

The adaptor protein complex AP-5 is stably associated with the hereditary spastic paraplegia proteins SPG11 and SPG15 in an apparently equimolar ratio to form a new type of coat. Results suggest that SPG15 facilitates the docking of the coat onto late endosomal/lysosomal membranes and that SPG11 (possibly together with SPG15) acts as a scaffold.
Abstract
The AP-5 complex is a recently identified but evolutionarily ancient member of the family of heterotetrameric adaptor proteins (AP complexes). It is associated with two proteins that are mutated in patients with hereditary spastic paraplegia, SPG11 and SPG15. Here we show that the four AP-5 subunits can be coimmunoprecipitated with SPG11 and SPG15, both from cytosol and from detergent-extracted membranes, with a stoichiometry of ∼1:1:1:1:1:1. Knockdowns of SPG11 or SPG15 phenocopy knockdowns of AP-5 subunits: all six knockdowns cause the cation-independent mannose 6-phosphate receptor to become trapped in clusters of early endosomes. In addition, AP-5, SPG11, and SPG15 colocalize on a late endosomal/lysosomal compartment. Both SPG11 and SPG15 have predicted secondary structures containing α-solenoids related to those of clathrin heavy chain and COPI subunits. SPG11 also has an N-terminal, β-propeller–like domain, which interacts in vitro with AP-5. We propose that AP-5, SPG15, and SPG11 form a coat-like complex, with AP-5 involved in protein sorting, SPG15 facilitating the docking of the coat onto membranes by interacting with PI3P via its FYVE domain, and SPG11 (possibly together with SPG15) forming a scaffold.
INTRODUCTION
AP-5 is the most recently identified and the least well characterized of the heterotetrameric adaptor protein (AP) complexes (Hirst et al., 2011). Its subunits share so little sequence identity with the subunits of the other AP complexes that they cannot be found using standard bioinformatics tools such as BLAST, and so for many years the existence of a fifth AP complex was unsuspected. However, structural prediction programs indicate that the subunits of AP-5 adopt similar folds to their counterparts in APs 1–4, and so their nomenclature follows the same convention: ζ and β5 for the two large subunits, μ5 for the medium subunit, and σ5 for the small subunit, encoded by the genes AP5Z1, AP5B1, AP5M1, and AP5S1, respectively. Like all of the AP complexes, AP-5 is evolutionarily ancient (Hirst et al., 2011) and ubiquitously expressed (http://biogps.org/, www.ncbi.nlm.nih.gov/UniGene/), although its expression profile in developing chick embryos (Hirst et al., 2013) suggests that it may be particularly important in neurons.
Characterization of AP-5 has been somewhat hampered by its low abundance (Hirst et al., 2013) and absence from a number of model organisms (Hirst et al., 2011). However, an important insight into its function came from the discovery by Słabicki et al. (2010) that AP-5 subunits could be coimmunoprecipitated with two proteins mutated in patients with hereditary spastic paraplegia (HSP), SPG11 and SPG15 (also known as spatacsin and spastizin/ZFYVE26/FYVE-CENT, respectively). HSP is a group of genetic disorders characterized by progressive spasticity in the lower limbs. Mutations in SPG11 and SPG15 are the major causes of HSP accompanied by thin corpus callosum and mental impairment (Boukhris et al., 2008), and patients with mutations in these two genes present with the same clinical features. In addition, morpholino knockdowns of SPG11 and SPG15 in zebrafish produce very similar phenotypes, affecting the development of motor neurons (Martin et al., 2012). Both observations are consistent with the two proteins acting together in the same pathway. Furthermore, Słabicki et al. (2010) discovered that mutations in AP5Z1 are also associated with HSP, although in this case the patients had a later onset of the disease.
Both SPG11 and SPG15 are large proteins (>250 kDa), and SPG15 has a FYVE domain that binds in vitro to phosphatidylinositol 3-phosphate (PI3P; Sagona et al., 2010). Little is known about the precise functions of the two proteins or how they associate with AP-5. In addition, there is some controversy over the localization of SPG11 and SPG15, with labeling reported in many different locations, including endoplasmic reticulum, endosomes, microtubules, mitochondria, nuclei, and the midbody of dividing cells (Hanein et al., 2008; Sagona et al., 2010; Murmu et al., 2011). In the present study, we use a combination of biochemistry and microscopy to begin to dissect the structural and functional relationship between AP-5, SPG11, and SPG15.
RESULTS
Stable association of SPG11/SPG15 with AP-5
We previously generated a HeLa cell line expressing green fluorescent protein (GFP)–tagged σ5 and showed by Western blotting that when cytosol from these cells is immunoprecipitated with anti-GFP, other AP-5 subunits coprecipitate (Hirst et al., 2011). To identify additional AP-5–associated proteins, we have analyzed our σ5-GFP immunoprecipitates by mass spectrometry and also carried out immunoprecipitations on cells expressing GFP-tagged ζ, SPG15, and SPG11.
In the σ5-GFP immunoprecipitates, we identified >50 proteins, but only 6 of these were specifically brought down in cells expressing σ5-GFP and not in control cells, and these were the ζ, β5, μ5, and σ5 subunits of AP-5, together with SPG11 and SPG15 (Supplemental Table S1). To investigate the interaction further, we used the Quantitative BAC InteraCtomics (QUBIC) method, which allows the sensitive and unbiased detection of protein–protein interactions (Hubner et al., 2010). Triplicate immunoprecipitations were performed on three BAC transgenic cell lines expressing GFP-tagged SPG11, SPG15, or AP-5 ζ from their endogenous promoters. Precipitated proteins were identified by mass spectrometry and compared with immunoprecipitations performed on a control cell line, using label-free quantification. Proteins specifically associated with the bait were thus readily distinguished from nonspecific background proteins (Figure 1A). The only proteins that were consistently and specifically coprecipitated with all three baits were the four AP-5 subunits, SPG11, and SPG15.
