

Abstract
Chemical reinvestigation of the aerial parts of Solanum schimperianum Hochst led to the isolation of ten compounds, lupeol (1), β-sitosterol (2), β-sitosterol glucoside (3), oleanolic acid (4), teferidin (5), teferin (6), ferutinin (7), 5-hydroxy-3,7,4′-trimethoxyflavone (8), retusin (9) and kaempferol-3-O-β-d-glucopyranoside (10). Compounds 5–7 were isolated for the first time from Solanaceae and compounds 1–4 and 8–9 for the first time from Solanum schimperianum. The structure elucidation of the isolated compounds was based on careful inspection of spectral data including 1D (1H and 13C NMR), 2D (1H–H COSY, HMQC and HMBC, ROESY), UV, MS and IR, in addition to, comparison with literatures. The antimicrobial activity of the extracts as well as the isolated compounds was tested. Only hexane extract showed activity against Bacillus subtilus and Staphylococcus aureus.
Keywords: Solanum schimperianum, Flavonoids, Sesquiterpene, Antimicrobial
1. Introduction
The genus Solanum is the largest genera of the family Solanaceae consisting of more than 1700 species distributed all over the world. In Saudi Arabia, the genus is represented by about 16 species, mainly in West and Southwest side of the country (Chaudhary, 2001; Collenette, 1999). Several species of genus Solanum are used in the folk medicine of different countries, Brazil, India, Taiwan, Germany, South Africa and Kenya, as remedy for various ailments such as hypoglycemic (Kar et al., 2006), hepatoprotective (Son et al., 2003), hepatotonic (De Silva et al., 2003), laxative, appetizer, cardiotonic (Mans et al., 2004), antispasmodic, renal pain, epilepsy (Perez et al., 2006; Schwarz et al., 2005), gastric, liver disorder (Antonio et al., 2004; Mesia-Velal et al., 2002), treatment of bronchitis, itches, body aches, cancer (Koduru et al., 2006; Oboh et al., 2005). Genus solanum is a rich source for many classes of compounds such as alkaloids (Emmanuel et al., 2006; Ndebia et al., 2007), steroids (Ferro et al., 2005; Jairo et al., 1998; Yoshimitsu et al., 2003) and phenolic compounds)El-Sayed and Hassan, 2006; Hodek et al., 2002; Sarmento Silva and Bezerra Nascimento, 2004). Solanum schimperianum Hochst grows throughout Southern region of Saudi Arabia. It is also widely distributed in the tropical Africa. The plant is locally known as Millyan and Nakhab (Chaudhary, 2001). Solanum schimperianum plant has no known folkloric usage. The literature survey showed the isolation of one coumarin, esculetin and four flavonols; astragalin, isoquercitin, 3-kaempferol-diglucoside and rutin, four glycoalkaloids; α-solamargine and β-solamargine, β-solanopubamine, and γ-solamarine (Al-Rehaily et al., 2011; Angenot, 1969; Coune and Denoel, 1975). The methanol extract of Solanum schimperianum was reported to have significant antitrypanosomal activity (Abdel-Sattar et al., 2009). The present study deals with the isolation and characterization of ten compounds, as well as the antimicrobial study of the extracts and all the pure compounds.
2. Materials and methods
2.1. Plant materials
The aerial parts of Solanum schimperianum Hochst were collected from Abha region in March 2005. The plant was identified by Dr. M. Atiqur Rahman Prof. of Taxonomy, College of Pharmacy, King Saud University. A voucher specimen (# 14903) was deposited at the herbarium in the College of Pharmacy at King Saud University (KSU).
2.2. General experimental procedure
HPLC/ESI MS is carried out using a Finnigan LCQ-DECA mass spectrometer connected to UV detector; EI MS were measured on Finnigan 8430 mass spectrometer; Melting points were determined on a Mettler FP 80 Central Processor supplied with a Mettler FP 81 MBC Cell Apparatus, and were uncorrected; Specific rotations were measured as solutions in methanol or chloroform, unless otherwise specified, on a Perkin–Elmer 241 Mc polarimeter, using a one-decimeter tube; Infra Red spectra were recorded on Perkin–Elmer FTIR model 1600 spectrophotometer, USA; 1H and 13C NMR spectra were recorded in CDCl3 and DMSO-d6 on a Bruker Avance DRX – 500 instrument (Central Lab. at the College of Pharmacy, KSU) at 500 MHz for protons and 125 MHz for carbons using the residual solvent signal as an internal standard and/or NMR measurements done by Prof. Dr. Peter Procksh at the institute of Organic Chemistry and Macromolecular Chemistry of Heinrich-Heine University, Düsseldorf. 1H and 13C NMR spectra were recorded at 300 K on Bruker DPX 300, ARX 400, 500 or AVANCE DMX 600 NMR spectrometers. All 1D and 2D spectra were obtained using the standard Bruker software.
2.3. Extraction and Isolation
The dried and grounded aerial parts of Solanum schimperianum (2.4 kg) were consecutively extracted at room temperature with n-hexane (3 × 5 L) and ethyl acetate (3 × 5 L) to yield after evaporation in vacuo A, 5.24 g and B, 25.32 g.
n-Hexane extract (A, 5 g) was chromatographed on a silica gel column. The elution was started with petroleum ether and ethyl acetate and polarity was increased up to 3:1. The collected fractions (100 ml each) were pooled according to their TLC behavior into 5 fractions. Fraction 1 (900.23 mg) gave 1 (153.2 mg, 3.06%) by crystallization in a mixture of chloroform and methanol. Fraction 2 (350.50 mg) was crystallized to afford 72.8 mg of compound 2 1.46%. Fraction 3 (879.76 mg) was rechromatographed over silica gel using petroleum ether and ethyl acetate (1:3) afforded 3 (100.3 mg, 2.01%). Repeated crystallization of fraction 4 (935.45 mg) from methanol afforded 4 (50.30 mg, 1.01%). Fraction 5 (1.52 g) was chromatographed over silica gel column and eluted using increased polarity of methanol in chloroform (0–10%) and resulted in four pooled subfractions (i–iv). Compounds 5 (17.23 mg, 0.34%) and 6 (12.31 mg, 0.34%) were isolated from subfractions (i) (200.53 mg) and (ii) (320.73 mg) were chromatographed over chromatotron (1 mm plate) using 5% and 10% ethyl acetate in hexane respectively. while subfraction (iii) (680.43 mg) was chromatographed over chromatotron (2 mm plate) using 20% ethyl acetate in hexane gave 7 (108.12 mg, 2.16%).
