

Abstract
Both the bacterial RecA protein and the eukaryotic Rad51 protein form helical nucleoprotein filaments on DNA that catalyze strand transfer between two homologous DNA molecules. However, only the ATP-binding cores of these proteins have been conserved, and this same core is also found within helicases and the F1-ATPase. The C-terminal domain of the RecA protein forms lobes within the helical RecA filament. However, the Rad51 proteins do not have the C-terminal domain found in RecA, but have an N-terminal extension that is absent in the RecA protein. Both the RecA C-terminal domain and the Rad51 N-terminal domain bind DNA. We have used electron microscopy to show that the lobes of the yeast and human Rad51 filaments appear to be formed by N-terminal domains. These lobes are conformationally flexible in both RecA and Rad51. Within RecA filaments, the change between the “active” and “inactive” states appears to mainly involve a large movement of the C-terminal lobe. The N-terminal domain of Rad51 and the C-terminal domain of RecA may have arisen from convergent evolution to play similar roles in the filaments.
The Escherichia coli RecA protein has served as a model for understanding protein-mediated genetic recombination (1, 2). RecA plays an important role in DNA repair, and studies of RecA continue to provide insight into how repair, replication, and recombination functions are intimately linked. RecA homologs, such as RadA, UvsX, Dmc1, and Rad51, have now been identified in many organisms. Evidence in support of a key role of Rad51 in recombination, repair (3, 4) and cancer (5) in humans has emerged over the past several years. Although RecA is not an essential gene in E. coli, it has been shown that RAD51 knockouts are lethal in both chicken and mammalian cell lines (6–8). Chromosome fragmentation occurs after RAD51 inactivation in chicken DT40 cells, showing that RAD51 is required for the repair of stalled or broken replication forks in proliferating cells (8).
Alignments of the RecA and Rad51 protein sequences (9, 10) have shown that, outside of the homologous core (containing the nucleotide binding site), RecA has a C-terminal extension that is absent in Rad51 and that the Rad51 proteins have an N-terminal extension that is absent in RecA. The Saccharomyces cerevisiae Rad51 (ScRad51) N-terminal extension is even longer than that found in the human protein (hRad51). The homologous core structure has also been found in the F1-ATPase (11) and in several helicases (12–15), suggesting that all of these proteins have diverged from a common ancestor. Although there is no apparent homology between the N-terminal domain of Rad51 and the C-terminal domain of RecA, it has been reported that the C-terminal domain of RecA binds double-stranded DNA (dsDNA) (16, 17) and that the N-terminal domain of hRad51 binds both single-stranded DNA (ssDNA) and dsDNA (18).
The active state of RecA appears to be a nucleoprotein filament formed on DNA (19, 20). The T4 UvsX protein (21), the ScRad51 protein (22), and the hRad51 protein (23) induce the same unusual conformation in DNA as that induced by the RecA protein: ≈5.1 Å rise per base pair (from 3.4 Å in B-DNA) and ≈18.6 bp per turn (from 10.5 in B-DNA). This extended filament is found with RecA bound to either ssDNA or dsDNA. In contrast, the extended filaments have been seen only with hRad51 filaments formed on dsDNA, whereas filaments formed on ssDNA were relatively compressed (23). We show in this paper that, under the appropriate conditions, extended hRad51 filaments can also be seen on ssDNA. Thus, RecA, UvsX, and Rad51, although they have relatively weak overall sequence similarity, change the pitch of DNA from ≈36 Å to ≈95 Å. It has been suggested that this unusual DNA conformation has been the basis for the conservation of these nucleoprotein filaments from bacteria to humans (22).
We have used a new approach to image analysis for looking at the filaments formed by RecA and Rad51. This approach has not only provided us with more detail, but has allowed us to visualize multiple conformational states of these filaments. We have been able to interpret the differences between these conformational states in terms of the domain structure of these proteins.
Methods
Preparation of RecA-DNA and hRad51-DNA Complexes.
