

Abstract
The aim of this review is to provide a broad perspective on intestinal absorption and the impact of intestinal first-pass metabolism on both clearance and drug–drug interaction prediction along with its historical perspectives. The review also considers abilities to bridge the gap between the increasing amount of intestinal in vitro data and the importance of intestinal first-pass metabolism in vivo. The significance of efflux transporters on the intestinal absorption is also discussed.
Keywords: Drug–drug interaction, Efflux transporters, Cytochrome P450, P-glycoprotein, Gut wall/mucosal enzymes, CYP3A4
1. Introduction
As most of the drugs are xenobiotics, they undergo metabolism by detoxification system of human body. It leads to terminating the effectiveness of moiety as a drug. The most desirable route of administration for therapeutic agents is oral; as it avoids pain and risk of infection associated with parenteral administration and thereby increases patient compliance. However most of the orally administered drugs display low systemic availability and diminished efficacy. The reasons are two fold viz. formulation related and those related with GI physiology. Factors that limit the drug release into the gastrointestinal lumen can be overcome with proper drug formulation but first-pass metabolism is an unavoidable obstacle, to achieve an optimal bioavailability. For some new drugs like HIV protease inhibitor saquinavir and the immunosuppressant tacrolimus, a mean oral bioavailability of <20% is accepted in the absence of a better alternative. Worst is the case for lidocaine, fentanyl opioids and insulin, which effectively preclude oral drug therapy.
The ability to successfully predict the pharmacokinetic properties plays a crucial role in the selection of candidate drugs and significantly reduces the number of potential failures in drug development. Current drug candidates typically show a very high affinity for the target receptors; however, the drawback is that many new lead compounds represent large, lipophilic molecules with low solubility, dissolution and/or permeability and consequently show poor absorption properties. In vitro–in vivo correlation prediction, application of in silico methods for clearance and drug–drug interaction predictions from hepatic cytochrome P450 data have been widely accepted by both pharmaceutical companies and academia, and have to meet certain regulatory requirements.
However, the application of these approaches to extrahepatic tissues, including the intestine, has proved to be challenging and less definitive. Estimates of intestinal clearance are not routinely incorporated into in vitro–in vivo strategies and this may partially explain the clearance under-prediction trend often observed.
The aim of this review is to provide a broad perspective, both from academia and industry, on intestinal absorption and the impact of intestinal first-pass metabolism on both clearance and drug–drug interaction predictions. The review also considers abilities to bridge the gap between the increasing amount of intestinal in vitro data and the importance of intestinal first-pass metabolism in vivo. Overviews of the biopharmaceutical and pharmacokinetic variables and their impact on the prediction of drug absorption from either chemical structure and/or in vitro data are presented, focusing in particular on the estimation and application of intestinal permeability. The role of uptake and efflux transporters and their interplay with the metabolic enzymes is becoming an increasingly important issue for drug candidates. Recently, drug elimination mechanisms (e.g., via metabolism, or coupled with uptake/efflux transporters or renal clearance) have been suggested as criteria to extend the Biopharmaceutical Classification System beyond the use of drug permeability/solubility characteristics to the prediction of drug disposition. The significance of efflux transporters on the intestinal absorption is also discussed.
1.1. Historical
Until 1997, pre-systemic elimination was fully credited to the liver. Also the attempts to define drug metabolism processes have been limited largely to various metabolic isoenzymes of the liver. Benet et al. (1996) and Wacher et al. (1996), were the first to hypothesize that for many drugs poor bioavailability could be due to the action of intestinal enzymes. They had performed certain cellular, animal and human studies and arrived at a conclusion that intestinal metabolic enzymes and efflux transporters working coordinately as a protective mechanism could be the cause for the poor bioavailability of certain drugs. Prior to that, this problem was addressed for a very restricted number of compounds (Ilett et al., 1990; Tam, 1993; Krisha and Klotz, 1994) and no attempt was made for their quantitative determination and extraction evaluation. This traditional view of intestinal metabolism has been reexamined by Thummel et al., 1997; Hall et al., 1999; Lin et al., 1999; Zhag et al., 1999 in light of finding that enzymes of the CYP3A sub-family (considered as major Phase I drug metabolizing enzymes in humans) are expressed at high levels in the premature villous tip enterocytes of the small intestine (Waziers et al., 1990; Watkins et al., 1987; Murray et al., 1988; Peters and Kerners, 1989; Kolars et al., 1992, 1994; Mckinnon et al., 1995).
Active secretion of the absorbed drug is now being recognized as a significant factor in oral drug bioavailability (Benet et al., 1996; Wacher et al., 1995). Of these multidrug efflux pumps; the MDR1 gene product p-glycoprotein (P-gp) is of particular interest. It was first recognized in tumor resistance (Leveque and Jehl, 1995).
Also CYP3A and P-gp share remarkable number of substrates as common among them. Dexamethasone, etoposide, rapamycin, taxol, and vinca alkaloids are a few of them (Wacher et al., 1995).
Since then, several workers had worked on different enzyme systems of the GIT. It was found that the intestinal monoamine oxidase primarily acts on most of the amine containing drugs. Sumatriptan and phenylephrine are a few of them (Warner et al., 1945; Lacey et al., 1995; Kanfer et al., 1993). Esterase and amidase activities are distributed widely in the human body including gastrointestinal tract (Leiweber1987). Alcohol dehydrogenase present in the liver as well as the intestine plays a major role in the metabolism of alcohol. Peptidases present in the intestinal and pancreatic secretions split amide linkages and thereby inactivate protein/polypeptide drugs. Bacterial microflora of the colon and the gut wall are Phase II metabolizing enzymes which also play a major role in the deactivation of drugs as studied by several workers. Thummel et al., 1996; Carrier et al., 2001 worked to quantitatively measure the extraction of drug by the intestinal system.
The present article reviews general principles and pharmacokinetic models to study drug metabolism by the enzymes in the gastrointestinal tract. It also focuses on variable drug responses due to variable enzyme expression and drug–drug, and drug–food interactions.
2. Anatomical considerations
Drug metabolizing enzymes in the liver and gut mucosa are present to limit the systemic exposure of foreign molecules that have been absorbed from the gastrointestinal tract. This is an evolutionary development in herbivorous or omnivorous animals. Any foreign molecule that is absorbed from the G.I. lumen travels across G.I. mucosa, capillary beds of small and large intestine, liver via portal circulation and is then transported to the rest of the body organs. Only exception to this is the molecules absorbed into the lymphatic system or distal rectum which effectively bypass the liver (Thummel et al., 1996; Shargel et al., 2006) (see Fig. 1).
Figure 1.
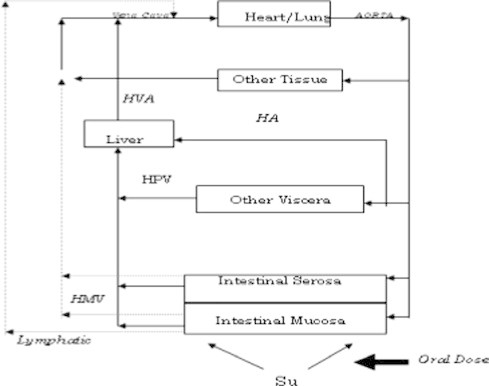
Drug absorption process in the GI tract.
The most optimal site for drug absorption is across the villi of the proximal small intestine. Columnar epithelia that form the surface barrier to gastrointestinal lumen contain high amounts of oxidative, conjugative and hydrolytic drug metabolizing enzymes. Foreign molecules, which gain access to the intestinal capillary bed by diffusion or transport across the luminal plasma membrane of the most mature enterocytes, must pass through this intracellular enzymatic barrier. Thus, if enzyme activities are sufficiently high, first pass metabolism at the mucosal epithelium can approach 100% extraction efficiency (Thummel et al., 1996; Bramhankar and Sunil, 2005) (see Fig. 2).
Figure 2.
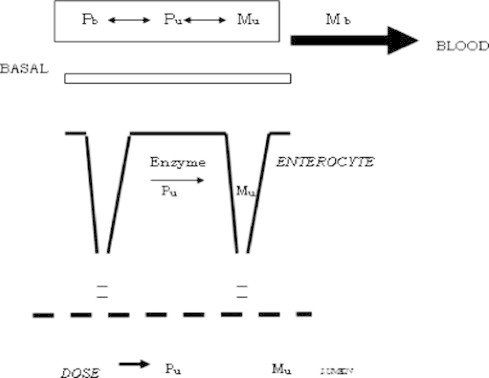
Architecture of the gastrointestinal mucosa.
The architecture of the gastrointestinal mucosa is exquisitely designed for xenobiotic absorption after oral intake. Drug absorption at this site can be described as a sequence of thermodynamically driven events. Disintegration of dosage form and dissolution of drug can be controlled by formulation but can be affected by peristaltic movement, luminal pH, and the release of bile salts and the presence of food. For most of the drugs (excluding some sustained release formulations) absorption occurs from duodenum and jejunum. Studies with Caco-2 cell monolayer cultures (a human colorectal cell line) suggest that transcellular absorption predominates for most of the lipophilic drugs, whereas the polar, hydrophilic compounds are taken up via paracellular mechanism (Thummel et al., 1996; Artursson, 1990; Artursson and Karlsson, 1991; Meunier et al., 1995; Collett et al., 1996; Watkins, 1994).
The movement of drug into the villous epithelium, positions it for intracellular enzyme-catalyzed metabolism. For many peptide or peptide-based drugs, extracellular enzyme-catalyzed degradation can also occur within the epithelial brush border and unstirred water layer (Meunier et al., 1995).
Drug in the intracellular or intercellular space will continue to diffuse across a concentration gradient into the interstitial space found between the epithelium basement membrane and capillary endothelium and subsequently diffuse across a ‘leaky’ endothelium for delivery into capillary blood. Mucosal capillary blood flow drains into the superior and inferior mesenteric veins, which converge to form the hepatic portal vein (Thummel et al., 1996).
3. GI enzymes for presystemic metabolism
GI enzymes, which contribute for presystemic metabolism of a drug, are categorized as (Bramhankar and Sunil, 2005; Watkins, 1997; Watkins, 1994) (see Fig. 3).