The ethyl acetate extract, (B, 10 g), was chromatographed over silica gel column using petroleum ether with increasing amount of ethyl acetate. Fractions eluted with 5% ethyl acetate in petroleum ether afforded 8 (12.11 mg, 0.12%). Fractions eluted with 10% ethyl acetate in petroleum ether afforded 9 (5.13 mg, 0.05%). The impure fractions were pooled together (5.21 g) and chromatographed over silica gel column using chloroform with increasing polarity with methanol. Fraction at 50% methanol in chloroform (1.53 g) was showing one major spot, which was further chromatographed over Sephadex column using 10% H2O in methanol to afford 63 mg of compound 10, 0.63%.
2.4. Acid hydrolysis
Acid hydrolysis of compound 10 afforded glucose as the sugar residue confirmed by co-TLC with authentic sample.
2.5. Antimicrobial assay
The extracts and all the pure compounds were tested for antimicrobial activity against Bacillus subtilus (ATCC 6633), Staphylococcus aureus (ATCC 292136), Escherichia coli (ATCC 25922), Pseudomonus aeruginosa (ATCC 15442), Candida albicans (ATCC 90023) and Mycobacterium smegmatis (ATCC 35797) microorganism using agar-cup diffusion method (Hugo and Russell, 1992). Briefly 20 ml of Nutrient Agar (Hi Media Pvt LTD) was poured into the Petri-dish and 8 mm well bored in the agar. 1 mg and 100 μg of each extract and pure compounds, respectively, were dissolved in 200 μL of dimethylsulfoxide and poured into the wells. The plates were incubated for 24 h at 37 °C and the zone of inhibition was measured in mm. DMSO was used as negative control, while Chloramphenicol was used as a positive control for Gram-positive.
3. Results
Ten compounds were isolated from the different extract of Solanum schimperianum. The structures of compounds 5–10 are presented in Fig. 1. The antimicrobial activities of hexane and ethyl acetate extracts as well as the isolated compounds were studied.
Figure 1.
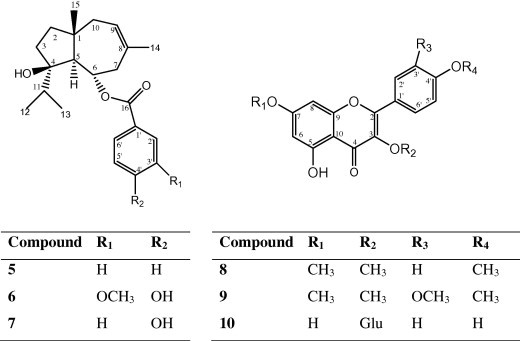
Structures of compounds 5–10.
3.1. The physical and spectral data of compounds 1–10
3.1.1. Compound 1
White powder; mp 210 °C; FTIR (KBr) 3235, 1640, 1382, 1185,1105 cm−1; UV λmax (MeOH) 200 nm; 1H NMR (500 MHz, CDCl3): δ 4.69, 4.56 (each 1H, m, H-29), 3.18 (1H, m, H-3), 1.69 (3H,s H-30), 1.04 (3H, s, H-26), 0.98 (3H, s, H-23), 0.97 (3H, s, H-27), 0.84, 0.79 and 0.77 (each 3H, s, H-25, 28, 24); 13C NMR (125 MHz, CDCl3): δ 150.8 (C-20), 109.3 (C-6), 78.9 (C-3), 55.2 (C-5), 50.3 (C-9), 48.2 (C-18), 47.8 (C-19), 42.9 (C-17), 42.5 (C-14) 40.8 (C-8) 39.9 (C-22), 38.8 (C-4), 38.5 (C-1), 38.0 (C-13), 37.1 (C-10), 35.4 (C-16), 34.2 (C-7), 29.8 (C-21), 27.9 (C-23), 27.4 (C-15), 27.1 (C-2), 25.0 (C-12), 20.9 (C-11), 19.2 (C-30), 18.2 (C-6), 17.8 (C-28), 16.1 (C-25), 15.9 (C-26), 15.3 (C-24), 14.5 (C-27); EI MS m/z 426 [M]+.
3.1.2. Compound 2
White powder; mp 136 °C; FTIR (KBr) 4330 cm−1 (OH), 1050 cm−1 (C–O), 2900, cm−1(for C–H), 1430 cm−1(C=C); 1H NMR (500 MHz, CDCl3): δ 5.38 (br s, H-6), 3.50 (1H, m, H-3), 0.99 (3H, s, H-19), 0.85 (3H, d, J = 7.5 Hz, H-26), 0.83 (3H, J = 7.5 Hz, H-27), 0.81 (3H, t, J = 7.5 Hz, H-29), 0.66 (3H, s, H-18); 13C NMR (125 MHz, CDCl3): δ 140.5 (C-5), 121.8 (C-6), 71.9 (C-3), 56.8 (C-14), 56.1 (C-17), 50.2 (C-9), 45.8 (C-24), 42.4 (C-13), 42.4 (C-4) 39.8 (C-12), 37.3 (C-1), 36.3 (C-10), 36.5 (C-20), 34.0 (C-22), 31.9 (C-7, 8), 31.7 (C-2), 29.2 (C-25), 28.3 (C-16), 26.1 (C-23), 24.4 (C-15), 23.1 (C-28), 21.2 (C-11), 19.9 (C-27), 19.4 (C-26), 19.1 (C-19), 18.8 (C-21), 12.0 (C-18), 11.9 (C-29); EI MS m/z 414 [M]+ for C29H50O.
3.1.3. Compound 3
White crystals (MeOH); mp 289–290 °C; {Rf; 0.31 (4% methanol in chloroform}; FTIR (KBr) 3500–3200 cm−1 (OH), 1026 cm−1 (C–O), 2920, 2820 cm−1(for C–H), 1460 cm−1(C=C); UV λmax (CHCl3) 243 nm; 1H NMR (500 MHz, DMSO-d6): δ 5.38 (br s, H-6), 4.26 (1H, d, J = 7 Hz, anomeric proton of glucose), 3.52 (1H, m, H-3), 1.05 (3H, s, H-19), 0.86 (6H, d, J = 7.5 Hz, H-26, H-27), 0.85 (3H, t, J = 7.5 Hz, H-29), 0.71 (3H, s, H-18); 13C NMR (125 MHz, DMSO-d6): δ 140.5 (C-5), 121.1 (C-6), 76.9 (C-3), 56.2 (C-14), 55.4 (C-17), 49.6 (C-9), 45.2 (C-24), 41.8 (C-13), 40.1 (C-4) 39.4 (C-12) 38.8 (C-1), 36.2 (C-10), 35.5 (C-20), 33.4 (C-22), 31.4 (C-7, 8), 29.3 (C-2), 28.8 (C-16, 25), 27.7 (C-23), 25.6 (C-15), 23.8 (C-28), 22.6 (C-11), 20.3 (C-27), 18.9 (C-26), 19.7 (C-19), 19.1 (C-21), 11.8 (C-18), 11.6 (C-29), 100.8 (C-1′), 76.8 (C-3′), 76.7 (C-5′), 73.5 (C-5′), 70.1 (C-4′), 61.1 (C-6′); EI MS m/z 576 [M]+ for C35H60O6, m/z 414 [M+-glucose].