The RecA protein was purified as described (24). Circular φX174 dsDNA (GIBCO/BRL) was linearized (25). RecA-dsDNA filaments were formed in 25 mM triethanolamine-HCl (Fisher) buffer (pH 7.2) during a 10-min incubation at 37°C, with a RecA concentration of 6 μM, RecA to linearized φX174 dsDNA ratio of 40:1 (wt/wt), 2.5 mM ATP-γ-S (Boehringer), 2 mM magnesium acetate (Sigma). The hRad51 protein was purified as described (26). Filaments of hRad51-ssDNA-ATP-γ-S were formed by incubation of 6 μM hRad51, M13 ssDNA (Sigma), and 2.5 mM ATP-γ-S (Boehringer) in 25 mM triethanolamine-HCl (Fisher) buffer (pH 7.2) at 37°C for 15 min. The ssDNA was present at a Rad51:ssDNA ratio of 80:1 (wt/wt). Filaments of hRad51-ssDNA-ADP-AlF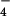 were formed by incubating 6 μM hRad51 in 25 mM triethanolamine-HCl (Fisher) buffer (pH 7.2) at 37°C for 5 min, with M13 ssDNA and 2.5 mM ATP (Sigma). The ssDNA was present at a Rad51:ssDNA ratio of 80:1 (wt/wt). Then NaF (Aldrich) and Al(NO3)2 (Aldrich) were added to a final concentration of 2.5 mM, and the reaction mixture was incubated at 37°C for an additional 15 min.
were formed by incubating 6 μM hRad51 in 25 mM triethanolamine-HCl (Fisher) buffer (pH 7.2) at 37°C for 5 min, with M13 ssDNA and 2.5 mM ATP (Sigma). The ssDNA was present at a Rad51:ssDNA ratio of 80:1 (wt/wt). Then NaF (Aldrich) and Al(NO3)2 (Aldrich) were added to a final concentration of 2.5 mM, and the reaction mixture was incubated at 37°C for an additional 15 min.
Electron Microscopy.
Samples were applied to carbon-coated grids and negatively stained with 1% uranyl acetate. Specimens were examined in a JEOL 1200 EXII electron microscope at an accelerating voltage of 80 keV and a nominal magnification of ×30,000. Negatives were densitometered with a Leaf 45 (Southborough, MA) scanner, using a raster of 4 Å/pixel.
N-Terminal Domain of Rad51.
Electron microscopy has been a powerful tool for visualizing the filaments formed by these recombination proteins. However, transmission electron microscopic images suffer from a poor signal-to-noise ratio and are very difficult to interpret because they result from projections of a three-dimensional structure onto two dimensions. Computational techniques have been essential in averaging images to generate reliable detail, as well as in reconstructing filaments in three dimensions. We have applied a new method of three-dimensional reconstruction of helical polymers (27) to images of filaments formed by the human and yeast Rad51 proteins and RecA on DNA. The method is based on an iterative real-space refinement of the helical geometry determined from large numbers of relatively short segments of these filaments. This method is capable of determining the average helical parameters to a very high degree of precision, as well as separating filament segments based on differences in pitch, twist, or subunit structure.