-
(1)
Luminal enzymes
-
(2)
Gut wall/mucosal enzymes
-
(3)
Bacterial enzymes.
Figure 3.
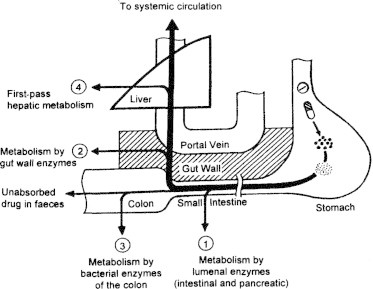
Role of GI enzymes in metabolism of xenobiotics.
3.1. Luminal enzymes
-
•
Introduction: These are the enzymes present in the gut fluids and include enzymes from pancreatic and intestinal secretions.
-
•
Peptidases: Insulin, calcitonin, teragastrin, thyrotropin releasing hormone, phenyl alanine, glycine are a few of the peptidyl drugs. But the oral bioavailability of these drugs is generally poor, since they are poorly absorbed and easily degraded by proteolytic enzymes in the gastrointestinal tract (Shargel et al., 2006; Lee and Yamamoto, 1990).
-
•
Esterases: Ester type drugs and prodrugs are hydrolyzed by esterases present in the intestinal tract (Crauste-Manciet et al. 1997) and mucosa (Odgers et al., 1995; Guerrero et al., 2006). A few studies have paid much attention to the metabolism of ester prodrug in the intestinal tract and mucosa (Feng et al., 2006; Augustijns et al., 1998).
4. Gut wall/mucosal enzymes
4.1. Introduction
These enzymes are mainly present in the stomach, intestine and colon. Alcohol dehydrogenase is an enzyme of stomach mucosa that inactivates ethanol. Intestinal mucosa contains both Phase I and Phase II enzymes. The colonic mucosa also contains both Phase I and Phase II enzymes. However the enzymes of the proximal small intestine are most active.
4.2. CYP450
4.2.1. The cytochrome P450 enzyme system
The CYP450 enzyme system is a key pathway for drug metabolism. Most of the lipophilic drugs must undergo biotransformation to more hydrophilic compounds to be excreted from the body. Drug biotransformation reactions consist of Phase I (e.g., oxidation, reduction) or Phase II (e.g., conjugation) reactions that occur primarily in the liver. The most common Phase I reaction is oxidation, which involves the insertion of an oxygen atom into the compound to form a polar hydroxyl group. Of the enzymes involved in the Phase I reactions, the CYP450 group is the most important38.
Cytochrome P-450 is a superfamily of hemoproteins which can be divided into families, subfamilies and/or single enzymes. The cytochrome P-450 enzymes act as a major catalyst for drug oxidation. To unify the nomenclature, a given gene family is defined as having >40% amino acid sequence homology and a subfamily as having >55% identical sequence homology. Using this nomenclature, the cytochrome P450 enzymes are designated by the letters CYP (representing cytochrome P450), followed by an Arabic numeral denoting the family, a letter representing the subfamily (when 2 or more exist) and another Arabic numeral designating the individual gene within the subfamily (e.g., CYP2D6) (see http://www.nursinglink.com).
Yan and Caldwell, 2001 have presented the status and given detailed descriptions of biotransformation, metabolic stability assays, identification of drug metabolizing P450 enzymes, prediction of pharmacokinetic parameters from in vitro metabolism data, structure elucidation of metabolites, CYP450 inhibition assays and CYP450 induction assays from a drug discovery perspective regarding the hepatic as well as the intestinal CYP450s (Yan and Caldwell, 2001).
Each enzyme of the CYP family is termed as an isoform (or isoenzyme) since it is derived from a different gene. An important subset of the cytochrome P450 family is the CYP3A4 isoenzyme, which accounts for nearly 60% of the total CYP450 in the liver and approximately 70% in the intestine. CYP3A4, which catalyzes the biotransformation of many drugs, is significantly expressed extrahepatically (Cheng-Chung et al., 2001). Extensive metabolism by CYP3A4 in the gastrointestinal tract contributes to the poor oral bioavailability of many drugs.
Many substrates, inhibitors and inducers of CYP3A4 have been identified. By definition, a substrate is a drug that is metabolized by an enzyme system. An inhibitor decreases the activity of the enzyme and may decrease the metabolism of substrates, generally leading to an increased drug effect. Inducers, however, may increase the metabolism of substrates and generally lead to a decreased drug effect.38
They are mainly microsomal and found attached to the cell endoplasmic reticulum. Only minimal amounts of CYP3A, 2E1, 2D6, 1A2 and 2C are found in the esophagus, stomach and colon (Thummel et al., 1996). In contrast cytochromes identified in human intestine include CYP1A1 (Wacher et al., 2001; Buchthal et al., 1995), CYP2, CYP2D6, CYP2E1 (trace amounts), CYP3A4 (Waziers et al., 1990; Kolars et al., 1992, 1994; Mckinnon et al., 1995) and more recently CYP3A5 (Kolars et al., 1994). By far the most important of these enzymes in drug metabolism is CYP3A4. Immunohistochemical studies have shown that the small intestinal concentrations of CYP3A4 are approximately, 80–100% of the CYP3A4 concentration in the liver, while CYP2C8–10 concentrations are only 5–10% and CYP2D6 concentrations are around 20% of their respective liver levels (Waziers et al., 1990). In addition, microsomes prepared from human jejunal enterocytes have demonstrated CYP3A4 protein concentrations and enzyme activities equal to or greater than those found in hepatic microsomes. In a study of 20 full-length human small intestines, Paine et al., 1997 found microsomal CYP3A4 expression to be continuous along the entire length of the small intestine, but variable from one region to the other. A mean value of 31 (<2–91), 23 (<2–98) and 17 (<2–60) pmol/mg protein was detected in the duodenum, distal jejunum and distal ileum respectively. Sequential analysis of each section of six intestines showed that the peak expression varied, but was generally highest within the first 10 feet distal to the pyloric valve. In the same study, the distribution pattern for NADH dependent cytochrome P450 reductase activity closely paralleled to that of CYP3A protein. In contrast mucosal microsomal cytochrome b5, protein content and b5 reductase activity tend to increase slightly in a proximal to distal direction (see Fig. 4).
Figure 4.

Expression of CYP3A5 and Cyt b5 reductase in the duodenum and ileum.
CYP3A5, which is expressed in only 25–30% of human livers (Wrighton et al., 1989), appears to be more commonly expressed in human intestine. It was found in intestinal biopsy samples of fourteen patients out of twenty, which is almost 70%. Most recently in 2001 Carrier et al., 2001 had used Caco-2 cells and primary cultures of pig enterocytes as a model for in vitro testing. They also found that CYP3A4 was most abundantly expressed in differentiated enterocytic cells. Although absolute CYP3A5 levels were not quantified in this study, they have found it much lower than CYP3A4 when enzyme and mRNA levels were measured in the small intestine from an organ donor. The intestine was treated with CYP3A inducers dexamethasone and phenytoin (Kolars et al., 1994). In another study, CYP3A5 mRNA was absent in the intestinal sample but was found in six of eleven colon samples (Peters and Kerners, 1989).
Most of the workers tried to compare CYP3A4 and CYP3A5 activities. Intestinal CYP3A5 appears not to be inducible (Wrighton et al., 1989; Wrighton et al., 1990; Schuetz et al., 1995) while CYP3A4 is induced by dexamethasone (Watkins, 1994) and rifampicin (Kolars et al., 1992). Although they are having similar substrate specificities, they significantly differ in catalytic activities (Wrighton et al., 1989; Wrighton et al., 1990; Schuetz et al., 1993; Aoyama et al., 1989; Gorski et al., 1994) for example, CYP3A4 and CYP3A5 show almost equal abilities to metabolize nifedipine, however CYP3A5 does not appear to catalyze erythromycin N-demethylation (Wrighton et al., 1990; Aoyama et al., 1989). Similarly, CYP3A4 is known to convert cyclosporine to three major metabolites (AM1, AM9, and AM4N) while CYP3A5 metabolizes cyclosporine to only one (AM9) (Aoyama et al., 1989). While CYP3A5 also demonstrates regioselective metabolism of midazolam, preferentially hydroxylating 1′- as compared to 4′-position by CYP3A4. And hence midazolam metabolite ratios have been suggested as a useful probe of in vivo CYP3A expression (Gorski et al., 1994).
4.2.2. In-vivo CYP-3A-dependent metabolism
Clinical evidence of significant Phase-I metabolism by the small intestine was available as early as 1977, in studies by Mahon et al., 1977 who instilled C-labeled flurazepam into the stomach of one patient and duodenum of two others. Thin layer chromatographic analysis of portal venous blood demonstrated the rapid appearance of significant levels of flurazepam metabolites, consistent with metabolism by the small intestinal mucosa. With the advent of time, contribution of intestinal enzymes to first-pass drug metabolism has become the focus of a great deal of research, as a large number of drugs were found to be substrates for CYP3A. CYP3A not only reduce the oral bioavailability of a compound but also acts as a major source of inter-individual variability in blood levels. Variable constitutive enzyme expression and drug–drug interactions in turn contribute to variable drug response.
4.2.2.1. Cyclosporine
Much of the work establishing intestinal metabolism as a major determinant of oral drug bioavailability has been conducted using the immunosuppressive agent cyclosporine. In a demonstration of CYP3A mediated intestinal metabolism, Kolars et al., 1991 instilled cyclosporine into the small bowel of two patients during the anhepatic phase of liver transplantation. Analysis of portal venous blood showed 25% and 51% of cyclosporine metabolites. It suggests that the extraction by intestine is more important than the liver in demonstrating cyclosporine bioavailability. Clinical pharmacokinetic studies have also provided strong evidence for extensive intestinal extraction of cyclosporine. In three separate studies i.v. and oral cyclosporine were administered to subjects either alone or together with the CYP3A4 inducer rifampin (Hebert et al., 1992) or the CYP3A4 inhibitors, ketoconazole (Gomez et al., 1995) and erythromycin (Gupta et al., 1988). The effect of the coadministered drug on the hepatic and intestinal metabolism of cyclosporine can be differentiated by treating the observed cyclosporine oral bioavailability (Fmeas.) as the product of the fraction absorbed intact from the intestinal lumen (Fabs) multiplied by the fraction reaching the portal blood unmetabolized by the intestine (Fg) multiplied by the fraction passing through the liver unmetabolized (Fhep). Fhep is calculated by i.v. cyclosporine clearance divided by hepatic blood flow. It is assumed as unaffected by the coadministered drug, and hence the equation becomes simple as Fabs × Fg (Hebert et al., 1992; Gomez et al., 1995; Chi-Yuan et al., 1995). A comparison of the effects of rifampin, ketoconazole, and erythromycin on oral cyclosporine pharmacokinetics is presented in Table 1.
