1993), ensuring a longer duration of GLUT4 residence in the PM. In vivo, increased GLUT4 on the cell surface of adipocytes via this pathway represents a mechanism by which glucose is removed from the blood in response to insulin after a meal (Abel et al., 2001; Dugani and Klip, 2005; Graham and Kahn, 2007; Chen et al., 2011a).
Although it is likely that AS160 GAP activity has more than one target (Miinea et al., 2005), RAB10 has emerged as the primary adipocyte Rab protein regulating GSV translocation downstream of AS160. First, it was found that the AS160 GAP domain has activity toward RAB10 (Miinea et al., 2005), prompting an investigation of the role of RAB10 in GLUT4 trafficking. Small interfering RNA (siRNA)–mediated knockdown of RAB10, but not other candidate Rab proteins, inhibits GLUT4 translocation in 3T3-L1 adipocytes (Sano et al., 2007, 2008) and rescues the AS160 knockdown phenotype of increased GLUT4 on the PM under basal conditions (Sano et al., 2007), providing strong evidence that RAB10 is the Rab protein normally maintained in the GDP-bound (inactive) state by AS160 to achieve GLUT4 basal retention (Eguez et al., 2005). RAB10 acts in the GSV trafficking pathway rather than in the regulation of general trafficking, as knockdown of RAB10 has no effect on TR trafficking (Sano et al., 2007), and recent studies suggest that GLUT4-positive, TR-negative vesicles that fuse with the PM in response to insulin (i.e., GSVs) are predominantly loaded with RAB10 (Chen et al., 2012). When RAB10 is inactivated, it must be reloaded with GTP for another round of activation; this is accomplished by the guanine nucleotide exchange factor (GEF) DENND4C, and knockdown of DENND4C inhibits GLUT4 translocation (Yoshimura et al., 2010; Sano et al., 2011). Finally, experiments using total internal reflection fluorescence (TIRF) microscopy suggest that RAB10 functions to recruit and/or dock GSVs near the PM rather than to regulate the final fusion step of GLUT4 translocation (Sano et al., 2007). In spite of this evidence, controversy remains regarding the role of RAB10 in this process. Of note, there are no reports that insulin stimulation promotes an increase in the proportion of RAB10 in the active, GTP-bound state, as expected if RAB10 were the target of the AS160 GAP activity that is inhibited by insulin signaling. In addition, RAB8A and not RAB10 appears to be the relevant target of AS160 in muscle cells (Ishikura et al., 2007; Ishikura and Klip, 2008; Randhawa et al., 2008; Sun et al., 2010). Given the importance of GLUT4 trafficking for normal glucose homeostasis (Abel et al., 2001; Dugani and Klip, 2005; Graham and Kahn, 2007; Chen et al., 2011a), a key regulatory factor such as RAB10 could represent an important drug target in the development of treatments for type 2 diabetes. Therefore it is essential that the role of the AS160-RAB10 signaling module be explored and defined.
Insulin regulates the various steps of GLUT4 trafficking independently (Gonzalez and McGraw, 2006; Martin et al., 2006; Bai et al., 2007; Fujita et al., 2010; Xiong et al., 2010). In addition to the AS160-RAB10 signaling module regulation of GSV accumulation at the plasma membrane, several other proteins have been identified to regulate other steps of GLUT4 traffic in adipocytes, including sorting of GLUT4 into GSVs (Jordens et al., 2010), transport of GSVs to the PM (Chen et al., 2007, 2012), functional engagement of GSVs with the PM (docking; Inoue et al., 2003; Gonzalez and McGraw, 2006; Chen et al., 2007; Lizunov et al., 2009; Xiong et al., 2010), fusion of GSVs with the PM (Kanda et al., 2005; Brandie et al., 2008; Smithers et al., 2008; Xie et al., 2011), and recycling of GLUT4 back into the endosome upon internalization (Guilherme et al., 2004; Shi et al., 2010; Li et al., 2012). To develop a detailed mechanistic understanding of the role of AS160-RAB10 in GLUT4 trafficking and examine the roles of other proteins reportedly involved in this process, we performed experiments investigating RAB10, as well as selected factors reported to participate in GLUT4 translocation at various steps. We found that RAB14 acts to regulate GLUT4 sorting into GSVs, whereas in our experimental system RalA, MUNC18C, and CDP138 did not have a role in the regulation of GSV trafficking. Our results demonstrate that by regulating GLUT4 sorting and GSV activity, RAB14 and RAB10 act sequentially to regulate the vital process of adipocyte glucose uptake.
RESULTS
AS160-RAB10 signaling module regulates GLUT4 translocation by controlling a prefusion vesicle accumulation step
In previous work we showed that overexpression of RAB10 increases GLUT4 in the plasma membrane of basal unstimulated adipocytes (Sano et al., 2007), and others and we have shown that RAB10 knockdown blunts insulin-stimulated GLUT4 translocation to the plasma membrane (Sano et al., 2007, 2008; Chen et al., 2012). Those results support the hypothesis that RAB10 is limiting in controlling the amount of GLUT4 in the plasma membrane of adipocytes. To test this hypothesis, we determined the relationship between the amount of GLUT4 in the plasma membrane and the amount of RAB10 expressed at the single-cell level. An siRNA-resistant FLAG-RAB10 construct, which rescues RAB10 knockdown (Supplemental Figure 1, A and B), was ectopically expressed in cells constitutively expressing a short hairpin RNA (shRNA) directed against RAB10 (RAB10 KD cells; Sano et al., 2007). GLUT4 trafficking in basal and insulin-stimulated conditions was compared with the level of RAB10 expression determined by quantitative anti-FLAG immunofluorescence. Cells with more FLAG-RAB10 showed a greater degree of GLUT4 translocation when stimulated with two different physiological concentrations of insulin (0.2 and 1 nM; Figure 1A). The data were binned into quartiles of RAB10 expression: RAB10 levels varied significantly between quartiles, and overall a large range of RAB10 levels was represented (Figure 1B). The effect appeared to saturate at the level of RAB10 expression achieved in Q3. GLUT4 translocation was somewhat blunted at the highest levels of RAB10 overexpression, perhaps due to high levels of ectopic overexpression disrupting the GTP–GDP cycling of RAB10. Alternatively, perhaps there is a limit to the number of molecules of RAB10 that can be inserted into the GSV membrane, and thus higher levels of RAB10 may not have an additional effect on GLUT4 translocation. Nonetheless, together these data suggest that RAB10 regulates GLUT4 translocation in single cells in a dose-dependent, nonlinear manner under both basal and insulin-stimulated conditions. Further, previous studies of RAB10 overexpression (Sano et al., 2007) may have underestimated this effect, as the mean increase over all quartiles would be less than that seen in the highest quartiles of RAB10 overexpression.
FIGURE 1:
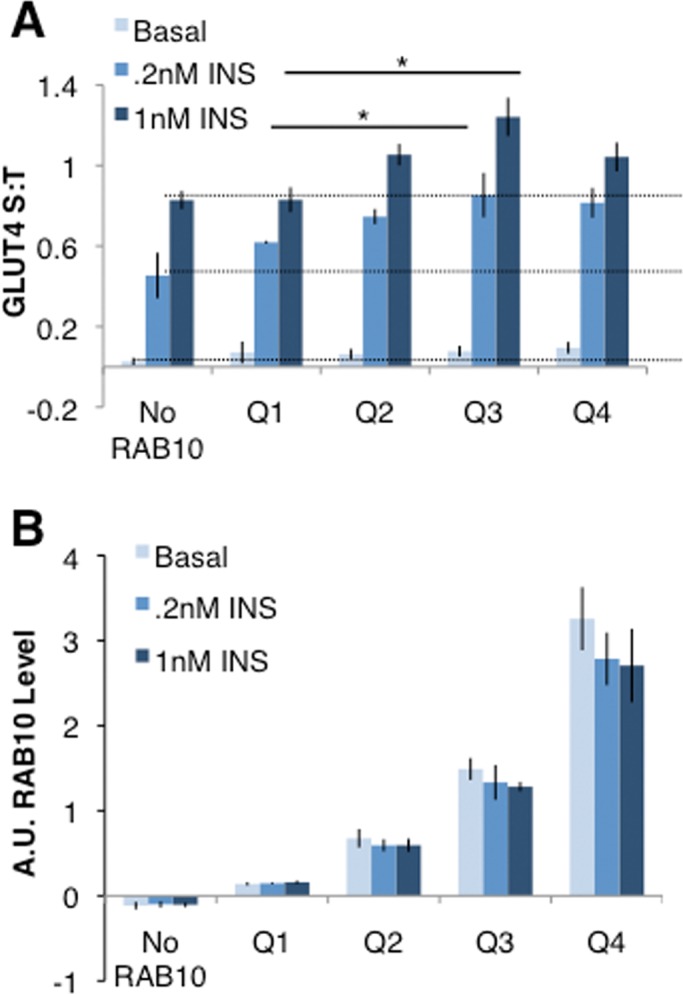
RAB10 overexpression increases GLUT4 translocation in single cells. (A) RAB10 KD cells electroporated with or without FLAG-RAB10 DNA were mixed and then plated for GLUT4 translocation assays. Cells were immunostained for FLAG expression to quantify RAB10 protein level, and data grouped accordingly. No RAB10, cells with FLAG staining below background. Within each experiment, S:T values for each cell were normalized to the mean S:T value for all cells in the 1 nM insulin condition. Mean normalized S:T values ± SEM, N = 3 independent assays. (B) RAB10 levels varied significantly between quartiles measured in A. Immunostaining for FLAG-RAB10 was quantified, and values for each cell were normalized to the average FLAG-RAB10 level measured in all cells quantified. Each quartile of RAB10 expression is significantly different from the quartile next to it; for example, Q4 has more RAB10 than Q3, and Q3 has more RAB10 than Q2. p > 0.05 in each case.
One element of the controversy surrounding RAB10 as a regulator of GLUT4 trafficking rests on the failure to show that the proportion of RAB10-GTP increases upon insulin stimulation (Sano et al., 2008). On the basis of our finding that RAB10 overexpression increases insulin-stimulated GLUT4 translocation, we reasoned that in unstimulated cells in which AS160 is depleted by constitutive expression of an AS160-directed shRNA (AS160 KD cells; Eguez et al., 2005), the proportion of GTP-bound RAB10 is presumably increased. Therefore in such cells, RAB10 overexpression could further increase surface GLUT4 levels even in the absence of insulin. This approach removes a portion of the normal inhibition on the system, allowing us to characterize the effect of RAB10 overexpression without the other effects of insulin. Overexpression of the functional pcDNA-RAB10 in unstimulated AS160 KD adipocytes resulted in a 9.5-fold increase of plasma membrane GLUT4 over that seen in control cells, which was significantly larger than the effects of AS160 knockdown alone or RAB10 overexpression in wild-type adipocytes (Figure 2A). These manipulations induced increases of 4.8- and 1.7-fold, respectively. When treated with 1 nM insulin, AS160 KD cells overexpressing RAB10 showed a trend toward increased GLUT4 translocation when compared with control cells. This was not statistically significant, however, perhaps because much of the GLUT4 present was already on the surface before insulin stimulation. In these studies RAB10 was overexpressed by about threefold (Figure 2B). The amount of TR in the plasma membrane, a marker of general endocytic traffic, was not affected by RAB10 overexpression in control or AS160 KD adipocytes (Figure 2C), establishing that the effect of RAB10 overexpression on GLUT4 is not due to effects on general membrane trafficking. Together these data strongly argue for the specific role of RAB10 in insulin regulation of GLUT4 translocation.