Fig. 1 shows images of two different states of hRad51 filaments formed on ssDNA. The average pitch of these filaments is 99 Å when they are formed with ADP-AlF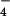 (Fig. 1a), but the average pitch is only 76 Å when these filaments are formed with ATP-γ-S (Fig. 1b). Both ADP-AlF
(Fig. 1a), but the average pitch is only 76 Å when these filaments are formed with ATP-γ-S (Fig. 1b). Both ADP-AlF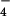 and ATP-γ-S are being used in this case as analogs for ATP, but it can clearly be seen that the filament structure depends on which analog is being used. Numerous studies have shown that aluminum fluoride can serve as a very good non-hydrolyzable analog for ATP (28), whereas it has been shown that ATP-γ-S is slowly hydrolyzed by proteins such as RecA (29–31). The smaller pitch filaments, resembling the “collapsed” state of the RecA filament formed in the absence of ATP (25), have previously been seen with hRad51 protein on ssDNA when ATP-γ-S was used as a cofactor (23). Applying the new method for reconstruction (27), the three dimensional information has been recovered, and surfaces are shown for the extended and collapsed hRad51-ssDNA filaments in Fig. 1 c and d, respectively. Both reconstructions display a highly polar structure, with one side of the deep helical groove having a relatively smooth surface, whereas the other side contains pendulous lobes of the protein. To help answer the question of what changes between these two structures as the nucleotide cofactor is varied, we have superimposed the two reconstructions in Fig. 1e. It can be seen that the main difference is a rotation of these pendulous lobes. The smooth helical backbone appears relatively unchanged. Whereas ATP-γ-S, even though is is slowly hydrolyzed, has been shown in many structural and biochemical studies with the RecA protein to be a good analog for ATP (32, 33), our observations indicate that ATP-γ-S and ATP induce two different filamentous states of the hRad51 protein on ssDNA. This finding is consistent with biochemical observations suggesting that ATP-γ-S and ATP behave differently with respect to the Rad51 and RecA proteins. For example, ATP-γ-S induces RecA protein's high affinity state for binding DNA, whereas ATP-γ-S does not induce this transition in Rad51 (34, 35). One possibility is that ATP-γ-S is not even being bound by hRad51 under the conditions that we use. We can dismiss this possibility, because we do not observe long filaments on ssDNA in the absence of nucleotide cofactor. Another possibility is that the ATP-γ-S is being rapidly hydrolyzed by hRad51 to ADP. This possibility appears unlikely, because the overall rate of ATP hydrolysis by hRad51 is significantly lower than RecA's rate. The most likely possibility is therefore that ATP-γ-S binds, but does not induce, the ATP state. In fact, the filaments formed by hRad51 on ssDNA in the presence of ADP (data not shown) are very similar to those formed in the presence of ATP-γ-S, consistent with the last possibility.
and ATP-γ-S are being used in this case as analogs for ATP, but it can clearly be seen that the filament structure depends on which analog is being used. Numerous studies have shown that aluminum fluoride can serve as a very good non-hydrolyzable analog for ATP (28), whereas it has been shown that ATP-γ-S is slowly hydrolyzed by proteins such as RecA (29–31). The smaller pitch filaments, resembling the “collapsed” state of the RecA filament formed in the absence of ATP (25), have previously been seen with hRad51 protein on ssDNA when ATP-γ-S was used as a cofactor (23). Applying the new method for reconstruction (27), the three dimensional information has been recovered, and surfaces are shown for the extended and collapsed hRad51-ssDNA filaments in Fig. 1 c and d, respectively. Both reconstructions display a highly polar structure, with one side of the deep helical groove having a relatively smooth surface, whereas the other side contains pendulous lobes of the protein. To help answer the question of what changes between these two structures as the nucleotide cofactor is varied, we have superimposed the two reconstructions in Fig. 1e. It can be seen that the main difference is a rotation of these pendulous lobes. The smooth helical backbone appears relatively unchanged. Whereas ATP-γ-S, even though is is slowly hydrolyzed, has been shown in many structural and biochemical studies with the RecA protein to be a good analog for ATP (32, 33), our observations indicate that ATP-γ-S and ATP induce two different filamentous states of the hRad51 protein on ssDNA. This finding is consistent with biochemical observations suggesting that ATP-γ-S and ATP behave differently with respect to the Rad51 and RecA proteins. For example, ATP-γ-S induces RecA protein's high affinity state for binding DNA, whereas ATP-γ-S does not induce this transition in Rad51 (34, 35). One possibility is that ATP-γ-S is not even being bound by hRad51 under the conditions that we use. We can dismiss this possibility, because we do not observe long filaments on ssDNA in the absence of nucleotide cofactor. Another possibility is that the ATP-γ-S is being rapidly hydrolyzed by hRad51 to ADP. This possibility appears unlikely, because the overall rate of ATP hydrolysis by hRad51 is significantly lower than RecA's rate. The most likely possibility is therefore that ATP-γ-S binds, but does not induce, the ATP state. In fact, the filaments formed by hRad51 on ssDNA in the presence of ADP (data not shown) are very similar to those formed in the presence of ATP-γ-S, consistent with the last possibility.