Table 1.
Comparison of effects of rifampin, ketoconazole, and erythromycin on oral cyclosporine pharmacokinetics.
| Substrate | Fa (%) | Fhep (%) | Fabs × Fg (%) |
|---|---|---|---|
| Cyclosporine | |||
| Alone | 27 (11) | 77 (4) | 33 (13) |
| +Rifampin | 10 (3) | 68 (8) | 14 (5) |
| Cyclosporine | |||
| Alone | 22 (5) | 75 (7) | 30 (8) |
| +Ketoconazole | 56 (12) | 86 (4) | 65 (11) |
| Cyclosporine | |||
| Alone | 36 (9) | 73 (10) | 49d |
| +Erythromycin | 62 (17) | 80 (4) | 78d |
In each case, the changes in Fmeas caused by coadministration of these drugs were not reflected by changes in hepatic extraction (Fhep) but were almost identical to changes in Fabs × Fg. The dosing regimen was such that no effect of inducers/inhibitors was expected on cyclosporine absorption. Thus the drug induced changes in cyclosporine oral bioavailability arose predominantly from their effects on intestinal cyclosporine metabolism (Fg) (Hebert et al., 1992; Gomez et al., 1995; Chi-Yuan et al., 1995). Boundary-condition analysis from this data demonstrated that at least 65% of an oral cyclosporine dose is absorbed, however this dose is more than 50% extracted by the intestine (Chi-Yuan et al., 1995). Fig. 5 presents mean estimates of Benet et al., 1996 for Fabs (86%), Fg (41%) and Fhep (76%) leading to the Fmeas value of 27% for cyclosporine formulation in healthy volunteers (see Fig. 5).
Figure 5.
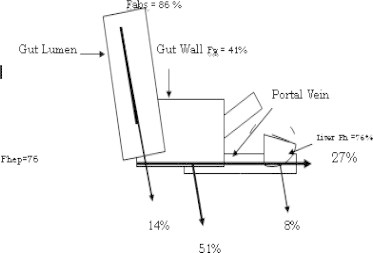
Extraction of cyclosporine through the gut (percentage).
Chi-Yuan et al. (1995) showed that approximately two thirds of cyclosporine metabolism occurs in the gut, with the liver responsible for only one third of metabolism. He also performed three drug interaction studies in which the pharmacokinetics of i.v. and orally administered cyclosporine was determined in blood, both before and after oral administration of a second known drug that alters the metabolism of cyclosporine. These studies of the effects of dosing rifampin (Hebert et al., 1992) (an inducer of cyclosporine metabolism), erythromycin (Gupta et al., 1989) and ketoconazole (Gomez et al., 1995) (inhibitors of cyclosporine metabolism) had served as the basis for the analysis to differentiate hepatic and gut metabolisms and to estimate the extent of absorption of cyclosporine. The results are shown in Table 2.
Table 2.
Quantitative metabolism of drugs in the liver and gut.
| Fabs (%) | ERG | XG |
|---|---|---|
| Rifampin | ||
| 100 | 0.65 | 1.31 |
| 86 | 0.59 | 1.40 |
| 77 | 0.54 | 1.49 |
| 65 | 0.46 | 1.68 |
| Ketoconazole | ||
| 100 | 0.71 | 0.49 |
| 86 | 0.66 | 0.36 |
| 77 | 0.62 | 0.25 |
| 65 | 0.55 | Indeterminate |
| Erythromycin | ||
| 100 | 0.51 | 0.44 |
| 86 | 0.43 | 0.23 |
| 77 | 0.37 | Indeterminate |
Angela Sansone-Parsons et al., 2007 analyzed the pharmacokinetic properties of the immuno-suppressants cyclosporine and tacrolimus when either is coadministered with oral posaconazole. Their findings suggest that the dosage of cyclosporine or tacrolimus should be reduced when posaconazole therapy is started and that plasma levels of the immunosuppressant should be monitored during and at the discontinuation of posaconazole therapy so that dosages are adjusted accordingly.
4.2.2.2. Midazolam
Kolars et al. (1994) had administered midazolam into the small bowel of two patients during the anhepatic phase of liver transplantation intravenously. They have compared the extent of metabolism and found approx. 12% metabolism for i.v. administration. It suggests that the intestinal metabolism plays a role in systemic as well as in pre systemic clearance. Thummel (1995) had conducted a series of studies in humans indicating that midazolam undergoes extensive intestinal first-pass metabolism. In a study of i.v. and oral midazolam pharmacokinetics in 20 healthy volunteers, indirect estimates of the intestinal and hepatic first pass extraction fractions were 44 ± 14% and 43 ± 0.4% respectively (Thummel et al., 1996). Direct verification of significance of the small intestine as a site of first pass metabolite extraction for midazolam was done in liver transplant patients (Paine et al., 1996). The difference between hepatic portal venous and arterial midazolam and 1′-hydroxymidazolam AUCs had given mean 1st pass intestinal extraction fraction of 43%.
Saquinavir was shown to increase the oral bioavailability of midazolam from 0.40 to 0.90, predominantly by increasing the intestinal availability (Palkama et al., 1999).
Tofisopam inhibits the pharmacokinetics of CYP3A4 substrate midazolam by inducing CYP3A enzymes (Mária et al., 2008).
Grape fruit juice has increased the oral bioavailability of midazolam from 0.24 to 0.35 with no significant alteration in AUC of intravenously administered midazolam (Kupferschimidt et al., 1995).
4.2.2.3. Other substrates
The metabolism of a number of drugs that undergo significant first pass extraction has been studied in vitro using mucosal homogenates, mucosal microsomes or intact mucosal membranes isolated from human intestine. CYP3A catalyzed biotransformations including flurazepam (Mahon et al., 1977), ethinyl estradiol (Rogers et al., 1987), erythromycin (Watkins et al., 1987), a cyclosporine analog SDZ IMM 125 (Vickers et al., 1995), tacrolimus (Lampen et al., 1995) and saquinavir (Fitzsimmons and Collins, 1997) when normalized for total nmol of P450, the intestinal microsomes proved metabolically superior, reflecting the greater contribution of the CYP3A fraction to total P450 content (Paine et al., 1997).
All of the above drugs have demonstrated a greater extent of intestinal metabolism as compared to hepatic metabolism. One exception to this general trend is the report of a much lower (∼1/30) rate of microsomal oxidation by intestinal microsomes in comparison to hepatic microsomes.
Based on these limited studies, the mean mucosal microsomal intrinsic clearance is likely to be within two to three fold of the corresponding hepatic microsomal intrinsic clearance for the most CYP3A substrates.
Day Y. Lee et al., 2007 had observed after oral administration of omeprazole to cirrhotic rats, the AUC difference (of 451% and 149%) was greater than that after intravenous administration. It was possibly due to a decrease in intestinal first-pass effect of omeprazole in addition to a decrease in hepatic metabolism in cirrhotic rats.
Van et al. studied the extrahepatic metabolism of sevoflurane by measuring the fluoride production in children receiving sevoflurane solely during the anhepatic phase of orthotopic liver transplantation. Out of two groups, in one group, isoflurane was replaced by sevoflurane as soon as the liver was removed from the patient and maintained until reperfusion of the new liver. Plasma fluoride concentrations were determined by ion-selective electrode. The fluoride concentration increased significantly as soon as sevoflurane was introduced; it remained stable in the group receiving isoflurane. The peak fluoride concentration was also significantly higher in the first group (mean ± SD: 5.5 ± 0.8 μM (sevoflurane group) versus 1.4 ± 0.5 μM (isoflurane group) P < 0.05). These results demonstrate the existence of an extrahepatic metabolism of sevoflurane in children with end-stage liver disease (Van et al., 2000) (see Table 3).
Table 3.
Predicted intestinal first-pass extraction ratios for selected CYP3A substrates.
| Drug | ER (%) |
|---|---|
| Midazolam | 38.3 |
| Nifedipine | 35.6 |
| Felodipine | 8.6 |
| Diltiazem (HCl) | 2.5 |
| S-verapamil (HCl) | 9.4 |
| Quinidine | 57.5 |
| Saquinavir | 99.3 |
| Indinavir | 11.8 |
| Carbamazepine | 0.2 |
| Tracrolimus | 14.2 |
| Cyclosporine | 3.3 |
| Terfenadine | 5.0 |
Poet et al. (2003) evaluated the role of intestinal metabolism in the first-pass elimination of CPF (chlorpyrifos) and DZN (diazinon). Similarly esterase-mediated metabolic profiles were demonstrated in isolated intestinal enterocytes. Compared on a per nmol P450 basis, the Vmax for CPF in enterocytes was 2–3 times higher than in liver microsomes for the production of CPF-oxon and TCP. These results suggest that intestinal metabolism may impact the first-pass metabolism of CPF and DZN, especially following low-dose oral exposures (Poet et al., 2003).
Most recently Hyun et al. (2006) observed that intestinal first-pass effect of Sildenafil was approx. 71% of oral dose in rats. Gracia et al. (2003) had done a PK study with intestinal elimination of albendazole sulfoxide. He found a clear and statistically significant interaction with verapamil with 43% increased AUC and 29% increased AUC with ketoconazole in rats respectively. The results are shown in Table 4.
Table 4.
Increased AUC of albendazole sulfoxide when co administered with verapamil, Ivermecitin, quinidine, ketoconazole in rats and sheep.
| Animal | Increased AUC when co administered with |
|||
|---|---|---|---|---|
| Verapamil | Ketoconazole | Quinidine | Ivermectin | |
| Rats | 43% | 29% | – | – |
| Sheep | 53.68% | – | 78.62% | 50.55% |
Walker et al. (2006) has done an inter-species comparison of liver and small intestinal microsomal metabolisms of Fluoranthene.