3.1.4. Compound 4
White powder; mp 196–198 °C (CHCl3–MeOH); {Rf; 0.59 (5% methanol in chloroform}; UV λmax (CHCl3) 205, 277, 329 nm; FTIR (KBr), 3429 (OH), 1695 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 5.16 (1H, m, H-12), 3.40 (1H, dd, 4.0, 14 Hz, H-3), 1.09 (3H, s, H-27), 0.90 (3H, s, H-26), 0.88 (9H, s, H-23, H-25, H-30), 0.72 (3H, s, H-29), 0.68 (3H, s, H-24); 13C NMR (125 MHz, DMSO-d6): δ 178.5 (C-28), 143.8 (C-13), 121.5 (C-12), 76.8 (C-3), 54.8 (C-5), 47.1 (C-9), 45.7 (C-19), 45.4 (C-17), 41.3 (C-14), 40.8 (C-18), 38.9 (C-8), 38.2 (C-4), 38.1 (C-1), 36.6 (C-10), 38.9 (C-8), 33.3 (C-21), 32.8 (C-29), 32.4 (C-7), 32.1 (C-22), 30.3 (C-20), 28.2 (C-23), 27.2 (C-15), 26.9 (C-2), 25.6 (C-27), 23.3 (C-30), 22.9 (C-11), 22.6 (C-16), 18.0 (C-6), 16.8 (C-26), 16.0 (C-24), 15.1 (C-25); EI MS m/z 456 [M+] for C30H48O3.
3.1.5. Compound 5
Yellow residue, [α]D +37.5° (c = 1.0, chloroform); UV λmax 258 nm; FTIR (KBr) 3285, 1698, 1609 cm−1; The EI MS [M]+ at m/z 342 for C22H30O3; 1H, 13C NMR (500 and 125 MHz, CDCl3) Table 1.
Table 1.
1H and 13C NMR data of compounds 5–7 in CDCl3.
| Position | Compound 5 |
Compound 6 |
Compound 7 |
|||
|---|---|---|---|---|---|---|
| δH (δ ppm, J = Hz) | δC | δH (δ ppm, J = Hz) | δC | δH (δ ppm, J = Hz) | δC | |
| 1 | – | 44.0 s | – | 44.0s | – | 44.0 s |
| 2 | 1.20 (1H, m), 1.49 (1H, m) | 41.3 t | 1.29 (1H, m), 1.57 (1H, m) | 41.3 t | 1.28(1H, m), 1.58 (1H, m) | 41.3 t |
| 3 | 1.56 (1H, m), 1.89 (1H, m) | 31.8 t | 1.60 (1H, m), 1.97 (1H, m) | 31.7 d | 1.66(1H, m), 1.97 (1H, m) | 31.6 t |
| 4 | – | 86.3 s | – | 86.3 s | – | 86.8 s |
| 5 | 1.95 (1H, d, J = 10.5) | 60.0 d | 1.99 (1H, m) | 60.1 d | 1.98 (1H, m) | 60.1 d |
| 6 | 5.23 (1H, d t, J = 3.0, 10.5) | 71.4 d | 5.29 (1H, dt, J = 2.5, 10.5) | 71.1 d | 5.30 (1H, t, J = 10.5) | 71.2 d |
| 7 | 2.23 (1H, dd, J = 2.5, 13.0) 2.50 (1H, t, J = 13.0) | 41.4 t | 2.32 (1H, dd, J = 2.5, 13.0)2.55 (1H, t, J = 13.0) | 41.4 t | 2.31 (1H, d, J = 13.0) 2.58 (1H, t, J = 13.0) | 41.4 t |
| 8 | – | 133.5 s | – | 133.6 s | – | 133.5 s |
| 9 | 5.49 (1H, br t) | 125.2 d | 5.58 (1H, br t) | 125.2 d | 5.58 (1H, br s) | 125.2 d |
| 10 | 1.92 (1H, d, J = 8.0) 2.02 (1H, d, J = 8.0) | 41.1 t | 1.98 (1H, m), 2.02 (1H, m) | 41.0 t | 1.98 (1H, m),2.02 (1H, m) | 41.0 t |
| 11 | 1.85 (1H, s ep, J = 6.5) | 37.2 d | 1.95 (1H, m) | 37.3 d | 1.88 (1H, m) | 37.1 d |
| 12 | 0.77 (3H, d, J = 7.0) | 17.4 q | 0.88 (3H, d, J = 6.5) | 17.5 q | 0.87 (3H, d,J = 6.5) | 17.5 q |
| 13 | 0.89 (3H, d, J = 7.0) | 18.5 q | 0.99 (3H, d, J = 6.5) | 18.5 q | 0.96 (3H, d, J = 6.5) | 18.5 q |
| 14 | 1.76 (3H, s) | 26.4 q | 1.85 (3H, s) | 26.4 q | 1.84 (3H, s) | 26.4 q |
| 15 | 1.04 (3H, s) | 20.2 q | 1.13 (3H, s) | 20.2 q | 1.12 (3H, s) | 20.2 q |
| 16 | – | 166.5 s | – | 166.3 s | – | 166.9 s |
| 1′ | – | 130.6 s | – | 122.7 s | – | 122.5 s |
| 2′ | 7.96 (1H, dd, J = 8.0, 1.0) | 129.6 d | 7.58 (1H, d, J = 2) | 111.9 d | 7.95 (1H, d, J = 8.0) | 132.0 d |
| 3′ | 7.39 (1H, t, J = 8.0) | 128.5 d | – | 146.3 s | 6.91 (1H, d, J = 8.0) | 115.4 d |
| 4′ | 7.51 (1H, t d, J = 8.0, 1.0) | 133.0d | – | 150.2 s | – | 160.6 s |
| 5′ | 7.39 (1H, t, J = 8.0) | 128.5 d | 6.98 (1H, d, J = 8) | 114.2 d | 6.91 (1H, d, J = 8.0) | 115.4 d |
| 6′ | 7.96 (1H, dd, J = 8.0, 1.0) | 129.6d | 7.63 (1H, dd, J = 2.0, 8.0) | 124.2 d | 7.95 (1H, d, J = 8.0) | 132.0 d |
| OCH3 | – | – | 3.97 (3H, s) | 56.0 q | – | – |
| OH | – | – | 6.08 | – | – | – |
3.1.6. Compound 6
White powder; mp 78–80 °C; [α]D +86.5° (c = 1.0, chloroform); UV λmax (CH3OH) 265 nm; FTIR (KBr) 3200–3600, 1695, 1620, 1595 and 1520 cm−1; The EI MS [M]+ at m/z 388 for C23H32O5; 1H, 13C NMR (500 and 125 MHz, CDCl3) Table 1.
