

Abstract
There is increasing evidence showing that the stromal cells surrounding cancer epithelial cells, rather than being passive bystanders, might have a role in modifying tumor outgrowth. The molecular basis of this aspect of carcinoma etiology is controversial. Some studies have reported a high frequency of genetic aberrations in carcinoma-associated fibroblasts (CAFs), whereas other studies have reported very low or zero mutation rates. Resolution of this contentious area is of critical importance in terms of understanding both the basic biology of cancer as well as the potential clinical implications of CAF somatic alterations. We undertook genome-wide copy number and loss of heterozygosity (LOH) analysis of CAFs derived from breast and ovarian carcinomas using a 500K SNP array platform. Our data show conclusively that LOH and copy number alterations are extremely rare in CAFs and cannot be the basis of the carcinoma-promoting phenotypes of breast and ovarian CAFs.
Molecular studies of carcinomas have predominantly focused on the elucidation of the events arising in the cancerous epithelial cells, and little attention has been given to the role of the surrounding stromal cell population. Recent evidence shows that stromal cells may not be passive bystanders but might have an important role in modifying tumor growth. Many functional studies have demonstrated the importance of fibroblasts in cancer initiation1, progression2–4 and metastases5. In animal models of prostate and breast cancers, oncogene-modified epithelial cells became malignant or showed enhanced growth when co-inoculated with fibroblasts that were either oncogene induced6 or derived from fresh human carcinoma tissues2,3 (that is, CAFs).
Of note, in these studies, the tumor-enhancing behavior of CAFs was stably maintained even without continued exposure to cancer cells. These observations implied that genetic or epigenetic alterations might underlie this stable phenotype, and invoked the provocative hypothetical interdependent co-evolution of somatic changes in cancer and CAFs7. Indeed, some studies have reported a high frequency of genetic aberrations such as gene copy number alterations, loss of heterozygosity, microsatellite instability and point mutations in tumor suppressor genes and oncogenes in CAFs derived from various human cancers8–11. Notably, in some studies, the frequency of LOH was reported to be similar to that observed in the epithelial components. For example, average marker-specific LOH frequencies of 59.7% and 28.4% have been reported in CAFs derived from BRCA1/2-related and sporadic breast carcinomas, respectively12,13. Moreover, a significant association between the CAF LOH signature and tumor grade and lymph node metastasis has been reported in women diagnosed with sporadic breast tumors14. In addition, up to 63% of ovarian CAFs have been reported to show LOH at 3p21 (ref. 15). The same investigators subsequently assessed copy number changes at 110 loci and observed that approximately 10.8 genes had undergone copy number changes in each ovarian CAF15,16.
However, not all studies have identified genetic alterations in CAFs; for example, one study did not find any clonally selected somatic genetic alterations in CAFs separated from fresh breast cancer biopsies using array comparative genomic hybridization (CGH) and SNP array analysis17, although these CAFs were epigenetically distinct from those from normal breast tissue, as demonstrated by subsequent genome-wide DNA methylation studies18.
The evidence for somatic genetic alterations as important mediators of the CAF phenotype is controversial and conflicting. We hypothesized that the contradictory data may in part be a reflection of inherent technical limitations of the various methodologies used. Therefore, we took advantage of innovative SNP array–based technologies19 to investigate in detail the genomic integrity of CAFs microdissected from fresh frozen primary human ovarian and breast cancers as well as short-term cultures of primary breast CAFs.
We assessed the sensitivity of the Affymetrix 500K SNP array platform to detect copy number and LOH in the context of normal DNA contamination in a mixing experiment using tumor epithelial cell DNA from a microdissected primary ovarian cancer that was mixed with various ratios of matched normal DNA. This tumor harbors a complex copy number profile on chromosome 17, including regions of high level copy number gain and regions of LOH with and without associated copy number loss. As shown in Supplementary Figure 1a online, the single copy number gain was clearly visible at 70% tumor DNA, and the high level gain was still discernible at 25% tumor DNA. LOH was also discernible at least down to 70% tumor as indicated by the divergence of the allele-specific copy number traces, consistent with previous validations of this array platform20,21.