FIGURE 1:

Stable association of SPG11 and SPG15 with AP-5. (A) QUBIC interaction proteomic analysis. GFP-tagged AP-5 ζ, SPG11, and SPG15 were stably expressed under the control of their endogenous promoters. Immunoprecipitations were performed with an anti-GFP antibody and compared by label-free quantitative mass spectrometry with immunoprecipitations (IPs) performed on a control cell line with no GFP bait protein. Every experiment was performed in triplicate. Data were analyzed with a t test to determine significant interactions (Hubner et al., 2010) and visualized in a “volcano plot.” For each identified protein, plots show the fold difference in abundance (bait IP vs. control IP; x-axis, log2 scale), as well as a p-value indicating robustness of the observed difference (y-axis, –log10 scale). Specific interactors have high fold differences and low p values (top right quadrant of the plot). The “volcano” lines indicate the significance cut-off that separates specific interactors from background. With every bait, all four AP-5 subunits, SPG11, and SPG15 are specifically coimmunoprecipitated. The SPG11 bait also coIPs a number of abundant cytoskeletal proteins, but since these proteins were not identified in the other two QUBIC experiments, it seems unlikely that these interactions are physiologically relevant. Furthermore, the SPG11 pull down has a greater scatter of background proteins than the AP-5 ζ and SGP15 pull downs, suggesting that it may be slightly less specific. (B) Stoichiometry analysis. Normalized peptide intensities were used to estimate the relative abundance of specific interactors identified in A (iBAQ method; Schwanhäusser et al., 2011). For each protein, the values from all triplicate repeats were plotted. Only coimmunoprecipitated proteins were included, since the bait protein tends to be overrepresented in immunoprecipitation experiments. The relative abundances of proteins were normalized to the median abundance of all proteins across each experiment (i.e., median set to 1.0). The data show that regardless of the bait protein, roughly equal molar amounts of AP-5 subunits, SPG11, and SPG15 are coprecipitated, which supports the existence of an equimolar hexameric complex consisting of AP-5, SPG11, and SPG15. The only exception is a substantially higher proportion of AP-5 σ precipitated with AP-5 ζ (top). Based on structural information on other AP complexes (Page and Robinson, 1995; Collins et al., 2002), these two subunits may form a stable subcomplex, and expression of tagged AP-5 ζ may thus stabilize and increase the recovery of AP-5 σ.
We also used the proteomic data to estimate the relative abundance of the precipitated proteins (Figure 1B). Our data indicate that all six proteins are present in equal copy numbers. In turn, this suggests that AP-5 is part of a stable hexameric complex consisting of one AP-5 tetramer and one copy each of SPG11 and SGP15.
The identification of AP-5 subunits in the SPG15-GFP immunoprecipitate was confirmed by Western blotting, which also showed that these interactions occur in cytosol, as well as on membranes (Figure 2A). The coprecipitation of SPG11 and SPG15 with cytosolic AP-5 indicates a very stable association because under the same conditions, clathrin does not coimmunoprecipitate with AP-1 or AP-2 (Figure 2B).
FIGURE 2:
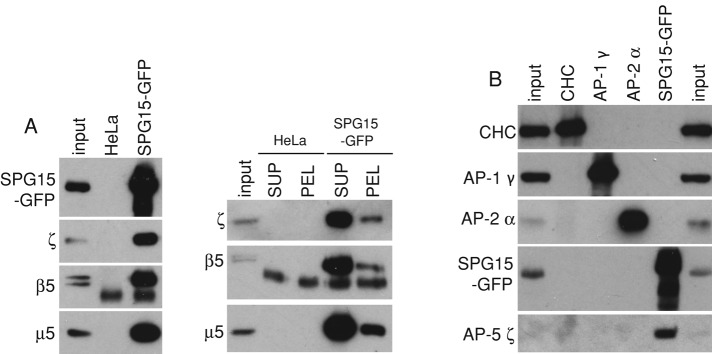
Western blots of immunoprecipitates. (A) Immunoprecipitations were carried out on either control HeLa cells or HeLa cells expressing SPG15-GFP using anti-GFP, and the blots were probed using antibodies against AP-5 subunits. AP-5 coprecipitates with SPG15-GFP in both a high-speed supernatant of homogenized cells (SUP) and a Triton X-100 extract of a high-speed pellet (PEL), indicating that the association occurs both in cytosol and on membranes. The lower–molecular weight band in the immunoprecipitates probed with anti-β5 appears to be nonspecific. (B) A cytosol fraction from SPG15-GFP–expressing cells was immunoprecipitated with the antibodies indicated at the top, and Western blots were probed with the antibodies indicated at the side. Although AP-5 coimmunoprecipitates with SPG15-GFP, AP-1 and AP-2 do not coimmunoprecipitate with clathrin heavy chain (CHC). The input is 2.5% relative to the IP for SPG15-GFP and 5% for CHC, AP-1, and AP-2.
SPG11 or SPG15 knockdown phenocopies AP-5 knockdown
If SPG11 and SPG15 are associated with AP-5, they might be expected to have similar knockdown phenotypes. We previously showed by immunofluorescence that knockdown of any of the AP-5 subunits results in perturbed trafficking of the cation-independent mannose 6-phosphate receptor (CIMPR), causing it to become trapped in clusters of endosomes that are positive for EEA1 and the retromer subunit Vps26 (Hirst et al., 2011; Figure 3). Knocking down either SPG11 or SPG15 produces a similar phenotype (Figure 3A), which we quantified by automated microscopy (Figure 3B; see Supplemental Figure S1 for Western blots of the knockdowns). In all three knockdowns, labeled structures appeared larger, brighter, and fewer, most likely due to endosomal clustering (Hirst et al., 2011). This phenotype could be observed not only with the small interfering RNA (siRNA) pool but also with single, nonoverlapping siRNAs (Supplemental Figure S2) and was slightly more pronounced for SPG15 than for SPG11. Knockdown of SPG15 (but not SPG11) also resulted in the tubulation of EEA1-positive endosomes (Supplemental Figure S2). Although the significance of the tubulation phenotype is unclear, knocking down another HSP protein, strumpellin, also causes endosomes to tubulate (Harbour et al., 2010).