FIGURE 2:

The AS160/RAB10 signaling module is a key regulator of GLUT4 translocation. (A) AS160 KD combined with RAB10 overexpression drives GLUT4 translocation without insulin. S:T values normalized to GLUT4 in 1 nM insulin–treated 3T3-L1 control cells determined in each individual experiment. Mean normalized S:T values ± SEM, N = 10 or 11 assays. (B) Western blot showing overexpression of RAB10 and knockdown of AS160 from cells assayed in A. (C) TR trafficking is not affected by RAB10 overexpression or AS160 knockdown. S:T values normalized to TR S:T in basal 3T3-L1 cells determined in each individual experiment. Mean normalized S:T values ± SEM, N = 3 assays. (D) DENND4C KD rescues the basal retention defect of AS160 KD cells to a similar extent as RAB10 KD. S:T values normalized to GLUT4 in basal AS160 KD cells determined in each individual experiment. Mean normalized S:T values ± SEM, N = 4–7 assays. Basal S:T, AS160 KD vs. AS160 KD + DENND4C KD, p = 0.011. *p < 0.05, two-tailed paired t test, nonnormalized raw data. NS, not significant with p > 0.05, two-tailed paired t test, nonnormalized raw data.
The GTP/GDP status of RAB10 is regulated not only by AS160, but also by the primary GEF responsible for activating RAB10 in adipocytes, DENND4C (Yoshimura et al., 2010; Sano et al., 2011). Previous reports suggest that DENND4C is constitutively active (i.e., not regulated by insulin; Sano et al., 2008), and the effect of RAB10 overexpression on GLUT4 trafficking in unstimulated AS160 KD adipocytes provides functional evidence that insulin regulation of RAB10 activity is principally via inhibition of the GAP rather than activation of the GEF. To investigate this possibility, we sought to remove DENND4C from AS160 KD cells: if the GEF for RAB10 is constitutively active rather than requiring activation by insulin, then knockdown of DENND4C should reduce the increased surface GLUT4 seen in basal AS160 KD cells, even in the absence of insulin. Using previously validated siRNA sequences (Sano et al., 2011), we found that DENND4C knockdown resulted in a blunting of GLUT4 translocation in cells stimulated by 1 nM insulin (Supplemental Figure 1C). Next, we knocked down DENND4C in AS160 KD cells. Supporting our hypothesis, knockdown of DENND4C in AS160 KD cells reduced the amount of GLUT4 in the plasma membrane of AS160 KD cells under basal conditions (Figure 2D). This reduction in plasma membrane GLUT4 was similar in magnitude to that achieved by knockdown of RAB10 in AS160 KD cells or rescue of AS160 KD cells with FLAG-tagged human AS160 (FLAG-AS160), providing evidence that DENND4C is the constitutively active, functional GEF for RAB10 in adipocytes.
Given the important, synergistic interaction of AS160 and RAB10, we next asked whether AS160 and RAB10 would act to control a similar step in GLUT4 translocation, as would be predicted by the role of AS160 as the GAP for RAB10 (Miinea et al., 2005; Sano et al., 2007). Using a variety of TIRF microscopy assays, it has been shown that AS160 is involved in the prefusion, accumulation/docking steps of translocation. Overexpression of an AS160 mutant that cannot be phosphorylated by AKT (AS160-4A) results in near-complete blockade of accumulation of GLUT4-containing vesicles near the PM (Zeigerer et al., 2004; Miinea et al., 2005; Bai et al., 2007; Sano et al., 2007; Xiong et al., 2010; Chen et al., 2012). If RAB10 is the functionally relevant target of AS160 in adipocytes, we predicted that RAB10 knockdown would impair prefusion steps of GLUT4 translocation, phenocopying the effect of AS160 knockdown.
In previous work, we showed that RAB10 knockdown inhibits GLUT4 accumulation at the PM in response to 170 nM insulin (Sano et al., 2007). To confirm that this result generalizes to more physiologically relevant conditions and to ask whether RAB10 also regulates insertion of GLUT4 into the PM, we performed TIRF microscopy assays to measure GLUT4 translocation, accumulation at the PM, and insertion into the PM in control and RAB10 KD cells (Figure 3, A and B). In these experiments, a GLUT4 construct with both a hemagglutinin (HA) tag and a green fluorescent protein (GFP; HA-GLUT4-GFP; see Materials and Methods) is electroporated into cells, just as for our standard GLUT4 translocation assays. After stimulation with insulin and fixation, four images are taken of the same cell: total GLUT4 (epifluorescence GFP), surface GLUT4 (epifluorescence anti-HA staining), accumulated GLUT4 (TIRF GFP), and inserted GLUT4 (TIRF anti-HA staining). Fluorescence is quantified from all four images and ratios calculated to measure the proportion of total GLUT4 on the surface (S:T), the proportion of total GLUT4 accumulated near the PM (A:T), the proportion of GLUT4 accumulated near the PM that is inserted into the PM (I:A), and the proportion of total GLUT4 that is inserted into the PM (I:T).
FIGURE 3:
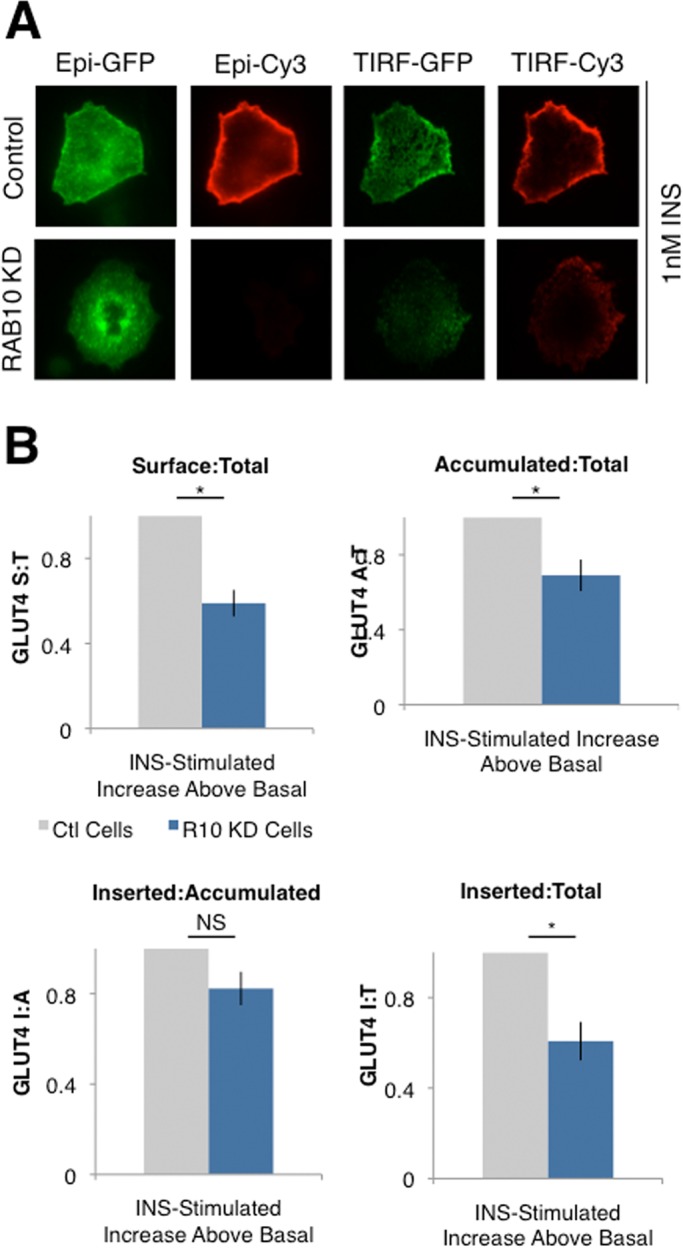
RAB10 regulates the accumulation/docking step of GLUT4 translocation. (A) TIRF microscopy was used to quantify total GLUT4 (Epi-GFP), surface GLUT4 (Epi-Cy3), GLUT4 accumulated in the TIRF zone (∼200 nm below the PM; TIRF-GFP), and GLUT4 inserted into the PM (TIRF-Cy3). Representative images of cells from the experiments in B. RAB10 KD cells with approximately equal amounts of total GLUT4 have less GLUT4 in the TIRF zone and on the cell surface after 1 nM insulin treatment. (B) Compared to control cells, RAB10 KD cells treated with 1 nM insulin have a lower proportion of total GLUT4 accumulated near the PM, resulting in a decreased proportion of GLUT4 on the surface/inserted into the PM. The proportion of total GLUT4 on the surface (Epi-Cy3/Epi-GLUT4; S:T, top left), the proportion of total GLUT4 accumulated near the PM (TIRF-GFP/Epi-GFP; Accumulated:Total, top left), the proportion of GLUT4 near the PM that is on the surface (TIRF-Cy3/TIRF-GFP, Inserted:Accumulated, bottom left), and the proportion of total GLUT4 inserted into the PM (TIRF-Cy3/Epi-GLUT4, Inserted:Total, bottom right) were calculated based on quantification of images such as those shown in A. Data are expressed as insulin-stimulated increase over basal conditions. RAB10 KD cell values are normalized to control cell values in each assay, mean ± SEM of N = 6 assays. *p < 0.05, two-tailed paired t test, nonnormalized raw data. NS, not significant with p > 0.05, two-tailed paired t test, nonnormalized raw data.
Using these methods, we found that the steady-state reduction of GLUT4 in the plasma membrane of insulin-stimulated RAB10 KD cells was accounted for largely by a decrease in the proportion of total GLUT4 accumulated near the PM (in the TIRF zone, ∼200 nm below the PM), whereas RAB10 knockdown did not have a statistically significant effect on the insulin-stimulated increase in GLUT4 insertion efficiency (the proportion of GLUT4 in the TIRF zone that is inserted into the PM; Figure 3C). There was a significant effect of RAB10 knockdown on the proportion of total GLUT4 inserted into the membrane, but this is to be expected, given that less GLUT4 accumulates near the PM in RAB10 KD cells (Figure 3C).
Together with previous work (Gonzalez and McGraw, 2006; Fujita et al., 2010; Xiong et al., 2010), the foregoing data support the conclusion that RAB10 activation downstream of AS160 inhibition controls the accumulation of GLUT4 vesicles near the PM.