For all these studies related to CYPs; reliable methods for determining enzyme activities are needed to characterize an individual CYP enzyme and to obtain a tool for evaluation of its role in drug metabolism in humans. Determining the role of CYP enzymes in the metabolism of a compound and evaluating the effect of NCEs on human CYP activities are key issues in pharmaceutical development as they may explain inter-subject variability, drug–drug interactions, non-linear pharmacokinetics and toxic effects. Different liquid chromatography tandem mass spectrometry methodologies have been developed recently by Lahoz et al. (2008) for the fast and routine analysis of major in vivo and in vitro CYPs enzyme activities. They have also reviewed most recent approaches to quickly assess CYPs activities using in vitro models as well as their application in early drug discovery (Lahoz et al., 2008).
5. P-gp
The MDR1 gene product, P-glycoprotein, a multidrug efflux pump, an ATP-binding cassette transmembrane transporter (ABC transporter), first recognized in tumor resistance (Leveque and Jehl, 1995); is expressed on the brush border membrane of enterocytes to excrete its substrates into the lumen (Ambudkar et al., 1999; Janice and Barry, 1997). Hence, in addition to CYP3A/3A4, we have to consider the role of P-gp in reducing oral bioavailability. Greiner et al. (1999) demonstrated this fact by examining the interaction between digoxin and rifampin in humans. Since, in humans, digoxin is a good substrate for P-gp, but not for CYP3A4, the role of P-gp can be directly demonstrated by this experiment. They determined AUC of digoxin before and after oral and intravenous administration before and after co administration with rifampin (Greiner et al., 1999). They have obtained a significant correlation between the AUC’s of digoxin after oral administration and P-gp expression levels in the intestine. Analytical reports also indicated that the administration of rifampin have prolonged the Tmax of digoxin; since rifampin is an inducer of P-gp. The role of liver in determining the oral bioavailability is negligible, since the hepatic extraction of digoxin after intravenous administration is minimal. Collectively these results suggest that intestinal metabolism may impact the first-pass metabolism of CPF P-gp is involved in the reduced bioavailability of digoxin following rifampin treatment. Recently, the role of P-gp in determining the oral bioavailability of digoxin in humans was also highlighted in relation to a single nucleotide polymorphism of the MDR1 gene. Hoff et al. (2000) demonstrated that a polymorph in exon 26 (C3435T) results in a reduced intestinal expression level of P-gp along with an increased bioavailability of digoxin.
The role of P-gp in reducing oral bioavailability of its substrates has also been suggested by comparing the disposition in normal and MDRl knock out mice. For digoxin, the intestinal excretion after IV administration was significantly lower in MDR (−/−) mice compared to normal mice (Mayer et al., 1996; Schinkel et al., 1997). Moreover, this intestinal excretion was abolished by oral PSC833, a potent P-gp inhibitor (Mayer et al., 1997). Same study was performed with paclitaxel (Sparrenboom et al., 1997) and HIV protease inhibitors (Kim et al., 1998) (indinavir, nelfinavir and saquinavir). The role of P-gp in the excretion of its substrates into the human intestinal lumen from the blood circulation has also been highlighted by the findings of Gramette and Oertel (1999). They demonstrated that talinolol, β-adrenergic receptor antagonist was excreted into the intestinal lumen of healthy volunteer and which was inhibited by R-verapamil.
Quinidine, P-gp substrate/inhibitor was found to interfere with etoposide (an anticancer agent) absorption as well as metabolism (Kolars et al., 1994). Hunter et al. (1997) have used verapamil and monoclonal antibody MRK6 as P-gp inhibitors increasing the absorptive flux when studied in Caco-2 cells. Some surfactants like Tween 80 and Cremophore EL, which exhibit their effects by inhibition of P-gp, were found to contribute in minimizing the oral bioavailability of peptides across the intestine (Augustins et al., 1993; Neurkar et al., 1996; Woodcock et al., 1992).
Moving further Jason et al., 2004 have studied the relationship between copolymer concentration and substrate hydrophobicity on P-glycoprotein efflux inhibition by amphiphilic diblock copolymers. Comparisons between the micelle association and Caco-2 cellular accumulation were evaluated using two structurally homologous P-gp substrates, the relatively hydrophobic R-6G and the hydrophilic R-123, over concentrations above and below the CMC for MePEG-b-PCL diblock copolymers. An approximately 3.75-fold enhancement of R-123 accumulation occurred with 2 mM MePEG17-b-PCL5, compared to approximately 1.25-fold for R-6G. The effective concentration range for surfactant mediated inhibition of P-gp appears to depend on P-gp substrate hydrophobicity (Jason et al., 2004; Norris et al., 1998).
Micuda et al. (2008) studied the effects of disease states on the metabolism of P-gp substrates. They explained the impaired elimination of P-gp substrates after short-term cholestasis that may commonly occur in clinical practice; by studying in vivo biliary and renal excretion of rhodamine 123 (Rho123), a P-glycoprotein (P-gp) substrate, in rats during either acute or chronic cholestasis induced by bile duct obstruction (BDO) (Micuda et al., 2008).
6. CYP3A & P-gp bisubstrates
Since the substrate specificity of CYP3A and P-gp overlap each other, these two proteins act synergistically in reducing the bioavailability of their substrates after oral administration. Xenobiotics are taken up by enterocytes and metabolized there by CYP enzymes. Drug molecules, which escape metabolic conversion, are eliminated from the cells into the lumen via P-gp. The drug molecules in the lumen may repeat the same cycle, resulting in repeated exposure to metabolic enzymes, further reducing intestinal bioavailability (Watkins, 1997; Wacher et al., 1998; Benet et al., 1999) (see Fig. 6).
Figure 6.
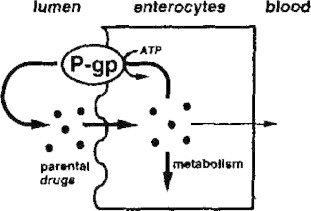
Mechanism of action of P-gp.
It is difficult to evaluate the relative role played by these two kinds of proteins in determining the oral bioavailability. Benet et al., 2004 discovered one of the first drug efflux–metabolism alliances in (Benet et al., 2004). They have used Caco-2 colon carcinoma cell line treated with sodium butyrate, phobal ester, and 12–0 teradecanoylphorbol-13-acetate (Cummins et al., 2003) (TPA) as a model for human intestinal drug absorption. They have examined the transport and metabolism of two P-gp and CYP3A4 co substrates (K77 and sirolimus) and two CYP3A4 substrates (midazolam and felodipine). They were dosed on the apical side of cells (mimicking human intestinal absorption) alone or in combination with the P-gp inhibitor GG918 or dual P-gp and CYP3A inhibitor cyclosporine. The results are shown in Table 5.
Table 5.
Extraction ratio of cyclosporine when coadministered with K77, sirolimus, midazolam and felodipine.
| Drug | Substrate for |
Efflux ratio B-A/A-B | Extraction Ratio present |
|||
|---|---|---|---|---|---|---|
| 3A4 | P-gp | Drug alone | Drug cyclosporine | Drug ± GG918 | ||
| K77 | Yes | Yes | 9 | 33 (3) | 5.7 (0.1) | 14 (1) |
| Sirolimus | Yes | Yes | 2.5 | 60 (5) | 15 (1) | 45 (1) |
| Midazolam | Yes | No | 1 | 25 (2) | 10 (1) | 23 (2) |
| Felodipine | Yes | No | 1 | 26 (1) | 14 (1) | 24 (2) |
These data support the role of P-gp in increasing the exposure of drugs to CYP3A4 in the intestine by allowing repeated cycling of drug via diffusion and active efflux. Cummins et al94 used CYP3A expressing Caco-2 cell model while CYP3A4 transfected Caco-2 cells were used along with sirolimus and midazolam by Cummins et al. (2004) and Lampen et al. (1998).
But Beverly et al. (2006) have theoretically stated that, in vitro approaches published to date, involving determination of ER, cannot dissect how P-gp affects CYP3A metabolism from the overall outcome in terms of intestinal epithelium or cell monolayer mediated extraction. Furthermore he suggested the use of pharmacokinetic theory, for understanding the relevance of P-gp/CYP3A interplay.
Zhang et al. (2007) evaluated whether curcumin could modulate P-glycoprotein (P-gp) and CYP3A expression, and in turn modify the pharmacokinetic profiles of P-gp and CYP3A substrates in male Sprague–Dawley rats. Regular curcumin consumption had caused the C(max) and area under the concentration–time curve (AUC(0–8) and total AUC) of peroral celiprolol (a P-gp substrate with negligible cytochrome P450 metabolism) at 30 mg/kg to increase, but the apparent oral clearance (CL(oral)) of the drug was reduced. Similarly, rats treated with curcumin for 4 consecutive days showed a higher AUC (AUC(0–4) and total AUC) and lower CL(oral) for peroral midazolam (a CYP3A substrate that does not interact with the P-gp) at 20 mg/kg in comparison with vehicle-treated rats (Zhang et al., 2007).
Lina and Eileen (2001) have examined the role of CYP450 1A1 and 1A2 in the metabolism of BG and identified the possible drug–drug interactions with DTIC. They have found that BG is a potent inhibitor of CYP1A1 and CYP1A2. BG and 8-oxoBG inhibit the conversion of DTIC to its active methylating agent. BG inhibition of DTIC conversion to an active methylating species suggests that the combination in human clinical trials should be approached with caution (Lina and Eileen, 2001).
Hirunpanich et al. (2008) evaluated the effects of docosahexaenoic acid (DHA) on the intestinal cytochrome P450 isoenzyme (CYP3A) and P-glycoprotein (P-gp) functions using midazolam and rhodamine-123 as specific substrates of CYP3A and P-gp, respectively. The intestinal extraction ratio (ER G) of midazolam was determined to be 0.43 and decreased significantly to 0.12, 0.07, and 0.06 in the presence of 50, 100, and 200 μM of DHA, respectively, in a concentration-dependent manner. The results from an in vitro study using rat intestinal microsomes demonstrated that DHA competitively inhibited the intestinal CYP3A activity with Ki of 15.7 and 27.1 μM for the formation of 1′-OH midazolam and 4-OH midazolam, respectively (Hirunpanich et al., 2008).