FIGURE 3:

Knockdown of SPG11 and SPG15 phenocopies AP-5 knockdown. (A) HeLa cells were treated with siRNAs as indicated and double labeled for the CIMPR and the retromer protein Vps26. In the siRNA-treated cells, the CIMPR clusters in Vps26-positive endosomes. There also appears to be increased colocalization of CIMPR and Vps26 in these cells. All of the images of siRNA-treated cells were taken at half the exposure time of the controls because of the increased brightness. Scale bar, 20 μm. (B) The knockdown phenotypes were quantified using an ArrayScan VTI microscope and Spot Detector V4 algorithm application for automated image collection and analysis. Means of CIMPR labeling in control and knockdown cells were compared using repeated-measures analysis of variance and the post hoc Tukey–Kramer significance test (*p < 0.05, **p < 0.01, ***p < 0.001). More than 1500 cells were scored per knockdown condition (two independent repeats). In every knockdown, there is an increase in the area and intensity of spots and a concomitant decrease in the number of spots (although the decrease in spot number could be a result of increased clustering rather than fewer structures).
Localization of SPG11 and SPG15
In our previous study on AP-5, we carried out immunolocalization studies on cells expressing either μ5-GFP or σ5-GFP and saw punctate labeling that partially overlapped with the late endosomal/lysosomal marker LAMP1. We also saw nuclear labeling for σ5-GFP but not for μ5-GFP. This is most likely due to excess nonassembled σ5-GFP, which is sufficiently small (<50 kDa) to diffuse freely into the nucleus (Hirst et al., 2011; Figure 4A). We found that GFP-tagged SPG15 and SPG11 had a similar pattern but without the nuclear background, including overlap with LAMP1 (Figure 4A and Supplemental Figure S3). There have been conflicting reports about the localization of SPG11 and SPG15, however, and there is always the danger that tagged constructs may be mislocalized. Therefore, we made monoclonal antibodies against both SPG11 and the AP-5 ζ subunit to investigate the distribution of the endogenous proteins. Antibodies against both proteins labeled puncta distributed throughout the cytoplasm in human fibroblasts and in SPG15-GFP–expressing HeLa cells (Figure 4, B and C, and Supplemental Figure S4), and double labeling for endogenous SPG11 and LAMP1 again showed substantial overlap (Figure 4B). This punctate labeling pattern was lost when the proteins were depleted using siRNA (Supplemental Figure S4), confirming the specificity of the antibodies. There was also substantial colocalization between SPG15-GFP and endogenous ζ and SPG11 (Figure 4C). To investigate whether there is any overlap with some of the other structures that have been reported to colocalize with SPG15 and/or SPG11, we labeled our cells expressing the tagged constructs with markers for early endosomes, centrosomes, and endoplasmic reticulum exit sites but did not see coincident labeling (Supplemental Figure S5).
FIGURE 4:

Immunofluorescence labeling of AP-5, SPG15, and SPG11. (A) Cells stably expressing either σ5-GFP or SPG15-GFP were fixed and double labeled with antibodies against GFP (to enhance the signal) and the late endosomal/lysosomal protein LAMP1. Cytosolic σ5-GFP was washed out by saponin before fixation, leaving nuclear staining (this construct is likely to diffuse freely in and out of the nucleus). The punctate GFP labeling throughout the cytoplasm is partially coincident with LAMP1. (B) Primary human fibroblasts were double labeled for endogenous SPG11 and LAMP1. The two antibodies show good colocalization. (C) Cells expressing SPG15-GFP were fixed and double labeled with anti-GFP and monoclonal antibodies against either SPG11 or ζ subunit of AP-5. The labeling patterns for tagged SPG15 and endogenous ζ or SPG11 are largely coincident. Scale bars, 20 μm.
We also carried out live-cell imaging on the SPG15-GFP–expressing cells and found that the puncta were dynamic in nature, with long-range as well as short-range movements (Figure 5A and Supplemental Movie S1). When the cells were incubated with either Lysotracker Red or Magic Red Cathepsin B (Figure 5B and Supplemental Movies S2 and S3), we found almost complete overlap with SPG15-GFP. This indicates that the SPG15 compartment is acidic and contains active hydrolases. In addition, localization of SPG15-GFP by immunogold electron microscopy (EM) showed labeling of structures that often contained membrane whorls (Figure 5C). Together, these data show that AP-5, SPG11, and SPG15 localize to organelles that can be morphologically, enzymatically, and biochemically defined as late endosomes/lysosomes.
FIGURE 5:

SPG15 localization. (A) Stills from a movie (Supplemental Movie S1) showing cells expressing SPG15-GFP. Cells were imaged every 10 s over 15 min. Motile structures can be seen moving over short (arrows) and long distances (circle). Scale bar, 20 μm. (See Supplemental Movie S1.) (B) Cells expressing SPG15-GFP were either incubated with Lysotracker Red, a vital stain for acidic organelles, and imaged immediately, or incubated with Magic Red Cathepsin B substrate, a vital stain for active lysosomal hydrolases, for 30 min and then imaged. SPG15-GFP colocalizes with both markers. Scale bar: 20 μm. (See Supplemental Movies S2 and S3.) (C) Immunogold labeling of SPG15-GFP–expressing cells. Because of the low abundance of the protein, labeling was sparse, but there was very little background. Gold particles can be seen associated with organelles containing membrane whorls, characteristic of late endosomes/lysosomes, but we did not find any label associated with budding profiles. Scale bar, 200 nm.
Dependence of AP-5 on SPG11 and SPG15
To investigate whether AP-5, SPG11, and SPG15 are dependent on each other for their localization and/or stability, we knocked down one protein and then looked for effects on the others by immunofluorescence microscopy. Knockdown of either SPG11 or SPG15 resulted in a dramatic loss of σ5-GFP punctate labeling, comparable to the loss of σ5-GFP labeling when other subunits of AP-5 are knocked down (Figure 6A). Western blotting indicated that AP-5 subunits may be destabilized when SPG15 is depleted (Supplemental Figure S1), so the loss of punctate AP-5 labeling could result from effects on stability, recruitment, or both. In contrast, knocking down AP-5 subunits produced little or no effect on the localization of GFP-tagged SPG15 or SPG11. We also observed that AP-5 ζ labeling was brighter in cells expressing SPG15-GFP (Figure 4C), suggesting that increasing the expression of SPG15 increases the membrane localization of AP-5. Thus AP-5 appears to depend on SPG11/SPG15 for its localization and/or stability but not vice versa.