Overexpression of AS160 and RAB10 enhances insulin-regulated trafficking of GLUT4 ectopically expressed in CHO cells
We used the approach of overexpression and reduced expression of AS160-RAB10 in adipocytes to identify the roles of these components in the specialized machinery that regulates GLUT4 traffic. A complementary approach is to determine whether these proteins, when expressed in non–insulin-responsive cells, create a robust, insulin-regulated trafficking pathway. The fibroblast-like CHO cells represent a good model for such studies. These cells do not normally express GLUT4, but when GLUT4 is ectopically expressed its traffic is regulated by insulin (compared with TR) but not to the same degree as in fat cells (Lampson et al., 2000). Of importance, when expressed in CHO cells, GLUT4 is sorted away from TR to specialized recycling vesicles that ferry it to the plasma membrane. Although in unstimulated CHO cells GLUT4 is retained intracellularly better than the TR, it is not retained as well as in adipocytes, leading to the hypothesis that differences in both basal retention and insulin responsiveness contribute to the differences in GLUT4 trafficking between CHO cells and adipocytes (Lampson et al., 2000). These differences could be caused by differential endogenous expression of AS160 and/or RAB10. We compared AS160 and RAB10 expression in CHO cells to that seen in 3T3-L1 adipocytes and found that 3T3-L1 adipocytes expressed 3-to 3.5-fold more of each protein than CHO cells (Supplemental Figure 2).
To test the hypothesis that the AS160-RAB10 signaling module can independently regulate GLUT4 trafficking, we transfected CHO cells with FLAG-AS160, pcDNA-RAB10, or both and performed translocation assays on these CHO cells. In these experiments, we expected CHO cells to be less insulin responsive than 3T3-L1 adipocytes (Lampson et al., 2000), and so we used a higher concentration of insulin (10 nM). We found that the addition of RAB10 to CHO cells modestly increased insulin-stimulated GLUT4 translocation over CHO cells transfected with only HA-GLUT4-GFP (Figure 4A). Addition of AS160 to CHO cells conferred basal retention to the CHO cells, lowering the proportion of GLUT4 on the surface under basal conditions (Figure 4A). The addition of both RAB10 and AS160 to CHO cells recapitulated both basal retention and insulin-stimulated GLUT4 translocation (Figure 4A). The basal retention seen in CHO cells with the addition of AS160 suggests that AS160 may be sufficient to mediate this aspect of normal GLUT4 trafficking.
FIGURE 4:
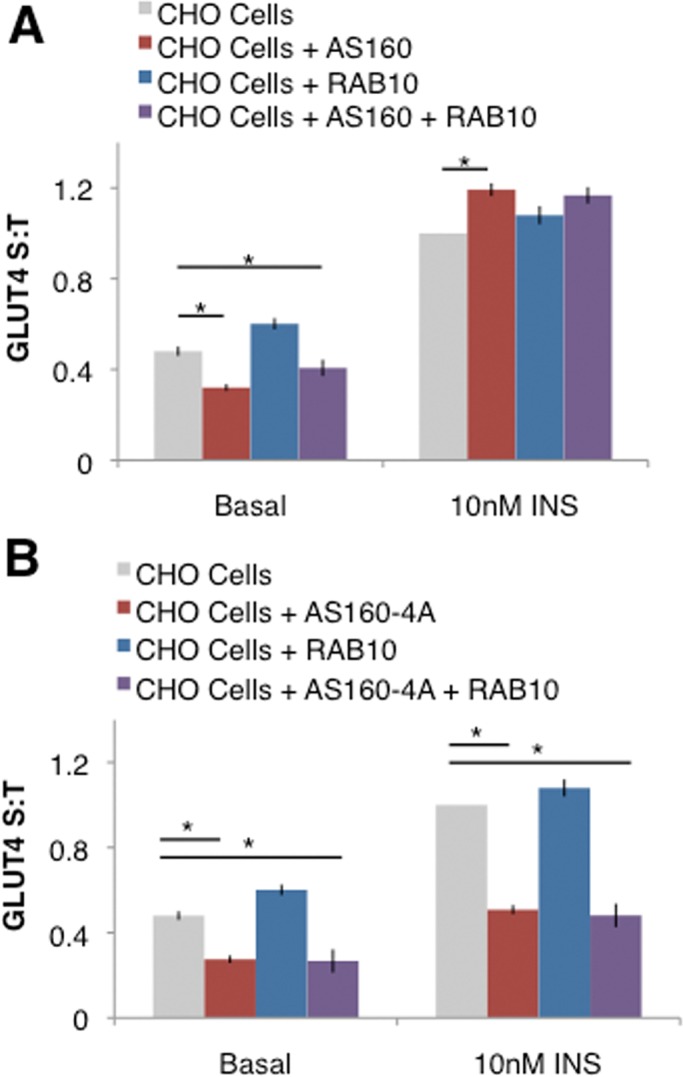
The AS160-RAB10 signaling module is partially sufficient for GLUT4 translocation. (A) Overexpression of FLAG-AS160 confers basal retention, and overexpression of pcDNA-RAB10 augments insulin-stimulated GLUT4 translocation in CHO cells. S:T values normalized to GLUT4 in insulin-stimulated CHO cells determined in each individual experiment. Mean normalized S:T values ± SEM, N = 4 assays. (B) Expression of constitutively active FLAG-AS160-4A in CHO cells confers basal retention but blocks insulin-stimulated GLUT4 translocation. S:T values normalized to GLUT4 in insulin-stimulated CHO cells determined in each individual experiment. Mean normalized S:T values ± SEM, N = 4–8 assays. *p < 0.05, one-tailed paired t test, nonnormalized raw data.
To confirm that the AS160 and RAB10 transfected into CHO cells in the preceding experiment are regulating GLUT4 in a similar manner to endogenous AS160 in 3T3-L1 adipocytes, we next transfected cells with the constitutively active AS160-4A, which is not inhibited by insulin stimulation due to the conversion of some of the AKT phosphorylation sites to alanines (Sano et al., 2003). 3T3-L1 adipocytes overexpressing AS160-4A show normal basal retention but lack insulin-stimulated GLUT4 translocation (Sano et al., 2003). We found a similar response in CHO cells transfected with AS160-4A: addition of AS160-4A to CHO cells conferred basal retention of GLUT4 to these cells; however, insulin-stimulated GLUT4 translocation was blocked (Figure 4B). This suggests that CHO cells can regulate GLUT4 translocation similarly to 3T3-L1 adipocytes when AS160 and RAB10 are added. Taken together, these experiments suggest that AS160 and RAB10 are partially sufficient for the normal regulation of GLUT4 translocation.
Regulation of GLUT4 traffic in addition to AS160-RAB10
The importance of the AS160-RAB10 signaling module in GLUT4 translocation is also supported by recent reports that the GSVs that translocate to and fuse with the PM in response to insulin are predominantly loaded with RAB10 and that, upon insulin simulation, these vesicles translocate to the PM and dock there in an AS160-dependent manner (Chen et al., 2012). Together with present and previous work (Sano et al., 2007, 2008; Chen et al., 2012), these data argue that the AS160-RAB10 signaling module is a principal regulator of GSV accumulation at the PM. Insulin regulates several independent steps in the GLUT4 translocation process (Gonzalez and McGraw, 2006; Fujita et al., 2010; Xiong et al., 2010), however, and several studies identified proteins in addition to AS160-RAB10 that regulate GLUT4 trafficking (Figure 5A). Indeed, the fact that the increase of GLUT4 in the plasma membrane of basal AS160 KD cells induced by overexpression of RAB10 is further increased by stimulation with 1 nM insulin (Figure 2A) suggests that although the AS160-RAB10 signaling module is crucial for normal insulin-stimulated GLUT4 translocation, additional factors are required for the full effect of insulin on GLUT4.
FIGURE 5:

RalA, MUNC18C, and CDP138 are not involved in GLUT4 translocation. (A) Schematic of regulated GLUT4 translocation. GLUT4 traffics to the PM via two pathways: a constitutive pathway shared with TR with a relatively fast rate constant under basal conditions and a modest increase in rate upon insulin stimulation, as well as a specialized GSV pathway with a slow rate of GLUT4 exocytosis under basal conditions but a fast rate when stimulated with insulin. The TELEY motif on GLUT4 promotes sorting of GLUT4 into GSVs, whereas the FQQI motif governs sorting of GLUT4 between the endosome and a TGN retention compartment. (B) Representative Western blot showing RalA knockdown via two different siRNA sequences, si5 and si9, or both in combination. Quantification suggests ∼73, 77, or 84% knockdown, respectively. (C) RalA knockdown does not affect GLUT4 translocation under basal conditions or in cells stimulated with 1 nM insulin. Mean normalized S:T values ± SEM, N = 3–5 assays. (D) RalA knockdown has no effect on GLUT4 translocation in control or RAB10 KD cells or in RAB10 KD cells rescued with FLAG-RAB10. Mean normalized S:T values ± SEM, N = 3 assays. (E) RalA knockdown does not rescue the basal retention defect of AS160 KD cells. Mean normalized S:T values ± SEM, N = 3 assays. (F) MUNC18C KD has no effect on insulin-stimulated GLUT4 translocation. Inset, representative Western blot showing MUNC18C KD. Quantification suggests ∼52% knockdown. Left, GLUT4 translocation in control and MUNC18C knockdown on day 7 of differentiation. Mean normalized S:T values ± SEM, N = 2 or 3 assays. Right, GLUT4 translocation in control and MUNC18C knockdown on day 10 of differentiation. Mean normalized S:T values ± SEM, N = 5 assays; 100 nM insulin, MUNC18C KD vs. control, *p < 0.05; two-tailed paired t test, nonnormalized raw data. (G) CDP138 KD has no effect on insulin-stimulated GLUT4 translocation. Mean normalized S:T values ± SEM, N = 12 assays. Inset, representative Western blot showing CDP138 KD accomplished with both a mix of siRNAs or one siRNA from that mix (si1717). Quantification suggests ∼90% knockdown in both cases.
We reasoned that such factors could act in one of three ways: 1) to control a GLUT4-vesicle population distinct from those regulated by RAB10, 2) to regulate the same population of GLUT4 vesicles but at a different step of GLUT4 trafficking, or 3) to regulate the same population of GLUT4 vesicles and at the same step of GLUT4 trafficking as AS160-RAB10 but via different effector molecules. To address this question, we investigated several factors reported to regulate GLUT4 trafficking in each of these three ways.