3.1.7. Compound 7
White powder, mp 121–122 °C; [α]D +66.1ο (c = 1.36, ethanol); UV λmax (CH3OH) 265 and 300 nm; FTIR (KBr) 3200–3600, 1690, 1530, 1610 cm−1; The EI MS [M]+ at m/z 358 for C22H30O4; 1H, 13C NMR (500 and 125 MHz, CDCl3) Table 1.
3.1.8. Compound 8
Yellow needles (methanol, chloroform); mp 144–146 °C; {Rf; 0.85, (20% ethyl acetate: n-hexane)}; UV λmax MeOH (254, 349), MeOH/NaOMe (268, 376), MeOH/AlCl3 (275, 290 sh., 355, 399), MeOH/AlCl3/HCl (264 sh., 276, 347, 497) MeOH/NaOAc (255, 351), MeOH/NaOAc/H3BO3 (254, 349); ESI MS [M+H]+ at m/z 329 for C18H16O6; 1H, 13C NMR (500 and 125 MHz, DMSO-d6) Table 2.
Table 2.
1H and 13C NMR data of compounds 8–10 in DMSO-d6.
| Position | Compound 8⁎ |
Compound 9⁎ |
Compound 10 |
|||
|---|---|---|---|---|---|---|
| δH (δ ppm, J = Hz) | δC | δH (δ ppm, J = Hz) | δC | δH (δ ppm, J = Hz) | δC | |
| 2 | – | 156.7 s | – | 156.1 s | – | 156.4 |
| 3 | – | 138.5 s | – | 137.0 s | – | 133.2 |
| 4 | – | 176.1 s | – | 175.7 s | – | 177.4 |
| 5 | – | 161.2 s | – | 160.9 s | – | 161.2 |
| 6 | 6.35 (1H, d, J = 1.9) | 98.0 d | 6.38 (1H, s) | 98.1 d | 6.21 (1H, d, J = 2.5) | 98.7 |
| 7 | – | 166.0 s | – | 163.0 s | – | 164.1 |
| 8 | 6.72 (1H, d, J = 1.9) | 92.2 d | 6.79 (1H, s) | 93.3 d | 6.43 (1H, d, J = 2.5) | 93.6 |
| 9 | – | 157.0 s | – | 157.2 s | – | 156.2 |
| 10 | – | 104.0 s | – | 104.0 s | – | 104.0 |
| 1′ | – | 122.6 s | – | 121.9 s | – | 120.9 |
| 2′ | 8.03 (1H, d, J = 9.2) | 129.9 d | 7.65 (1H, s) | 115.1 d | 8.04 (1H, d, J = 8.5) | 130.8 |
| 3′ | 7.11 (1H, d, J = 9.2) | 114.0 d | – | 147.7 s | 6.88 (1H, d, J = 8.5) | 115.1 |
| 4′ | – | 161.9 s | – | 149.6 s | – | 159.9 |
| 5′ | 7.11 (1H, d, J = 9.2) | 114.0 d | 7.15 (1H, d, J = 8.5) | 115.9 d | 6.88 (1H, d, J = 8.5) | 115.1 |
| 6′ | 8.03 (1H, d, J = 9.2) | 129.9 d | 7.72 (1H, dd, J = 8.5, 1.9) | 119.9 d | 8.04 (1H, d, J = 8.5) | 130.8 |
| 1′′ | – | – | – | – | 5.45 (1H, d, J = 7.0) | 100.9 |
| 2′′ | – | – | – | – | 3.22 (1H, m) ⁎ | 74.2 |
| 3′′ | – | – | – | – | 3.08 (1H, m) ⁎ | 77.4 |
| 4′′ | – | – | – | – | 3.18(1H, m) ⁎ | 76.4 |
| 5′′ | – | – | – | – | 3.10 (1H, m) ⁎ | 69.9 |
| 6′′ | – | – | – | – | 3.35 (1H, br s), 3.84 (1H, t, J = 3.0) | 60.9 |
| OH | 12.50 (1H, s) | – | 12.60 (1H, s) | – | 12.61 (1H, s) | – |
| 3-OCH3 | 3.79 (3H, s) | 59.5 q | 3.81 (3H, s) | 60.6 | – | – |
| 7-OCH3 | 3.84 (3H, s) | 55.9 q | 3.85 (3H, s) | 56.4 | – | – |
| 4′-OCH3 | 3.84 (3H, s) | 55.5 q | 3.85 (3H, s) | 56.5 | – | – |
| 3′-OCH3 | – | – | 3.85 (3H, s) | 56.2 | – | – |
Carbons assignments based on HMBC spectrum and comparison with literature.
3.1.9. Compound 9
Yellow needles (methanol, chloroform); mp 152–154 °C; {Rf 0.71, (20% ethyl acetate: n-hexane)}; UV λmax MeOH (254, 345), MeOH/NaOMe (250, 327 sh., 382), MeOH/AlCl3 (266, 280 sh., 300 sh., 377), MeOH/AlCl3/HCl (264, 284, 366, 400 sh.) MeOH/NaOAc (253, 341), MeOH/NaOAc/H3BO3 (254, 341); FTIR (KBr) 3421, 1631, and 1603 cm−1; ESI MS at m/z 359 [M+1]+ for C19H18O7; 1H, 13C NMR (500 and 125 MHz, DMSO-d6) Table 2.
3.1.10. Compound 10
Yellow needles (methanol, chloroform); mp 228 °C; Rf value; 0.8 [Butanol: acetic acid: Water, 24:10;10]; UV λmax; MeOH (266, 351), MeOH/NaOMe (275, 327 sh., 399), MeOH/AlCl3 (274, 304 sh., 352, 397), MeOH/AlCl3/HCl (273, 303 sh., 342, 393 sh.) MeOH/NaOAc (274, 315 sh., 390), MeOH/NaOAc/H3BO3 (266, 351);The ESI MS [M++H] at m/z 449 for C21H20O11, 287 [M+ - glu + H], m/z 153 [C7H5O4] and 121 [C7H5O2]; 1H, 13C NMR (500 and 125 MHz, DMSO-d6) Table 2.