FIGURE 6:

Localization of AP-5 depends on SPG11/SPG15 and is sensitive to wortmannin. (A) Cells stably expressing σ5-GFP, SPG15-GFP, or SPG11-GFP were treated with siRNAs and then labeled with anti-GFP. The σ5-GFP–expressing cells were treated with saponin before fixation to wash out cytosolic proteins. The punctate labeling of σ5-GFP is lost when ζ, SPG11, or SPG15 is depleted. In contrast, the punctate labeling of SPG15-GFP or SPG11-GFP is not lost when ζ is depleted. SPG15 labeling becomes diffuse when SPG11 is depleted, however, and SPG11 labeling becomes diffuse when SPG15 is depleted. In both cases, siRNAs targeting the construct itself (plus the endogenous version of the protein) strongly reduce the total fluorescence. (B) Cells stably expressing either SPG15-GFP or σ5-GFP were treated with 5 μg/ml brefeldin A (BFA) for 5 min or 100 nM wortmannin for 1 h and then fixed. The σ5-GFP–expressing cells were treated with saponin to wash out cytosolic proteins before fixation. The punctate labeling of both proteins is insensitive to brefeldin A but is lost upon treatment with wortmannin. Scale bars, 20 μm.
A number of vesicle coat proteins, including AP-1, AP-3, and AP-4, require the small GTPase ARF1 to localize to membranes, and become cytosolic when cells are treated with the drug brefeldin A, which inhibits guanine nucleotide exchange factors for ARF1. Neither AP-5 nor SPG15 is affected by brefeldin A (Hirst et al., 2011; Figure 6B), however, indicating that their localization to membranes is ARF1 independent. The plasma membrane adaptor AP-2 is also ARF1 independent, but it requires a specific phosphoinositide for its localization, phosphatidylinositol 4,5-bisphosphate, which is mainly generated on the plasma membrane (Beck and Keen, 1991; Honing et al., 2005). SPG15 has been shown to bind in vitro to another phosphoinositide, PI3P (Sagona et al., 2010), which is found mainly on endosomes. To investigate the importance of this interaction in vivo, we treated cells with the phosphoinositide (PI) 3-kinase inhibitor wortmannin. The punctate patterns of both SPG15-GFP and σ5-GFP were lost under these conditions (Figure 6B), indicating that PI3P is required for the recruitment of both SPG15 and AP-5, most likely by interacting with the FYVE domain of SPG15.
AP-5 interacts with the N-terminal domain of SPG11
We previously showed that AP-5 does not colocalize with clathrin and cannot be detected in clathrin-coated vesicle-enriched fractions (Hirst et al., 2011), indicating that if it is a component of a vesicle coat, it must use some other type of scaffold. It is intriguing that both SPG11 and SPG15 are predicted to contain α-helical solenoids, similar to those of clathrin heavy chain, the α and β′ subunits of the COPI coat, and the Sec31 subunit of the COPII coat (Devos et al., 2004). In clathrin, COPI, and COPII, the α-solenoid is preceded by a β-propeller, and SPG11 also has a predicted N-terminal, β-propeller–like fold. In addition, HHpred (Söding et al., 2005) identifies clathrin heavy chain, α-COP, and β′-COP as matches for both the α-solenoid and the β-propeller regions (Figure 7, A and B).
FIGURE 7:

Domain organization of SPG15 and SPG11. (A) The domain organization of SPG15 was predicted by DOMpred, and then homology searching with each domain was carried out using HHpred (http://toolkit.tuebingen.mpg.de/hhpred). More information about the HHpred hits is available in Supplemental Table S2. PSIpred was used to carry out a secondary structure prediction for each residue. The α-helices are in magenta and β-strands in cyan. The height of each colored vertical line is proportional to the confidence of the secondary structure prediction (McGuffin et al., 2000). (B) A similar analysis was carried out on SPG11. (C) GST alone or the N-terminal domain of SPG11 coupled to GST was incubated with HeLa cell cytosol, and bound AP-5 ζ was detected by Western blotting. The N-terminal domain of SPG11 (GST-SPG11N) pulls down AP-5 ζ from cytosol. We estimate, however, that no more than ∼10% of the total AP-5 ζ was pulled down by the SPG11 construct, probably because most of the AP-5 already has SPG11 stably associated with it, so the pull down only captures “unoccupied” AP-5. As controls, blots of the cytosol and pull downs were also probed with antibodies against the AP-1 γ and AP-2 α subunits. Although both of these proteins are much more abundant in cytosol than AP-5 ζ, neither was detected in the GST-SPG11N pull down.
The β-propeller of clathrin heavy chain is a major hub for protein–protein interactions (Lemmon and Traub, 2012), with binding partners including AP-1, AP-2, and several “alternative adaptors.” To determine whether the β-propeller–like domain of SPG11 also acts as a binding platform, we carried out a glutathione S-transferase (GST) pull down on HeLa cell cytosol, using residues 1–500 of SPG11 as bait. We were able to detect the ζ subunit of AP-5 by Western blotting (Figure 7C), indicating that a similar type of interaction attaches AP-5 to SPG11. As controls, we probed for the γ subunit of AP-1 and the α subunit of AP-2. Even though AP-1 and AP-2 are ∼30- and ∼70-fold more abundant in HeLa cells than AP-5 (Hirst et al., 2013), respectively, they could not be detected in the pull down.
DISCUSSION
By analogy with other AP complexes, it seems likely that the role of AP-5 is to act as a cargo adaptor for a novel type of coat. Other components of the coat may include proteins that form a docking site to facilitate recruitment onto membranes and proteins that can assemble into some sort of scaffold. We propose that SPG15 and SPG11 function as a docking site and scaffold, respectively.