Initially, our goal was to pair knockdown of previously reported factors with knockdown of RAB10 and/or AS160, reasoning that this epistasis analysis would allow us to investigate whether such factors were acting downstream of RAB10 in the same pathway or separately to control other steps of GLUT4 trafficking or separate populations of GLUT4 vesicles. Before testing for additivity, however, we wanted to confirm that these proteins were indeed involved in GLUT4 trafficking, as measured under our experimental conditions that induce robust, insulin-stimulated GLUT4 translocation while recapitulating insulin levels within the physiological range. Further, using ratiometric measurement of surface to total GLUT4 allows for the detection of a graded response to insulin (Gonzalez and McGraw, 2006; Martin et al., 2006; Blot and McGraw, 2008b).
The small GTPase RalA has been reported to play a role in the regulation of GLUT4 translocation. Previous studies suggest that RalA resides on GLUT4 vesicles that traffic to the PM in response to insulin, in part due to the interaction of RalA with the motor protein Myo1c (Chen et al., 2007). RalA also interacts with subunits of the Exocyst complex (Chen et al., 2007), which may play a role in docking GSVs near the PM in response to insulin (Inoue et al., 2003; Lizunov et al., 2009). This is analogous to roles proposed for RAB10, as recent data suggest that RAB10 interacts with the motor protein MyoVa (Chen et al., 2012) and functional data suggest a role for AS160-RAB10 in accumulating GSVs near the PM (Sano et al., 2007; Xiong et al., 2010; Chen et al., 2012; Figure 2). Further, like another small GTPase, insulin-stimulated activation of RalA is accomplished via a mechanism analogous to that of activation of RAB10: AKT2-mediated phosphorylation of RGC, the GAP for RalA, leads to a decrease in the GAP activity of RGC and, in turn, an increase in active RalA (Chen et al., 2011c). Given previous reports supporting a role for RalA in the regulation of GSV transport and docking, we hypothesized that this small GTPase and its cognate GAP, RGC, could form a signaling module analogous to AS160-RAB10 and therefore constitute an additional signaling module acting either sequentially with RAB10—for example, RAB10 tethers the GSV to the PM and then RalA docks it there—or in parallel, each acting redundantly to control docking of GSVs at the PM.
To investigate the roles of RalA and AS160-RAB10 in GLUT4 translocation, we began by using two different siRNA sequences either alone or in combination to achieve efficient knockdown of RalA (Figure 5B). In our experimental conditions, transient RalA knockdown in 3T3-L1 cells had no effect on GLUT4 translocation (Figure 5C). This is in contrast to previous studies, which reported a decrease in surface GLUT4 stimulated by 170 nM insulin in RalA knockdown cells (Chen et al., 2007). Because of the body of evidence supporting a role for RalA in GLUT4 translocation, we further tested RalA function by studying cells in which both RalA and RAB10 were simultaneously knocked down. We reasoned that knockdown of RAB10 might make the cells more sensitive to the loss of RalA if these two pathways were in fact operating in parallel at the step of GSV accumulation near the PM in response to insulin. In this case, we would expect to see a further decrease in GLUT4 translocation when both factors were removed from the system. RalA knockdown had no additional effect on GLUT4 translocation in RAB10 KD cells, and reexpression of RAB10 in these cells fully restored translocation (Figure 5D). We also asked whether RalA knockdown would reduce the increased surface GLUT4 seen in AS160 KD cells. As discussed earlier, RAB10 knockdown in AS160 KD cells partially rescues the AS160 KD cell phenotype (Sano et al., 2007; Figure 2D), however RalA knockdown did not rescue the increased basal surface GLUT4 in AS160 KD cells (Figure 5E). These data suggest that RalA is not acting in the same pathway and at the same step as AS160-RAB10. Although it is possible that our experiments have not captured the effect of RalA on GLUT4 translocation, under our experimental conditions we find that RalA is not required.
MUNC18C and CDP138 have both been reported to have roles in the fusion of docked GSVs with the PM, accomplishing final insertion of GLUT4 into the cell membrane. As such, these proteins are candidates that might regulate GSV translocation downstream of the accumulation/docking step regulated by AS160-RAB10. Previous studies showed that in the basal state, MUNC18C inhibits the fusion of GLUT4-containing vesicles with the PM by binding to the exocytosis protein syntaxin4, blocking formation of the exocytosis complex of VAMP2/SNAP-23/syntaxin4, an interaction necessary for vesicle fusion. On insulin stimulation, tyrosine phosphorylation repositions MUNC18C on syntaxin4, allowing syntaxin4 to achieve the open conformation necessary for VAMP2 binding, vesicle fusion, and GLUT4 insertion into the PM (Tamori et al., 1998; Thurmond et al., 1998; D'Andrea-Merrins et al., 2007; Brandie et al., 2008; Smithers et al., 2008; Jewell et al., 2011). We tested the necessity of MUNC18C for GLUT4 trafficking using our standard protocol (see Materials and Methods) with doses of insulin within the physiological range (1 nM insulin). Using these methods, we were unable to see an effect of MUNC18C knockdown on GLUT4 translocation (Figure 4F, left). In our hands, however, the previously published siRNA sequences achieved only ∼50% knockdown of MUNC18C (Figure 5F, inset). To see whether this level of knockdown is sufficient to impair GLUT4 translocation under other experimental conditions, we sought to replicate the published protocol. We tested 3T3-L1 adipocytes electroporated with control or MUNC18C knockdown siRNA and treated with 100 nM insulin on day 10 of differentiation. Under these conditions of supraphysiological doses of insulin, we did see a significant reduction in insulin-stimulated GLUT4 translocation in MUNC18C knockdown cells (Figure 5F, right). MUNC18C, however, cannot be responsible for the non–AS160-RAB10-dependent effects of insulin on GLUT4 translocation at more physiological concentrations of insulin doses.
Recently the previously uncharacterized C2 domain–containing protein CDP138 was also reported as a factor downstream of AKT2 involved in regulating the fusion of GSVs with the PM in response to insulin (Xie et al., 2011). SiRNA-mediated knockdown of CDP138 resulted in decreased plasma membrane GLUT4 in response to insulin due to a specific defect in GSV fusion, as the accumulation of GLUT4 near the PM remains intact (Xie et al., 2011). However, knockdown of CDP138 had no effect on GLUT4 translocation using our methods. We tested both the previously published mix of siRNAs (Xie et al., 2011) and the one siRNA from that mix that accomplished efficient CDP138 knockdown on its own (si1717; Figure 5G, inset) and found that in both cases CDP138 knockdown did not alter GLUT4 translocation (Figure 5G). Thus our results suggest that neither MUNC18C nor CDP138 is the factor that regulates GSV fusion or another step of GSV translocation to the plasma membrane downstream of insulin receptor activation.
RAB14 regulates GLUT4 sorting independently of the AS160-RAB10 signaling module but not a separate population of GLUT4-containing vesicles
In muscle cells, RAB14 is a target of the GAP activity of AS160 (Ishikura et al., 2007), and knockdown of RAB14 in muscle cells blunts insulin-stimulated GLUT4 translocation (Ishikura and Klip, 2008). Recently it was reported that adipocyte GLUT4 translocation is partly accounted for by the actions of RAB14 on GLUT4-containing endosomal compartments (Chen et al., 2012): upon insulin stimulation, these GLUT4-, TR-, and RAB14-positive vesicles translocate to and fuse with the PM. Further, knockdown of RAB14 inhibits some portion of overall GLUT4 translocation (Chen et al., 2012). In other cell types, RAB14 has been identified as a regulator of specialized trans-Golgi network (TGN) and endosomal sorting for various cargoes, including the ADAM-family protease ADAM10 (Linford et al., 2012), fibroblast growth factor receptor (Ueno et al., 2011), and vasoactive intestinal polypeptide (Kitt et al., 2008).
To investigate the role of RAB14 in adipocyte GLUT4 translocation, we first confirmed that siRNA-mediated knockdown of RAB14 results in a blunting of insulin-stimulated GLUT4 translocation (Figure 6A). We also confirmed that this defect does not result from a defect in insulin signaling: 3T3-L1 adipocytes lacking RAB14 have comparable insulin-stimulated AKT phosphorylation as control cells (Figure 6B). On the basis of these data and previously published findings (Chen et al., 2012), we initially hypothesized that perhaps this population of GLUT4 that is ferried back to the PM with TR and is regulated by RAB14 represents an independent population of GLUT4 vesicles not controlled by AS160-RAB10.
FIGURE 6:

RAB14 acts to regulate GLUT4 translocation via the GSV pathway at a step downstream of insulin signaling to AKT but upstream of AS160 regulation. (A) Top, representative Western blot showing knockdown of RAB14. Quantification suggests ∼95% knockdown. Bottom, RAB14 knockdown in 3T3-L1 cells blunts insulin-stimulated GLUT4 translocation. S:T values normalized to GLUT4 in 1 nM insulin–treated 3T3-L1 control cells determined in each individual experiment. Mean normalized S:T values ± SEM, N = 7 or 8 assays. (B) Representative Western blot showing normal AKT phosphorylation in response to 1 nM insulin treatment in 3T3-L1 adipocytes lacking RAB14. (C) RAB14 KD has no additive effect when combined with RAB10 KD. S:T values normalized to GLUT4 in 1 nM insulin–treated 3T3-L1 control cells determined in each individual experiment. Mean normalized S:T values ± SEM, N = 5 assays. (D) Overexpression of RAB14 in RAB10 KD cells does not rescue the GLUT4 translocation defect in RAB10 KD cells. S:T values normalized to GLUT4 in 1 nM insulin–treated RAB10 KD cells rescued with RAB10 reexpression determined in each individual experiment. Mean normalized S:T values ± SEM, N = 4 assays. (E) Representative Western blot showing expression of RAB10 and RAB14 in adipocytes electroporated with RAB10- and/or RAB14-directed siRNA sequences. Knockdown of one Rab protein does not result in a compensatory change in the expression of the other Rab. (F) Representative Western blot showing expression of RAB10 and RAB14 in adipocytes electroporated with DNA encoding FLAG-RAB10 or FLAG-RAB14. Overexpression of one Rab protein does not result in a compensatory change in the expression of the other Rab. (G) RAB14 KD does not rescue the basal retention defect of AS160 KD cells. Mean normalized S:T values ± SEM, N = 5 assays. S:T values normalized to GLUT4 in basal AS160 KD cells determined in each individual experiment. *p < 0.05, two-tailed paired t test, nonnormalized raw data.
To test this hypothesis, we studied 3T3-L1 adipocytes lacking both RAB10 and RAB14. If RAB14 does in fact regulate a pool of GLUT4 vesicles separate from the pool regulated by RAB10, we would expect that knockdown of both proteins would result in an additive decrease in insulin-stimulated GLUT4 translocation. However, transient knockdown of both RAB10 and RAB14 resulted in a similar degree of GLUT4 translocation as seen with RAB10 knockdown alone (Figure 6C). This strongly suggests that although RAB14 may play a role in the regulation of GLUT4 translocation, it is unlikely to act on a pool of GLUT4 vesicles independent from the GSV pool regulated by RAB10. Rather, RAB14 may act in the same pathway as RAB10, perhaps to regulate the same step of GLUT4 translocation.