For all cases, only areas of stroma that were located within 5 mm distance of the cancer epithelial compartment were needle microdissected from 10 μm thick fresh frozen sections. A typical example of an ovarian and breast cancer specimen before and after microdissection is shown in Figure 1a and 1b, respectively. As stroma can comprise many different cell types, microdissected areas were assessed by two independent pathologists to estimate the percentage of fibroblast cells in each preparation. The microdissected areas of stroma from 20 of 25 of the ovarian cancers were estimated to contain ≥70% fibroblast (Supplementary Table 1 online), whereas the microdissected areas of stroma from all ten of the breast cancers were estimated to contain ≥75% fibroblast (Supplementary Table 2 online).
Figure 1.

Examples of manual microdissection from hematoxylin and eosin stained sections. (a,b) Serous ovarian cancer IC131 (a) and breast medullary carcinoma P5077 (b) shown before and after microdissection. The tumor epithelial cells (T) and CAFs (F) that were within 5 mm distance from tumor epithelia were microdissected and analyzed with 500K SNP arrays and MSMs. Scale bar, 5 mm in a; 2 mm in b.
Using SNP array analysis, we identified a high frequency of copy number changes and LOH in each of the 10 breast tumor and 25 ovarian epithelia (examples shown in Figs. 2 and 3). By contrast, none of the breast CAF preparations showed any evidence of copy number gain or loss, or LOH on any chromosome (Fig. 2b). Similarly, 24 of 25 of the ovarian CAF preparations showed a normal genomic profile (Fig. 3b). One ovarian CAF sample (from case IC307) did show LOH, coupled with copy number loss of the entire chromosome 22 (Fig. 4a). Of note, the tumor epithelium also showed loss of chromosome 22; however, there was a small region of copy number gain near the centromere. This suggests that the loss observed in the CAF component was not simply due to contamination from the epithelial component. To validate the loss of chromosome 22 in this CAF preparation, we microdissected a total of four tumor foci and six CAF foci taken from the original tumor block, as well as additional three independent tumor blocks (examples shown in Fig. 4b,c), and analyzed the DNA for allelic imbalance using microsatellite markers (MSMs). Analysis with D22S1169 showed that every tumor focus showed loss of the larger allele (Fig. 4d). We examined a total of six CAF foci and found that three showed allelic imbalance, but of the allele opposite to that of the tumor foci. The other three CAF foci retained heterozygosity with D22S1169. Notably, on the same block, one CAF focus (F2b) showed allelic imbalance, whereas the other focus (F2a) retained heterozygosity (Fig. 4d). An identical pattern of allelic imbalance was observed with D22S1174 (data not shown).
Figure 2.
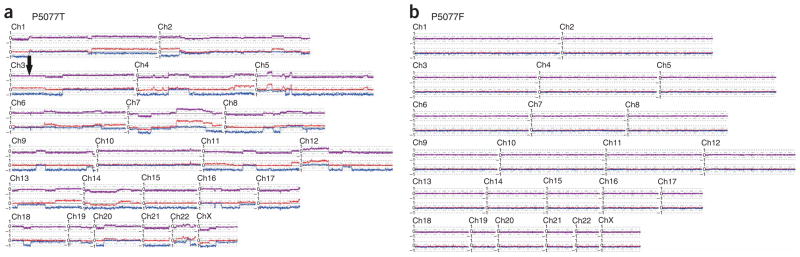
Genome-wide copy number and LOH analysis plot of different cell types within medullary breast cancer sample P5077. DNA of epithelial and stromal cell components were independently analyzed on 500K SNP arrays, and the results were normalized to corresponding normal blood DNA. Purple line is a plot of a 20-SNP moving average of the log2 copy number ratios. Blue and red lines represent allele-specific log2 copy numbers with a value of 0 being equivalent to 1 copy, and a value of –1 equivalent to complete loss of the allele. Note the region of copy number–neutral LOH on chromosome 3p in tumor P5077T (black arrow) as indicated by a near diploid copy number (purple line), whereas the allele-specific copy number shows loss of one allele and a corresponding gain of the other allele. (a) Epithelial cells. (b) Fibroblasts.