The interaction between AP-5 and SPG11/SPG15 was initially demonstrated by immunoprecipitation. We extended these observations to determine whether the proteins are associated with each other in cytosol as well as on membranes, determine their stoichiometry, and look for other binding partners. We found that the four AP-5 subunits, SPG11, and SPG15 invariably coimmunoprecipitate with each other, without pulling down any other proteins, and can be coimmunoprecipitated from cytosol, as well as from membrane extracts. This is in contrast to AP-1/AP-2 and clathrin, which only interact on membranes and do not efficiently coimmunoprecipitate even from membrane extracts. Thus AP-5 and SPG11/SPG15 are more like the COPI coat in this respect, where there is a relatively stable complex, called the coatomer, which can be dissociated into two subcomplexes (Pavel et al., 1998). SPG11 and SPG15 appear to be in an equimolar ratio with the four AP-5 subunits, which again is reminiscent of coatomer, where all seven subunits are stoichiometric with each other.
In addition to coimmunoprecipitating with AP-5, SPG11 and SPG15 have similar knockdown phenotypes to the AP-5 subunits. In every case, the CIMPR becomes trapped in membrane clusters that are positive for EEA1 and Vps26, indicating that they are early endosomal compartments. AP-5, SPG11, and SPG15 also have very similar subcellular distributions, localizing to a late endosomal/lysosomal compartment. This pattern can be seen with antibodies against endogenous proteins, as well as with tagged constructs, and it is strongly reduced when the proteins are depleted with siRNA, demonstrating that the labeling is specific. The identity of the compartment is based on several lines of evidence: the label shows substantial overlap with LAMP1; it colocalizes with both Lysotracker Red, a vital stain for acidic organelles, and Magic Red Cathepsin B, a vital stain for organelles containing active hydrolases; and by immunogold EM it is associated with structures containing membrane whorls. Thus, although knocking down the proteins produces changes in an early endosomal compartment, the proteins themselves localize (at least primarily) to a later compartment.
The connection between SPG11/SPG15/AP-5 and HSP indicates that a loss of these proteins is particularly deleterious to neurons with long axons because these are the cells that are primarily affected. Late endosomes and lysosomes are found mostly in the neuronal cell body, but some are present in axons, where they are transported mainly in the retrograde direction (Tsukita and Ishikawa, 1980; Cai et al., 2010). Whether mutations in AP-5, SPG11, or SPG15 affect axonal trafficking of these organelles or cause HSP for some other reason (e.g., by impairing axonal maintenance) remains to be determined.
The PI 3-kinase inhibitor wortmannin causes AP-5, SPG11, and SPG15 to appear cytosolic rather than membrane associated, indicating that the phosphoinositide PI3P acts as a membrane identity marker, most likely by binding to the FYVE domain of SPG15. Although PI3P is usually regarded as marker for an early endosomal compartment, there is at least one other protein, sorting nexin 16 (Snx16), that binds to PI3P (via a PX domain) but localizes to a late endosomal compartment (Brankatschk et al., 2011). In the case of Snx16, another domain also contributes to localization (Hanson and Hong, 2003), and it seems likely that additional interactions will be found to facilitate the binding of SPG11/SPG15 to late endosomes, similar to the “coincidence detection” mechanism used to recruit AP-2 onto membranes (Haucke, 2005).
Although knockdown of SPG11 or SPG15 affects the localization of AP-5, knocking down AP-5 does not affect the localization of SPG11 or SPG15. Most organisms have either both AP-5 and SPG11/SPG15 or neither, but there are a few exceptions, including Drosophila, that have SPG11/SPG15 but not AP-5 (Hirst et al., 2011). Thus it is possible that SPG11/SPG15 may be able to function in the absence of AP-5. The observation that patients with mutations in AP-5 ζ/AP5Z1 have a later onset of HSP than patients with mutations in SPG11 or SPG15 (Słabicki et al., 2010) is consistent with this possibility. However, only two AP5Z1-deficient patients from a single family have been identified, so more examples will be needed before firm conclusions can be drawn. Fifty-two different loci associated with HSP have been identified, but the causative genes have been found for only 31 of these (Finsterer et al., 2012), and there are likely to be other, as-yet-unidentified loci. Whole-genome or whole-exome sequencing may be the most efficient way of identifying additional HPS-causing mutations (Züchner, 2010; Bettencourt et al., 2013; Gonzalez et al., 2013). AP5B1, AP5M1, and AP5S1 are promising candidates for some of the other HPS causative genes, especially since mutations in all four of the AP-4 subunit genes have been shown to cause a form of “complex” HSP (reviewed in Hirst et al., 2013).
Unlike SPG15, SPG11 does not have any obvious functional domains; however, its predicted secondary structure, consisting of a β-propeller–like N-terminal domain followed by an α-solenoid, together with its homology to other coat components, including clathrin, suggests that it may form some sort of scaffold. Clathrin uses its N-terminal β-propeller domain to interact with AP complexes, and our pull-down experiments suggest that the same is true for the interaction between SPG11 and AP-5. It is interesting that this domain is missing in SPG11 from insects, which also lack AP-5. The α-solenoids of SPG11 and SPG15 may interact with each other to form a scaffold, similar to the clathrin, COPI, and COPII coats. It is not clear, however, what the morphology of such a scaffold might be because the labeling we observe for tagged SPG15 does not appear to be associated with budding profiles, suggesting that the AP-5/SPG11/SPG15 complex may be more analogous to the flat, bilayered clathrin/ESCRT-0 coats on early endosomes (Raiborg et al., 2002), which are believed to hold cargo proteins in place rather than package them into vesicles.
If AP-5 is indeed involved in cargo selection, what cargo proteins does it sort? We had hoped to find candidates in our immunoprecipitates, but the only proteins that were clearly being brought down specifically were AP-5 subunits, SPG11, and SPG15. This result is not entirely unexpected because coat–cargo interactions are very transient and often difficult to capture. We have recently been able to use the “knocksideways” technique for rapid protein inactivation, followed by subcellular fractionation and comparative proteomics, to identify >50 cargo proteins that are dependent on AP-1 and/or GGA2 for efficient packaging into CCVs (Hirst et al., 2012). By using a similar approach on AP-5, SPG11, and SPG15, we hope to be able to establish the precise functions of each of these proteins.