4. Discussion
4.1. Identification of the isolated compounds
Compounds 1–4 were isolated from n-hexane extract and gave positive Liebermann’s and Salkowiski’s tests indicating its; steroidal and triterpenoidal nature respectively. The compounds showed exact TLC and co-TLC Rf values with that of reference compounds obtained from pharmacognosy department, College of pharmacy, KSU. Compounds 1–4 were identified as lupeol, β-sitosterol, β-sitosterol glucoside and oleanolic acid, respectively by direct comparison of its physical and spectroscopic data, namely, mp; mmp and 1H NMR, with authentic samples.
Compound 5: The UV spectrum of 5 showed an absorption band at 258 nm indicated the presence of an aromatic ring. The FTIR spectrum displayed absorption bands at 3285 cm−1 for hydroxyl group, 1698 cm−1 for ester carbonyl functionality and 1609 cm−1 for benzene ring, respectively. The EI MS spectrum of compound 5 showed a molecular ion peak [M]+ at m/z 342, which is in agreement with the molecular formula C22H30O3. The 1H NMR and 13C NMR spectral data (Table 1) were close to that type of compound reported (Miski et al., 1983) suggesting that 5 had the same sesquiterpene moiety as that for jaeschkeanadiol. The 13C NMR spectra of compound 5 demonstrated 22 signals, 15 of which were very similar to those reported for the jaeschkeanadiol moiety of ferutinin (Chen et al., 2000). The remaining signals were attributed to an acyl moiety. A diagnostic feature in the 1H NMR spectrum of 5 was the presence of one proton septet at δH 1.85 (H-11) and two methyl doublets at δH 0.77 (3H-12) and δH 0.89 (3H-13) for an isopropyl group. In addition, the spectrum showed one olefinic proton at δH 5.49 (IH, br t) was connected to carbon appeared at δC 125.2 (C-9) and showed COSY relation with both CH2-10 (Hα at δH 1.92, d, J = 8.0 Hz) and (Hβ at δH 2.02, d, J = 8.0 Hz). One deshielded methine proton resonating at δH 5.23 (δC 71.4) suggesting acylation at this position. Furthermore, three signals were clearly observed in the aromatic region of the 1H NMR spectrum at δH 7.96 (2H, dd, J = 1.0, 8.0 Hz), 7.51 (1H, td, J = 1.0, 8.0 Hz) and 7.39 (2H, t, J = 8.0 Hz) indicated the presence of monosubstitueted benzene ring in the compound. The identification and connectivity of compound 5 was deduced via HMBC (Fig. 3). The aromatic protons at δH 7.96, which integrated for two protons (H-2′/H-6′) showed three bond correlations with ester carbonyl carbon at δC 166.5 (C-16), C-4′ (δC 133.0) and C-2′/C-6′ (δC 129.6), while the aromatic proton at δH 7.51 (H-4′) exhibited three bond correlations with C-2′/6′ (δC 129.6). In addition, the aromatic protons at δH 7.39 (H-3′/H-5′) displayed three bond correlations with C-1′ (δC 130.6) and C-3′/C-5′ (δC 128.5). These finding proved the presence of monosubstituted aromatic ring. Furthermore, the oxygenated proton at δH 5.23 (H-6) displayed three bond correlation with ester carbonyl carbon at δC 166.5 (C-16) verified the acylation at C-6 (δC 71.4). Based on the analysis of the spectral data, compound 5 was identified as daucane sesquiterpene, jaeschkeanadiol benzoate (teferidin) [Saidkhodzhaev and Nikonov 1976; Miski et al., 1983; Chen et al., 2000]. This is first time to isolate teferidin from the family Solanaceae.
Figure 3.
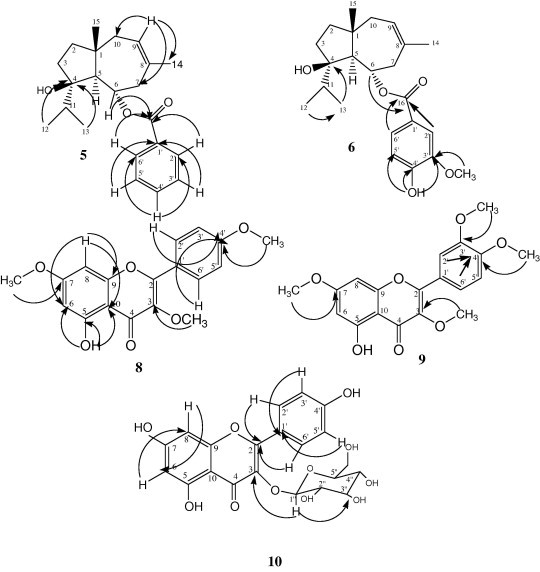
Key HMBC correlations of compounds 5, 6, 8, 9 and 10.
Compound 6 was obtained as white powder; mp 78–80 °C; [α]D +86.5° (c 1.0, chloroform). The UV spectrum (CH3OH) exhibited absorption maxima at 265 nm for the presence of the aromatic ring in the molecule. The IR spectrum of 6 has absorption bands at 3200–3600 cm−1 for hydroxyl groups, 1695 cm−1 for carbonyl ester, and bands at 1620, 1595 and 1520 cm−1 for aromatic nucleus. The EIMS spectrum of compound 6 exhibited a molecular ion peak [M]+ at m/z 388, which is in agreement with the molecular formula C23H32O5. The 1H and 13C NMR spectral data are presented in Table 1. The 13C NMR spectra displayed 23 signals, which accounted for five methyls, four methylenes, seven methines and seven quaternary carbons. These NMR data are very close to those of teferidin and suggesting that 6 had exactly the same sesquiterpene moiety as of 5 and differ only in the acyl moiety. Identification the nature of acyl moiety was easily determined as vanillic acid through analysis of COSY and HMBC spectra. In COSY spectrum, the aromatic proton at δH 7.63 (dd, J = 2.0, 8.0 Hz, H-6′) showed an ortho coupling with the proton at δH 6.98 (d, J = 8.0 Hz, H-5′) and meta coupling with the proton at δH 7.58 (d, J = 2.0 Hz, H-2′), which proved the presence of tri-substituted aromatic ring. In HMBC correlations (Fig. 3), the methyl protons of the methoxy group at δH 3.97 showed three bond correlation with the carbon at δC 146.3 (C-3′) while the hydroxyl proton exhibited three bond correlations with the carbons at δC 146.3 (C-3′), δC 114.2 (C-5′) and two bond correlation with the carbon at δC 150.2 (C-4′) confirmed the position of the methoxy group at C-3′ and the hydroxyl function at C-4′. Thus, compound 6 was identified as teferin [Kh. Khasanov et al., 1974; Ahmed, 1998]. This is first time to isolate teferin from the family Solanaceae.
