

The role of tau in axonal transport disruption during early-stage Alzheimer disease is controversial. The amyloid-β oligomers markedly impair BDNF transport in primary wild-type and tau-knockout neurons. This occurs by nonexcitotoxic activation of calcineurin, and inhibition of calcineurin rescues transport defects independent of tau.
Abstract
Disruption of fast axonal transport (FAT) is an early pathological event in Alzheimer's disease (AD). Soluble amyloid-β oligomers (AβOs), increasingly recognized as proximal neurotoxins in AD, impair organelle transport in cultured neurons and transgenic mouse models. AβOs also stimulate hyperphosphorylation of the axonal microtubule-associated protein, tau. However, the role of tau in FAT disruption is controversial. Here we show that AβOs reduce vesicular transport of brain-derived neurotrophic factor (BDNF) in hippocampal neurons from both wild-type and tau-knockout mice, indicating that tau is not required for transport disruption. FAT inhibition is not accompanied by microtubule destabilization or neuronal death. Significantly, inhibition of calcineurin (CaN), a calcium-dependent phosphatase implicated in AD pathogenesis, rescues BDNF transport. Moreover, inhibition of protein phosphatase 1 and glycogen synthase kinase 3β, downstream targets of CaN, prevents BDNF transport defects induced by AβOs. We further show that AβOs induce CaN activation through nonexcitotoxic calcium signaling. Results implicate CaN in FAT regulation and demonstrate that tau is not required for AβO-induced BDNF transport disruption.
INTRODUCTION
Fast axonal transport (FAT) is essential for neuronal function and survival. Disruption of FAT is an early pathological event in several neurodegenerative diseases, including amyotrophic lateral sclerosis, Parkinson's disease, and Alzheimer's disease (AD; Goldstein, 2012; Millecamps and Julien, 2013). Amyloid-β oligomers (AβOs), increasingly considered proximal neurotoxins in AD, interact with glutamate receptors at the dendritic membrane, induce abnormal calcium influx and oxidative stress, block long-term potentiation (LTP), and facilitate long-term depression, ultimately leading to synapse failure (Ferreira and Klein, 2011; Benilova et al., 2012). Of importance, AβOs impair FAT in cultured neurons and in AD mouse models (Goldstein, 2012; Millecamps and Julien, 2013). Although FAT disruption is implicated in AD pathogenesis, mechanisms underlying this process are poorly understood.
Tau, an axonal microtubule-associated protein, is believed to mediate AβO toxicity and FAT disruption. AβOs induce hyperphosphorylation of tau (p-tau), promoting its dissociation from microtubules and aggregation into neurofibrillary tangles (NFTs; De Felice et al., 2008). AβOs also induce tau proteolysis by calpains and caspases, generating fragments that aggregate independently of hyperphosphorylation (Reifert et al., 2011). Despite the accumulation of p-tau in affected neurons, it is controversial whether tau is required for AβO toxicity. Tau−/− neurons are resistant to Aβ-induced death (King et al., 2006); by contrast, NFTs persist in viable neurons possessing intact microtubule networks until late-stage AD (Castellani et al., 2008). Recent studies suggest that p-tau inhibits FAT by interacting directly with motor-cargo complexes or initiating aberrant signaling cascades that alter FAT dynamics (LaPointe et al., 2009; Kanaan et al., 2011), and that tau reduction prevents AβO-induced defects in mitochondria and neurotrophin receptor TrkA transport (Vossel et al., 2010). Conversely, axonal transport is not affected by tau overexpression or suppression in vivo (Yuan et al., 2008). We previously reported that AβOs block FAT of dense core vesicles (DCVs) and mitochondria in cultured neurons through an N-methyl-d-aspartate receptor (NMDAR)–dependent mechanism that is mediated by a tau kinase, glycogen synthase 3β (GSK3β; Decker et al., 2010). Of note, we did not observe concomitant microtubule destabilization, suggesting that the microtubule-binding capacity of tau did not affect transport of these cargoes.
Aberrant calcium signaling is implicated in AD (Berridge, 2010) and may contribute to FAT disruption. AβOs dysregulate calcium influx through NMDARs, leading to cytosolic calcium elevation and activation of the calcium/calmodulin-dependent phosphatase, calcineurin (CaN). Although CaN mediates AβO-induced synapse failure (Jurgensen et al., 2011; Reese and Taglialatela, 2011), a role for CaN in FAT regulation has not been investigated. Activated CaN relieves inhibition of protein phosphatase 1 (PP1), which activates GSK3β (Peineau et al., 2007), suggesting that CaN might be involved in FAT disruption.
Here we show that AβOs impair transport of DCVs containing brain-derived neurotrophic factor (BDNF) in hippocampal neurons from both wild type (tau+/+) and tau-knockout (tau−/−) mice, suggesting that tau is not required for transport disruption. FAT inhibition is not accompanied by microtubule destabilization or cell death. Significantly, AβOs impair BDNF transport by overactivating CaN through nonexcitotoxic calcium signaling, and inhibition of CaN rescues transport defects. Furthermore, inhibition of PP1 and GSK3β, downstream targets of CaN, prevents BDNF transport defects independent of tau. These findings implicate CaN in FAT regulation and challenge the requirement for tau in AβO-induced transport disruption.
RESULTS
AβO-induced disruption of axonal transport is not accompanied by tau hyperphosphorylation at microtubule-binding sites, spatial redistribution, or fragmentation
AβOs induce p-tau (De Felice et al., 2008), which may inhibit FAT by destabilizing microtubules. To determine whether p-tau correlates with transport disruption in cultured hippocampal neurons (Decker et al., 2010), we assessed tau phosphorylation at Ser-396 and Ser-404, residues that are characteristically hyperphosphorylated in AD, and at Thr-231 and Ser-262, located in the microtubule-binding domain of tau, by semiquantitative immunocytochemistry and immunoblotting. In agreement with previous results for the AD-related epitope PHF-1 (Ser-396/Ser-404; De Felice et al., 2008), we found a twofold increase in p-tau at Ser-396 and Ser-404 after 4 h of exposure to 500 nM AβOs (Figure 1A). It is noteworthy, however, that no perturbation of axonal transport was detected within this time frame (Decker et al., 2010). After 18 h of exposure to AβOs, when transport is markedly reduced (Decker et al., 2010), a threefold to fivefold increase in p-tau (at the Ser-396 and Ser-404 residues) was observed (Figure 1A), similar to PHF-1. Of interest, however, p-tau at Thr-231 and Ser-262 showed no differences between AβO-treated and control neurons (Figure 1A), suggesting that the association of tau with microtubules was unaffected. Total tau protein levels were also unchanged by AβO treatment (Figure 1A). These results show that AβOs do not induce tau phosphorylation at residues associated with microtubule binding during the time frame in which the transport defect is initiated.
FIGURE 1:

AβO-induced disruption of axonal transport is not accompanied by tau hyperphosphorylation at microtubule-binding sites, spatial redistribution, or fragmentation. (A) Quantification of p-tau immunocytochemistry. After 4 and 18 h, AβOs increase p-tau at AD-related sites (Ser-396 and Ser-404) but do not elevate p-tau at residues located at the microtubule-binding domain of tau (Thr-231and Ser-262). Inset: AβOs do not change total tau levels. Graph shows mean ± SEM. A minimum of 24 cells from three independent cultures were analyzed per condition; *p < 0.05 and **p < 0.005 relative to controls. The dashed line represents the control condition. (B) Representative images of p-tau immunocytochemistry (Ser-396). AβOs do not disrupt the spatial gradient of axonal p-tau. A similar staining pattern was observed for Ser-404 (Supplemental Figure S1). Star indicates cell body, arrowhead indicates proximal axon, and arrows indicate distal axon. Scale bar, 100 μm. Quantitation of phospho-tau immunofluorescence comparing the ratio of fluorescence signals between initial and distal portions of the axon. A minimum of 14 cells from two independent cultures were analyzed per condition. a.u., arbitrary units. (C) AβOs do not induce cleavage of full-length tau (50–70 kDa) and generation of a 17-kDa fragment after 18 h of treatment. Quantification of PHF p-tau in control and AβO-treated neuronal lysates. AβOs induce a twofold increase in p-tau. Graph shows mean ± SEM from three independent cultures.