Figure 3.
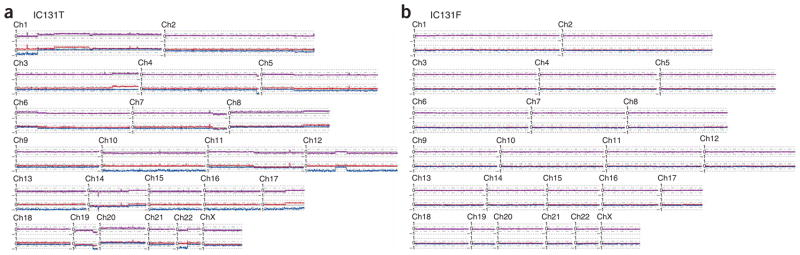
Genome-wide copy number and LOH analysis plot of different cell types within serous ovarian cancer sample IC131. Analysis and labeling were done as in Figure 2. (a) Epithelial cells. (b) Fibroblasts.
Figure 4.

Allelic imbalance analysis of chromosome 22 in fibroblast and epithelial foci of ovarian cancer sample IC307. (a) Copy number plot of chromosome 22 in tumor epithelial (T1) and fibroblast DNA (F1a) visualized using CNAG. Note that the small region of amplification in the epithelia (indicated by black arrow) is absent from the F1a component. (b) Hematoxylin and eosin–stained section of the original microdissected block 1 showing one tumor epithelia (T1) and two CAF regions (F1a and F1b) included in this study. Note that T1 and F1a were analyzed with both SNP chips and two chromosome 22 MSMs. CAF region F1b was analyzed with MSMs only. (c) Hematoxylin and eosin–stained section of an independent frozen block shows examples of additional regions of tumor epithelia and CAF samples used for microsatellite analysis. (d) Allelic imbalance analysis of DNA extracted from the microdissected tumor and CAF regions illustrated in b and c using D22S1169 MSM. Both analyzed tumor foci (T1 and T2) showed loss of the larger allele (indicated by red arrow). Three analyzed CAFs (F1a, F1b and F2b) showed loss of the smaller allele (illustrated by red asterisk). The other analyzed CAF (F2a) retained heterozygosity, compared with matching normal DNA (see ‘Normal’ panel, left chromatogram). The allelic imbalance ratio for each tumor and stroma is shown under each chromatogram. Scale bar, 2 mm in b; 5 mm in c.
We analyzed DNA from seven short-term cultured breast CAF populations for copy number alterations. Six of the fibroblast cultures showed no evidence of copy number alterations or LOH (for example, IDC-14; Fig. 5a). One fibroblast culture (IDC-1819) showed gain of chromosomes 7 and 10 (Fig. 5b) with a mean copy number of approximately 2.3. The data suggests that this CAF culture is comprised of a mixture of predominantly diploid cells plus a small subpopulation of cells containing three or possibly more copies of chromosomes 7 and 10. Matching normal DNA was not available for this case, but given the extent of the alteration and the fact that it was present only in a small proportion of the cells, it is unlikely that this could represent a germline variation. Therefore, it probably represents a somatic event that either was present in the original primary fibroblast cells or, more likely, arose during the short-term culture process.
Figure 5.
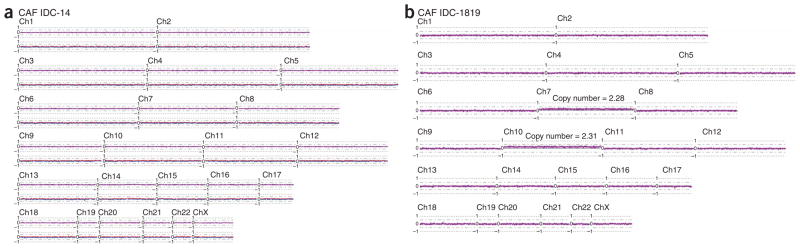
Genome-wide copy number plots and LOH plot of two primary breast CAFs. DNA of both primary cultured fibroblasts was analyzed on the 250K NspI SNP array. (a) CAFs IDC-14 shows that both the overall copy number (purple line) and the allele-specific copy number values (blue and red lines) seem to be normal. (b) CAFs IDC-1819 shows approximately 0.2 (log2 scale) of copy number gain across whole chromosome 7 and 10 with the estimated copy number (2.28 and 2.31, respectively) in linear scale across entire chromosome shown. Only the smoothed overall copy number plot is shown (purple line) for IDC-1819 because of the absence of its matching normal.