Ogasawara et al. (2007) evaluated the intestinal first-pass effect of midazolam (MDZ) and fexofenadine (FEX), typical substrates for CYP3A and P-glycoprotein (P-gp), respectively, with ketoconazole (KTZ) as a potent dual CYP3A/P-gp inhibitor in cynomolgus monkeys. When MDZ or FEX was administered i.v. at doses of 0.3 or 1 mg/kg, respectively, the plasma concentration–time profiles were not influenced by p.o. coadministration of KTZ (20 mg/kg). On the other hand, when MDZ or FEX was administered p.o. at doses of 1 or 5 mg/kg, respectively, concomitant with p.o. KTZ (20 mg/kg), significant increases were observed in the area under the plasma concentration–time curves of MDZ or FEX (22-fold in MDZ and 3-fold in FEX). These findings indicate that both CYP3A and P-gp play a key role in the intestinal barrier and that the inhibition of intestinal CYP3A/P-gp activities contributes exclusively toward the drug–drug interactions (DDI) with KTZ. These results also suggest that monkeys may be an appropriate animal species for evaluating the intestinal first-pass effect of p.o. administered drugs and for predicting the intestinal DDI related to CYP3A4 and P-gp in humans (Ogasawara et al., 2007).
Wacher et al. (1995) had given some of the overlapping substrates between cytochromes 3A and P-gp as – CYP3A substrates that also interact with P-gp. They are shown in Table 6.
Table 6.
Substrates common between cytochrome3A and P-gp.
| P-gp (−) | P-gp substrate | P-gp substrate and (−) |
|---|---|---|
| Amiodarone | Dexamethasone | Cortisol |
| Erythromycin | Etoposide | Cyclosporine |
| Felodipine | Rapamycin | Diltiazem |
| Itraconazole | Taxol | Nicardipine |
| Ketoconazole | Vinca alkaloids | Verapamil |
| Lidocaine | ||
| Nifedipine | ||
| Nitrendipine | ||
| Progesterone | ||
| Quinidine | ||
| Tamoxifen |
7. Phase II metabolism in intestine
7.1. Glucuronosyl transferase – (GT)
Studies of human intestinal Phase II metabolism have been directed toward several drugs that undergo significant 1st pass metabolism as well as isozyme specific substrate probes (Benet and Cummins, 2001). Gut activity is reported for estradiol and 17β-estradiol (Diczfaluzy et al., 1962), ethinyl estradiol (Back et al., 1981), acetaminophen (Ueda et al., 1992), p-nitrophenol, bilirubin (Peters et al., 1991), morphine (Pacifici et al., 1986; Cappiello et al., 1991) and propofol (Raoof et al., 1996).
In comparison with the liver, scientists are not having unique opinion. For example, maximal conjugation rates for morphine and ethinyl estradiol were approx. 1/10th to 1/5th for ileal tissue in comparison with the liver, when data were expressed in terms of per gram tissue wt (Cappiello et al., 1991). In contrast the maximal rate of morphine glucoronidation (per gram protein) in the liver and intestinal mucosal homogenates differed by only two fold (Pacifici et al., 1986). In another study, microsomes isolated primarily from human jejunal and duodenal mucosa catalyzed propofol conjugation at a maximal rate that was ∼89% of the corresponding liver microsomal rate.
Trevor et al. have used cultures of the human colorectal carcinoma line, HT29, to assess the susceptibility to glucuronidation of the cytostatic, immunosuppressive drug mycophenolic acid (MPA) and 19 of its analogs. They suggested that cultures of human colorectal carcinoma cell lines may provide a rapid and convenient means of assessing the susceptibility of novel synthetic compounds to both Phase I and Phase II metabolic conversions (Franklin et al., 1997).
7.2. Sulfotransferase
Cytosolic sulfotransferase activity is distributed in several organs of the body, including those of the gastrointestinal tract. Multiple isozymes have been identified and four types are detected as estrogen sulfotransferase, dehydropiandrosterone sulfotransferase, M form of phenol sulfotransferase (Pacifici et al., 1993a, Pesola and Walle, 1993), as well as thermolabile monoamine metabolizing surfotransferase (Pacifici et al., 1993b). Human intestinal mucosa catalyzes the sulfation of ethinyl estradiol (Back et al., 1981; Pacifici et al., 1993a), isoproterenol (Pacifici et al., 1993a), and terbutaline (Pacifici et al., 1993b). In case of terbutaline cytosol of small intestinal mucosa exhibits a greater Vmax activity than liver cytosol (Pacifici et al., 1993b) suggesting a key role for the gut wall in the pronounced first-pass elimination of orally administered drugs. But a dose ranging study with intact mucosa (using chamber) and acetaminophen as a substrate suggests that intestinal sulfotransferase activity is readily saturated (Pacifici et al., 1993b).
Meinl et al. (2008) found differentiation-dependent expression of SULT in the Caco-2 and TC7 cultured cells reflecting the in vivo situation, where SULT expression is focused on differentiated enterocytes. This is concluded from the study of benzo[a]pyrene metabolism.
7.3. N-Acetyl transferase
N-Acetyl transferase indicates an extensive presystemic acetylation activity in both the liver and intestine (Ilett et al., 1990) for the metabolites of prodrug sulfasalazine (s-aminosalicylic acid and sulfapyridine).
In contrast to regional differences in P450 expression, N-acetyl transferase activities appear to be of similar magnitude throughout the entire small intestine (Hickman et al., 1995).
7.4. S-Methyl transferase
S-Methyl transferase exhibits the same level of metabolic activity as that of the liver (Pacifici et al., 1991, 1993b). It was found to be responsible for extensive metabolism of 6-mercaptopurine.
7.5. Thiopurine methyl transferase
Thiopurine methyl transferase shows similar metabolic pattern as that of s-methyl transferase. Its most recognized substrate is captopril (Hickman et al., 1995; Pacifici et al., 1991).
7.6. Glutathione s-transferase
Three forms of Glutathione s-transferase α, μ and π have been identified in human small and large intestines (Dewaziers et al., 1990; Mckay et al., 1993; Her et al., 1996; Prueksaritanont et al., 1996). But a significant role of this enzyme in reducing oral bioavailability of drugs remains undefined.
7.7. Variability
Unfortunately, an enzymatic metabolic barrier that limits oral bioavailability often brings with it the problem of significant inter-individual variability in systemic blood concentrations of drug. It is the consequence of variability in metabolic enzyme expression (Burton et al., 1993). Understanding the biochemical and physiological basis for variability in the extent of loss of orally administered drug is an essential step in optimizing oral drug therapy.
To cite the importance of variability in absolute bioavailability, the graphical interpretation indicating relationship between absolute bioavailability (F) and inter-subject variability (CV) is shown in Fig. 7).
Figure 7.
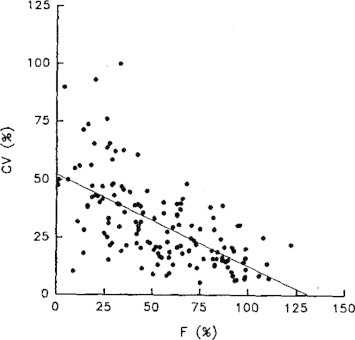
Relationship between absolute bioavailability (F) and inter-subject variability (CV).
Also because of unique anatomical location, enzymes of the gut wall represent an important and highly sensitive site for metabolically based interactions of orally administrated drugs. Again inter individual variability may make it impossible to predict the likelihood of an interaction in any given patient. Hopefully, newer models for studying human gut wall metabolic extraction will provide the means to predict the average extraction ratio and maximum first pass availability of substrates.123 With these models it has been possible to examine the sources of inter individual variability in drug bioavailability including variable constitutive enzyme expression (both genetic and environmentally determined), enzyme induction by drugs, disease and diet and intrinsic/acquired differences in plasma protein binding and organ blood flow (food and drug effects).
7.7.1. Problems in modeling intestinal extraction
As enlisted by Thummel et al. (1996) some problems in modeling intestinal extraction are enlisted below.
-
(1)
There is no uniform perfusion of the intestine by drug after oral administration. Also diffusional movement of drug across the apical epithelial cell and into the splanchnic capillaries competes with intracellular metabolic clearance.
-
(2)
All potential metabolic sites are not exposed to drug at the same time since drug distribution is not uniform across the entire intestine.
-
(3)
The intestinal transit time creates a source of intra and inter individual variability.
-
(4)
Presence and distribution of P-gp and other efflux transporters interfere with intestinal extraction.
7.7.2. Well-stirred model of extraction (Thummel et al., 1996)
This model relates the biochemical variable i.e. unbound intrinsic clearance to the organ extraction ratio. It is based on the assumption that:
-
(a)
There is a homogeneous distribution of enzymes for all regions.
-
(b)
Blood flow to mucosa is a fraction of total arterial blood flow to the small intestine.
-
(c)
Drug distributes rapidly between the mucosal apical epithelium and the splanchnic capillaries.
The model states that,
where EI, intestinal extraction ratio; Clin. u, unbound intrinsic clearance; QSMA, total blood flow to the superior mesenteric artery present in mucosa and fv, fraction of total arterial flow.
Chen and Pang (1997) successfully used the well stirred intestinal model to demonstrate the influence of saturability of intestinal metabolism for highly extracted 4-methyl umbeliferone on sequential biotransformation within the liver and intestine. They have used a vascularly perfused system for drug delivery to the enzyme rather than luminal administration.
7.7.3. Drawback
Predicting the effect of any splanchnic blood flow modulator on gut wall metabolism is difficult by this formula (like strenuous physical activity, ingestion of food, vascular acting co medication, posture, exercise etc.).
Galetin (2007) has shown comparable intestinal and hepatic catalytic activities (when expressed per pmol of P450 enzyme) of the major P450 enzymes. Hence he concluded that once normalized for the relative abundance of the enzyme investigated, hepatic or recombinant data would be equally useful for the prediction of intestinal clearance once an appropriate mechanistic model is applied (Galetin, 2007).