We next investigated the possibility of a functional redundancy between RAB10 and RAB14 by asking whether overexpression of RAB14 could rescue the RAB10 KD cell defect in GLUT4 translocation. We found no evidence for this. RAB10 KD cells overexpressing RAB14 showed a similar degree of insulin-stimulated GLUT4 translocation as RAB10 KD cells alone (Figure 6D). To confirm that knockdown or overexpression of one of these two Rab proteins does not result in a compensatory change in the expression levels of the other protein, we determined by Western blot the levels of each Rab after knockdown or overexpression of the other Rab (Figure 6, E and F). These results suggest that although RAB14 may act in the same pathway as RAB10, these RABs function independently of one another.
Given that RAB14 acts on the same population of GLUT4 vesicles as RAB10 but not at the same step, we next asked whether RAB14 also functions downstream of AS160. This possibility seemed likely, given that RAB14 is a target of AS160 in muscle cells (Ishikura et al., 2007). To test this, we knocked down RAB14 in AS160 KD cells to determine whether this would rescue the AS160 KD cell phenotype. We found that cells lacking both AS160 and RAB14 showed an increase in surface GLUT4 similar to AS160 KD cells alone (Figure 6G), suggesting that RAB14 does not act downstream of AS160 in a similar manner to RAB10 (Figure 2D). Although our initial experiments support a role for RAB14 in the process of insulin-stimulated GLUT4 translocation, our experiments investigating the nature of this role are inconsistent with previous reports suggesting that RAB14 regulates an endosomal pool of GLUT4: we found no evidence of additivity with RAB10 and RAB14 knockdown (Figure 6C), nor does it seem that RAB14 is involved in the AS160-RAB10 pathway either redundantly with RAB10 (Figure 6D) or downstream of AS160 (Figure 6G). Instead, our results suggest that RAB14 regulates the same population of GLUT4-containing vesicles as AS160-RAB10 but at a different step of GLUT4 translocation upstream of AS160.
On the basis of these findings, we next explored the possibility that RAB14 could regulate GLUT4 trafficking by regulating GLUT4 sorting steps. To test this hypothesis, we tested the effect of RAB14 knockdown on the trafficking of GLUT4 mutants that disrupt GLUT4 sorting. The F5A mutation in the FQQI trafficking motif of GLUT4 (FA-GLUT4) causes a shift in GLUT4 distribution from the perinuclear retention compartment to the endosome, resulting in more GLUT4 on the cell surface in both basal and insulin-stimulated states, as FA-GLUT4 travels to the PM via both the GSV and the TR pathways (Blot and McGraw, 2008a). The E499A/E501A mutations in the TELEY trafficking motif (EE-GLUT4) shift the distribution of GLUT4 from GSVs to the endosome, resulting in a modest increase in surface GLUT4 in the basal state but a blunting of insulin-stimulated GLUT4 translocation due to a depleted GSV pool (Blot and McGraw, 2008a). When we electroporated 3T3-L1 adipocytes with RAB14-directed siRNA and wild type (WT)-, FA-, or EE-GLUT4, we found that RAB14 knockdown blunted translocation of WT as well as FA-GLUT4 but had no effect on the insulin-stimulated translocation of EE-GLUT4 (Figure 7A). This suggests that RAB14 normally acts at a similar step as the TELEY trafficking motif of GLUT4, namely sorting GLUT4 into GSVs. When examined by epifluorescence microscopy of HA-GLUT4-GFP in control or RAB14 knockdown cells, the lack of RAB14 does not appear to result in a gross mislocalization of GLUT4 (Figure 7B). It is possible, however, that changes in GLUT4 localization that are not detectable at this level of analysis do occur in RAB14 knockdown cells. Together these data support the hypothesis that RAB14 acts at a GLUT4 sorting step presumably upstream from AS160-RAB10 (Figure 4A).
FIGURE 7:
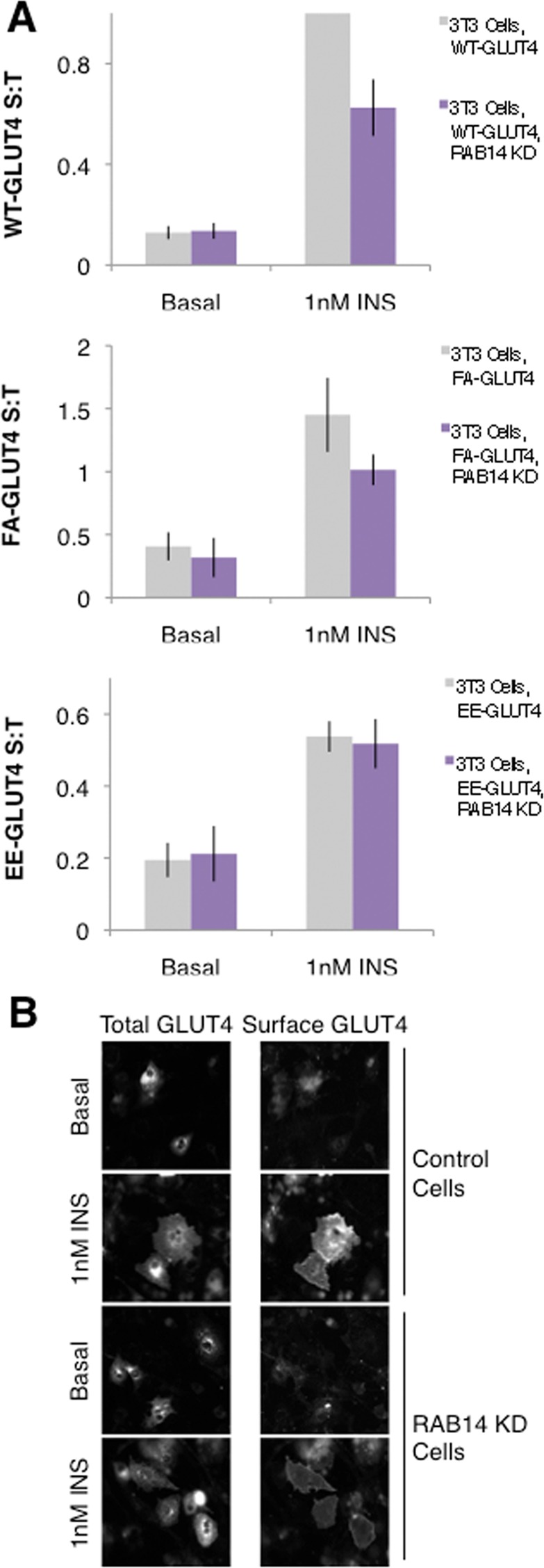
RAB14 controls a GLUT4 sorting step. (A) RAB14 KD blunts insulin-stimulated translocation of WT-GLUT4 and FA-GLUT4 but not EE-GLUT4. S:T values normalized to WT GLUT4 in 1 nM insulin–treated 3T3-L1 cells determined in each individual experiment. Mean normalized S:T values ± SEM, N = 3–5 assays. p < 0.05, two-tailed paired t test, nonnormalized raw data. NS, not significant with p > 0.05, two-tailed paired t test, nonnormalized raw data. (B) RAB14 KD does not induce a gross redistribution of GLUT4, as measured by epifluorescence microscopy. Images of total GLUT4 (HA-GLUT4-GFP intrinsic fluorescence, left) and surface GLUT4 (anti-HA immunostaining, right) of both basal and insulin-treated control and RAB14 KD cells.
DISCUSSION
Surface expression of adipocyte GLUT4 is necessary for normal glucose homeostasis in vivo (Abel et al., 2001), and alterations of the AS160-RAB10 signaling module itself can impair whole-body glucose regulation, even when the alteration occurs only in adipocytes (Chen et al., 2011a). In light of the important role for GLUT4 trafficking in glucose homeostasis and the development of type 2 diabetes, we sought to expand our understanding of the factors regulating adipocyte GLUT4 translocation. We show that RAB10 is not only necessary for normal GLUT4 trafficking in 3T3-L1 adipocytes, but that endogenous RAB10 levels in these cells dose dependently regulate the degree of GLUT4 translocation. Further, our data suggest that AS160-RAB10 is partially sufficient for GLUT4 translocation. Together with previous work, our experiments show that RAB10 acts to regulate accumulation of GLUT4 near the PM in response to insulin and that the principal GEF for RAB10, DENND4C, is not regulated by insulin but instead is constitutively active. As a whole, these data support a role for the AS160-RAB10 signaling module as the principal factor regulating insulin-stimulated GLUT4 translocation.
We find that elimination of either part of the AS160-RAB10 signaling module blocks maximal insulin-stimulated GLUT4 translocation; however, siRNA-mediated knockdown of other factors previously reported to be important for this process showed, in our experimental system, no defect in surface GLUT4 expression in response to insulin. Further, overexpression of RAB10 in individual 3T3-L1 adipocytes as well as adipocytes lacking AS160 increases GLUT4 translocation above levels seen in control cells in response to the same dose of insulin, suggesting that RAB10 levels act cell autonomously to regulate GLUT4 and that endogenous levels of RAB10 are limiting for GLUT4 trafficking. We also show that addition of the AS160-RAB10 signaling module to CHO cells—normally lacking specialized GLUT4 trafficking machinery—confers adipocyte-like GLUT4 behavior, suggesting that this signaling module is at least partially sufficient for normal GLUT4 regulation. Extending the role of RAB10 in GLUT4 trafficking, we confirm that, like AS160 (Zeigerer et al., 2004; Xiong et al., 2010), RAB10 acts primarily at an accumulation step of GLUT4 translocation under normal physiological conditions. We hypothesize that this step would be to regulate the docking of GSVs at the PM, but we cannot at this time eliminate a role for RAB10 in regulating GSV transport to the PM or GLUT4 sorting into GSVs, other prefusion trafficking steps that affect accumulation of GLUT4 near the PM. Although we did see a trend toward a decrease in insertion of GLUT4 into the PM in RAB10 KD cells, this difference was not statistically significant, supporting previous findings (Gonzalez and McGraw, 2006; Martin et al., 2006; Bai et al., 2007; Fujita et al., 2010; Xiong et al., 2010) that accumulation and insertion are independently regulated by insulin.
Our data support a model in which insulin regulation of RAB10 occurs solely through its GAP, AS160. AS160 knockdown results in an increase in surface GLUT4 levels in the absence of insulin stimulation, presumably due to a lack of repression on RAB10 activity (Zeigerer et al., 2004; Eguez et al., 2005; Larance et al., 2005). Knockdown of RAB10 in addition to AS160 rescues this defect, presumably by normalizing levels of active RAB10 (Sano et al., 2007). We provide evidence that the principal GEF for RAB10, DENND4C (Yoshimura et al., 2010; Sano et al., 2011), is constitutively active, that is, not regulated by insulin: knockdown of DENND4C in AS160 KD cells phenocopies RAB10 knockdown in these cells under basal conditions, suggesting that DENND4C promotes RAB10 activity even in the absence of insulin and is therefore likely to be constitutively active as a GEF for RAB10.