Compound 7: The UV spectra of 7 showed absorption maxima at 265 and 300 nm. Its FTIR spectra exhibited absorption bands at 3200–3600 cm−1 for hydroxyl groups, 1690 cm−1 for carbonyl ester and 1530, 1610 cm−1 for aromatic nucleus. The EIMS spectrum showed a molecular ion peak [M]+ at m/z 358 corresponding to the molecular formula C22H30O4. The 1H NMR and 13C NMR spectral data of 7 (Table 1) suggested that 7 had the same sesquiterpene moiety as teferidin and teferin and differ only in the acyl moiety. The aceyl moiety was easily identified as p-hydroxybenzoic acid by observing two doublets in the 1H NMR spectrum at δH 7.95 (2H, J = 8.0 Hz) and 6.91 (2H, J = 8.0 Hz), which displayed the para orientation of the hydroxyl group. Compound 7 was identified as ferutinin (jaeschkeanadiol p-hydroxybenzoate) in agreement to its physical and spectral data and by comparison with published data (Chen et al., 2000). This is first time to isolate ferutinin from the family Solanaceae.
Compound 8: The UV spectral data with different shifting reagents indicated that this compound is a flavonoid with free hydroxyl group at C-5 (Mabry et al., 1970; Markham, 1982). The molecular formula of the compound 8 C18H16O6, was obtained from the ESI-MS [M+H]+ which showed a molecular ion peak at m/z 329. The 1H NMR data (Table 2) are in agreement with the above data through the presence of three signals of methoxy group at δH 3.79 (3-OCH3), 3.84 (7-OCH3) and 3.84 (4′-OCH3). In addition, the 1H NMR showed AA′ BB′ spin system for p-disubstituted ring B arise from two doublets at δH 7.11 (H-3′ and H-5′) and δH 8.03 (H-2′ and H-6′) with (J = 9.2 Hz) and two meta coupled aromatic proton resonances for ring A recognized as doublets (J = 1.9 Hz) at δH 6.35 and 6.72, assignable to H-6 and H-8, respectively. The position of the various substituents at the flavone skeleton were further determined via HMBC (Fig. 3), in which the methoxy group at δH 3.79 (3-OCH3) showed three bond correlation with C-3 at δC 138.5 and the methoxy group at δH 3.84 (7-OCH3) exhibited three bond correlation with C-7 at δC 166.0, while the methoxy group at δH 3.84 (4′-OCH3) displayed three bond correlations with C-4′ (δC 161.9). The aromatic proton at δH 6.72 (H-8) showed three bond correlations with C-6 (δC 98.0) and C-10 (δC 104.0) and two bond correlations with C-9 (δC 157.0) and C-7 (δC 166.0) confirming the position of one methoxy group at C-7. In addition, The aromatic protons at δH 8.03 (H-2′/H-6′) exhibited three bond correlations with C-4′ (δC 161.9) and C-2′/C-6′ (δC 129.9) and two bond correlation with C-3′/C-5′ (δC 114.0) while the aromatic protons at δH 7.11 (H-3′/H-5′) displayed three bond correlation with C-1′ (δC 122.6) and two bond correlation with C-4′ (δC 161.9) confirming the position of second methoxy group at C-4′. Analyzing the ROESY correlations (Fig. 2) further supporting these finding through the appearance of correlations between the methoxy group at δH 3.84 (7-OCH3) and H-8 (δH 6.72), H-6 (δH 6.35) and correlations between the methoxy group at δH 3.84 (4′-OCH3) and H-2′/H-3′. In addition the interaction between the methoxy group at δH 3.79 (3-OCH3) and H-5′/H-6′. Careful review of the literature on flavone chemistry confirmed that the physical and spectral data of compound 8 were in full agreement with those reported for the 5-hydroxy-3,7,4′-trimethoxyflavone (Sunder et al., 1974; Voirin, 1983).
Figure 2.
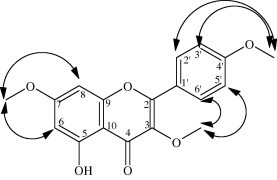
Important ROESY correlations of compound 8.
Compound 9: The UV spectral data with different shifting reagents indicated that this compound is a flavonoid with free hydroxyl group at C-5 (Mabry et al., 1970; Markham, 1982). The IR spectrum of 9 displayed bands at 3421, 1631, and 1603 cm−1 for OH, C=O and C=C functionalities, respectively. The molecular formula of 9 was assigned as C19H18O7 by molecular ion peak at m/z 359 [M+1]+ in ESI MS suggesting that compound 9 is very close to compound 8 with an additional methoxy group. This was supported by 1H NMR data of compound 9 (Table 2), which showed typical signals for H-6 and H-8 of ring A at δH 6.38 and 6.79, respectively. In addition, two singlet signals attributed to one methoxy group at δH 3.81 (3-OCH3) and three methoxy groups at δH 3.85 (9H, s) for 3′-OCH3, 4′-OCH3 and 7-OCH3. Besides that, the B-ring aromatic protons appeared as an ABX splitting system at δH 7.65 (br s, H-2′), 7.15 (d, J = 8.5 Hz, H-5′) and 7.72 (dd, J = 8.5, 1.9 Hz, H-6′). This analysis suggested that ring B was 3′,4′ disubstituted with two methoxy group. The positions of these methoxy groups were determined via HMBC experiment. The methoxy group at δH 3.81 should be located at C-3, based on its long range coupling with C-3 (δC 137.0). On the other hand, the HMBC correlations (Fig. 3) showed that the other three methoxy groups centered at δH 3.85 should be located at C-3′ (δC 147.7), C-4′ (δC 149.6) and/or C-7 (δC 163.0). The aromatic proton at δH 6.38 (H-6) showed three bond correlations with C-8 (δC 93.3), C-10 (δC 104.0) and two bond correlations with C-5 (δC 160.9) and C-7 (δC 163.0). Likewise, the aromatic proton at δH 6.79 (H-8) exhibited three bond correlations with C-10 (δC 104.0) and two bond correlations with C-7 (δC 166.0) and C-9 (δC 157.2), which confirmed the location of one methoxy resonating at δH 3.85 to C-7. In addition, the aromatic proton at δH 7.65 (H-2′) exhibited three bond correlations with C-2 (δC 156.1), C-4′ (δC 149.6) and two bond correlations with C-1′ (δC 121.9) and C-3′ (δC 147.7). While the aromatic proton at δH 7.15 (H-5′) displayed two bond correlation with C-4′ (δC 149.6) and the aromatic proton at δH 7.72 (H-6′) showed three bond correlation with C-4′ (δC 149.6) confirming the presence of other two methoxy groups at C-3′ and C-4′. Thus, compound 9 was identified as retusin (5-hydroxy-3,7,3′,4′-tetramethoxyflavone) by comparing its physical and spectral data with those reported (Silva, 2009). This is the first time to isolate 5-hydroxy-3,7,3′,4′-tetramethoxyflavone (retusin) from Solanum shimperianum.