To further assess axonal cytoskeletal integrity and its potential role in transport disruption induced by AβOs, we evaluated the proximodistal gradient of p-tau, important for axonal development and function (Mandell and Banker, 1996). Excessive phosphorylation of tau and its subsequent detachment from microtubules may perturb this gradient and impair transport (Dixit et al., 2008). To determine the spatial distribution of axonal p-tau, we used an approach that extracts soluble tau while stabilizing tau associated with microtubules (Mandell and Banker, 1996). Semiquantitative immunocytochemistry revealed no changes in the Ser-396 or the Ser-404 p-tau gradient after 18 h of AβO treatment compared with control cells (Figure 1B and Supplemental Figure S1). These findings suggest that changes in tau solubility and association with the axonal cytoskeleton may not contribute significantly to transport impairment.
Independently of phosphorylation, AβOs induce tau fragmentation through activation of calpain and caspases, rendering tau prone to aggregation and potentially contributing to AβO toxicity (Reifert et al., 2011). To determine whether tau fragmentation contributes to AβO-induced transport impairment, we probed control and AβO-treated hippocampal cell lysates for PHF-1 p-tau and total tau. In agreement with immunocytochemistry data (Figure 1A), we detected a twofold increase in p-tau after 18 h of exposure to AβOs (Figure 1D). We did not, however, detect a reduction in full-length tau (50–70 kDa) or appearance of a 17-kDa tau fragment (Reifert et al., 2011; Figure 1D). Moreover, calpain and caspase-3, known to induce tau fragmentation, were not activated under our experimental conditions (Supplemental Figure S3). Collectively these results suggest that p-tau and tau fragmentation do not mediate AβO-induced BDNF transport disruption.
AβOs disrupt BDNF transport independent of tau
Recent studies suggest that tau impairs transport by promoting motor protein dissociation from microtubules (Seitz et al., 2002) or disrupting motor protein–cargo interactions (Kanaan et al., 2011). To determine whether tau is required for AβO-induced FAT disruption, tau+/+ and tau−/− neurons expressing BDNF–monomeric red fluorescent protein (mRFP), a DCV cargo, were imaged 18 h after exposure to 500 nM AβOs (Figure 2A and Supplemental Movie S1). Irreversible AβO binding to dendrites was confirmed retrospectively by immunocytochemistry (Figure 2A and Supplemental Figure S4) using an oligomer-specific antibody (NU-4; Lambert et al., 2007). Representative kymographs illustrate differences between BDNF transport in control (vehicle-treated) and AβO-treated neurons (Figure 2B). Total axonal flux was similarly and markedly reduced by AβOs both in the presence and absence of tau (59 and 62% decrease, respectively; Figure 2, B and C, and Supplemental Table S1). AβOs also significantly decreased anterograde average velocity by ∼12% in both tau+/+ and tau−/− neurons, whereas a reduction in anterograde average run length was detected only in AβO-treated tau+/+ neurons (18%; Supplemental Table S1). A correlation between average velocity and run length was not observed (Supplemental Figure S1). Results show that AβOs perturb BDNF transport similarly in tau+/+ and tau−/− neurons and suggest that alternative mechanisms regulate transport in the absence of tau.
FIGURE 2:

Tau is not required for AβO-induced disruption of BDNF transport. (A) Expression of soluble BFP and BDNF-mRFP in an AβO-treated tau−/− neuron (from left to right). Overlay of BFP and AβO images shows binding of AβOs exclusively to dendrites. Immunocytochemistry shows that AβOs remain oligomeric after 18 h in culture. Arrows indicate axon; arrowheads indicate dendrites. Scale bar, 50 μm (B) Representative kymographs of BDNF transport in control and 500 nM AβO-exposed tau+/+ and tau−/− neurons. Green trace indicates anterograde transport; red trace indicates retrograde transport. (C) Effects of 500 nM AβO treatment on BDNF flux. Flux is markedly reduced in both tau+/+ and tau−/− neurons treated with AβOs. A minimum of 20 cells from three different cultures were analyzed per condition; ***p < 0.001 relative to controls. (D) BDNF transport disruption is dose and time dependent. Treatment with 100 nM AbOs induces significant transport defects by 72 h. A minimum of 15 cells from three independent cultures were analyzed per condition. Complete statistical evaluation is presented in Supplemental Tables S1 and S2.
AβO-induced disruption of BDNF transport is dose and time dependent
Next we investigated whether transport defects are induced by AβOs in a dose- and time-dependent manner. We treated tau+/+ and tau−/− neurons with 100 nM AβOs and assessed BDNF transport after 18, 48, and 72 h. Bidirectional transport was markedly reduced in both tau+/+ and tau−/− neurons after 72 h of AβO exposure (Figure 2D). In addition, we found significant reductions in average anterograde and retrograde run lengths in tau+/+ and tau−/− neurons (∼22 and 23% respectively), along with significant decrease in average velocity in tau−/− neurons (∼15%; Supplemental Table S2). These results differ from those obtained with 500 nM AβOs at 18 h (e.g., a decrease only in anterograde run length; Supplemental Table S1), suggesting that the selective mechanisms for BDNF transport disruption depend on the conditions of AβO exposure.
BDNF transport defects occur independent of changes in microtubule stability and tubulin posttranslational modifications
To verify that tau-independent transport defects do not result from microtubule destabilization, we assessed tubulin stability in control and AβO-treated tau+/+ and tau−/− neurons by gently extracting soluble tubulin while stabilizing polymerized tubulin (Black et al., 1996). Immunoblots of these protein fractions revealed no change in the ratio of soluble to polymerized tubulin between control and AβO-treated tau−/− neurons, indicating that AβOs do not induce tubulin depolymerization in the absence of tau (Figure 3A). Similar results for tau+/+ neurons were obtained as reported in Decker et al. (2010).
FIGURE 3:

BDNF transport defects occur in the absence of changes in microtubule stability and tubulin posttranslational modifications. (A) Immunoblots of tubulin from tau−/− neurons extracted in PHEM buffer. No change in the ratio of soluble to polymerized tubulin between control and AβO-treated neurons was observed. Similar results were obtained for tau+/+ neurons (data not shown). (B) Immunoblots of acetylated, tyrosinated, and detyrosinated tubulin. AβOs did not alter levels of these modifications in control and AβO-treated tau+/+ and tau−/− neurons. Graphs show means ± SEM from three independent cultures. (C, D) Representative images and quantification of acetylated tubulin immunocytochemistry. AβO treatment does not induce significant changes in tubulin acetylation. Graphs show average total fluorescent pixel counts, normalized against the tau+/+ control. Error bars represent SEM. A minimum of 20 cells from three independent cultures were analyzed per condition. Scale bar, 25 μm.