Figure 1.

Electron micrograph of hRad51 after incubation with ssDNA in the presence of ADP and AlF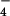 (used as a non-hydrolyzable analog of ATP; a) and after incubation with ssDNA and ATP-γ-S (b). Average from filaments of hRad51 formed on ssDNA in the presence of ADP and AlF
(used as a non-hydrolyzable analog of ATP; a) and after incubation with ssDNA and ATP-γ-S (b). Average from filaments of hRad51 formed on ssDNA in the presence of ADP and AlF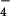 (c) is compared with average of hRad51 filaments on ssDNA in the presence of ATP-γ-S (d). The average in d was generated from 4,199 segments and has a pitch of 76 Å. The symmetry of this filament, 6.43 subunits per turn, is quite close to the symmetry of the 99-Å pitch filament in c, 6.39 subunits per turn. When the two structures are superimposed (e), it can be seen that the main difference is a rotation of the subunit lobes (red double arrow). Because of the large difference in pitch between the ADP-AlF
(c) is compared with average of hRad51 filaments on ssDNA in the presence of ATP-γ-S (d). The average in d was generated from 4,199 segments and has a pitch of 76 Å. The symmetry of this filament, 6.43 subunits per turn, is quite close to the symmetry of the 99-Å pitch filament in c, 6.39 subunits per turn. When the two structures are superimposed (e), it can be seen that the main difference is a rotation of the subunit lobes (red double arrow). Because of the large difference in pitch between the ADP-AlF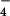 filament (shown as a glass surface) and the ATP-γ-S filament (in yellow), the comparison shown in e is meaningful only for the subunit labeled with the arrow. The scale bar in a is 1,000 Å.
filament (shown as a glass surface) and the ATP-γ-S filament (in yellow), the comparison shown in e is meaningful only for the subunit labeled with the arrow. The scale bar in a is 1,000 Å.
To compare the hRad51 filaments with the ScRad51 and RecA filaments, we have applied the same method of image analysis to these other proteins. The averaged reconstructions for hRad51 (Fig. 2a) and ScRad51 (Fig. 2b) may be compared with a reconstruction of E. coli RecA (Fig. 2c). What is immediately apparent is that the lobes of hRad51 protrude into the deep helical groove of the filament in a manner similar to that of the C-terminal lobes of the RecA filament (22, 25, 36–38). The ScRad51 filament, on the other hand, contains larger, less prominent lobes, but displays the same polar structure: a smooth backbone on one surface, with a modulation of the density because of subunits on the opposing surface. Because the C-terminal extension of RecA that is responsible for the lobes in the RecA filament is absent in hRad51 and ScRad51, we have attempted to see whether they could be explained by the N-terminal extensions present in hRad51 and ScRad51 that are absent in RecA.
Figure 2.

Surfaces of reconstructions of filaments formed by hRad51 (a), ScRad51 (b), and E. coli RecA (c) on DNA. The hRad51 filaments were formed on ssDNA in the presence of ADP and AlF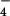 , and 7,620 filament segments were used to generate the reconstruction. The ScRad51 filaments were formed on dsDNA in the presence of ATP-γ-S, and 10,757 segments were used. The RecA filaments were formed on dsDNA with ATP-γ-S, and 8,635 segments were used. The helical parameters that were determined from the real-space refinement (27) for the hRad51 were a pitch of 99 Å and 6.39 subunits/turn. The parameters for the ScRad51 were a pitch of 94 Å with 6.28 subunits per turn, and the parameters for RecA were a pitch of 91 Å with 6.16 subunits/turn.