Previous studies have reported the existence of CAF mutation hot spots with specific MSMs showing LOH frequencies of up to 63% on chromosome 3 in ovarian cancer15 and up to 50% on chromosome 11 in breast cancer13,14. Even though the SNP array data indicated a 0% frequency of LOH or copy number loss at these loci, we nevertheless sought to verify this by analyzing the allelic imbalance status using five MSMs identical to those previously reported and one additional MSM located within the hot-spot region. DNA was available from 12 primary ovarian CAFs, eight primary breast CAFs and four cultured breast CAFs for this analysis. No evidence of allelic imbalance was shown in any of the ovarian CAFs, but allelic imbalance was readily detected on chromosome 3 in 9 of 12 (75%) paired ovarian tumor epithelia, an example of which is shown in Figure 6a. Furthermore, none of the 12 breast CAF populations showed any evidence of allelic imbalance, whereas three of ten (30%) available paired breast tumor epithelia showed allelic imbalance (Fig. 6b).
Figure 6.

Assessment of allelic imbalance at stroma-specific mutation hot spots in ovarian cancer sample IC293 and breast cancer sample P5077. The CNAG copy number plots of the 500K SNP array data for the tumor and fibroblast components are shown at the top of each figure, and the MSM chromatograms are shown below. Locations of the MSMs relative to the CNAG plot are indicated by arrows. The allelic imbalance ratio for each tumor and stroma sample was calculated in comparison with corresponding normal DNA and is listed under each chromatogram. Allelic imbalance is considered significant when the allelic imbalance ratio was ≤0.67 or ≥1.5 (indicated by red asterisk). (a) Allelic imbalance analysis of ovarian cancer sample IC293 using three chromosome 3 MSMs: D3S3640, D3S1613 and D3S1744. Allelic imbalance is evident in the tumor component for all 3 MSMs, which is consistent with the SNP array data. There is no evidence of allelic imbalance in the fibroblast preparations with any MSMs. (b) Allelic imbalance analysis of breast cancer 5077 using three chromosome l1 MSMs: D11S1999, D11S2365 and D11S2002. The tumor component shows allelic imbalance with MSMs D11S1999 and D11S2002, but retention of heterozygosity with D11S2365 that is consistent with the prediction of 500K SNP array data (see allele-specific copy number plot, blue and red lines). There is no evidence of allelic imbalance in the fibroblast samples with any MSMs.
In our view, previous studies reporting either the absence or frequent occurrence of genetic alterations in CAFs had significant technical limitations. In particular, studies reporting exceptionally high frequencies of alterations have relied exclusively on microdissection from formalin-fixed paraffin-embedded (FFPE) cancer tissues followed by highly multiplexed PCR-based microsatellite analysis. It is well established that the quality and quantity of DNA accessible from FFPE tissues may be suboptimal for reliable assessment of allelic imbalance22–24.
Consequently, we undertook a careful examination of CAFs from breast and ovarian cancers using tissues and techniques that minimize the potential of false-positive and false-negative data. First, we exclusively used fresh frozen cancer tissues. Second, we used micro-dissection to obtain CAF populations that were largely free of nonfibroblast cells (Supplementary Tables 1 and 2) and that were within 5 mm distance of the epithelial compartment of the carcinoma. It is reasonable to assume the presence of some adjacent non-neoplastic and nonfibroblast cells within the 5 mm distance of the epithelial compartment of the carcinoma, but this problem would be shared by all published results, including this work. Third, we used ultra high–resolution SNP arrays, which can simultaneously detect copy number alterations and LOH events with high precision and sensitivity19,20.