We next examined whether AβOs might alter posttranslational tubulin modifications involved in microtubule stability (Henriques et al., 2010) and recruitment or control of motor proteins (Janke and Kneussel, 2010). We compared tubulin acetylation and detyrosination as indicators of microtubule stability. We also assessed levels of tyrosinated tubulin, an indicator of microtubule destabilization (Janke and Kneussel, 2010). AβOs did not alter levels of acetylation, detyrosination, and tyrosination in either control or AβO-treated tau+/+ and tau−/− neurons (Figure 3B). In addition, we evaluated these tubulin modifications by semiquantitative immunocytochemistry (Figure 3, C and D). No significant differences in acetylated tubulin immunofluorescence were observed between control and AβO-treated tau+/+ and tau−/− neurons (Figure 3, C and D). Similar results were obtained for detyrosinated and tyrosinated tubulin (Supplemental Figure S2). Together, these findings indicate that tau-independent transport defects do not result from microtubule destabilization and changes in tubulin modifications.
Calcineurin inhibition rescues AβO-induced transport defects
We previously showed that AβOs reduce DCV transport through an NMDAR-dependent mechanism, which is mediated by GSK3β (Decker et al., 2010). Dysregulated calcium influx through NMDARs leads to elevation of resting intracellular calcium (LaPointe et al., 2009; Demuro et al., 2010) and activation of the calcium/calmodulin-dependent phosphatase, calcineurin (CaN; Reese and Taglialatela, 2011). Activated CaN relieves inhibition of PP1, which in turn activates GSK3β (Peineau et al., 2007). Thus, we investigated whether CaN mediates AβO-induced transport disruption. We exposed tau+/+ and tau−/− neurons to 500 nM AβOs for 18 h and subsequently treated them with 1 μM FK506, a highly specific, potent inhibitor of CaN (Schreiber and Crabtree, 1992). Remarkably, inhibition of CaN reversed AβO-induced transport defects in tau+/+ and tau−/− neurons within 1–3 h of treatment (Figure 4 and Supplemental Movie S1). FK506 rescued both anterograde and retrograde flux (Figure 4, A–C). Average DCV velocity and run length were also restored to control levels (Supplemental Table S1). These data support a key role for CaN in FAT disruption.
FIGURE 4:
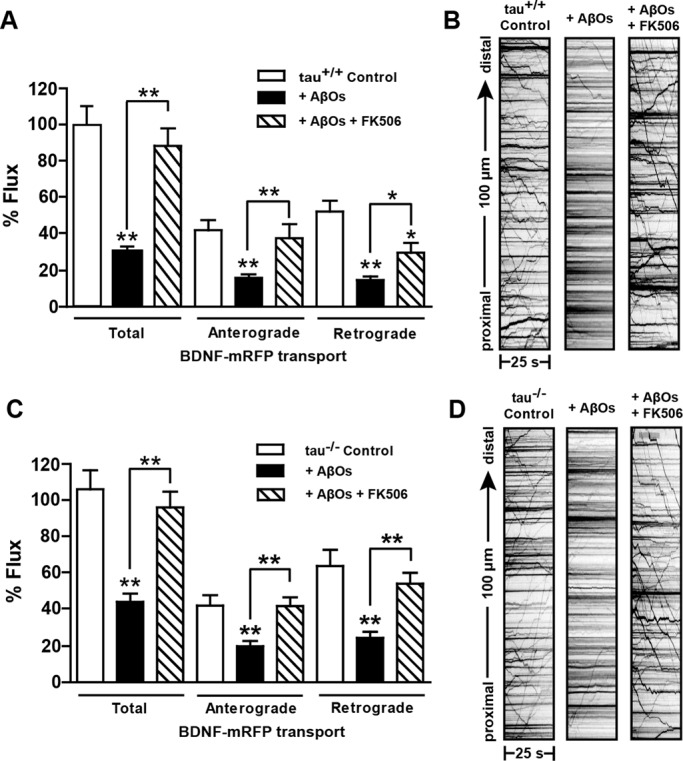
Inhibition of calcineurin by FK506 rescues AβO-induced transport defects independent of tau. (A, B) Effects of AβOs and FK506 treatment on BDNF flux in tau+/+ and tau−/− neurons. Flux is markedly reduced in cells treated with AβOs and is restored 1–3 h after treatment with 1 μM FK506. (C, D) Representative kymographs comparing BDNF transport in control and AβO-treated neurons. A minimum of 15 cells from three different cultures were analyzed per condition; *0.001 < p < 0.05, and **p < 0.001 relative to controls. Scale bar, 25 μm. Complete statistical evaluation is presented in Supplemental Table S1. Recordings of BDNF transport disruption and rescue in tau−/− neurons are shown in Supplemental Movie S1.
Calcineurin activity and protein phosphatase inhibitor-1 dephosphorylation are elevated by AβOs and normalized by FK506
To determine whether AβO-induced transport disruption involved changes in CaN activity, we used an in vitro phosphatase assay based on colorimetric detection of RII substrate dephosphorylation. We treated tau+/+ and tau−/− neurons with AβOs and FK506 and measured total phosphatase activity, CaN activity, and the combined activity of PP1/PP2A in their lysates (Figure 5, A and B). Total phosphatase activity was significantly elevated by AβOs (29%), significantly reduced by FK506 alone (55%), and restored to control levels in the presence of both agents (Figure 5, A and B). These effects were largely attributed to changes in CaN activity, which followed analogous trends (Figure 5, A and B). No significant differences in the combined activities of PP1/PP2A were detected by this method, attesting to RII substrate specificity for CaN. We confirmed these changes in CaN activity and assessed their potential effect on downstream PP1 activity by probing tau+/+ and tau−/− neuronal lysates for phospho-inhibitor-1 (I-1; Figure 5, C and D). CaN directly inactivates I-1 by dephosphorylation at Thr-35; by this mechanism, CaN relieves inhibition of PP1 (Peineau et al., 2007). In both tau+/+ and tau−/− neurons, I-1 phosphorylation was significantly reduced by AβOs (40%), significantly elevated by FK506 alone (100%), and restored to control levels in the presence of both agents (Figure 5, C and D). Collectively, these findings demonstrate that AβOs impair BDNF transport by activating CaN and suggest that PP1 is also involved in transport disruption.
FIGURE 5:

Calcineurin activity is elevated by AβOs and normalized by FK506. (A, B) In vitro phosphatase assays on tau+/+ and tau−/− primary neuronal lysates. Total phosphatase activity is significantly elevated by AβOs, significantly reduced by FK506 alone, and restored to control levels in the simultaneous presence of both agents. These effects are largely attributed to changes in CaN activity, which follows analogous trends. AβOs do not induce significant changes in the combined activity of PP1 plus PP2A. (C, D) Protein phosphatase inhibitor-1 (I-1) dephosphorylation in tau+/+ and tau−/− neurons. Dephosphorylation of I-1 (29 kDa) is significantly elevated by AβOs, significantly reduced by FK506 alone, and normalized in the presence of both agents. Tubulin immunoblots indicate equal sample loading. (E, F) Calpain-dependent cleavage of calcineurin in tau+/+ and tau−/− neurons. No significant differences in the ratios of truncated CaN (57 and 48 kDa) to full-length CaN (61 kDa) are detected between control and AβO-treated neurons. Tubulin immunoblots indicate equal sample loading. Graphs show mean ± SEM from three independent cultures; *p < 0.05, and **p < 0.001 relative to controls.