, and 7,620 filament segments were used to generate the reconstruction. The ScRad51 filaments were formed on dsDNA in the presence of ATP-γ-S, and 10,757 segments were used. The RecA filaments were formed on dsDNA with ATP-γ-S, and 8,635 segments were used. The helical parameters that were determined from the real-space refinement (27) for the hRad51 were a pitch of 99 Å and 6.39 subunits/turn. The parameters for the ScRad51 were a pitch of 94 Å with 6.28 subunits per turn, and the parameters for RecA were a pitch of 91 Å with 6.16 subunits/turn.
The N-terminal portion of hRad51 is known to be an independently folding domain (18), and sequence alignments suggest that this N-terminal extension should be the main difference in the comparison with the RecA core. These findings imply that the pendulous lobes in the hRad51 filament are due to the N-terminal domain. Further support for this interpretation comes from the ability to place the hRad51 N-terminal domain, solved by NMR spectroscopy (18), into the pendulous lobes of the hRad51 reconstruction (Fig. 3). It can be seen that the fit is excellent, and that the lobes in the hRad51 filament are of a size expected for this domain. Unfortunately, at the available resolution, the asymmetry in the domain is small enough that there is not a unique orientation of the domain in the electron microscopy reconstruction. Despite the ambiguity about rotation, all of the best fits place the domain into the reconstruction so that the residues (nos. 61–69) identified as involved in binding DNA (18) are oriented at the bottom of the lobe.
Figure 3.

An atomic structure for the N-terminal domain of hRad51 (18) can be easily fit into the lobe of the hRad51 reconstruction (glass surface). Although an ambiguity exists about the rotational orientation of this domain within the filament, the residues (nos. 61–69) involved in binding DNA are located at the bottom of the lobe.
We expect that the N-terminal domain in the ScRad51 should be even larger than the corresponding domain in the hRad51, because of the longer N-terminal extension present in ScRad51. It was previously suggested that this entire domain was not visualized in a helical reconstruction, because of disorder (22). Our assignment of the N-terminal domain in hRad51 leads to a slightly different interpretation. Within the ScRad51 reconstruction, large lobes can also be seen pointing into the helical groove (Fig. 2b), but these are rounder and less prominent than the lobes in the hRad51 filament (Fig. 2a). A superimposition of the ScRad51 surface upon the RecA core (data not shown) suggests that these larger lobes are due to the N-terminal overhang in ScRad51. Nevertheless, it is likely that part of this N-terminal domain in ScRad51 is disordered and not fully visualized.
C-Terminal Domain of RecA.
The new insight into the N-terminal lobes of Rad51 filaments provides motivation for a more detailed examination of the C-terminal lobes of the RecA filament. We find that the position of these lobes can be quite variable. Image analysis of RecA filaments using the single particle approach (27) has led to the surprising finding that two discrete states of the filament can be found on dsDNA in the presence of ATP-γ-S (Fig. 4). One state (Fig. 4b) is very similar in appearance to the conformation of “inactive” RecA crystal self-polymers in the absence of DNA and ATP (ref. 37; Fig. 4a), whereas the second state (Fig. 4 c and d) is in the conformation previously described as “active” (25). Because these filaments were formed with the slowly hydrolyzable ATP analog ATP-γ-S (29, 30), the simplest explanation is that the segments in the inactive conformation result from the hydrolysis of the bound nucleotide. This interpretation is consistent with the fact that, on the time scale of the incubations used (≈15 min), a significant fraction (≈15%) of RecA subunits may have hydrolyzed ATP-γ-S to ADP, given the observed kcat for this reaction of 0.01 min−1 (29, 30). This possibility is discussed more fully below.
Figure 4.