Compound 10 The UV spectrum (MeOH) showed absorption bands at λmax 266 and 351 nm, suggesting the flavonol nature of the compound (Mabry et al., 1970; Markham, 1982). Addition of sodium methoxide shifting reagent resulted in bathochromic shift of band I, which indicated the presence of free OH at C-4′. While, the bathochromic shift of +46 nm with aluminum chloride confirmed the existence of free hydroxyl group at C-5 and/or C-3. On the other hand, the presence of bathochromic shift in band II by 14 nm with sodium acetate indicated the presence of free hydroxyl group at C-7. The ESI-MS spectrum of compound 10 disclosed a molecular ion [M++H] at m/z 449, which is in agreement with the molecular formula C21H20O11. The mass spectrum also showed a fragment ion peak at m/z 286 indicated loss of glucose moiety. The 1H and 13C NMR spectral data are presented in Table 2. The 13C NMR data showed 19 signals, which were accounted for one methylene, eleven methines and nine quaternary carbon atoms. The 1H NMR data and COSY spectrum showed two meta-coupled protons at δH 6.21 and 6.43 (J = 2.5 Hz) suggested the 5,7-disubstituted A ring of flavonoid and assigned to H-6 and H-8, respectively. In addition, there were AA′ BB′ spin system for p-disubstituted ring B arise from two doublets at δH 8.04 (H-2′ and H-6′) and δH 6.88 (H-3′ and H-5′) with (J = 8.5 Hz). Exchangeable proton appeared as sharp singlet at δH 12.61 was assigned to 5-OH. Furthermore, the 1H and 13C NMR spectra exhibited five methines and one methylene attributed to the glucose moiety. The linkage of the glucose moiety was found to be at C-3 from HMBC correlations (Fig. 3), as the anomeric proton at δH 5.45 (H-1′′) showed three bond correlations with C-3 (δC 133.2). Based on the above data and reported records, compound 10 was identified as Kaempferol-3-O-glucopyranoside (Astrgalin) (Subramanian and Nair, 1970).
4.2. Biological activity
4.2.1. Antimicrobial activity
The antimicrobial activity of Solanum schimperianum extracts (1 mg/ml) are presented in Table 3. The hexane extract has only antimicrobial activity against Bacillus subtilis and Staphylococcus aureus while ethyl acetate extract has no antimicrobial activity, as well as the isolated compounds (100 μg/ml) were also devoid of any activity.
Table 3.
Results of antimicrobial screening of successive extracts (1 mg/ml) of S. schimperianum.
| Micro-organism | Inhibition zone | |
|---|---|---|
| Hexane extract | Ethyl acetate extract | |
| Bacillus subtilis | + | − |
| Staphylococcus aureus | + | − |
| Escherichia coli | − | − |
| Pseudomonas aeruginosa | − | − |
| Mycobacterium smegmatis | − | − |
| Candida albicans | − | − |
Chloramphenicol was used as positive control at a concentration of 4 μg/ml.
DMSO was used as negative control.
+ (The size of inhibition is >10 mm in diameter).
− (The size of inhibition is <10 mm in diameter).
Acknowledgements
The authors are thankful to Prof. Ghada Shaker, Department of pharmaceutics and microbiology, College of Pharmacy, King Saud University for microbiology work. The authors extend their appreciation to Prof. Proksch Institute of pharmaceutical biology, Heinrich Heine University, Dusseldorf, Germany for NMR analysis of some flavonoids. This research project was supported by a grant from the research center of the center for female scientific and medicinal colleges in King Saud University.
Footnotes
Peer review under responsibility of King Saud University.

References
- Abdel-Sattar E., Harraz M.F., Al-Ansari M.S., El-Mekkawy S., Chikara I., Hiroaki K., Kazuhiko O., Satoshi O., Haruki Y. Antiplasmodial and antitrypanosomal activity of plants from the kingdom of Saudi Arabia. Natural Med. 2009;63:232–236. doi: 10.1007/s11418-008-0305-5. [DOI] [PubMed] [Google Scholar]
- Ahmed A.A. Sesquiterpene coumarins and sesquiterpene from Ferula sinaica. Phytochemistry. 1998;50:109–112. [Google Scholar]
- Al-Rehaily J.A., Ahmad M.S., Mustafa J., Al-Oquil M., Hassan W.H., Khan S.I., Lhan I.A. Solanopubamine, a rare steroidal alkaloid from Solanum schimperianum: synthesis of some new alkyl and acyl derivatives, their anticancer and antimicrobial evaluation. J. Saudi Chem. Soc. 2011 [Google Scholar]
- Angenot L. Spectrophotometric and chromatographic study of flavone and coumarin derivatives of Solanum schimperianum. Plantes Medicinales et Phytotherapie. 1969;3:243–254. [Google Scholar]
- Antonio J.M., Gracioso J.S., Toma W., Lopez L.C., Oliveira F., Souza Brito S.A. Antiulcerogenic activity of ethanol extract of Solanum variabile (false “ jurubeba”) J. Ethnopharmacol. 2004;93:83–88. doi: 10.1016/j.jep.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Chaudhary, S.A., 2001. Flora of the Kingdom of Saudi Arabia. Ministry of Agriculture and Water, Kingdom of Saudi Arabia, Volume 2 (Part 2).
- Chen B., Teranishi R., Kawazoe K., Takaishi Y., Honda G., Itoh M., Takeda Y., Kodzhimatov O.K. Sesquiterpenoids from Ferula kuhistanica. Phytochemistry. 2000;54:717–722. doi: 10.1016/s0031-9422(00)00197-7. [DOI] [PubMed] [Google Scholar]
- Collenette, S., 1999. Wildflowers of Saudi Arabia. National Commission for wildlife Conservation and Development, Kingdom of Saudi Arabia, pp. 703-707.