Much of the controversy surrounding the role of RAB10 in GLUT4 translocation centers around the differences between the role of RAB10 in muscle and fat cells: specifically, it appears that RAB10 is not involved in myocyte GLUT4 translocation but instead that RAB8A acts as the primary Rab regulating GLUT4 trafficking in these cells (Ishikura et al., 2007; Ishikura and Klip, 2008; Sun et al., 2010). Although we did not directly address this discrepancy here, our data strongly support RAB10 as the key factor regulating insulin-stimulated GLUT4 translocation in adipocytes. Significant variation in this process from cell type to cell type is not only plausible but likely, given that RAB10 has been proposed to regulate additional processes besides GLUT4 translocation in other cell types (Babbey et al., 2010; Shi et al., 2010; Wang et al., 2010; English and Voeltz, 2013). We hypothesize that RAB10 and other Rab proteins mediate their myriad roles through interactions with different effectors in different cell types or contexts.
Recently it was reported that RAB10 regulates endoplasmic reticulum (ER) dynamics in COS-7 cells: RAB10 knockdown in these cells causes defects in ER tubule fusion, resulting in disrupted ER morphology (English and Voeltz, 2013). Although it is possible that the defects in insulin-stimulated GLUT4 translocation that we observe in 3T3-L1 adipocytes are secondary to disrupted ER morphology, we find this possibility unlikely. First, RAB10 knockdown has no effect on TR trafficking (Sano et al., 2007), and we show here that RAB10 overexpression also does not affect this general trafficking pathway. If knockdown of RAB10 did indeed disrupt ER function in adipocytes as it does in COS-7 cells, we would expect an effect on the expression of other proteins, such as the TR, in the plasma membrane, contrary to what we observe. Second, as mentioned earlier, RAB10 plays a role in processes other than GLUT4 translocation in nonadipocyte cells (Babbey et al., 2010; Shi et al., 2010; Wang et al., 2010; English and Voeltz, 2013), and we consider it plausible that there exist species or cell-type differences in the role of RAB10: COS-7 cells are derived from monkey kidney and therefore might use RAB10 very differently from 3T3-L1 adipocytes. Finally, it is possible that RAB10 does indeed regulate ER dynamics in 3T3-L1 adipocytes but that effector specificity and/or differential localization would enable RAB10 to play multiple roles in the same cell. We hypothesize that only a portion of the total cellular RAB10 is used to regulate GLUT4 trafficking: locally activated RAB10 could promote GLUT4 accumulation at the PM, whereas RAB10 localized to the ER could perform other functions. This is supported by the finding that only ∼5% of total cellular RAB10 is found on GLUT4 vesicles (Sano et al., 2008). Indeed, this would provide a potential explanation for one of the remaining controversies concerning RAB10’s role in GLUT4 trafficking: perhaps it has been difficult to measure an insulin-induced increase in GTP-RAB10 because only a small fraction of total cellular RAB10 is regulated by insulin. Further, it would seem that in both contexts, RAB10 acts to stabilize the association of two membrane compartments: GSVs with the PM, or two growing ER tubules.
Our initial experiments investigating RAB10 hinted that there were other factors in addition to the AS160-RAB10 signaling module that contribute to regulation of GLUT4 trafficking in adipocytes. Several such proteins have been reported (Inoue et al., 2003; Guilherme et al., 2004; Kanda et al., 2005; Chen et al., 2007, 2012; Brandie et al., 2008; Smithers et al., 2008; Lizunov et al., 2009; Jordens et al., 2010; Shi et al., 2010; Xie et al., 2011; Li et al., 2012). Owing to variations in methodology, however, we decided to test candidate factors under our experimental conditions. We examined four proteins, each reported to act on a separate population of GLUT4-containing vesicles than that regulated by RAB10, on the same population of GLUT4 vesicles but at a different step of GLUT4 trafficking than AS160-RAB10, or on the same population of GLUT4 vesicles and at the same step of GLUT4 trafficking as AS160-RAB10, potentially via a distinct signaling cascade. RalA was proposed as another small GTPase regulating docking of GSVs near the PM (Inoue et al., 2003; Chen et al., 2007, 2011b, c; Chen and Saltiel, 2011), whereas MUNC18C and CDP138 have been reported to regulate the final fusion step of GS translocation (Tamori et al., 1998; Thurmond et al., 1998, 2000; Thurmond and Pessin, 2000; Khan et al., 2001; Macaulay et al., 2002; Oh et al., 2005; D'Andrea-Merrins et al., 2007; Brandie et al., 2008; Aran et al., 2011; Jewell et al., 2011; Xie et al., 2011). In contrast to previous reports, in our experiments siRNA-mediated knockdown of each of these three proteins had no detectable effect on GLUT4 translocation stimulated by physiological levels of insulin. We do not know the specific reason for these differences, although one obvious technical difference between our assays and those reported previously is that we used 1 nM insulin to stimulate GLUT4 translocation, whereas other groups used higher, supraphysiological doses.
Recently it was found that in adipocytes, RAB14 resides on GLUT4- and TR-positive vesicles and was proposed to regulate a non-GSV population of GLUT4 (Chen et al., 2012). Although our experiments support RAB14 as a small GTPase necessary for normal GLUT4 trafficking, our data suggest that RAB14 acts at an endosomal GLUT4 sorting step. Our finding that RAB14 knockdown when combined with RAB10 knockdown has no additional effect on GLUT4 translocation compared with RAB10 knockdown alone suggests that rather than regulating a separate population of GLUT4 vesicles, RAB14 in fact regulates the same population of GLUT4 as RAB10, namely GSVs. The lack of rescue of the AS160 knockdown phenotype by RAB14 knockdown further suggests that while acting on the same GLUT4 population, RAB14 regulates a step of translocation upstream from AS160 and therefore not the same step as RAB10. Finally, RAB14 knockdown has no effect on the trafficking of EE-GLUT4, suggesting that RAB14 regulates some aspect of the sorting of GLUT4 into GSVs.
Despite our different conclusion regarding the role of RAB14, our data are consistent with previous reports. We do replicate the primary finding (Chen et al., 2012) of a defect in GLUT4 translocation with RAB14 knockdown, and localization of RAB14 to TR- and GLUT4-containing vesicles is not mutually exclusive with a sorting role of RAB14. Perhaps RAB14 acts as a vesicle-localized signal to sort TR- and GLUT4-containing vesicles into the GSV pathway. In the absence of RAB14, vesicles containing both TR and GLUT4 would be sorted into the TR trafficking pathway rather than the specialized GSV pathway, resulting in a net decrease in surface GLUT4 in response to insulin due to the decreased insulin responsiveness of the TR pathway relative to the GSV pathway. Our results differ from those of Chen et al. (2012) in that we do not find an additive effect of knocking down both RAB10 and RAB14. In this case, however, different assays were used to assess GLUT4 translocation, potentially explaining the discrepancy in our results.
We defined two Rab proteins as regulators of GLUT4 translocation. While playing roles in other cellular processes, these Rabs are harnessed by the adipocyte GLUT4 trafficking pathway to regulate independent steps. We identified RAB14 as a regulator of GLUT4 sorting and gave strong evidence that the AS160-RAB10 signaling module acts at a GSV accumulation step as a key factor regulating GLUT4 translocation in adipocytes. Unambiguously identifying the proteins involved in regulating adipocyte GLUT4 trafficking is of great importance, as these factors represent potential drug targets in the development of treatments for type 2 diabetes.
MATERIALS AND METHODS
Cell lines and culture
As previously described (Zeigerer et al., 2002), 3T3-L1 fibroblasts were maintained in culture and differentiated into adipocytes. For some experiments, 3T3-L1 cell lines stably expressing shRNA sequences directed against target transcripts were used: AS160 KD cells (Eguez et al., 2005) and RAB10 KD cells (Sano et al., 2007) were described previously. For experiments involving CHO cells, cells were maintained in culture in F12 media supplemented with 10% fetal bovine serum, antibiotics for selection, 13.5 mM sodium bicarbonate, and 11.1 mM D+ glucose.
DNA and siRNA constructs
The HA-GLUT4-GFP, HA-FA-GLUT4-GFP, HA-EE-GLUT4-GFP, TR, FLAG-AS160, and FLAG-AS160-4A DNA plasmids have been described (Lampson et al., 2001; Kane et al., 2002; Sano et al., 2003, 2007; Zeigerer et al., 2004; Chen et al., 2006; Itoh et al., 2006; Blot and McGraw, 2008a). AS160 plasmids contained sequences for human AS160 and were therefore resistant to knockdown by mouse AS160-directed siRNA sequences.
For Rab10 DNA constructs, full-length murine RAB10 was amplified by PCR using a 5′ primer containing the BamH1 site followed by the Kozac sequence and the FLAG tag sequence, and a 3′ primer containing the EcoR1 site. The fragment was then subcloned between the BamH1 and EcoR1 sites of pcDNA3.1(+). FLAG-RAB10 plasmids contained mouse RAB10 sequences that were wobbled against the specific siRNA or shRNA sequence used in both RAB10 KD cells and in experiments involving transient electroporation of RAB10-directed siRNAs. The pcDNA-RAB10 construct was created by cloning the wild-type mouse RAB10 sequence from the FLAG-RAB10 construct into the pcDNA plasmid without the FLAG sequences. Before use, plasmids were functionally validated for their ability to rescue the knockdown phenotype of the given protein (see Supplemental Figure S1A).
The siRNA constructs were, wherever possible, as previously published. Sequences were as follows:
RAB10: si251 = GCAUCAUGCUAGUGUAUGA (same sequence as shRNA expressed by RAB10 KD cells; Sano et al., 2007)
RAB14: si1 = ACGCAAGGAAUCUCACCAA; si4 = GGUGUUGAAUUUGGUACAA (Chen et al., 2012)
MUNC18C: TACAAAGCAGCGTATATATACTTCA (Oh and Thurmond, 2009)
CDP138: si1717 = ATTCAGGCAAGGTTATGTCGATTAA; mix = CCAGGCATAATTTCTGTATTACAAC + UAAGTAGACTCCTGTGGCCGACGCT + TTAGTCTGACCACTCTACTGACGT + si1717 (Xie et al., 2011)
RalA: si5 = GGAAGAAGUGCAGAUCGACAUCUUA; si9 = UUCAGGGAGCAGAUUUUAAGAGUAA
DENND4C: GGCCGUCACUGAUAUCUGU (Sano et al., 2011).
When only one siRNA sequence was used to achieve efficient knockdown, 2 nmol of that siRNA was electroporated. When two or more siRNA sequences were used, equal parts of each siRNA were combined, and a total of 2 nmol of siRNA was electroporated. For DENND4C knockdown, 4 nmol of siRNA was used. All siRNA sequences were confirmed by Western blot to achieve efficient knockdown of the target protein before use in subsequent assays.