A comparison of a low-resolution rendering of the RecA crystal structure (ref. 37; a) with reconstructions of the “inactive” RecA filament (b) and two states of the “active” filament (c and d). Whereas the inactive filament, in general, has a shorter pitch than the active filament, the pitch of the two states can overlap substantially. The filament in b (averaged from 3,014 segments) has a pitch of 82 Å with 6.09 subunits/turn. The reconstruction of the active RecA filament in Fig. 1c (from 8,635 segments) had a pitch of 91 Å with 6.16 subunits/turn. However, smaller averages can be obtained of filaments in this state with very different values for the pitch. The filament in c (averaged from 1,294 segments) has a pitch of 84 Å with 5.97 subunits/turn, whereas the filament in d (averaged from 1,551 segments) has a pitch of 97 Å with 6.20 subunits/turn. When the inactive filament in b is displayed as a glass surface and superimposed on the RecA crystal structure (e), it can be seen that there is a large rotation (≈10°-15°) of the C-terminal lobe between the two (red double arrow). A comparison (f) of the inactive 82 Å pitch filament (b), shown as a glass surface, and the active 84 Å pitch filament (c), displayed in blue, shows that the main difference is due to a large shift in the C-terminal domain (arrow).
Numerous reports have described the difference in helical pitch between the inactive (≈65–85 Å) and active (≈95 Å) forms of the RecA protein (25, 39–44), but single particle analysis has revealed that there is considerable overlap in pitch between these two states. For example, a large subset of filaments in the active conformation can be found with an average pitch of 84 Å (Fig. 4c), and a correspondingly large subset of inactive filaments may be found with a pitch larger than this (data not shown). Thus, pitch may not be the main determinant of activity, as has been previously suggested (41, 45).
A comparison between the RecA crystal structure and the inactive RecA conformation found on dsDNA with ATP-γ-S (Fig. 4e) shows that the subunits in the crystal are shifted to a slightly larger radius. Whereas the backbone of the two structures (containing the conserved nucleotide-binding core) can be aligned fairly well, it is clear that there is a large rotation (≈10°) between the two of the C-terminal lobes (red double arrow, Fig. 4e). A comparison between the inactive RecA-dsDNA-ATP-γ-S filament and the active filament prepared under the same conditions shows that the main difference here (arrow, Fig. 4f) is also due to a shift in the C-terminal lobe. These comparisons show that the C-terminal domain of RecA can exist in different conformational states, and that the main transition within RecA associated with activation appears to be a large shift in the C-terminal domain.
A further comparison can be made (Fig. 5) between the RecA filaments that we have reconstructed and an in situ crystal of RecA and DNA that has been observed to form after SOS induction in E. coli (46). It was shown that the axially projected density in this in situ crystal was very similar to that in the in vitro crystal of RecA protein alone, with the major difference being that, in the in vitro crystal, there is no density along the filament axis (arrow, Fig. 5a), whereas a large peak of density was found at the corresponding location within the in situ crystal (Fig. 5b). It was suggested that this difference would be due to the presence of DNA and the DNA-binding loops of the protein, both of which are missing in the in vitro crystal structure (46). We can directly compare model crystals formed from the two different states of the RecA filament on dsDNA that we observe (Fig. 4) with the crystal formed in situ. In Fig. 5 c and e, we show axial projections of such model crystals generated from the inactive and active filament conformations, respectively. Lower resolution filtered images of these projections are shown in Fig. 5 d and f, and it can be seen that a crystal formed from the inactive conformation of the RecA filament (Fig. 6d) provides a very good match to what has actually been observed in situ (Fig. 5b), whereas the crystal made from the active conformation of the filament (Fig. 5f) provides a poor match at even very low resolution. This comparison shows that the filaments found in the in situ crystal can be matched quite well by RecA filaments formed in vitro on dsDNA that are in an “ADP-like” state, and that the RecA filaments visualized in situ have a C-terminal domain that is in a different conformation from that seen in vitro for RecA-ATP filaments.
Figure 5.