Inhibition of protein phosphatase-1 and glycogen synthase kinase 3β prevents AβO-induced transport defects
AβOs activate several phosphatases and kinases downstream of CaN, including PP1 and GSK3β (Krafft and Klein, 2010; Braithwaite et al., 2012). Activated CaN dephosphorylates inhibitor-1, which leads to activation of PP1 (Mulkey et al., 1994). PP1 activates GSK3β by dephosphorylation of Ser-9 (Morfini et al., 2004; Lee et al., 2005; Szatmari et al., 2005; Peineau et al., 2007). This cascade is invoked during long-term depression (LTD) induction (Peineau et al., 2008) and might also contribute to AβO-induced synaptotoxicity (Berridge, 2010; Benilova et al., 2012). Moreover, PP1-GSK3β signaling has been shown to disrupt motor protein–cargo binding in squid axoplasm models of AD (Morfini et al., 2002; LaPointe et al., 2009; Kanaan et al., 2011). Thus, we investigated whether PP1 and GSK3β mediate AβO-induced transport defects in tau+/+ and tau−/− neurons. First, we induced expression of I-2, an inhibitory subunit of PP1 (Zhang et al., 2003), for 24 h and subsequently treated neurons with 500 nM AβOs for 18 h (Figure 6A). Inhibition of PP1 by this mechanism prevented reductions in bidirectional flux and restored average velocity and run length to control levels (Figure 6B and Supplemental Table S3). To determine whether GSK3β dysregulates transport, we pretreated tau+/+ and tau−/− neurons with a selective cell-permeant chemical inhibitor (inhibitor VIII) or expressed a dominant-negative form of GSK3β (K85A) for 24 h before AβO treatment. Using both approaches, we found that AβO-induced BDNF transport defects were prevented in both tau+/+ and tau−/− neurons (Figure 6B and Supplemental Table S4). Conversely, transfection with a constitutively active form of GSK3β (S9A; Stambolic and Woodgett, 1994) significantly inhibited transport in both tau+/+ and tau−/− neurons, even in the absence of AβOs (Supplemental Table S4). Collectively, these findings suggest that PP1-GSK3β signaling downstream of CaN mediates AβO-induced transport disruption.
FIGURE 6:

Inhibition of protein phosphatase-1 and glycogen synthase kinase 3β prevents AβO-induced transport defects. (A) Expression of myc-tagged I-2 and BDNF-mRFP in an AβO-treated tau−/− neuron (from left to right). Overlay of BDNF-mRFP and AβO images shows binding of AβOs exclusively to dendrites. Retrospective immunocytochemistry was used to confirm I-2 expression and AβO binding. Arrows indicate axon; arrowheads indicate dendrites. Scale bar, 50 μm. (B) Effects of PP1 and GSK3β inhibition on BDNF flux in AβO-treated tau+/+ and tau−/− neurons. Bidirectional flux is markedly reduced by AβOs, and transport defects are prevented by expression of I-2 for 24 h, pretreatment with 5 μM inhibitor VIII for 30 min, and expression of kinase-dead GSK3β (K85A) for 24 h. Flux is similarly impaired by expression of kinase-active GSK3β (S9A). (C) Representative kymographs comparing BDNF transport in control, AβO-treated, and inhibitor-treated tau+/+ and tau−/− neurons. A minimum of 15 cells from three different cultures were analyzed per condition; ***p < 0.001 relative to controls and +++p < 0.001 relative to AβO-treated cells. Complete statistical evaluation is presented in Supplemental Tables S3 and S4.
AβO-induced activation of calcineurin and disruption of transport are not mediated by excitotoxic calcium signaling
Finally, we investigated whether AβOs activate CaN through excitotoxic calcium signaling by probing tau+/+ and tau−/− neuronal lysates for truncated forms of CaN, generated by calpain-induced cleavage (Liu et al., 2005). In glutamate-induced excitotoxicity, activated calpain cleaves full-length CaN, producing constitutively active 57- and 48-kDa truncated forms (Figure 5E). However, no differences in the ratios of truncated, active CaN to full-length CaN (61 kDa) were detected between control and AβO-treated lysates (Figure 5E). We further measured levels of nonerythroid α-II spectrin (280 kDa), a well-defined physiological substrate of calpain, in AβO-exposed neurons. Immunoblots revealed that calpain-mediated cleavage of spectrin, which generates a characteristic 150-kDa fragment, was not increased by AβOs (Supplemental Figure S3). These results demonstrate that AβOs do not activate CaN through excitotoxic, calpain-mediated fragmentation.
During excitotoxicity, intracellular calcium is sequestered into the mitochondrial matrix, decreasing the electrochemical gradient generated by the electron transport chain, thereby reducing ATP synthesis. The concurrent accumulation of intramitochondrial calcium and reduced ATP synthesis is a primary cause of cell death. To rule out the possibility that transport disruption under our conditions resulted from these events, we measured ATP levels in control and AβO-treated tau+/+ and tau−/− neurons using an in vitro luciferase-based ATP assay and observed no changes in ATP levels (Supplemental Figure S3). Furthermore, caspase-3, a key mediator of excitotoxic cell death, was not cleaved and activated (Supplemental Figure S3). Collectively, these results show that transport disruption does not result from excitotoxic CaN activation and cell death, suggesting that a mechanism downstream of CaN signaling triggers transport defects induced by AβOs.
DISCUSSION
FAT disruption is an early pathological manifestation that leads to loss of synapse function and axonal degeneration in AD (De Vos et al., 2008). AβOs are central to AD pathology and impair FAT. AβOs are known to induce tau hyperphosphorylation (De Felice et al., 2008), although it is unclear whether tau is required for AβO-mediated FAT disruption (Castellani et al., 2008). Here, through direct assessment of FAT at high temporal and spatial resolution in living neurons, we demonstrate that AβO-induced defects in axonal BDNF transport persist in the absence of tau and cannot be attributed to microtubule destabilization or cell death. Our findings support the notion that tau is not essential for normal transport (Yuan et al., 2008; Vossel et al., 2010), and suggest that additional intracellular signaling mechanisms negatively regulate FAT in AβO-exposed neurons. We combined multiple approaches, including live imaging, in vitro phosphatase assays, and immunoblotting, to demonstrate that inhibition of CaN by FK506 completely rescues BDNF transport defects, and that AβOs impair transport by overactivating CaN through nonexcitotoxic calcium signaling. Collectively, our work implicates CaN in FAT regulation and challenges a requirement for tau in AβO-induced transport disruption in primary neurons.
Considerable evidence implicates tau in AβO toxicity, and a variety of proposed mechanisms explain how tau reduction might prevent or ameliorate it. Such mechanisms include altered microtubule stability (Rapoport et al., 2002; King et al., 2006), elimination of a toxic tau fragment (Park and Ferreira, 2005), regulation of neuronal activity (Shipton et al., 2011), and changes in subcellular localization of the Src kinase, Fyn (Ittner et al., 2010). Intriguingly, we found that tau ablation does not prevent AβO-induced FAT defects (Figure 2). This suggests that the role of tau in AβO-induced toxicity depends on differences in model systems and relevant stages of AD progression examined or to temporal differences in activation of distinct signaling mechanisms that lead to toxicity. For instance, tau reduction ameliorates cognitive deficits in human amyloid precursor protein (APP)–overexpressing mice. These mice exhibit extensive plaque formation, mimicking chronic Aβ accumulation during late-stage AD, when abnormal phosphorylation, distribution, and signaling properties of tau are prevalent and likely contribute significantly to LTP impairment and toxicity (Morris et al., 2011; Shipton et al., 2011). In contrast, we assessed FAT in neurons that were acutely exposed to AβOs. These conditions are likely more relevant to early-stage AD, when hyperphosphorylated and fragmented tau have not reached detrimental concentrations (Morris et al., 2011), and when evidence of transport defects exists in animal models (Kim et al., 2011). We cannot rule out the possibility that p-tau ultimately accumulates and inhibits FAT by interacting directly with motor–cargo complexes or initiating aberrant signaling cascades that eventually alter FAT dynamics. Our findings imply that tau, although critical for AβO toxicity during late-stage AD, is probably not essential for initiating transport defects in early-stage AD.