8. Conclusion
Thus over recent years a number of studies have assessed the catalytic activity of intestinal metabolic enzymes in comparison with the liver, focusing predominantly on CYP3A4/CYP3A5 as well as a range of glucuronidating (UGT) enzymes. Although intuitively one would expect the activity of enzymes in both organs to be comparable, a number of in vitro as well as in vivo studies have reported differential importance of the intestine relative to the liver. In addition, inter-individual variability in P450/UGT expressions in both the liver and intestine and their relative abundance in the corresponding organs have generally not been taken into account. Although this approach is applicable to P450 enzymes, certain specific issues need to be addressed in case of Phase II enzymes. For example, the relative UGT expression levels in vivo are not clearly defined.
9. Future trends
Examining the effects of uptake transporters, efflux transporters and metabolizing enzymes on metabolic processes is an exciting area for future research because, a number of other transporter metabolism pairs exhibit similar phenomena as that of MDRl–CYP3A4. MPR2 and UGTs are one of them. Concerning the synergistic role of conjugation enzymes/carboxyesterases and efflux transporters, little information is available for the human small intestine, although some reports have been published on the disposition in experimental animals and Caco-2 cells. For more precise analysis, transporter molecules responsible for the cellular extrusion of these conjugated metabolites need to be cloned and studied. A recent study by Cao et al. indicated a 3-fold higher expression of UGTs relative to CYP3A4 in human duodenum. However, as the expression of intestinal metabolic enzymes, and transporters, follows certain gradient patterns along the intestine and within the villi, this will not necessarily reflect the UGT:P450 abundance ratio along the whole length of the gut. In addition, the identification of appropriate extrahepatic scaling factors represents a major challenge. Only recently, has a consensus on such numbers been reached for the scaling of hepatic data, and there is no such comprehensive data available for the analysis of the intestinal microsomal recovery. Also the factors like drug concentrations, site-dependence of metabolic enzyme expression, induction or inhibition of enzyme site expression and interactions with physiologic luminal contents or co-administered drugs or exipients require more research in order to develop a full understanding of the absorption and poor bioavailability of drugs.
Contributor Information
Yogeshkumar Nanasaheb Gavhane, Email: gavhaneyogesh@rediffmail.com.
Adhikrao Vyankatrao Yadav, Email: avyadav03@rediffmail.com.
References
- Ambudkar S. Biochemical, cellular and pharmacological aspects of multidrug transporter. Annu. Rev. Phrmacol. Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- Aoyama T. Cyt. P 450 hPCN3, a novel cytochrome P-450 3A gene product that is differentially expressed in human adult liver. J. Biol. Chem. 1989;264:10295–10388. [PubMed] [Google Scholar]
- Artursson P. Epithelial transport of drugs in cell cultures. I: a model for studying the passive diffusion of drugs over intestinal absorptive (CaCo-2) cells. J. Pharm. Sci. 1990;79:476–482. doi: 10.1002/jps.2600790604. [DOI] [PubMed] [Google Scholar]
- Artursson P., Karlsson J. Correlation between oral drug absorption in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991;175:880–885. doi: 10.1016/0006-291x(91)91647-u. [DOI] [PubMed] [Google Scholar]
- Augustijns P. Drug absorption studies of prodrug esters using the Caco-2 model: evaluation of ester hydrolysis and transepithelial transport. Int. J. Pharm. 1998;166(1):45–53. [Google Scholar]
- Augustins P. Evidence for a polarized efflux system in Caco-2 cells capable of modulation of cyclosporine transport. Biochem. Biophys. Res. Commun. 1993;197:360–365. doi: 10.1006/bbrc.1993.2487. [DOI] [PubMed] [Google Scholar]
- Back D. The in vitro metabolism of ethinyl oestradiol, mestranol and levonorgestrel by human jejunal mucosa. Br. J. Clin. Pharmacol. 1981;11:275–278. doi: 10.1111/j.1365-2125.1981.tb00534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benet L., Cummins C. The drug efflux metabolism alliance: biochemical aspects. Adv. Drug Deliv. Rev. 2001;50:53–511. doi: 10.1016/s0169-409x(01)00178-8. [DOI] [PubMed] [Google Scholar]
- Benet L. Intestinal drug metabolism and antitransport processes: a potential paradigm shift in oral drug delivery. J. Control Release. 1996;39:139–143. [Google Scholar]
- Benet L. Intestinal MDR Transport proteins and P450 enzyme as barriers to oral drug delivery. J. Control Release. 1999;62:25–31. doi: 10.1016/s0168-3659(99)00034-6. [DOI] [PubMed] [Google Scholar]
- Benet L. Unmasking the dynamic interplay between efflux transporters and metabolic enzymes. Int. J. Pharm. 2004;277:3–9. doi: 10.1016/j.ijpharm.2002.12.002. [DOI] [PubMed] [Google Scholar]
- Beverly k. Deconvulating the effects of P-GP on intestinal CYP3A a major challenge. Curr. Opin. Pharmacol. 2006;6:528–532. doi: 10.1016/j.coph.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Biopharmaceutics and pharmacokinetics: A Treatise D. M. Bramhankar, Sunil B. Jaiswal Published by- M. K. Jain (Vallabh Prakashan) 1st ed. 1995, reprint – 2005.
- Buchthal J. Induction of cytochrome P450 1A by smoking or omeprazole in comparison with UDP glucoronosyl transferase in biopsies of human duodenal mucosa. Eur. J. Clin. Pharmacol. 1995;47:431–435. doi: 10.1007/BF00196857. [DOI] [PubMed] [Google Scholar]
- Burton P. Evidence for a polarized efflux for peptides in the apical membrane of Caco-2 cells. Biochem. Biophys. Res. Commun. 1993;190:760–766. doi: 10.1006/bbrc.1993.1114. [DOI] [PubMed] [Google Scholar]
- Cappiello M. Distribution of UDP-glucoronyltransferase and its endogenous substrate uridine 5′-diphosphoglucuronic acid in human tissues. Eur. J. Clin. Pharmacol. 1991;41:345–350. doi: 10.1007/BF00314965. [DOI] [PubMed] [Google Scholar]
- Carrier V. Intestinal responses to xenobiotics. Toxicol. In vitro. 2001;15:373–378. doi: 10.1016/s0887-2333(01)00039-x. [DOI] [PubMed] [Google Scholar]
- Chen J., Pang K. Effect of flow on first-pass metabolism of drugs: single pass studies on 4-methylumbelliferone conjugation on the serially perfused rat intestine and liver preparations. J. Pharmacol. Exp. Ther. 1997;280:24–31. [PubMed] [Google Scholar]
- Cheng-Chung T. Identification and localization of five CYP2Cs in murine extrahepatic tissues and their metabolism of arachidonic acid to regio- and stereoselective products. Pharmacol. Exp. Ther. 2001;299(1):39–47. [PubMed] [Google Scholar]
- Chi-Yuan W. Differentiation of absorption and first pass gut and hepatic metabolism in humans; studies with cyclosporine. Clin. Pharmacol. Ther. 1995;58:492–497. doi: 10.1016/0009-9236(95)90168-X. [DOI] [PubMed] [Google Scholar]
- Collett A. Comparison of HT 29-18 C and Caco-2 cell lines as models for studying intestinal paracellular drug absorption. Pharm. Res. 1996:13216–13221. doi: 10.1023/a:1016082829111. [DOI] [PubMed] [Google Scholar]
- Crauste-Manciet S. Cefpodoxime-proxetil hydrolysis and food effects in the intestinal lumen before absorption: in vitro comparison of rabbit and human material. Int. J. Pharm. 1997;157(2):153–161. doi: 10.1016/s0378-5173(97)00227-5. [DOI] [PubMed] [Google Scholar]
- Cummins C. In vivo modulation of intestinal cyp3A metabolism by P-gP: studies using rat single pass intestinal perfusion model. J. Pharmacol. Exp. Ther. 2003;305:306–314. doi: 10.1124/jpet.102.044719. [DOI] [PubMed] [Google Scholar]
- Cummins C. CYP3A 4 – transfected Caco-2 cells as a tool for understanding biochemical absorption barriers. Studies with sirolimus and midazolam. J. Pharmacol. Exp. Ther. 2004;308:143–155. doi: 10.1124/jpet.103.058065. [DOI] [PubMed] [Google Scholar]
- Dewaziers I. Cytochrome P450 isozymes, epoxide hydrolase and glutathione transferases in rat and human hepatic and extrahepatic tissues. J. Pharmacol. Exp. Ther. 1990;255:387–394. [PubMed] [Google Scholar]
- Diczfaluzy E. Formation of oestrone glucosiduronate by the human intestinal tract. Acta Endocrinol. 1962;40:537–551. doi: 10.1530/acta.0.0400537. [DOI] [PubMed] [Google Scholar]
- Feng C. Prodrugs of scutellarin: ethyl, benzyl and N,N-diethylglycolamide ester synthesis, physicochemical properties, intestinal metabolism and oral bioavailability in the rats. Eur. J. Pharm. Sci. 2006;29(5):385–393. doi: 10.1016/j.ejps.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Fitzsimmons M., Collins J. Selective biotransformation of the human immunodeficiency virus protease inhibitor saquinavir by human small-intestinal cytochrome P450 3A4. Drug Metab. Dispos. 1997;25:256–266. [PubMed] [Google Scholar]
- Franklin T. Human colorectal carcinoma cells in vitro as a means to assess the metabolism of analogs of mycophenolic acid. Drug Metab. Dispos. 1997;25(3):367–370. [PubMed] [Google Scholar]
- Galetin A. Intestinal first-pass metabolism – bridging the gap between in vitro and in vivo (prediction of intestinal first-pass metabolism. Curr. Drug Metab. 2007;6(7):643–644. doi: 10.2174/138920007782109779. (2) [DOI] [PubMed] [Google Scholar]
- Gomez D. The effects of ketoconazole on the intestinal metabolism and bioavailability of cyclosporine. Clin. Pharmacol. Ther. 1995;58:15–19. doi: 10.1016/0009-9236(95)90067-5. [DOI] [PubMed] [Google Scholar]
- Gorski J. Regioselective biotransformation of midazolam by members of the cytochrome P450 3A (CYP3A) subfamily. Biochem. Pharmacol. 1994;47:1643–1653. doi: 10.1016/0006-2952(94)90543-6. [DOI] [PubMed] [Google Scholar]
- Gracia M. Intestinal elimination of albendazole sulfoxide: Pharmacokinetic effects of inhibitors. Int. J. Pharm. 2003;263:123–132. doi: 10.1016/s0378-5173(03)00369-7. [DOI] [PubMed] [Google Scholar]
- Gramette T., Oertel R. Intestinal secretion of intravenous talinol is inhibited by luminal R-verapamil. Clin. Pharmacol. Ther. 1999;66:239–245. doi: 10.1016/S0009-9236(99)70031-7. [DOI] [PubMed] [Google Scholar]
- Greiner E. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 1999;104:147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero F. Role of MDR and esterase-mediated metabolism in pyrethroid-resistant populations of Haematobia irritans (Diptera: Muscidae) in Brazil. J. Med. Entomol. 2006;43(5):896–901. doi: 10.1603/0022-2585(2006)43[896:rokaem]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Gupta S. Erythromycin enhances the absorption of cyclosporine. Br. J. Clin. Pharmacol. 1988;25:401–402. doi: 10.1111/j.1365-2125.1988.tb03320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. Cyclosporine–erythromycin interaction in renal transplant patients. Br. J. Clin. Pharmacol. 1989;27:475–481. doi: 10.1111/j.1365-2125.1989.tb05396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall S. Molecular and physical mechanisms of first pass extraction. Drug Metab. Dispos. 1999;27:161–166. [PubMed] [Google Scholar]
- Hebert M. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin. Pharmcol Ther. 1992;52:453–455. doi: 10.1038/clpt.1992.171. [DOI] [PubMed] [Google Scholar]
- Her C. Human jejunal estrogen sulfotransferase and dehydro epiandrosterone sulfotransferase. Immunochemical characterization of interindividual variation. Drug Metab. Dispos. 1996;24:1328–1335. [PubMed] [Google Scholar]
- Hickman, D. et al. (1995). Longitudinal distribution of arylanine N-acetyl transferases along the human small intestine. 155 8 250.