Projections down the filament axis from the crystal of RecA alone (ref. 37; a) and from an in situ crystal of RecA that has been shown to contain DNA (ref. 46; b). The arrow in a indicates the lack of any density (black) near the filament axis in the RecA crystal. The two different states of the RecA filament described in this paper have very different axial projections. Pseudocrystalline lattices of these filaments are shown in c for the ADP-state and e for the ATP-state. The lattices are not exactly crystalline, because the filaments do not have 6.0 subunits/turn, but have 6.09 subunits/turn (c) and 6.20 subunits/turn (e). Low-pass filtered images of c and e are shown in d and f, respectively. It can be seen that the lattice generated from the ADP-dsDNA-RecA filaments provides a very good match to the in situ crystal (b), but the ATP-dsDNA-filament lattice (f) would be easily distinguishable, even at very low resolution. Further, the density because of the DNA and the putative DNA-binding loops is most likely responsible for the large difference in axial density between the RecA crystal (a) and the RecA-dsDNA filaments (c and d). [a and b are reproduced with permission from ref. 46 (Copyright 2000, PNAS).]
Discussion
We now know that the nucleotide-binding core, first observed in the RecA protein (37), also exists in the F1-ATPase (11) and helicases (47). The conservation of structure from RecA to the F1-ATPase is so strong that ≈120 α-carbons of one may be superimposed upon the other with less than 2.0 Å rms deviation (11). Using sequence analysis, it has been estimated that 134 genes in S. cerevisiae (≈2% of the total number of genes) encode helicases (48), showing that a significant fraction of all proteins in the cell may have had the same origin as RecA and share a common nucleotide-binding core. When RecA is compared with its eukaryotic homologs, such as Rad51 (9) and Dmc1 (49), it is only this nucleotide-binding core that is conserved at the sequence level.
Structural studies of helicases have shown that domains other than the RecA-like nucleotide-binding core are not conserved at all. For example, the N-terminal domain of the E. coli DnaB helicase (50) has no structural similarity to the N-terminal domain of the E. coli rho transcription termination helicase (51, 52), and neither of these has homology to any of the domains of the mammalian F1-ATPase (11), Bacillus stearothermophilus PcrA (12) or E. coli Rep (13). The same is true for the domains associated with the helicase core in UvrB (53–56). Further, these associated domains may be highly mobile with respect to this nucleotide-binding core. In the Rep helicase, an associated domain has been observed to rotate by ≈130° between two forms of the subunit (13). It has also been postulated that the N-terminal domain of DnaB undergoes a large rotation (50) to explain the dimerization of subunits that can be observed within the DnaB hexamer (57, 58). We suggest that the domains associated with the nucleotide-binding core in the RecA-like filaments may have a similar diversity of origin and a similar mobility.
Remarkably, both the C-terminal domain of RecA (16, 17) and the N-terminal domain of Rad51 (18) have been shown to bind DNA. As with the helicases, these domains associated with the nucleotide-binding core have no structural homology (18, 37). This lack of homology would be the result of convergent evolution of these unrelated domains. An additional similarity with helicases is that the main conformational plasticity in these filaments appears to be due to large rotations of these associated domains. The data presented here suggest that the N-terminal lobe of Rad51 and the C-terminal lobe of RecA are mobile. We have shown in this paper that the N-terminal lobe of hRad51 undergoes a large rotation between an extended ATP state on ssDNA and a compressed state of the filament formed by hRad51 on ssDNA in the presence of ATP-γ-S. We have also been able to show that the main conformational change associated with the transition from the active to the inactive filament in RecA appears to be a large movement of this C-terminal domain.