AβOs instigate aberrant calcium signaling before excessive tau hyperphosphorylation, NFT formation, cognitive decline, and other late-stage hallmarks of AD (Stutzmann, 2007). In cultured neurons and AD mouse models, AβOs persistently elevate resting cytosolic calcium by allowing dysregulated calcium influx through NMDARs and subsequent release of calcium from internal stores (De Felice et al., 2007; Lopez et al., 2008; Demuro et al., 2010; Paula-Lima et al., 2011). Inhibition of NMDARs prevents FAT defects (Decker et al., 2010), rendering aberrant calcium signaling a plausible mechanism for AβO-induced FAT disruption in the absence of tau. Our findings (e.g., Figures 4 and 5) corroborate numerous reports of detrimental CaN signaling during early AD pathogenesis (Supnet and Bezprozvanny, 2010). Within minutes of AβO treatment, a progression of calcium/calmodulin-dependent CaN activation is observed, first in dendritic spines and minutes to hours later in the cell body (Wu et al., 2012). Subsequent activation of downstream kinases, such as GSK3β, can lead to glutamate receptor internalization, spine retraction, LTP blockage, and LTD facilitation (Reese and Taglialatela, 2011). AβOs can further increase CaN signaling through nonapoptotic caspase-3 activation in dendritic spines (D'Amelio et al., 2011; Hyman, 2011). These observations are characteristic of early AD pathogenesis, as they occur in very young APP-overexpressing mice before Aβ plaque deposition, NFT formation, and significant neuronal loss (de Calignon et al., 2009, 2010; D'Amelio et al., 2011). CaN activation is predominantly dependent on calcium/calmodulin in our studies and similarly precedes apoptotic caspase-3 activation and cell death (Figures 1 and 5 and Supplemental Figure S3). Taken together, these results emphasize roles for calcium dysregulation and CaN activation in AβO toxicity during early AD pathogenesis.
There are several mechanisms by which AβOs might disrupt axonal BDNF transport independently of tau. One mechanism could involve CaN-dependent inhibition of motor protein activity, mediated by GSK3β. GSK3β is implicated in many aspects of AD pathogenesis (Hooper et al., 2008) and negatively regulates axonal transport of APP in Drosophila embryos (Weaver et al., 2013). Negative regulation of kinesin-1 (KIF5) and cytoplasmic dynein is accomplished by reducing the number of motors that are bound to microtubules. A second mechanism might comprise disruption of motor protein–cargo binding. Phosphorylation of kinesin light chain-1 (KLC1) by PP1-GSK3β signaling (Morfini et al., 2002) and casein kinase 2 (Pigino et al., 2009) dissociates KIF5 from vesicular cargoes in a squid axoplasm model of AD. Moreover, axonal trafficking of calsyntenin-1, a membrane protein that mediates the attachment of KIF5 to APP-containing vesicles, is impaired by phosphorylation of KLC1 (Vagnoni et al., 2011). Likewise, it is possible that CaN activation leads to GSK3β-mediated impairment of axonal BDNF transport by a similar mechanism(s).
Our results demonstrate that AβOs impair bidirectional BDNF transport and that PP1 and GSK3β inhibition prevents reductions in anterograde and retrograde flux. It is possible that GSK3β largely governs anterograde transport; however, several recent studies have elucidated regulatory mechanisms that are coordinated through opposing motors, whereby disrupting one motor impairs bidirectional transport (Ally et al., 2009; Uchida et al., 2009; Welte, 2009; Jolly and Gelfand, 2011). In AD models, bidirectional transport of mitochondria (Rui et al., 2006; Vossel et al., 2010), amyloid precursor protein (Weaver et al., 2013), and organelles contained in squid axoplasm (Pigino et al., 2009) and primary hippocampal neurons (Hiruma et al., 2003) are similarly perturbed. Although this mechanism is commonly observed, subtler changes in axonal transport have been detected for different AβO treatments and cargoes (Tang et al., 2012).
Transport regulation is specific to the motor proteins and cargoes involved. Intriguingly, tau reduction prevents AβO-induced defects in axonal transport of mitochondria and TrkA (Vossel et al., 2010). The apparent difference between our present results and those of Vossel et al. (2010) might be attributed to different mechanisms of motor protein regulation. BDNF and other DCV cargoes are transported primarily by KIF1A (Park et al., 2008; Lo et al., 2011), whereas mitochondria and TrkA are transported primarily by KIF5. KIF5 interacts with the calcium-sensitive mitochondrial protein Miro, which inhibits KIF5 motility in a calcium-dependent manner (Wang and Schwarz, 2009). This switch in KIF5 activity and subsequent inhibition of mitochondrial transport are observed within minutes of intracellular calcium elevation. Thus, imaging tau-deficient neurons at finer temporal resolution might have revealed a similar, temporary arrest in mitochondrial transport immediately following AβO application and coinciding with transient intracellular calcium elevation (Vossel et al., 2010). It is possible that transport defects observed in wild-type neurons within 1 h of AβO treatment might indeed be primarily attributed to tau. Moreover, micromolar concentrations of AβOs (as used in previous studies) might induce robust tau hyperphosphorylation, initiate its detachment from microtubules, and expose a phosphatase-activating domain within the N-terminus of tau that activates PP1-GSK3β signaling and impedes transport (Kanaan et al., 2011). By contrast, nanomolar concentrations of AβOs used here might permit observation of calcium-mediated transport disruption, while the relative contribution of pathogenic tau is minimal. Typically, high calcium is required for physiological cessation of transport (Schlager and Hoogenraad, 2009); specific calcium-dependent mechanisms that modulate KIF1A motility have yet to be characterized.
On the basis of our present findings and other current models of AD pathogenesis, we propose the following mechanism for AβO-induced disruption of axonal BDNF transport (Figure 7). AβOs elevate resting cytosolic calcium by allowing dysregulated calcium influx through NMDARs and calcium-induced calcium release from internal stores. Calmodulin binds free calcium ions and subsequently activates CaN, dephosphorylating I-1 and relieving inhibition of PP1. On activation by PP1, GSK3β may disrupt BDNF transport by directly phosphorylating and inhibiting motor proteins and/or disrupting motor–DCV interactions, representing an early transport deficit in the progression of AD (Goldstein, 2012). These events occur before excessive tau hyperphosphorylation, NFT formation, and microtubule destabilization. Furthermore, transport defects are not secondarily influenced by cellular toxicity despite prolonged, irreversible AβO binding at low nanomolar concentrations. Of note, reduced levels of BDNF correlate with the progression of AD (Diniz and Teixeira, 2011), and this reduction could be related to transport and release of this neuropeptide. Finally, prevention or reversal of FAT defects and consequent axonal degeneration might help to ameliorate neuronal loss and cognitive decline in AD.
FIGURE 7:

Proposed mechanism for fast axonal transport disruption in an AD model. At dendrites, AβOs aberrantly activate NMDARs and induce calcium influx, elevating cytosolic calcium. Activated calcineurin relieves inhibition of PP1, which activates GSK3β. GSK3β may inhibit motor protein activity (1) and/or disrupt motor-cargo interactions (2) and impede transport in the presence of AβOs.