- Hirunpanich V. Inhibitory effect of docosahexaenoic acid (DHA) on the intestinal metabolism of midazolam: in vitro and in vivo studies in rats. Int. J. Pharm. 2008;3; 351(1–2):133–143. doi: 10.1016/j.ijpharm.2007.09.037. 3. [DOI] [PubMed] [Google Scholar]
- Hoff M. Functional polymorphism of the human multidrug resistance genes, multiple sequence variations and correlation of one allele with P-gp expression and activity in vivo. Proc. Natl. Acad. Sci. USA. 2000;97:3473–3478. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J. Intestinal secretion of drugs. The role of P-glycoprotein and related drug efflux systems in limiting oral drug absorption. Adv. Drug Deliv. Rev. 1997;25(2–3):129–157. [Google Scholar]
- Hyun S. Pharmacokinetics of sildenafil after intravenous and oral administration in rats. Hepatic and intestinal first pass effects. Int. J. Pharm. 2006;320:64–70. doi: 10.1016/j.ijpharm.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Ilett K. Metabolism of drugs and other xenobiotics in the gut lumen and wall. Pharmacol. Ther. 1990;46:93–97. doi: 10.1016/0163-7258(90)90036-2. [DOI] [PubMed] [Google Scholar]
- Ilett K. Metabolism of drugs and other xenobiotics in the gut lumen and wall. Pharmacol. Ther. 1990;46:67–93. doi: 10.1016/0163-7258(90)90036-2. [DOI] [PubMed] [Google Scholar]
- Janice H., Barry H. Intestinal secretion of drugs: the role of P-gp and related drug efficacy systems in limiting oral drug absorption. Adv. Drug Deliv. Rev. 1997;25:129–157. [Google Scholar]
- Jason A., et al. (2004) P-glycoprotein efflux inhibition by amphiphilic diblock copolymers: relationship between copolymer concentration and substrate hydrophobicity, 21 (8) 1489–1497. [DOI] [PubMed]
- Kanfer I. Pharmacokinetics of oral decongestants. Pharmacotherapy. 1993;13:1165–1285. [PubMed] [Google Scholar]
- Kim R. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J. Clin. Invest. 1998;101:289–294. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolars J. First pass metabolism of cyclosporine by the gut. Lancet. 1991;338:1488–1490. doi: 10.1016/0140-6736(91)92302-i. [DOI] [PubMed] [Google Scholar]
- Kolars J. Identification of rifampin- inducible P450III3A4 (CYP3A4) in human small bowel enterocytes. J. Clin. Invest. 1992;90:1871–1888. doi: 10.1172/JCI116064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolars J. CYP3A gene expression in human gut epithelium. Pharmacogenetics. 1994;4:247–259. doi: 10.1097/00008571-199410000-00003. [DOI] [PubMed] [Google Scholar]
- Krisha D., Klotz U. Extrahepatic metabolism of drugs in humans. Clin. Pharmacokinet. 1994;26:144–160. doi: 10.2165/00003088-199426020-00007. [DOI] [PubMed] [Google Scholar]
- Kupferschimidt H. Interaction between grapefruit juice and midazolam in humans. Clin. Pharmacol. Ther. 1995;58:20–28. doi: 10.1016/0009-9236(95)90068-3. [DOI] [PubMed] [Google Scholar]
- Lacey L. Single dose pharmacokinetics of sumatriptan in healthy volunteers. Eur. J. Clin. Pharmacol. 1995;47:543–548. doi: 10.1007/BF00193709. [DOI] [PubMed] [Google Scholar]
- Lahoz A. Strategies to in vitro assessment of major human cyp enzyme activities by using liquid chromatography tandem mass spectrometry. Curr. Drug Metab. 2008;9(1):12–19. doi: 10.2174/138920008783331112. [DOI] [PubMed] [Google Scholar]
- Lampen A. Metabolism of the immunosuppressant tacrolimus in the small intestine: cytochrome P450, drug interactions, and interindividual variability. Drug Metab. Dispos. 1995;23:1315–1324. [PubMed] [Google Scholar]
- Lampen A. Metabolism and transport of the macrolide immunosuppressant sirolimus in the small intestine. J. Pharmacol Exp. Ther. 1998;23:1104–1112. [PubMed] [Google Scholar]
- Lee V., Yamamoto A. Penetration and enzymatic barriers to peptide and protein absorption. Adv. Drug Deliv. Rev. 1990;4:171–207. [Google Scholar]
- Lee D. Pharmacokinetics of omeprazole after intravenous and oral administration to rats with liver cirrhosis induced by dimethylnitrosamine. Indian J. Pharm. 2007;330(1–2):37–44. doi: 10.1016/j.ijpharm.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Leiweber F. Possible physiological roles of carboxylic acid hydrolases. Drug Metab. Dispos. 1987;18:379–439. doi: 10.3109/03602538708994129. [DOI] [PubMed] [Google Scholar]
- Leveque D., Jehl F. P-glycoprotein and pharmacokinetics. Anticancer Res. 1995;15:331–336. [PubMed] [Google Scholar]
- Lin J. Is the role of small intestine in first pass metabolism overemphasized? Pharmacol. Rev. 1999;51:135–157. [PubMed] [Google Scholar]
- Lina L., Eileen M. Role of cytochrome P450 isoenzymes in metabolism of O6-benzylguanine : implications for dacarbazine activation. Clin. Cancer Res. 2001;7:4239–4244. [PubMed] [Google Scholar]
- Mahon W. Metabolism of flurazepam by small intestine. Clin. Pharmacol. Ther. 1977;22:228–233. doi: 10.1002/cpt1977222228. [DOI] [PubMed] [Google Scholar]
- Mária T. Tofisopam inhibits the pharmacokinetics of CYP3A4 substrate midazolam. Eur. J. Clin. Pharmacol. 2008;64:93–94. doi: 10.1007/s00228-007-0397-y. Number 1 Publisher Springer Berlin / Heidelberg. [DOI] [PubMed] [Google Scholar]
- Mayer U. Substantial excretion of digoxin via the intestinal mucosa and prevention of long-term digoxin accumulation in the brain by the mdr la P-gp. Br. J. Pharmacol. 1996;119:1008–1044. doi: 10.1111/j.1476-5381.1996.tb15775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer U. Full blockade of intestinal P-gp and expensive inhibition of blood brain barrier P-gp by oral treatment of mice with PsC 833. J. Clin. Invest. 1997;100:2430–2436. doi: 10.1172/JCI119784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckay J. Xenobiotic metabolizing enzyme expression in colonic neoplasia. Gut. 1993;34:1234–1239. doi: 10.1136/gut.34.9.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckinnon R. Characterization of CYP3A gene subfamily expression in human gastrointestinal tissues. Gut. 1995;36:259–267. doi: 10.1136/gut.36.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinl W. Sulfotransferase forms expressed in human intestinal Caco-2 and TC7 cells at varying stages of differentiation and role in benzo[a]pyrene metabolism. Drug Metab. Dispos. 2008;36(2):276–283. doi: 10.1124/dmd.107.018036. [DOI] [PubMed] [Google Scholar]
- Meunier V. The human Intestinal epithelial cell line CaCo-2; pharmacological and pharmacokinetic applications. Cell Biol. Toxicol. 1995;11:187–194. doi: 10.1007/BF00756522. [DOI] [PubMed] [Google Scholar]
- Micuda S. P-glycoprotein function and expression during obstructive cholestasis in rats. Eur. J. Gastroenterol. Hepatol. 2008;20(5):404–412. doi: 10.1097/MEG.0b013e3282f471bf. [DOI] [PubMed] [Google Scholar]
- Murray G. The immunocytochemical localization and distribution of Cytochrome P450 in normal human hepatic and extrahepatic tissues with a monoclonal antibody to human cytochrome P-450. Br. J. Pharmacol. 1988;25:465–475. doi: 10.1111/j.1365-2125.1988.tb03331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurkar M. The use of surfactants to enhance the permeability of peptides through Caco-2 cells by inhibition of an apically polarized efflux system. Pharm. Res. 1996;13(4):528–534. doi: 10.1023/a:1016033702220. (7) [DOI] [PubMed] [Google Scholar]
- Norris D. The effect of physical barriers and properties on the oral absorption of particulates. Adv. Drug Deliv. Rev. 1998;34:135–154. doi: 10.1016/s0169-409x(98)00037-4. [DOI] [PubMed] [Google Scholar]
- Odgers W. Nucleotide polymorphism in the 5′ promoter region of esterase 6 in Drosophila melanogaster and its relationship to enzyme activity variation. Genetics. 1995;141(1):215–222. doi: 10.1093/genetics/141.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara A. Effect of oral ketoconazole on intestinal first-pass effect of midazolam and fexofenadine in cynomolgus monkeys. Drug Metab. Dispos. 2007;35(3):410–418. doi: 10.1124/dmd.106.011288. [DOI] [PubMed] [Google Scholar]
- Pacifici G. Presystemic glucuronidation of morphine in humans and rhesus monkeys: sub cellular distribution of the UDP-glucoronyltransferase in the liver and intestine. Xenobiotica. 1986;16:123–128. doi: 10.3109/00498258609043514. [DOI] [PubMed] [Google Scholar]