MATERIALS AND METHODS
Antibodies and constructs
Antibodies used in this study include in-house antibodies against clathrin and AP-1 (Simpson et al., 1996) and commercial antibodies against EEA1 (E41120; BD Transduction Labs, Lexington, KY), LAMP1 (sc18821; Santa Cruz Biotechnology, Santa Cruz, CA), GFP (ab6556; Abcam, Cambridge, MA), CIMPR (ab2733; Abcam), C-NAP1 (BD611375; BD Biosciences, San Diego, CA), AP-2 α (610502; BD Biosciences), and AP5Z1 (KIAA0415; sc139260; Santa Cruz Biotechnology). Rabbit anti-GFP and sheep anti-SEC16A were kind gifts from Matthew Seaman (Cambridge Institute for Medical Research, Cambridge, United Kingdom) and David Stephens (University of Bristol, Bristol, United Kingdom), respectively. Horseradish peroxidase–labeled secondary antibodies were purchased from Sigma-Aldrich (St. Louis, MO) and fluorescently labeled secondary antibodies (species and/or isotype specific) from Invitrogen (Carlsbad, CA). Monoclonal antibodies were raised against peptides from AP5Z1 (1H1) and SPG11 (4C10, 4L15, 3I13) by Abmart (Shanghai, China). Sixteen peptide immunogens for either SPG11 or ζ were overexpressed in Escherichia coli, purified by Ni-affinity chromatography, and injected into BALB/C mice. Spleen cells were fused with SP2/0 myeloma cells, and selected clonal cell lines were used to produce ascites fluid, from which antibodies were purified by protein A/G affinity chromatography. Where known, the epitope is indicated: 3I13 (KDHAKTSDPG), 4L15 (PVQNYKTKEG), and 4C10 (PQELQGSKQE). The isotypes of the mouse monoclonal antibodies made for this study are immunoglobulin G2b (IgG2b; 1H1 and 4C10), IgG2a (4L15), and IgG3 (3I13). The mouse monoclonals against EEA1, C-NAP1, and LAMP1 are all IgG1, and the mouse anti-GFP is IgG2a.
For pull-down experiments, a cDNA encoding residues 1–500 of SPG11 was cloned into pGEX4T-1 for expression of GST-SPG11N, and the resulting fusion protein (which was partially insoluble and degraded) was purified using glutathione-Sepharose, as specified by the manufacturer (GE Healthcare, Piscataway, NJ).
RNA interference
Knockdowns were performed using the following On-Target Plus siRNA reagents from Dharmacon, Lafayette, CO) or a nontargeting SMARTpool siRNA (D-001810-10) as a control. The siRNAs were as follows: μ5 (C14orf108), J-015523-09, J-015523-10; ζ (KIAA0415), L-025284-01; SPG11 (FLJ21439), L-017138-00; SPG15 (ZFYVE26), J-031136-09, J-031136-10, J-031136-11, J-031136-12; all used at a concentration of 25 nM. Knockdowns were performed with a single-hit 72-h protocol using Oligofectamine (Invitrogen) and Opti-Mem (Invitrogen) following the manufacturer's instructions.
Tissue culture
HeLaM cells (Tiwari et al., 1987) were grown in DMEM (Sigma-Aldrich) supplemented with 10% (vol/vol) fetal calf serum (Sigma-Aldrich), 2 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. A stable clonal cell line expressing σ5-GFP (Hirst et al., 2011) was derived by G418 selection. HeLa cells stably expressing GFP-tagged SPG11, SPG15, and KIAA0415 (ζ) have been previously described (Słabicki et al., 2010). Because of loss of expression over time in culture, the cells were sorted by flow cytometry for medium to high expression of GFP and maintained in G418-containing medium.
Fluorescence microscopy
For immunofluorescence microscopy, cells were plated into glass-bottom dishes (MatTek, Ashland, MA) and treated where indicated with 5 μg/ml brefeldin A for 5 min, 100 nM wortmannin for 1 h, or 0.05% (wt/vol) saponin in phosphate-buffered saline (PBS) for 1 min. The cells were then fixed with 3% formaldehyde, permeabilized with 0.1% Triton X-100, and labeled as indicated. The cells were imaged with a Zeiss Axiovert 200 inverted microscope (Carl Zeiss, Jena, Germany) using a Zeiss Plan Achromat 63× oil immersion objective (numerical aperture 1.4), an OCRA-ER2 camera (Hamamatsu, Hamamatsu, Japan), and Improvision Openlab software (PerkinElmer, Waltham, MA).
For live-cell microscopy, cells were plated into glass-bottom dishes (MatTek) and incubated in CO2-independent media with 50 nM Lysotracker Red DND-99 (Invitrogen) or Magic Red Cathepsin B substrate (AbD Serotec, Raleigh, NC), following manufacturer's instructions. The cells were imaged on a Zeiss LSM710 confocal microscope with Zeiss ZEN software. Movie images were captured every 10 s for a period of up to 15 min.
To quantify knockdown phenotypes, we used an automated ArrayScan VTI microscope (Cellomics/Thermo Fisher, Pittsburgh, PA) and the SpotDetector V4 assay algorithm. Cells were plated onto 96-well PerkinElmer microplates and stained with anti-CIMPR, followed by Alexa Fluor 488–donkey anti-mouse IgG and whole-cell stain (Invitrogen). The cells were imaged with a modified Zeiss Axiovert 200M inverted microscope, a Zeiss 40×/0.5 Achroplan objective, and a Hamamatsu OCRA-ER camera, and >1500 cells quantified for each condition using ARRAYSCAN software.
Electron microscopy
For immunogold electron microscopy, a clonal line of cells expressing SPG15-GFP was derived, permeabilized by immersion in liquid N2, and fixed by adding an equal volume of freshly prepared 8% paraformaldehyde/0.2% gluteraldehyde in 0.1 M phosphate buffer, pH 7.4. After 5 min the solution was removed and cells were postfixed in 4% paraformaldehyde/0.1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, for 1 h at room temperature and further processed as previously described (Hirst et al., 2009). Ultrathin sections were labeled with the commercial GFP antibody (see previous description), followed by protein A conjugated to colloidal gold (Utrecht University, Utrecht, Netherlands), and viewed using a Phillips CM 100 transmission electron microscope (Philips Electron Optics, Cambridge, United Kingdom) at an operating voltage of 80 kV.