MATERIALS AND METHODS
Hippocampal cell culture and expression of transgenes
Primary hippocampal neuronal cultures from E16 wild-type (tau+/+) and tau-knockout (tau−/−) mice (Jackson Laboratory, Bar Harbor, ME) were prepared as described by Kaech and Banker (2006) and kept in Neurobasal/B27 (Invitrogen, Carlsbad, CA) or PNGM primary neuron growth media (Lonza, Basel, Switzerland). At 10–12 d in vitro, cells were cotransfected with pβ-actin-BDNF-mRFP and pmUBa–enhanced blue fluorescent protein (BFP; from Gary Banker, Oregon Health and Sciences University, Portland, OR) using Lipofectamine (Invitrogen). Cells expressed the plasmids for 24–36 h before live imaging. Experiments assessing a role for GSK3β and PP1 used the following plasmids and were transfected as described: pcDNA3 HA-GSK3β S9A and pcDNA3 HA-GSK3β K85A (gifts from Jim Woodgett, University of Toronto; plasmids 14754 and 14755; Addgene, Cambridge, MA) and I-2/pCS2 (plasmid 16317; Addgene). The absence of tau in tau−/− mice was confirmed by immunoblotting with the antibodies PHF-1 and tau-46 (Supplemental Figure S1). All experiments with animals were approved by and followed the guidelines set out by the Simon Fraser University Animal Care Committee, Protocol 943-B05.
AβO, FK506, and GSK3β inhibitor VIII treatments
Soluble, full-length Aβ 1-42 peptides (AβOs; American Peptide, Sunnyvale, CA) were prepared exactly according to the method of Lambert et al. (1998) and applied to cells at a final concentration of 500 nM for 18 h or 100 nM for up to 72 h. After AβO or vehicle exposure, cells were incubated with 1 μM FK506 (Sigma-Aldrich, St. Louis, MO) or equivalent volumes of vehicle (ethanol) for 1–3 h before imaging of transport. Cells were treated with 5 μM GSK3b inhibitor (Calbiochem, La Jolla, CA) 30 min before AbO exposure.
Live imaging and analysis of BDNF-mRFP transport
BDNF-mRFP transport was analyzed using a standard wide-field fluorescence microscope equipped with a cooled charge-coupled device camera and controlled by MetaMorph (Molecular Devices, Sunnyvale, CA) according to Kwinter et al. (2009). All imaging—typically 100 frames—was recorded by the “stream acquisition module” in MetaMorph. Briefly, cells were sealed in a heated imaging chamber, and streaming recordings were acquired from double transfectants at an exposure time of 250 ms for 90 s. This captured dozens of transport events per cell in 100-μm segments of the axon. Axons were initially identified based on morphology and confirmed retrospectively by immunostaining against MAP2, a dendritic cytoskeletal protein. Soluble BFP detection was necessary to determine the orientation of the cell body relative to the axon and thus to distinguish between anterograde and retrograde transport events. Vesicle flux, velocity, and run lengths were obtained through tracing kymographs in MetaMorph. Vesicle flux was defined as the total distance traveled by vesicles standardized by the length and duration of each movie (in micron-minutes), 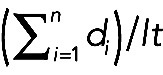 , where di are the individual DCV run lengths, l is the length of axon imaged, and t is the duration of the imaging session. A vesicle was defined as undergoing a directed run if it traveled a distance of ≥2 μm. This distance was determined as a safe estimate of the limit of diffusion based on the assumption that root-mean-squared displacement equals
, where di are the individual DCV run lengths, l is the length of axon imaged, and t is the duration of the imaging session. A vesicle was defined as undergoing a directed run if it traveled a distance of ≥2 μm. This distance was determined as a safe estimate of the limit of diffusion based on the assumption that root-mean-squared displacement equals 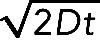 , where D is the diffusion coefficient (D = 0.01 μm2/s for DCVs) and t is the duration of the imaging period (t = 50 s; Abney et al., 1999). A run was defined as terminating if the vesicle was found to remain in the same position for at least four consecutive frames. Percentage flux represents the flux in treated neurons normalized to controls (100%).
, where D is the diffusion coefficient (D = 0.01 μm2/s for DCVs) and t is the duration of the imaging period (t = 50 s; Abney et al., 1999). A run was defined as terminating if the vesicle was found to remain in the same position for at least four consecutive frames. Percentage flux represents the flux in treated neurons normalized to controls (100%).
Immunocytochemistry
Neurons were fixed in 4% paraformaldehyde and blocked with 0.5% fish-skin gelatin (Kwinter et al., 2009). To confirm AβO binding to dendrites and verify qualitatively that AβOs remained oligomeric after 18 h in culture, cells were stained with an Aβ oligomer–specific antibody (NU-4, 1:1000; from W. L. Klein, Northwestern University, Evanston, IL) or 6E10 (1:1000; Covance, Berkeley, CA) and anti-MAP2 (1:2000; Millipore, Billerica, MA). To assess p-tau and tubulin modifications, neurons were stained with anti–p-tau PHF1 (1:300; from P. Davies, Albert Einstein School of Medicine, New York, NY), anti–p-tau Ser-262, Thr-231, Ser-396, Ser-404 (1:300; Sigma-Aldrich; from D. Vocadlo, Simon Fraser University), anti–acetylated tubulin (1:2000; 6-11B-1, Sigma-Aldrich), anti–detyrosinated tubulin (1:2000; Millipore), or anti–tyrosinated tubulin (1:1000; TUB-1A2, Sigma-Aldrich). The presence of HA-GSK3β and myc-I-2 was confirmed retrospectively by antibody staining against the hemagglutinin (1:100; Roche, Indianapolis, IN) or myc (1:500; Sigma-Aldrich) epitopes. Neurons were subsequently incubated with compatible secondary antibodies conjugated to Cy3 (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA), Alexa 488 (1:500, Invitrogen), or Cy5 (1:500; Jackson ImmunoResearch Laboratories). To quantify tau phosphorylation and tubulin modifications, histograms were generated using ImageJ (National Institutes of Health, Bethesda, MD) from the fluorescence intensity of each pixel across several images, and the average intensity and SE of the mean were calculated. Appropriate thresholds were applied to eliminate background signal before histogram analysis, as described in De Felice et al. (2008). Phospho-tau axon gradient staining for each epitope, Ser-396 and Ser-404, was performed and quantified exactly according to Mandell and Banker (1996).
Immunoblotting
Neurons were treated with vehicle, AβOs, and/or FK506 as described. To induce caspase 3 activation, neurons were treated with 5 μM staurosporine (EMD Millipore, Darmstadt, Germany). Neurons were lysed in RIPA buffer containing Complete Protease Inhibitor Cocktail (Roche) and Halt Phosphatase Inhibitor Cocktail (Thermo Scientific, Waltham, MA). Samples (5-10 μg) were resolved on 10–12% SDS–polyacrylamide gels and transferred to polyvinylidene fluoride membranes. Membranes were incubated with the following primary antibodies overnight at 4°C: anti–p-tau PHF-1 (1:1000), antixtau-5 and tau-46 (1:1000; from C. Krieger, Simon Fraser University), anti–acetylated tubulin (1:2000), anti–detyrosinated tubulin (1:1000), anti–tyrosinated tubulin (1:1000), anti–phospho-I1 Thr-35 (Santa Cruz Biotechnology, Santa Cruz, CA; 1:500, preadsorbed on adult mouse whole-brain homogenate), anti–CaN-A (1:1000; Enzo Life Sciences, Plymouth Meeting, PA), anti–α-II spectrin (1:1000; AA6, Millipore), anti–caspase-3 (1:1000; 8G10, New England BioLabs, Ipswich, MA), and anti-tubulin (1:2000; DM1A, Millipore). Immunoreactive bands were visualized using enhanced chemiluminescent substrate (ECL; Thermo Scientific) for detection of peroxidase activity from horseradish peroxidase–conjugated antibodies. Densitometric scanning and quantitative analysis were carried out using ImageJ.