Electroporation and GLUT4 and TR translocation assays
Indicated cell lines were electroporated with 0–90 μg of DNA, typically including 45–55 μg of HA-GLUT4-GFP and/or 2–5nmol of siRNA as previously described (Zeigerer et al., 2002). Assays were performed 1–3 d after electroporation, as needed to achieve knockdown of the targeted protein as validated by Western blot. On the day of the assay, GLUT4 or TR translocation assays were performed as described previously (Lampson et al., 2001; Zeigerer et al., 2002, 2004; Sano et al., 2007; Xiong et al., 2010). Briefly, cells were washed and incubated in media without sera for 2 h to eliminate the effects of any insulin in serum. Cells were then stimulated with the indicated final concentrations of insulin for 30 min to achieve steady-state surface GLUT4 levels. Cells were fixed and an anti-HA antibody was used, without permeabilization, to label HA-GLUT4-GFP on the cell surface. HA staining was visualized with fluorescently tagged secondary antibodies, and total HA-GLUT4-GFP was visualized by direct fluorescence, as described later (Lampson et al., 2000; Zeigerer et al., 2004). Mouse anti-HA was purchased from Covance (Princeton, NJ), and secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA) and Invitrogen (Grand Island, NY).
For experiments involving CHO cells, cells were transfected with 3.5 of μg DNA using Lipofectamine (Invitrogen). Assays were performed 2 d after transfection. GLUT4 translocation assays were performed as described for 3T3-L1 adipocytes.
For experiments involving the single-cell measurement of FLAG-RAB10 expression, RAB10 KD cells were electroporated with 0, 45, or 90 μg of FLAG-RAB10 DNA and then pooled to create a heterogeneous population expressing different levels of FLAG-RAB10. GLUT4 translocation assays were performed as described, followed by immunofluorescence staining with rabbit anti-FLAG antibodies (F-7425; Sigma-Aldrich, St. Louis, MO). Secondary labeling of both FLAG and HA was quantified per cell, and data were binned within each experiment by FLAG-RAB10 expression level.
Microscopy, image quantification, and statistics
All epifluorescence images were collected on an inverted microscope at room temperature using a 20× air objective (Leica Microsystems, Jena, Germany) and a cooled charge-coupled device 12-bit camera. Exposure times (Xiong et al., 2010) and image quantification (Lampson et al., 2001) using MetaMorph image processing software (Universal Imaging, West Chester, PA) were performed as previously described. Fluorescence signals were background corrected, and the surface:total GLUT4 (or TR) ratio was calculated for each cell. The S:T values were normalized within each assay to the average S:T value for the indicated condition to allow for averaging results from identical conditions across multiple biological repeat assays. Paired Student's t tests were performed on raw (nonnormalized) S:T average values from multiple assays. One-tailed t tests were only used when a reasonable expectation could be formed from previous results as to the direction of the effect being measured.
For TIRF microscopy, TIRF and epifluorescence images were acquired on an Olympus IX 70 (Thornwood, NY) with a 60×/1.45 numerical aperture oil-immersion objective using dual-color TIRF imaging, as previously described (Sano et al., 2007; Xiong et al., 2010). Exposure times, image quantification, and data processing were performed as described.
Western blots and associated antibodies
Western blot experiments were performed using standard protocols. Cellular protein extracts were collected from 3T3-L1 differentiated adipocytes or indicated cell lines expressing indicated siRNA or DNA constructions. Most presented Western blots were performed using extracts made from the same cells tested in the presented functional assays. Actin was always measured as a loading control. Antibodies used were as follows: RAB10, 1:500 (4262S; Cell Signaling, Danvers, MA); AS160, 1:2000 (07-741; Millipore, Temecula, CA); MUNC18C, 1:5000 (a gift from Deborah Thurmond, Indiana University, Indianapolis, IN; Thurmond et al., 1998); CDP138, 1:2000 (a gift from Zhen Jiang, Sanford Burnham Medical Research Institute, Orlando FL; Xie et al., 2011); RalA, 1:10,000 (610222; BD Transduction Laboratories, San Jose, CA); RAB14, 1:2000 (R-0781; Sigma-Aldrich); actin, 1:10,000 (AAN01-A; Cytoskeleton, Denver, CO); AKT, 1:5000 (9272S; Cell Signaling); and P-AKT S473, 1:2000 (9271S; Cell Signaling).
Quantitative PCR
Measurement of DENND4C knockdown was performed by quantitative PCR. 3T3-L1 adipocytes were electroporated with DENND4C-directed siRNAs as described. At 72 h after electroporation, cells were harvested, and RNA was extracted using the RNeasy kit from Qiagen (Germantown, MD). From extracted RNA, cDNA was made using the Sprint RT Complete Oligo(dT) Kit from Clontech (Mountain View, CA), and quantitative PCR was performed using a qSTAR primer pair obtained from OriGene (Rockville, MD).
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health R01 Grant DK5285 to T.E.M and an American Diabetes Association Mentored Postdoctoral Fellowship Award (7-12-MN-54). L.A.S. is supported by National Institutes of Health F32 NRSA DK095532. J.B. is supported by National Institutes of Health Medical Scientist Training Program Grant GM007739. We thank Gus Lienhard and Eva Gonzalez for comments on the manuscript and the members of the McGraw laboratory for helpful discussion.
Note added in proof. During the preparation of the revisions of this article a report appeared presenting data supporting a role for RAB14 in the intracellular sorting of GLUT4 to insulin-responsive compartments, in agreement with our findings (Reed et al., 2013).
Abbreviations used:
- GAP
GTPase-activating protein
- GEF
guanine exchange factor
- GSVs
GLUT4 storage vesicles
- HA
hemagglutinin
- PM
plasma membrane
- shRNA
small hairpin RNA
- siRNA
small interfering RNA
- TIRF
total internal reflection fluorescence
- TR
transferrin receptor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-02-0103) on June 26, 2013.
*Present address: Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, Yunnan 650201, China.
The authors declare that they have no conflict of interest.
REFERENCES
- Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- Aran V, Bryant NJ, Gould GW. Tyrosine phosphorylation of Munc18c on residue 521 abrogates binding to syntaxin 4. BMC Biochem. 2011;12:19. doi: 10.1186/1471-2091-12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbey CM, Bacallao RL, Dunn KW. Rab10 associates with primary cilia and the exocyst complex in renal epithelial cells. Am J Physiol Renal Physiol. 2010;299:F495–F506. doi: 10.1152/ajprenal.00198.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Wang Y, Fan J, Chen Y, Ji W, Qu A, Xu P, James DE, Xu T. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metabolism. 2007;5:47–57. doi: 10.1016/j.cmet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Blot V, McGraw TE. Molecular mechanisms controlling GLUT4 intracellular retention. Mol Biol Cell. 2008a;19:3477–3487. doi: 10.1091/mbc.E08-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot V, McGraw TE. Use of quantitative immunofluorescence microscopy to study intracellular trafficking: studies of the GLUT4 glucose transporter. Methods Mol Biol. 2008b;457:347–366. doi: 10.1007/978-1-59745-261-8_26. [DOI] [PubMed] [Google Scholar]
- Brandie FM, Aran V, Verma A, McNew JA, Bryant NJ, Gould GW. Negative regulation of syntaxin4/SNAP-23/VAMP2-mediated membrane fusion by Munc18c in vitro. PLoS One. 2008;3:e4074. doi: 10.1371/journal.pone.0004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wasserman DH, MacKintosh C, Sakamoto K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab. 2011a;13:68–79. doi: 10.1016/j.cmet.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XW, Inoue M, Hsu SC, Saltiel AR. RalA-exocyst-dependent recycling endosome trafficking is required for the completion of cytokinesis. J Biol Chem. 2006;281:38609–38616. doi: 10.1074/jbc.M512847200. [DOI] [PubMed] [Google Scholar]
- Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell. 2007;13:391–404. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Chen XW, et al. Exocyst function is regulated by effector phosphorylation. Nat Cell Biol. 2011b;13:580–588. doi: 10.1038/ncb2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XW, Leto D, Xiong T, Yu G, Cheng A, Decker S, Saltiel AR. A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol Biol Cell. 2011c;22:141–152. doi: 10.1091/mbc.E10-08-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XW, Saltiel AR. Ral's engagement with the exocyst: breaking up is hard to do. Cell Cycle. 2011;10:2299–2304. doi: 10.4161/cc.10.14.16524. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Zhang J, Deng Y, Jiang L, Song E, Wu XS, Hammer JA, Xu T, Lippincott-Schwartz J. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J Cell Biol. 2012;198:545–560. doi: 10.1083/jcb.201111091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech MP, Buxton JM. Insulin action on the internalization of the GLUT4 glucose transporter in isolated rat adipocytes. J Biol Chem. 1993;268:9187–9190. [PubMed] [Google Scholar]
- Dugani CB, Klip A. Glucose transporter 4: cycling, compartments and controversies. EMBO Rep. 2005;6:1137–1142. doi: 10.1038/sj.embor.7400584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugani CB, Randhawa VK, Cheng AW, Patel N, Klip A. Selective regulation of the perinuclear distribution of glucose transporter 4 (GLUT4) by insulin signals in muscle cells. Eur J Cell Biol. 2008;87:337–351. doi: 10.1016/j.ejcb.2008.02.009. [DOI] [PubMed] [Google Scholar]
- D'Andrea-Merrins M, Chang L, Lam AD, Ernst SA, Stuenkel EL. Munc18c interaction with syntaxin 4 monomers and SNARE complex intermediates in GLUT4 vesicle trafficking. J Biol Chem. 2007;282:16553–16566. doi: 10.1074/jbc.M610818200. [DOI] [PubMed] [Google Scholar]
- Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- English AR, Voeltz GK. Rab10 GTPase regulates ER dynamics and morphology. Nat Cell Biol. 2013;15:169–178. doi: 10.1038/ncb2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Hatakeyama H, Watanabe TM, Sato M, Higuchi H, Kanzaki M. Identification of three distinct functional sites of insulin-mediated GLUT4 trafficking in adipocytes using quantitative single molecule imaging. Mol Biol Cell. 2010;21:2721–2731. doi: 10.1091/mbc.E10-01-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, McGraw TE. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol Biol Cell. 2006;17:4484–4493. doi: 10.1091/mbc.E06-07-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, McGraw TE. Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Acad Sci USA. 2009;106:7004–7009. doi: 10.1073/pnas.0901933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TE, Kahn BB. Tissue-specific alterations of glucose transport and molecular mechanisms of intertissue communication in obesity and type 2 diabetes. Horm Metab Res. 2007;39:717–721. doi: 10.1055/s-2007-985879. [DOI] [PubMed] [Google Scholar]