Immunoprecipitation and GST pull-down experiments
For immunoprecipitations from whole-cell lysates, cells stably expressing σ5-GFP or SPG15-GFP were solubilized in PBS containing 1% Triton X-100 and insoluble material removed before incubation with GFP-Trap (ChromoTek, Martinsried, Germany), according to the manufacturer's instructions. For analysis by mass spectrometry, proteins were processed by filter-aided sample preparation solution digest (Wis´niewski et al., 2009), and the sample was analyzed by liquid chromatography–tandem mass spectrometry in an Orbitrap mass spectrometer (Thermo Scientific; Waltham, MA; Antrobus and Borner, 2011). For immunoprecipitations from cytosol and membrane fractions, cells stably expressing SPG15-GFP were scraped in PBS and lysed by six passages through a 21-gauge needle/5-ml syringe. Nuclei and unbroken cells were removed by centrifugation at 4000 × g for 5 min, and then membranes were recovered at 50,000 × g for 1 h. The membrane pellet was solubilized in PBS containing 1% Triton X-100 and clarified by centrifugation. Triton X-100 was also added to the supernatant to a concentration of 1%, and then both samples were incubated with GFP-Trap (ChromoTek) according to the manufacturer's instructions.
For GST pull-down experiments, cells were solubilized in PBS containing 1% NP40, and insoluble material was removed by centrifugation at 20,000 × g for 30 min. Samples containing 5 mg of starting lysate were precleared with 50 μg/ml GST, followed by glutathione–Sepharose. The lysates were then incubated with 50 μg/ml GST-SPG11N, followed by glutathione–Sepharose, and washed with PBS containing 1% NP40, followed by PBS. Bound proteins were eluted with SDS–PAGE loading buffer.
Several cell lines were analyzed by QUBIC. QUBIC is a recent proteomics method for unbiased and sensitive identification of protein–protein interactions (Hubner and Mann, 2011). It is based on the generation of stable cell lines that express a GFP-tagged, full-length bait protein under control of its endogenous promoter. The tagged bait protein is expressed at near-physiological levels and can be immunoprecipitated with an anti-GFP antibody. Quantitative mass spectrometric analysis of immunoprecipitates from bait and control cell lines reveals proteins specifically associated with the bait. QUBIC was performed essentially as described by Hubner et al. (2010). Anti-GFP immunoprecipitations of BAC and control cell lines were performed in triplicate. The precipitated proteins were analyzed by mass spectrometry and compared using label-free quantification. The following cell lines were analyzed. BAC cell lines: SGP11-GFP, AP-5 ζ-GFP (Słabicki et al., 2010), SPG15-GFP (originally established by Słabicki et al., 2010; we selected a clonal cell line from this); control cell line: derived from the SPG15-GFP cell line; we selected cells that had lost expression of the SPG15-GFP bait (as determined by immunofluorescence microscopy and Western blotting). This control cell line therefore closely corresponds to the parental HeLa cells used to generate all of the BAC cell lines.
To gauge the stoichiometry of identified proteins, we used intensity-based absolute quantification (iBAQ) estimation of protein abundance (Schwanhäusser et al., 2011; implemented in the MaxQuant package by Cox and Mann, 2008). iBAQ sums the intensities of all identified peptides for each protein and normalizes the total intensity to the number of theoretically obtainable tryptic peptides of the protein. Unlike the original publication, we omitted a spike-in standard and assumed proportionality between the iBAQ intensity and protein molarity. iBAQ can be used to estimate the relative abundance of subunits in a protein complex (Arike et al., 2012). Although the accuracy of the method is limited, it can clearly distinguish between stoichiometric (1:1) and substoichiometric (<<1:1) interactions. For each individual pull down, iBAQ values of coimmunoprecipitated proteins were first normalized to the iBAQ value of the bait protein. Data from pull downs with the same bait (triplicate repeats) were then combined and iBAQ values normalized to the median iBAQ value of the set (excluding the bait protein). Values were then log-transformed and plotted (Figure 1B).
Supplementary Material
Acknowledgments
We thank Damien Devos for help with structural predictions, Nick Bright for initial electron microscope characterization, Matthew Seaman and David Stephens for antibodies, Evan Reid for helpful advice, and Mark Bowen for help with confocal microscopy.
Abbreviations used:
- AP
adaptor protein
- CIMPR
cation-independent mannose 6-phosphate receptor
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HSP
hereditary spastic paraplegia
- PI3P
phosphatidylinositol 3-phosphate
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-03-0170) on July 3, 2013.