- Pacifici G. Methylation of captopril in human liver, kidney and intestine. Xenobiotica. 1991;21:1107–1112. doi: 10.3109/00498259109039550. [DOI] [PubMed] [Google Scholar]
- Pacifici G. (+) and (−) terbutaline are sulphated at a higher rate in human intestine than in liver. Eur. J. Clin. Pharmacol. 1993;45:483–487. doi: 10.1007/BF00315522. [DOI] [PubMed] [Google Scholar]
- Pacifici G. S-methyl transferases in human intestine: different distribution of the microsomal thiol methyl transferase and cytosolic thiopurine methyltransferase along the human bowel. Xenobiotica. 1993;23:671–679. doi: 10.3109/00498259309059404. [DOI] [PubMed] [Google Scholar]
- Paine M. First pass metabolism of midazolam by the human intestine. Clin. Pharmacol. Ther. 1996;60:14–24. doi: 10.1016/S0009-9236(96)90162-9. [DOI] [PubMed] [Google Scholar]
- Paine M. Characterization of inter and intra intestinal differences in human CYP-3A dependent metabolism. J. Pharmacol. Exp. Ther. 1997;283:1552–1562. [PubMed] [Google Scholar]
- Palkama V. Effect of saquinavir on the pharmacokinetics and pharmacodynamics of oral and intravenous midazolam. Clin. Pharmacol. Ther. 1999;66:33–39. doi: 10.1016/S0009-9236(99)70051-2. [DOI] [PubMed] [Google Scholar]
- Pesola G., Walle T. Stereoselective sulfate conjugation of isoproterenol in humans: comparison of hepatic, intestinal and platelet activity. Chirality. 1993;5:602–609. doi: 10.1002/chir.530050807. [DOI] [PubMed] [Google Scholar]
- Peters W., Kerners P. Cytochromes P-450 in the intestinal mucosa of man. Biochem. Pharmacol. 1989;38:1535–1538. doi: 10.1016/0006-2952(89)90194-9. [DOI] [PubMed] [Google Scholar]
- Peters W. Biotransformation enzymes in human intestine: critical low levels in the colon. Gut. 1991;32:408–412. doi: 10.1136/gut.32.4.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poet T. In vitro rat hepatic and intestinal metabolism of the organophosphate pesticides chlorpyrifos and diazinon. Toxicol. Sci. 2003;72(2):193–200. doi: 10.1093/toxsci/kfg035. [DOI] [PubMed] [Google Scholar]
- Prueksaritanont T. Comparative studies of drug metabolizing enzymes in dog, monkey and human small intestines and in caco-2 cells. Drug Metab. Dispos. 1996;24:634–642. [PubMed] [Google Scholar]
- Raoof A. Extrahepatic glucuronidation of propofol in man: possible contribution of gut wall and kidney. Eur. J. Clin. Pharmacol. 1996;50:91–96. doi: 10.1007/s002280050074. [DOI] [PubMed] [Google Scholar]
- Rogers S. Intestinal metabolism of ethinyl oestradiol and paracetamol in vitro studies using chambers. Br. J. Clin. Pharmacol. 1987;23:727–734. doi: 10.1111/j.1365-2125.1987.tb03108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone-Parsons A. Effect of oral posaconazole on the pharmacokinetics of cyclosporine and tacrolimus. Pharmacotherapy. 2007;27(6):825–834. doi: 10.1592/phco.27.6.825. [DOI] [PubMed] [Google Scholar]
- Schinkel A. Normal variability and altered pharmacokinetics in mice lacking mdr 1 type (drug transporting) P-glycoprotein. Proc. Natl. Acad. Sci. USA. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz E. Regulation of human liver cytochrome P 450 family 3A in primary and continuous culture of human hepatocytes. Hepatology. 1993;18:1254–1262. [PubMed] [Google Scholar]
- Shargel L. sixth ed. Lippincott Williams& Wilkins; 2006. Comprehensive Pharmacy Review Publication. 84. [Google Scholar]
- Sparrenboom A. Limited oral bioavailability and active epithelial excretion of paclitaxel caused by P-gp in the intestine. Proc. Natl. Acad. Sci. USA. 1997;94:2031–2035. doi: 10.1073/pnas.94.5.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam Y. Individual variation in 1st pass metabolism. Clin. Pharmacokinet. 1993;25:300–328. doi: 10.2165/00003088-199325040-00005. [DOI] [PubMed] [Google Scholar]
- Thummel, K., (1995). Modelling hepatic and intestinal metabolism of the CYP3A probe midazolam from in vitro data. AAPS symposium on prediction of metabolic clearance, bioavailability and drug interactions from in vitro-derived data. AAPS Tenth Annual Meeting and exposition. Miami Beach, FL.
- Thummel K. Oral first pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin. Pharmacol. Ther. 1996;59:491–502. doi: 10.1016/S0009-9236(96)90177-0. [DOI] [PubMed] [Google Scholar]
- Thummel K. Enzyme catalyzed processes of first pass hepatic metabolism and intestinal drug extraction. Adv. Drug Deliv. Rev. 1997;27:99–127. doi: 10.1016/s0169-409x(97)00039-2. [DOI] [PubMed] [Google Scholar]
- Ueda K. Human P-gp transports cortisol, aldosterone, and dexamethasone, but not progesterone. J. Biol. Chem. 1992;267:24248–24252. [PubMed] [Google Scholar]
- Van O. Extrahepatic metabolism of sevoflurane in children undergoing orthotopic liver transplantation. Anesthesiology. 2000;92(3):683–686. doi: 10.1097/00000542-200003000-00011. [DOI] [PubMed] [Google Scholar]
- Vickers A. Sites of biotransformation for the cyclosporine derivatives SDZ IMM 125 using human liver and kidney slices and intestine. Drug Metab. Dispos. 1995;23:327–333. [PubMed] [Google Scholar]
- Wacher V. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol. Carcinog. 1995;13:129–134. doi: 10.1002/mc.2940130302. [DOI] [PubMed] [Google Scholar]
- Wacher V. Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv. Drug Deliv. Rev. 1996;20:99–112. doi: 10.1016/s0169-409x(00)00126-5. [DOI] [PubMed] [Google Scholar]
- Wacher V. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J. Pharm sci. 1998;87:1322–1330. doi: 10.1021/js980082d. [DOI] [PubMed] [Google Scholar]
- Wacher V. Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv. Drug Deliv. Rev. 2001;46:89–102. doi: 10.1016/s0169-409x(00)00126-5. [DOI] [PubMed] [Google Scholar]
- Walker S. Inter-species comparison of liver and small intestinal microsomal metabolism of fluoranthene. Food Chem. Toxicol. 2006;44:380–387. doi: 10.1016/j.fct.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Warner P. Sumatriptan absorption from different regions of the human gastrointestinal tract. Pharm. Res. 1945;12:138–143. doi: 10.1023/a:1016211409315. [DOI] [PubMed] [Google Scholar]
- Watkins P. Non invasive tests of CYP3A enzymes. Pharmacogenetics. 1994;4:171–184. doi: 10.1097/00008571-199408000-00001. [DOI] [PubMed] [Google Scholar]
- Watkins P.B. The barrier functions of CYP3A4 and P-gp in the small bowel. Adv. Drug Deliv. Rev. 1997;27:161–170. doi: 10.1016/s0169-409x(97)00041-0. [DOI] [PubMed] [Google Scholar]
- Watkins P. Identification of glucocorticoid inducible cytochromes P-450 in the intestinal mucosa of rats and man. J. Clin. Invest. 1987;80:1029–1036. doi: 10.1172/JCI113156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waziers D. Cytochrome P450 isoenzymes, epoxide hydrolases and glutathione transferases in rat and human hepatic and extrahepatic tissues. J. Pharmacol. Exp. Ther. 1990;253:387–394. [PubMed] [Google Scholar]
- Woodcock D. Reversal of multidrug resistance by surfactants. Br. J. Cancer. 1992;66:62–68. doi: 10.1038/bjc.1992.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighton S. Identification of a polymorphically expressed member of the human cytochrome P 450 family. Mol. Pharmacol. 1989;36:97–105. [PubMed] [Google Scholar]
- Wrighton S. Studies on the expression and metabolic capabilities of human liver cytochrome P 400 345. Mol. Pharmcol. 1990;38:207–213. [PubMed] [Google Scholar]
- http://www.nursinglink.com/.../320-clinically-significant-drug-interaction-with-the-cytochrome-p450-.
- Yan Z., Caldwell G. Metabolism profiling and cytochrome P450 inhibition & induction in drug discovery. Curr. Top. Med. Chem. 2001;1(5):403–425. doi: 10.2174/1568026013395001. [DOI] [PubMed] [Google Scholar]
- Zhag Q. Characterization of human small intestinal cytochromes P 450. Drug Metab. Dispos. 1999;27:804–809. [PubMed] [Google Scholar]
- Zhang W. Impact of curcumin-induced changes in P-glycoprotein and CYP3A expression on the pharmacokinetics of peroral celiprolol and midazolam in rats. Drug Metab. Dispos. 2007;35(1):110–115. doi: 10.1124/dmd.106.011072. [DOI] [PubMed] [Google Scholar]