In contrast to previous studies in ovarian cancer15,16 and breast cancer13,14, our study did not find any evidence of frequent somatic genetic alterations in CAFs from either cancer type. The sole confirmed somatic genetic alteration was detected in an endometrioid ovarian cancer, which showed copy number loss of the entire chromosome 22. This case underscores the fact that somatic alterations can be readily detected in CAFs using the SNP array technology but also highlights the reality that such alterations are extremely rare. The chromosome 22 loss in this case does not necessarily impute a cancer-promoting phenotype for this CAF population, as it might be due to a random and benign alteration that occurred in a fibroblast stem cell.
Our study also included seven primary CAF cultures, for which the phenotypes can potentially be examined. According to our findings, only one of seven primary CAF cultures showed copy number alterations of chromosome 7 and 10 in a subpopulation of cells. Notably, we found that four of the CAF cultures without any detectable somatic changes promoted tumor growth more efficiently than normal breast stromal fibroblasts in xenograft assays (Supplementary Table 3 online). Tumor fibroblast cultures were tested for the methylation and expression of Cxorf12, a gene we had previously found to be abnormally methylated and silenced in breast tumor stroma compared to normal breast stroma18. All samples showed different methylation and expression patterns compared to their normal counterparts (when applicable), although variation among samples existed (Supplementary Fig. 2 online and Supplementary Table 3). Thus, the results of these assays confirmed the abnormal phenotype of the CAF cultures in the absence of somatic genetic changes.
In conclusion, we have conducted the highest resolution analysis of breast and ovarian CAFs reported to date. We have used tissues, technologies and methodologies that overcome the potential weaknesses of previous investigations that have produced sharply conflicting evidence for the existence of somatic genetic alterations in CAFs. Our data show that although somatic alterations can occur in CAFs, they are exceedingly rare and are unlikely to be responsible for the stable cancer-promoting attributes of CAFs. However, our findings cannot exclude the possibility that other mechanisms, such as epigenetic changes, have a role in the tumor-promoting phenotype of CAFs18,25.
METHODS
Human tissue samples
We obtained 25 fresh frozen ovarian carcinoma biopsies from women undergoing surgery for primary epithelial ovarian cancer at hospitals in the south of England26, and ten fresh frozen breast carcinoma biopsies from the Peter MacCallum Cancer Centre tissue bank or the Victorian Breast Cancer Research Consortium tissue bank. We extracted matching normal DNA for both the breast and ovarian cancers cases from peripheral blood lymphocytes or normal breast tissue. All the biopsies were collected with informed consent, and this study was approved by the Peter MacCallum Cancer Centre Human Research Ethics Committee. Clinicopathological details of the ovarian and breast cancer subjects are described in Supplementary Tables 1 and 2, respectively.
Tissue microdissection and DNA extraction
We stained 7–20 sequential 10 μm thick sections from each fresh frozen tumor biopsy with hematoxylin and eosin. All cases were reviewed by two pathologists to confirm the cancer diagnosis and to identify tumor epithelial and CAF regions suitable for microdissection. Tissue sections were then visualized under a dissecting microscope, and tumor epithelia and surrounding stroma were microdissected using an 18 gauge needle as described previously26. Only CAFs that were located within 5 mm distance from the tumor epithelial compartment were microdissected. We extracted DNA using the Blood and Tissue DNeasy kit (Qiagen) according to manufacturer’s instructions.
Primary culture of breast CAF samples
We purified six breast CAFs from human tumors essentially as previously described17. Primary cultures of fibroblasts were initiated and maintained in DMEM medium (Invitrogen) supplemented with 10% iron-fortified bovine calf serum (Hyclone) and penstrep (Invitrogen). One breast CAF culture was prepared independently as previously described27. Details on the source of the CAF cultures are provided in Supplementary Table 3.