Biochemical quantification of tubulin
Control and AβO-treated neurons were extracted with PHEM buffer (60 mM 1,4-piperazinediethanesulfonic acid, pH 6.9, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 10 mM ethylene glycol tetraacetic acid [EGTA], 2 mM MgCl2, 10 μM Taxol, 0.2% Triton X-100, and protease and phosphatase inhibitors), a microtubule-stabilizing buffer, as described previously (Black et al., 1996). Equal protein amounts of Triton X–soluble and –insoluble fractions were analyzed by immunoblotting using anti-tubulin DM1A (1:2000; Sigma-Aldrich).
In vitro phosphatase activity assays
Neurons were treated with vehicle, AβOs, and/or FK506 as described. Total phosphatase activity, CaN activity, and the combined activity of PP1 plus PP2A were measured using a colorimetric assay based on RII substrate dephosphorylation (Calcineurin Cellular Activity Assay Kit; Calbiochem). Briefly, cells were scraped in lysis buffer, and protein extracts were collected by high-speed centrifugation and desalted using chromatography columns (GE Healthcare, Piscataway, NJ). To measure total phosphatase activity, lysates were incubated with RII according to manufacturer's instructions. To discriminate between CaN and PP1 plus PP2A activity, lysates were additionally incubated with 10 mM EGTA and 500 nM okadaic acid. RII was omitted from a parallel set of reactions to assess background phosphatase activity. Human recombinant calcineurin served as a positive control. After incubation at 30°C for 30 min, reactions were terminated with the GREEN color indicator and developed at room temperature for 30 min. Absorbance was measured at 620 nm using a Spectramax M2 microplate reader (Molecular Devices), and the number of nanomoles of phosphate released was determined from a standard curve. CaN activity was calculated by subtracting phosphatase activity in the presence of EGTA from total phosphatase activity. Similarly, PP1 plus PP2A activity was calculated by subtracting phosphatase activity in the presence of okadaic acid from total phosphatase activity.
ATP assay
ATP levels in control and AβO-treated neurons were assessed by luciferase-based detection of ATP, according to the manufacturer's protocol (CellTiter-Glo Luminescent Cell Viability Assay; Promega, Madison, WI). After vehicle and AβO exposures, neurons were washed briefly in phosphate-buffered saline (PBS). Neurons were incubated in 200 μl of CellTiter-Glo luminescent reagent and 200 μl of PBS for 10 min at room temperature. Luminescence was measured using a microplate reader (Molecular Devices).
Statistical analyses
Statistical analyses were performed using Excel (Microsoft, Redmond, WA) or Prism (GraphPad, La Jolla, CA). Data are presented as mean ± SEM. Significant differences between treatments were analyzed by t tests with equal or unequal variance at a 95% confidence interval. For live-imaging experiments, a minimum of 15 cells from three independent cultures (n = 3) were analyzed. For immunoblots, in vitro phosphatase assays, and ATP assays, neuronal lysates from at least three independent cultures were analyzed. To determine the strength of relationship between velocity and run length, the Spearman's rank correlation coefficient was calculated. Spearman's r between 0.3 and 0.5 indicates a moderate-to-low correlation between the two variables.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Science and Engineering Research Council of Canada (327100-06), the Canadian Foundation for Innovation (12793), and the Canadian Institutes of Health Research (90396) to M.A.S. We thank Lesley Chen and Cathleen Pfefferkorn for their expert technical assistance, C. D. Link and T. Tomiyama for their critical reading of the manuscript, and V. Gelfand for essential discussions. K.J.G. is funded by a C.D. Nelson Memorial Graduate Scholarship from Simon Fraser University and a National Science and Engineering Research Council of Canada Postgraduate Scholarship. H.D. thanks the Brazilian agency Coordenação de Aperfeiçoamento de Pessoal de Nível Superior for fellowship support. S.T.F. is supported by grants from the National Institute of Translational Neuroscience, Conselho Nacional de Desenvolvimento Científico e Tecnológico, and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (Brazil). We also thank the Simon Fraser University Animal Care Services staff for their essential assistance in our experiments.
Abbreviations used:
- AβOs
amyloid-β oligomers
- AD
Alzheimer's disease
- BDNF
brain-derived neurotrophic factor
- BFP
blue fluorescent protein
- CaN
calcineurin
- DCV
dense core vesicle
- FAT
fast axonal transport
- GSK3β
glycogen synthase 3 β
- NMDAR
N-methyl-d-aspartate receptor
- PP1
protein phosphatase 1
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0858) on June 19, 2013.
*Co–first authors.
†Present address: Jungers Center for Neurosciences Research, Oregon Health and Science University, Portland, OR 97239.
E.M.R., K.J.G., and E.Y.F. characterized transport defects in tau−/− neurons. E.M.R. detected tau fragmentation and activation of calpain and caspase-3, assessed tubulin polymerization and modifications by immunoblotting, and investigated the dose and time dependence of AβO-induced transport defects. K.J.G. assessed tubulin modifications by immunocytochemistry, designed and conducted CaN and GSK3β inhibition and transport rescue experiments, and measured CaN activation. E.Y.F. performed the PP1 inhibition and transport rescue experiments. H.D. quantified p-tau and analyzed its spatial distribution by immunocytochemistry. M.M.S. performed the ATP assays and assisted in data analysis of transport. E.M.R. analyzed her data and contributed to writing of the article. K.J.G., S.T.F., and M.A.S. analyzed data and wrote and revised the article. M.A.S. conceived and supervised the study.