- Coune C., Denoel A. Chromatographic study of Solanum schimperianum Glucoalkaloids. Plantes Medicinales et Phytotherapie. 1975;9:14–20. [Google Scholar]
- De Silva H.A., Saparamadu P.A., Thabrew M.I., Pathmeswaran A., Fonseka M.M., De Silva H.J. Liv-52 in alcoholic liver disease: a prospective, controlled trial. J. Ethnopharmacol. 2003;84:47–50. doi: 10.1016/s0378-8741(02)00263-5. [DOI] [PubMed] [Google Scholar]
- El-Sayed Z.I., Hassan W.H. Polymethoxylated flavones from Solanum abutiloides, grown in Egypt (Solanaceae). Zagazig. J. Pharm. Sci. 2006;15:53–59. [Google Scholar]
- Emmanuel S., Ignacimuthu S., Perumalsamy R., Amalraj T. Anti-inflammatory activity of Solanum trilobatum. Fitoterapia. 2006;77:611–612. doi: 10.1016/j.fitote.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Ferro E.A., Alvarenga N.L., Ibarrola D.A., Hellion-Ibarrola M.C., Ravelo A.G. Anew steroidal saponin from Solanum sisymbriifolium roots. Fitoterapia. 2005;76:577–579. doi: 10.1016/j.fitote.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Hodek P., Trefil P., Stiborova M. Flavonoids-potent and versatile biologically active compound interacting with cytochromes P450. Chem. Biol. Interact. 2002;139:1–21. doi: 10.1016/s0009-2797(01)00285-x. [DOI] [PubMed] [Google Scholar]
- Hugo W.B., Russell A.D. 5th ed. Blackwell Scientific Publications; Oxford, London: 1992. Pharmaceutical Microbiology. pp. 258-297. [Google Scholar]
- Jairo S., Cardona W., Espinal D., Blai S., Mesa J., Bocar M., Jossang A. Five new steroids from Solanum nudum. Tetrahedron. 1998;54:10771–10778. [Google Scholar]
- Kar D.M., Maharana L., Pattnaik S., Dash G.K. Studies on hypoglycaemic activity of Solanum xanthocarpum Schrad. & Wendl, Fruit extract in rats. J. Ethnopharmacol. 2006;108:251–256. doi: 10.1016/j.jep.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Kh. Khasanov T., Saidkhodzhaev A.I., Nikonov G.K. Structure of teferin, a new ester from the roots ofFerula tenuisecta. Chem. Nat. Compd. 1974;10(4):542. [Google Scholar]
- Koduru S., Grierson D.S., Aderoga M.A., Eloff J.N., Afolayan A.J. Antioxidant activity of Solanunm aculeastrum (Solanaceae) berries. International Journal of Pharmacology. 2006;2:262–264. [Google Scholar]
- Mabry T.J., Markham K.R., Thomas M.B. Springer-Verlag; New York, Heidlberg, Berlin: 1970. The Systemic Identification of Flavonoids. pp. 266, 354. [Google Scholar]
- Mans D.R.A., Toelsie J., Mohan S., Jurgens S., Muhringen M., Illes S., Macnack R., Bipat R. Spasmogenic effects of a Solanum melongena leaf extract on guinea pig tracheal chains and its possible mechanism. J. Ethnopharmacol. 2004;95:329–333. doi: 10.1016/j.jep.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Markham K.R. Academic Press; London: 1982. Techniques of flavonoids identification. pp. 38–39. [Google Scholar]
- Mesia-Velal S., Santos M.T., Souccar C., Lima-Landman M.T.R., Lapal A.J. Solanum paniculatum L. (Jurubeba): Potent inhibitor of gastric acid secretion in mice. Phytomedicine. 2002;9:508–514. doi: 10.1078/09447110260573137. [DOI] [PubMed] [Google Scholar]
- Miski M., Ulubelen M., Mabry T.J. Six sesquiterpene alcohol esters from Ferula elaeochytris. Phytochemistry. 1983;22:2231–2233. [Google Scholar]
- Ndebia E., Kamgang R., Nkeh-Chungag B.N. Analgesic and anti-inflammatory properties of aqueous extract from leaves of Solanum torvum (solanaceae). African Journal of Traditional. Complementary and Alternative Medicines. 2007;4:240–244. doi: 10.4314/ajtcam.v4i2.31214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oboh G., Ekperigin M.M., Kazeem M.I. Nutritional and haemolytic properties of egg plants (Solanum macrocarpum) leaves. J. Food Compos. Anal. 2005;18:153–160. [Google Scholar]
- Perez A.C., Franca V., Daldegan V.M., Duarte I.D. Effect of Solanum lycocarpum St. Hill on various haematological parameters in diabetic rats. J. Ethnopharmacol. 2006;106:442–444. doi: 10.1016/j.jep.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Saidkhodzhaev A.I., Nikonov G.K. The structure of teferidin, a new ester from Ferula tenuisecta fruit. Chem. Nat. Compd. 1976;12:96–97. [Google Scholar]
- Sarmento Silva T.M., Bezerra Nascimento R.J. Distribution of flavonoids and N-trans-caffeoyl-tyramine in Solanum subg. Leptostemonum. Biochem. Syst. Ecol. 2004;32:513–516. [Google Scholar]
- Schwarz A., Soares M.R., Flόrio J.C., Bernardi M.M., Spinosa H.S. Rats exposed to Solanum lycocarpum fruit in utero and during lactation: Neurochemical, behavioral and histopathological effects. Nerotoxicology and Teratology. 2005;27:861–870. doi: 10.1016/j.ntt.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Silva B.A. Spectroscopy study on structural elucidation of flavonoids from Solanum jabrense and S. paludosum. Quim. Nova. 2009;32:1119–1128. [Google Scholar]
- Son Y.O., Kim J., Lim J.C., Chung Y., Chung G.H., lee J.C. Ripe fruits ofSolanum nigrum L. inhibit cell growth and induce apoptosis in MCF-7 cells. Food Chem. Toxicol. 2003;41:1421–1428. doi: 10.1016/s0278-6915(03)00161-3. [DOI] [PubMed] [Google Scholar]
- Subramanian S.S., Nair A.G.R. Chlorogenin and kaempferol glycosides from the flowers of Agave americana. Phytochemistry. 1970;9:2582–2589. [Google Scholar]
- Sunder R., Ayengar K.N.N., Rangaswami S. Crystalline chemical components of Cheilanthes longissima. Phytochemistry. 1974;13:1610. [Google Scholar]
- Voirin B. UV spectral differentiation of 5-hydroxy- and 5-hydroxy-3-methoxy flavones with mono-(4′)-di-(3′,4′) or tri (3′,4′,5′)- substituted B-ring. Phytochemistry. 1983;22:2107. [Google Scholar]
- Yoshimitsu H., Nishida M., Nohara T. Steroidal glycosides from the fruits of Solanum abutilosides. Phytochemistry. 2003;64:1361–1366. doi: 10.1016/j.phytochem.2003.08.008. [DOI] [PubMed] [Google Scholar]