Human mapping 500K SNP array analyses
High-throughput microarray genotyping analysis was done for each tumor epithelia, corresponding stroma, and matched normal DNA using GeneChip Human Mapping 250K Arrays NspI and/or StyI (Affymetrix) following manufacturer’s instructions and as described previously19. We analyzed 28 of ovarian and breast fresh frozen samples with both chips, and the primary breast CAFs on the NspI array only. The standard probe generation assay utilizes 250 ng of input DNA per array, but this provides an amount of labeled probe that is well in excess of what is required. Therefore, in 30 of 193 (15.5%) of analyzed cases where DNA was limiting, we used an input of 75 ng of DNA instead. In these cases, we carried out the restriction digestion and ligation in three-tenths volume of the standard protocol. In both circumstances, all subsequent steps followed the standard protocol.
Microarray analysis
Data were first analyzed in GTYPE v4.0 (Affymetrix) to generate genotype information. The mapping algorithm call threshold setting was set at 0.26. Copy number information was derived after first normalizing each tumor to its matched normal in CNAG v2.0 (ref. 28). For samples that did not have a matching normal, we generated the copy number plot by normalizing them to five unmatched normal references, which gave a <0.3 standard deviation. For samples analyzed on both the 250K StyI and 250K NspI arrays, we combined data from the two arrays. Regions of copy number gains and losses were visually identified and confirmed by interrogation of the raw copy number log2 ratios from CNAG v2.0 (ref. 28). The datasets were also analyzed using DNA-chip analyzer (dCHIP) to assess LOH and copy number changes29.
Microsatellite analysis
We used eight microsatellite markers (MSMs) spanning 3p14.3, p21.3 and 3q24 (D3S1613, D3S3640 and D3S1744), 11p15.1, 11q11 and q14.1 (D11S1999, D11S2365 and D11S2002) and 22q11.23 and q13.32 (D22S1174 and D22S1169) to analyze allelic imbalance. The primer sequences were obtained from The Human Genome Database and are listed in Supplementary Table 4 online. We amplified the target sequences in a total volume of 10 μl, containing 1× PCR reaction buffer, 0.2 mM dNTPs, 0.25 μM forward and reverse primers and 0.25 U HotStar Taq polymerase (Qiagen). Fragment analysis was done using an ABI 3130 DNA analyzer (Applied Biosystems). The genotyping results were analyzed by automated fluorescence detection using ABI GeneMapper v3.7 (Applied Biosystems). Allelic imbalance is considered significant when allelic ratio is ≤0.67 or ≥1.5, as described previously14.
Quantitative methylation-specific PCR (qMSP) and quantitative RT-PCR (qRT-PCR)
Genomic DNA was bisulfite-treated and purified, and quantitative MSP and RT-PCR amplifications were done as previously described18,30. A list of all primers used is available in Supplementary Table 4.
Supplementary Material
Acknowledgments
We thank the Peter MacCallum Cancer Centre Tissue Bank, and particularly L. Devereux, for their assistance in accessing tumor samples. We also thank the Peter MacCallum Cancer Centre Microarray and Bioinformatics core facilities for their assistance in the SNP array analysis. This work was supported by a grant (ID 400107) from the National Health and Medical Research Council of Australia (NHMRC). W.Q. is supported by NHMRC Dora Lush Postgraduate Scholarship. E.R.T. is supported by National Breast Cancer Foundation Postgraduate Scholarship. M.R. is supported by Melbourne Research Scholarship from University of Melbourne, Australia. I.H. is supported by US Department of Defense WXH1-1-06-0643 and the Komen for the Cure grants. This work was supported in part by US National Institutes of Health (CA116235) and American Cancer Society (RSG-05-154-01-MGO) grants to K.P.
Footnotes
AUTHOR CONTRIBUTIONS
I.G.C., I.H. and W.Q. designed the study and wrote the paper. W.Q. undertook the bulk of the experimental work including tissue microdissection, SNP genotyping and microsatellite analysis. K.P. provided cell lines, academic support and assisted in manuscript preparation. A.S., E.R.T., M.R. and K.L.G. assisted in SNP genotyping. M.H. and M.S. provided breast CAF DNA. M.H. undertook expression and promoter methylation analysis. K.O. and S.F. provided pathological review of tissue samples. M.T. performed immunohistochemistry analysis.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
Note: Supplementary information is available on the Nature Genetics website.