REFERENCES
- Abney JR, Meliza CD, Cutler B, Kingma M, Lochner JE, Scalettar BA. Real-time imaging of the dynamics of secretory granules in growth cones. Biophys J. 1999;77:2887–2895. doi: 10.1016/S0006-3495(99)77120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ally S, Larson AG, Barlan K, Rice SE, Gelfand VI. Opposite-polarity motors activate one another to trigger cargo transport in live cells. J Cell Biol. 2009;187:1071–1082. doi: 10.1083/jcb.200908075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Calcium signalling and Alzheimer's disease. Neurochem Res. 2010;36:1149–1156. doi: 10.1007/s11064-010-0371-4. [DOI] [PubMed] [Google Scholar]
- Black MM, Slaughter T, Moshiach S, Obrocka M, Fischer I. Tau is enriched on dynamic microtubules in the distal region of growing axons. J Neurosci. 1996;16:3601–3619. doi: 10.1523/JNEUROSCI.16-11-03601.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC. Protein phosphatases and Alzheimer's disease. Prog Mol Biol Transl Sci. 2012;106:343–379. doi: 10.1016/B978-0-12-396456-4.00012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Nunomura A, Lee HG, Perry G, Smith MA. Phosphorylated tau: toxic, protective, or none of the above. J Alzheimers Dis. 2008;14:377–383. doi: 10.3233/jad-2008-14404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amelio M, et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Spires-Jones TL, Pitstick R, Carlson GA, Hyman BT. Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009;68:757–761. doi: 10.1097/NEN.0b013e3181a9fc66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- De Felice FG, et al. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA. Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010;30:9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz BS, Teixeira AL. Brain-derived neurotrophic factor and Alzheimer's disease: physiopathology and beyond. Neuromol Med. 2011;13:217–222. doi: 10.1007/s12017-011-8154-x. [DOI] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol Learn Mem. 2011;96:529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS. Axonal transport and neurodegenerative disease: can we see the elephant. Prog Neurobiol 99, 186–190. 2012 doi: 10.1016/j.pneurobio.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques AG, Vieira SI, da Cruz ESEF, da Cruz ESOA. Abeta promotes Alzheimer's disease-like cytoskeleton abnormalities with consequences to APP processing in neurons. J Neurochem. 2010;113:761–771. doi: 10.1111/j.1471-4159.2010.06643.x. [DOI] [PubMed] [Google Scholar]
- Hiruma H, Katakura T, Takahashi S, Ichikawa T, Kawakami T. Glutamate and amyloid beta-protein rapidly inhibit fast axonal transport in cultured rat hippocampal neurons by different mechanisms. J Neurosci. 2003;23:8967–8977. doi: 10.1523/JNEUROSCI.23-26-08967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT. Caspase activation without apoptosis: insight into Abeta initiation of neurodegeneration. Nat Neurosci. 2011;14:5–6. doi: 10.1038/nn0111-5. [DOI] [PubMed] [Google Scholar]
- Ittner LM, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Janke C, Kneussel M. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010;33:362–372. doi: 10.1016/j.tins.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Jolly AL, Gelfand VI. Bidirectional intracellular transport: utility and mechanism. Biochem Soc Trans. 2011;39:1126–1130. doi: 10.1042/BST0391126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensen S, Antonio LL, Mussi GE, Brito-Moreira J, Bomfim TR, De Felice FG, Garrido-Sanabria ER, Cavalheiro EA, Ferreira ST. Activation of D1/D5 dopamine receptors protects neurons from synapse dysfunction induced by amyloid-beta oligomers. J Biol Chem. 2011;286:3270–3276. doi: 10.1074/jbc.M110.177790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, Andreadis A, Fu Y, Brady ST, Binder LI. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci. 2011;31:9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Choi IY, Michaelis ML, Lee P. Quantitative in vivo measurement of early axonal transport deficits in a triple transgenic mouse model of Alzheimer's disease using manganese-enhanced MRI. Neuroimage. 2011;56:1286–1292. doi: 10.1016/j.neuroimage.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol. 2006;175:541–546. doi: 10.1083/jcb.200605187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafft GA, Klein WL. ADDLs and the signaling web that leads to Alzheimer's disease. Neuropharmacology. 2010;59:230–242. doi: 10.1016/j.neuropharm.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Kwinter DM, Lo K, Mafi P, Silverman MA. Dynactin regulates bidirectional transport of dense-core vesicles in the axon and dendrites of cultured hippocampal neurons. Neuroscience. 2009;162:1001–1010. doi: 10.1016/j.neuroscience.2009.05.038. [DOI] [PubMed] [Google Scholar]
- Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, et al. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100(1):23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YI, Seo M, Kim Y, Kim SY, Kang UG, Kim YS, Juhnn YS. Membrane depolarization induces the undulating phosphorylation/dephosphorylation of glycogen synthase kinase 3beta, and this dephosphorylation involves protein phosphatases 2A and 2B in SH-SY5Y human neuroblastoma cells. J Biol Chem. 2005;280:22044–22052. doi: 10.1074/jbc.M413987200. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- Lo KY, Kuzmin A, Unger SM, Petersen JD, Silverman MA. KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci Lett 491, 168–173. 2011 doi: 10.1016/j.neulet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Lopez JR, Lyckman A, Oddo S, Laferla FM, Querfurth HW, Shtifman A. Increased intraneuronal resting [Ca2+] in adult Alzheimer's disease mice. J Neurochem. 2008;105:262–271. doi: 10.1111/j.1471-4159.2007.05135.x. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. 1996;16:5727–5740. doi: 10.1523/JNEUROSCI.16-18-05727.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–176. doi: 10.1038/nrn3380. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Paula-Lima AC, Adasme T, SanMartin C, Sebollela A, Hetz C, Carrasco MA, Ferreira ST, Hidalgo C. Amyloid beta-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid Redox Signal. 2011;14:1209–1223. doi: 10.1089/ars.2010.3287. [DOI] [PubMed] [Google Scholar]
- Park JJ, Cawley NX, Loh YP. A bi-directional carboxypeptidase E-driven transport mechanism controls BDNF vesicle homeostasis in hippocampal neurons. Mol Cell Neurosci. 2008;39:63–73. doi: 10.1016/j.mcn.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci. 2005;25:5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Bradley C, Taghibiglou C, Doherty A, Bortolotto ZA, Wang YT, Collingridge GL. The role of GSK-3 in synaptic plasticity. Br J Pharmacol. 2008;153(Suppl 1):S428–S437. doi: 10.1038/bjp.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci USA. 2009;106:5907–5912. doi: 10.1073/pnas.0901229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci USA. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese LC, Taglialatela G. A role for calcineurin in Alzheimer's disease. Curr Neuropharmacol. 2011;9:685–692. doi: 10.2174/157015911798376316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifert J, Hartung-Cranston D, Feinstein SC. Amyloid beta-mediated cell death of cultured hippocampal neurons reveals extensive Tau fragmentation without increased full-length tau phosphorylation. J Biol Chem. 2011;286:20797–20811. doi: 10.1074/jbc.M111.234674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager MA, Hoogenraad CC. Basic mechanisms for recognition and transport of synaptic cargos. Mol Brain. 2009;2:25. doi: 10.1186/1756-6606-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- Seitz A, Kojima H, Oiwa K, Mandelkow EM, Song YH, Mandelkow E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002;21:4896–4905. doi: 10.1093/emboj/cdf503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipton OA, et al. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303:701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE. The pathogenesis of Alzheimers disease—is it a lifelong “calciumopathy”? Neuroscientist. 2007;13:546–559. doi: 10.1177/1073858407299730. [DOI] [PubMed] [Google Scholar]
- Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. 2010;47:183–189. doi: 10.1016/j.ceca.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari E, Habas A, Yang P, Zheng JJ, Hagg T, Hetman M. A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem. 2005;280:37526–37535. doi: 10.1074/jbc.M502699200. [DOI] [PubMed] [Google Scholar]
- Tang Y, Scott DA, Das U, Edland SD, Radomski K, Koo EH, Roy S. Early and selective impairments in axonal transport kinetics of synaptic cargoes induced by soluble amyloid beta-protein oligomers. Traffic. 2012;13:681–693. doi: 10.1111/j.1600-0854.2012.01340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A, Alami NH, Brown A. Tight functional coupling of kinesin-1A and dynein motors in the bidirectional transport of neurofilaments. Mol Biol Cell. 2009;20:4997–5006. doi: 10.1091/mbc.E09-04-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnoni A, Rodriguez L, Manser C, De Vos KJ, Miller CC. Phosphorylation of kinesin light chain 1 at serine 460 modulates binding and trafficking of calsyntenin-1. J Cell Sci. 2011;124:1032–1042. doi: 10.1242/jcs.075168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver C, Leidel C, Szpankowski L, Farley NM, Shubeita GT, Goldstein LS. Endogenous GSK-3/shaggy regulates bidirectional axonal transport of the amyloid precursor protein. Traffic. 2013;14:295–308. doi: 10.1111/tra.12037. [DOI] [PubMed] [Google Scholar]
- Welte MA. Bidirectional transport: matchmaking for motors. Curr Biol. 2009;20:R410–R413. doi: 10.1016/j.cub.2010.03.018. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Uemura K, Fan ZY, Berezovska O, Grosskreutz CL, Bacskai BJ, Hyman BT. Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-beta revealed by a novel fluorescence resonance energy transfer assay. J Neurosci. 2012;32:5298–5309. doi: 10.1523/JNEUROSCI.0227-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci. 2008;28:1682–1687. doi: 10.1523/JNEUROSCI.5242-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.