- Guilherme A, Soriano NA, Furcinitti PS, Czech MP. Role of EHD1 and EHBP1 in perinuclear sorting and insulin-regulated GLUT4 recycling in 3T3-L1 adipocytes. J Biol Chem. 2004;279:40062–40075. doi: 10.1074/jbc.M401918200. [DOI] [PubMed] [Google Scholar]
- Huang S, Lifshitz LM, Jones C, Bellve KD, Standley C, Fonseca S, Corvera S, Fogarty KE, Czech MP. Insulin stimulates membrane fusion and GLUT4 accumulation in clathrin coats on adipocyte plasma membranes. Mol Cell Biol. 2007;27:3456–3469. doi: 10.1128/MCB.01719-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422:629–633. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- Ishikura S, Bilan PJ, Klip A. Rabs 8A and 14 are targets of the insulin-regulated Rab-GAP AS160 regulating GLUT4 traffic in muscle cells. Biochem Biophys Res Commun. 2007;353:1074–1079. doi: 10.1016/j.bbrc.2006.12.140. [DOI] [PubMed] [Google Scholar]
- Ishikura S, Klip A. Muscle cells engage Rab8A and myosin Vb in insulin-dependent GLUT4 translocation. Am J Physiol Cell Physiol. 2008;295:C1016–C1025. doi: 10.1152/ajpcell.00277.2008. [DOI] [PubMed] [Google Scholar]
- Itoh T, Satoh M, Kanno E, Fukuda M. Screening for target Rabs of TBC (Tre-2/Bub2/Cdc16) domain-containing proteins based on their Rab-binding activity. Genes Cells. 2006;11:1023–1037. doi: 10.1111/j.1365-2443.2006.00997.x. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Oh E, Ramalingam L, Kalwat MA, Tagliabracci VS, Tackett L, Elmendorf JS, Thurmond DC. Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. J Cell Biol. 2011;193:185–199. doi: 10.1083/jcb.201007176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhun BH, Rampal AL, Liu H, Lachaal M, Jung CY. Effects of insulin on steady state kinetics of GLUT4 subcellular distribution in rat adipocytes. Evidence of constitutive GLUT4 recycling. J Biol Chem. 1992;267:17710–17715. [PubMed] [Google Scholar]
- Jordens I, Molle D, Xiong W, Keller SR, McGraw TE. Insulin-regulated aminopeptidase is a key regulator of GLUT4 trafficking by controlling the sorting of GLUT4 from endosomes to specialized insulin-regulated vesicles. Mol Biol Cell. 2010;21:2034–2044. doi: 10.1091/mbc.E10-02-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H, Tamori Y, Shinoda H, Yoshikawa M, Sakaue M, Udagawa J, Otani H, Tashiro F, Miyazaki J, Kasuga M. Adipocytes from Munc18c-null mice show increased sensitivity to insulin-stimulated GLUT4 externalization. J Clin Invest. 2005;115:291–301. doi: 10.1172/JCI22681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, Lienhard GE. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- Khan AH, Thurmond DC, Yang C, Ceresa BP, Sigmund CD, Pessin JE. Munc18c regulates insulin-stimulated glut4 translocation to the transverse tubules in skeletal muscle. J Biol Chem. 2001;276:4063–4069. doi: 10.1074/jbc.M007419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitt KN, Hernandez-Deviez D, Ballantyne SD, Spiliotis ET, Casanova JE, Wilson JM. Rab14 regulates apical targeting in polarized epithelial cells. Traffic. 2008;9:1218–1231. doi: 10.1111/j.1600-0854.2008.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Racz A, Cushman SW, McGraw TE. Demonstration of insulin-responsive trafficking of GLUT4 and vpTR in fibroblasts. J Cell Sci. 2000;113:4065–4076. doi: 10.1242/jcs.113.22.4065. [DOI] [PubMed] [Google Scholar]
- Lampson MA, Schmoranzer J, Zeigerer A, Simon SM, McGraw TE. Insulin-regulated release from the endosomal recycling compartment is regulated by budding of specialized vesicles. Mol Biol Cell. 2001;12:3489–3501. doi: 10.1091/mbc.12.11.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larance M, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- Li J, Malaby AW, Famulok M, Sabe H, Lambright DG, Hsu VW. Grp1 plays a key role in linking insulin signaling to glut4 recycling. Dev Cell. 2012;22:1286–1298. doi: 10.1016/j.devcel.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linford A, Yoshimura S, Nunes Bastos R, Langemeyer L, Gerondopoulos A, Rigden DJ, Barr FA. Rab14 and its exchange factor FAM116 link endocytic recycling and adherens junction stability in migrating cells. Dev Cell. 2012;22:952–966. doi: 10.1016/j.devcel.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizunov VA, Lisinski I, Stenkula K, Zimmerberg J, Cushman SW. Insulin regulates fusion of GLUT4 vesicles independent of Exo70-mediated tethering. J Biol Chem. 2009;284:7914–7919. doi: 10.1074/jbc.M806460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay SL, Grusovin J, Stoichevska V, Ryan JM, Castelli LA, Ward CW. Cellular munc18c levels can modulate glucose transport rate and GLUT4 translocation in 3T3L1 cells. FEBS Lett. 2002;528:154–160. doi: 10.1016/s0014-5793(02)03279-9. [DOI] [PubMed] [Google Scholar]
- Martin OJ, Lee A, McGraw TE. GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. J Biol Chem. 2006;281:484–490. doi: 10.1074/jbc.M505944200. [DOI] [PubMed] [Google Scholar]
- Martin S, Millar CA, Lyttle CT, Meerloo T, Marsh BJ, Gould GW, James DE. Effects of insulin on intracellular GLUT4 vesicles in adipocytes: evidence for a secretory mode of regulation. J Cell Sci. 2000;113:3427–3438. doi: 10.1242/jcs.113.19.3427. [DOI] [PubMed] [Google Scholar]
- Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391:87–93. doi: 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Spurlin BA, Pessin JE, Thurmond DC. Munc18c heterozygous knockout mice display increased susceptibility for severe glucose intolerance. Diabetes. 2005;54:638–647. doi: 10.2337/diabetes.54.3.638. [DOI] [PubMed] [Google Scholar]
- Oh E, Thurmond DC. Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes. 2009;58:1165–1174. doi: 10.2337/db08-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa VK, Ishikura S, Talior-Volodarsky I, Cheng AW, Patel N, Hartwig JH, Klip A. GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. J Biol Chem. 2008;283:27208–27219. doi: 10.1074/jbc.M804282200. [DOI] [PubMed] [Google Scholar]
- Reed SE, Hodgson LR, Song S, May MT, Kelly EE, McCaffrey MW, Mastick CC, Verkade P, Tavaré JN. A role for Rab14 in the endocytic trafficking of GLUT4 in 3T3-L1 adipocytes. J Cell Sci. 2013;126:1931–1941. doi: 10.1242/jcs.104307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 2007;5:293–303. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- Sano H, Peck GR, Kettenbach AN, Gerber SA, Lienhard GE. Insulin-stimulated GLUT4 protein translocation in adipocytes requires the Rab10 guanine nucleotide exchange factor Dennd4C. J Biol Chem. 2011;286:16541–16545. doi: 10.1074/jbc.C111.228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE. Rab10 in insulin-stimulated GLUT4 translocation. Biochem J. 2008;411:89–95. doi: 10.1042/BJ20071318. [DOI] [PubMed] [Google Scholar]
- Shi A, Chen CC, Banerjee R, Glodowski D, Audhya A, Rongo C, Grant BD. EHBP-1 functions with RAB-10 during endocytic recycling in Caenorhabditis elegans. Mol Biol Cell. 2010;21:2930–2943. doi: 10.1091/mbc.E10-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithers NP, Hodgkinson CP, Cuttle M, Sale GJ. Insulin-triggered repositioning of munc18c on syntaxin-4 in GLUT4 signalling. Biochem J. 2008;410:255–260. doi: 10.1042/BJ20070802. [DOI] [PubMed] [Google Scholar]
- Sun Y, Bilan PJ, Liu Z, Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc Natl Acad Sci USA. 2010;107:19909–19914. doi: 10.1073/pnas.1009523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamori Y, Kawanishi M, Niki T, Shinoda H, Araki S, Okazawa H, Kasuga M. Inhibition of insulin-induced GLUT4 translocation by Munc18c through interaction with syntaxin4 in 3T3-L1 adipocytes. J Biol Chem. 1998;273:19740–19746. doi: 10.1074/jbc.273.31.19740. [DOI] [PubMed] [Google Scholar]
- Thurmond DC, Ceresa BP, Okada S, Elmendorf JS, Coker K, Pessin JE. Regulation of insulin-stimulated GLUT4 translocation by Munc18c in 3T3L1 adipocytes. J Biol Chem. 1998;273:33876–33883. doi: 10.1074/jbc.273.50.33876. [DOI] [PubMed] [Google Scholar]
- Thurmond DC, Kanzaki M, Khan AH, Pessin JE. Munc18c function is required for insulin-stimulated plasma membrane fusion of GLUT4 and insulin-responsive amino peptidase storage vesicles. Mol Cell Biol. 2000;20:379–388. doi: 10.1128/mcb.20.1.379-388.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond DC, Pessin JE. Discrimination of GLUT4 vesicle trafficking from fusion using a temperature-sensitive Munc18c mutant. EMBO J. 2000;19:3565–3575. doi: 10.1093/emboj/19.14.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H, Huang X, Tanaka Y, Hirokawa N. KIF16B/Rab14 molecular motor complex is critical for early embryonic development by transporting FGF receptor. Dev Cell. 2011;20:60–71. doi: 10.1016/j.devcel.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Wang D, Lou J, Ouyang C, Chen W, Liu Y, Liu X, Cao X, Wang J, Lu L. Ras-related protein Rab10 facilitates TLR4 signaling by promoting replenishment of TLR4 onto the plasma membrane. Proc Natl Acad Sci USA. 2010;107:13806–13811. doi: 10.1073/pnas.1009428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, et al. C2 domain-containing phosphoprotein CDP138 regulates GLUT4 insertion into the plasma membrane. Cell Metab. 2011;14:378–389. doi: 10.1016/j.cmet.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Jordens I, Gonzalez E, McGraw TE. GLUT4 is sorted to vesicles whose accumulation beneath and insertion into the plasma membrane are differentially regulated by insulin and selectively affected by insulin resistance. Mol Biol Cell. 2010;21:1375–1386. doi: 10.1091/mbc.E09-08-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura S, Gerondopoulos A, Linford A, Rigden DJ, Barr FA. Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J Cell Biol. 2010;191:367–381. doi: 10.1083/jcb.201008051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigerer A, Lampson MA, Karylowski O, Sabatini DD, Adesnik M, Ren M, McGraw TE. GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol Biol Cell. 2002;13:2421–2435. doi: 10.1091/mbc.E02-02-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigerer A, McBrayer MK, McGraw TE. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell. 2004;15:4406–4415. doi: 10.1091/mbc.E04-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.