References
- 1.Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117:1495–1502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- 2.Olumi AF, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 4.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–1601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 5.Grum-Schwensen B, et al. Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1) gene. Cancer Res. 2005;65:3772–3780. doi: 10.1158/0008-5472.CAN-04-4510. [DOI] [PubMed] [Google Scholar]
- 6.Kuperwasser C, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci USA. 2004;101:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Littlepage LE, Egeblad M, Werb Z. Coevolution of cancer and stromal cellular responses. Cancer Cell. 2005;7:499–500. doi: 10.1016/j.ccr.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 8.Kurose K, et al. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32:355–357. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto N, Yoshida T, Okayasu I. High epithelial and stromal genetic instability of chromosome 17 in ulcerative colitis-associated carcinogenesis. Cancer Res. 2003;63:6158–6161. [PubMed] [Google Scholar]
- 10.Macintosh CA, Stower M, Reid N, Maitland NJ. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res. 1998;58:23–28. [PubMed] [Google Scholar]
- 11.Moinfar F, et al. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000;60:2562–2566. [PubMed] [Google Scholar]
- 12.Weber F, et al. Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am J Hum Genet. 2006;78:961–972. doi: 10.1086/504090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukino K, et al. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004;64:7231–7236. doi: 10.1158/0008-5472.CAN-04-2866. [DOI] [PubMed] [Google Scholar]
- 14.Fukino K, Shen L, Patocs A, Mutter GL, Eng C. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. J Am Med Assoc. 2007;297:2103–2111. doi: 10.1001/jama.297.19.2103. [DOI] [PubMed] [Google Scholar]
- 15.Tuhkanen H, et al. Genetic alterations in the peritumoral stromal cells of malignant and borderline epithelial ovarian tumors as indicated by allelic imbalance on chromosome 3p. Int J Cancer. 2004;109:247–252. doi: 10.1002/ijc.11733. [DOI] [PubMed] [Google Scholar]
- 16.Tuhkanen H, et al. Frequent gene dosage alterations in stromal cells of epithelial ovarian carcinomas. Int J Cancer. 2006;119:1345–1353. doi: 10.1002/ijc.21785. [DOI] [PubMed] [Google Scholar]
- 17.Allinen M, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 18.Hu M, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37:899–905. doi: 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- 19.Gorringe KL, et al. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin Cancer Res. 2007;13:4731–4739. doi: 10.1158/1078-0432.CCR-07-0502. [DOI] [PubMed] [Google Scholar]
- 20.Huang J, et al. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genomics. 2004;1:287–299. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto G, et al. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet. 2007;81:114–126. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sieben NL, ter Haar NT, Cornelisse CJ, Fleuren GJ, Cleton-Jansen AM. PCR artifacts in LOH and MSI analysis of microdissected tumor cells. Hum Pathol. 2000;31:1414–1419. [PubMed] [Google Scholar]
- 23.Kern SE, Winter JM. Elegance, silence and nonsense in the mutations literature for solid tumors. Cancer Biol Ther. 2006;5:349–359. doi: 10.4161/cbt.5.4.2551. [DOI] [PubMed] [Google Scholar]
- 24.Winter JM, Brody JR, Kern SE. Multiple-criterion evaluation of reported mutations: a proposed scoring system for the intragenic somatic mutation literature. Cancer Biol Ther. 2006;5:360–370. doi: 10.4161/cbt.5.4.2552. [DOI] [PubMed] [Google Scholar]
- 25.Fiegl H, et al. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 2006;66:29–33. doi: 10.1158/0008-5472.CAN-05-2508. [DOI] [PubMed] [Google Scholar]
- 26.Jiang X, et al. Microsatellite analysis of endometriosis reveals loss of heterozygosity at candidate ovarian tumor suppressor gene loci. Cancer Res. 1996;56:3534–3539. [PubMed] [Google Scholar]
- 27.Lebret SC, Newgreen DF, Thompson EW, Ackland ML. Induction of epithelial to mesenchymal transition in PMC42-LA human breast carcinoma cells by carcinoma-associated fibroblast secreted factors. Breast Cancer Res. 2007;9:R19. doi: 10.1186/bcr1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nannya Y, et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 29.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.