REFERENCES
- Antrobus R, Borner GHH. Improved elution conditions for native co-immunoprecipitation. PLoS One. 2011;23:e18218. doi: 10.1371/journal.pone.0018218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arike L, Valgepea K, Peil L, Nahku R, Adamberg K, Vilu R. Comparison and applications of label-free absolute proteome quantification methods on Escherichia coli. J Proteomics. 2012;75:5437–5448. doi: 10.1016/j.jprot.2012.06.020. [DOI] [PubMed] [Google Scholar]
- Beck KA, Keen JH. Interaction of phosphoinositide cycle intermediates with the plasma membrane-associated clathrin assembly protein AP-2. J Biol Chem. 1991;266:4442–4447. [PubMed] [Google Scholar]
- Bettencourt C, et al. Exome sequencing is a useful diagnostic tool for complicated forms of hereditary spastic paraplegia. Clin Genet. 2013 doi: 10.1111/cge.12133. doi: 10.1111/cge.12133. [DOI] [PubMed] [Google Scholar]
- Boukhris A, et al. Hereditary spastic paraplegia with mental impairment and thin corpus callosum in Tunisia: SPG11, SPG15, and further genetic heterogeneity. Arch Neurol. 2008;65:393–402. doi: 10.1001/archneur.65.3.393. [DOI] [PubMed] [Google Scholar]
- Brankatschk B, Pons V, Parton RG, Gruenberg J. Role of SNX16 in the dynamics of tubulo-cisternal membrane domains of late endosomes. PLoS One. 2011;6:e21771. doi: 10.1371/journal.pone.0021771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, Sheng ZH. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron. 2010;68:73–86. doi: 10.1016/j.neuron.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ. Molecular architecture and functional model of the endocytic AP2 complex. Cell. 2002;109:523–535. doi: 10.1016/s0092-8674(02)00735-3. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Devos D, Dokudovskaya S, Alber F, Williams R, Chait BT, Sali A, Rout MP. Components of coated vesicles and nuclear pore complexes share a common molecular architecture. PLoS Biol. 2004;2:e380. doi: 10.1371/journal.pbio.0020380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 2012;318:1–18. doi: 10.1016/j.jns.2012.03.025. [DOI] [PubMed] [Google Scholar]
- Gonzalez MA, Lebrigio RF, Van Booven D, Ulloa RH, Powell E, Speziani F, Tekin M, Schüle R, Züchner S. GEnomes Management Application (GEM.app): a new software tool for large-scale collaborative genome analysis. Hum Mutat. 2013;34:842–846. doi: 10.1002/humu.22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanein S, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet. 2008;82:992–1002. doi: 10.1016/j.ajhg.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson BJ, Hong W. Evidence for a role of SNX16 in regulating traffic between the early and later endosomal compartments. J Biol Chem. 2003;278:34617–34630. doi: 10.1074/jbc.M300143200. [DOI] [PubMed] [Google Scholar]
- Harbour ME, Breusegem SY, Antrobus R, Freeman C, Reid E, Seaman MNJ. The cargo-selective retromer complex is a recruiting hub for protein complexes that regulate endosomal tubule dynamics. J Cell Sci. 2010;123:3703–3717. doi: 10.1242/jcs.071472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haucke V. Phosphoinositide regulation of clathrin-mediated endocytosis. Biochem Soc Trans. 2005;33:1285–1289. doi: 10.1042/BST0331285. [DOI] [PubMed] [Google Scholar]
- Hirst J, Barlow LD, Francisco GC, Sahlender DA, Seaman MNJ, Dacks JB, Robinson MS. The fifth adaptor protein complex. PLoS Biol. 2011;9:e1001170. doi: 10.1371/journal.pbio.1001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Borner GHH, Antrobus R, Peden AA, Hodson NA, Sahlender DA, Robinson MS. Distinct and overlapping roles for AP-1 and GGAs revealed by the “knocksideways” system. Curr Biol. 2012;22:1711–1716. doi: 10.1016/j.cub.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Irving C, Borner GHH. Adaptor protein complexes AP-4 and AP-5: new players in endosomal trafficking and progressive spastic paraplegia. Traffic. 2013;14:153–164. doi: 10.1111/tra.12028. [DOI] [PubMed] [Google Scholar]
- Hirst J, Sahlender DA, Choma M, Sinka R, Harbour ME, Parkinson M, Robinson MS. Spatial and functional relationship of GGAs and AP-1 in Drosophila and HeLa cells. Traffic. 2009;10:1696–1710. doi: 10.1111/j.1600-0854.2009.00983.x. [DOI] [PubMed] [Google Scholar]
- Honing S, Ricotta D, Krauss M, Spate K, Spolaore B, Motley A, Robinson M, Robinson C, Haucke V, Owen DJ. Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol Cell. 2005;18:519–531. doi: 10.1016/j.molcel.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J Cell Biol. 2010;189:739–754. doi: 10.1083/jcb.200911091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner NC, Mann M. Extracting gene function from protein-protein interactions using Quantitative BAC InteraCtomics (QUBIC) Methods. 2011;53:453–459. doi: 10.1016/j.ymeth.2010.12.016. [DOI] [PubMed] [Google Scholar]
- Lemmon SK, Traub LM. Getting in touch with the clathrin terminal domain. Traffic. 2012;13:511–519. doi: 10.1111/j.1600-0854.2011.01321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Yanicostas C, Rastetter A, Naini SM, Maouedj A, Kabashi E, Rivaud-Péchoux S, Brice A, Stevanin G, Soussi-Yanicostas N. Spatacsin and spastizin act in the same pathway required for proper spinal motor neuron axon outgrowth in zebrafish. Neurobiol Dis. 2012;48:299–308. doi: 10.1016/j.nbd.2012.07.003. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Murmu RP, et al. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol Cell Neurosci. 2011;47:191–202. doi: 10.1016/j.mcn.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Page LJ, Robinson MS. Targeting signals and subunit interactions in coated vesicle adaptor complexes. J Cell Biol. 1995;131:619–630. doi: 10.1083/jcb.131.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavel J, Harter C, Wieland FT. Reversible dissociation of coatomer: functional characterization of a beta/delta-coat protein subcomplex. Proc Natl Acad Sci USA. 1998;95:2140–2145. doi: 10.1073/pnas.95.5.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol. 2002;4:394–398. doi: 10.1038/ncb791. [DOI] [PubMed] [Google Scholar]
- Sagona AP, Nezis IP, Pedersen NM, Liestøl K, Poulton J, Rusten TE, Skotheim RI, Raiborg C, Stenmark H. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol. 2010;12:362–371. doi: 10.1038/ncb2036. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Simpson F, Bright NA, West MA, Newman LS, Darnell RB, Robinson MS. A novel adaptor-related protein complex. J Cell Biol. 1996;133:749–760. doi: 10.1083/jcb.133.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Słabicki M, et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 2010;8:e1000408. doi: 10.1371/journal.pbio.1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari RK, Kusari J, Sen GC. Functional equivalents of interferon-mediated signals needed for induction of an mRNA can be generated by double-stranded RNA and growth factors. EMBO J. 1987;6:3373–3378. doi: 10.1002/j.1460-2075.1987.tb02659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Ishikawa H. The movement of membranous organelles in axons. Electron microscopic identification of anterogradely and retrogradely transported organelles. J Cell Biol. 1980;84:513–530. doi: 10.1083/jcb.84.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wis´niewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Züchner S. Peripheral neuropathies: whole genome sequencing identifies causal variants in CMT. Nat Rev Neurosci. 2010;6:424–425. doi: 10.1038/nrneurol.2010.108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.