Previously, we had shown that removal of 18 C-terminal residues from RecA leads to a significant conformational change within the filament (24), suggesting that there are large allosteric effects within RecA associated with the most C-terminal residues. Other evidence for this effect comes from biochemistry and genetics. It has been shown that deletion of ≈20 C-terminal residues from the C terminus of the Proteus mirabilis RecA protein enhanced in vitro binding to dsDNA, but left binding to ssDNA unchanged (59). Removal of ≈45 residues from the E. coli protein had a similar effect (60). However, it was shown that a RecA protein missing 25 C-terminal residues had enhanced binding to both dsDNA and ssDNA (61). It is difficult to imagine a gain of function (binding to dsDNA) induced by the loss of these C-terminal residues unless the truncated protein is in a conformation that no longer inhibits nucleation on dsDNA. This inhibition exists under normal conditions for the wild-type protein, and it was directly observed that the RecA protein missing 25 C-terminal residues nucleated much more readily on dsDNA than the wild type (61). Activation of RecA in the cell proceeds from the binding of RecA to ssDNA (62, 63), and thus the binding to dsDNA is normally suppressed. That is why the expression of a C-terminal truncated or mutated protein in the cell can lead to constitutive expression of the SOS response (64, 65).
An alternate explanation to allosteric effects is that the C-terminal region of RecA is part of an interfilament contact region that stabilizes an inactive, storage form of RecA in the cell (37). Disruption of this region through mutations or truncation might destabilize this storage form and make the polymerization more favorable. There are several observations that argue against this explanation. The length of a RecA filament will be related to the relative rates of nucleation versus polymerization. When nucleation is suppressed relative to polymerization, there will be a few long filaments, whereas there will be many short filaments when nucleation is comparable in rate to polymerization. The fact that a C-terminal truncated RecA was directly observed to nucleate more readily on dsDNA than the wild-type protein and form many short filaments (61) suggests that the rate-limiting step being affected is the nucleation event itself, and not destabilization of a storage form. For the interfilament contact region argument to apply, one would need to show that the inactive RecA in vitro is contained in such a crystalline array, and this situation has not been found under normal conditions. In fact, a crystalline arrangement of RecA filaments has been observed in cells, but this observation is after SOS induction, and not before it occurs (46). Remarkably, the packing of filaments in these large crystals appears to be extremely similar to the packing of pure RecA protein in crystals, with the difference being that DNA is present within the crystals and appears to be located near the filament axis. We would expect that this is dsDNA, because the amount of ssDNA present in the cell is very small. This finding leads to a seeming paradox, because wild-type RecA can bind to ssDNA in either the inactive or active state (with respect to ATP hydrolysis), but can bind to dsDNA only in the active state (25, 66–69). It is therefore difficult to imagine how and why the cell would massively bundle dsDNA in a crystal that is actively hydrolyzing ATP. Resolution of this paradox comes both from previous in vitro kinetic data as well as our own electron microscopy observations presented here.
It has been noted that ATP hydrolysis within a RecA filament may not be directly coupled to dissociation of subunits from the filament, and that neighboring subunits may stabilize subunits within the filament in an ADP-state (2, 70–72). Some of the earliest kinetic data suggested that such changes of state within a filament must be cooperative (73). We have previously observed that structural changes associated with such hydrolysis occur with a high degree of cooperativity within RecA filaments (29). These observations would explain why we can now find segments of filaments, in the presence of the slowly hydrolyzable ATP analog ATP-γ-S, that can be found in one state or the other. In the absence of cooperativity, isolated subunits within a filament would switch states, with a random distribution of such subunits in any filament. The segments of RecA filaments that we observe in different states are consistent with the “active cluster hypothesis” that was originally advanced to account for the cooperativity of the RecA ATPase (74). We can now explain why RecA will not form filaments on dsDNA in the presence of ADP, but why segments of filaments can be trapped in an ADP-like state when such filaments are initially formed on dsDNA with ATP-γ-S. Thus, the in vivo crystals of RecA and DNA (46), which have the same packing as the in vitro crystal of RecA alone (37), likely represent the same inactive conformation of RecA that we can see in filament segments after the hydrolysis of ATP-γ-S. The role of such structures may be to protect the DNA (46).
In summary, electron microscopy and image analysis suggest that the N-terminal domain of the Rad51 proteins forms a lobe protruding into the helical groove of these filaments that is very similar to the lobe formed by the C-terminal domain of RecA. Previous observations show that both of these domains bind DNA, indicating that their roles in the RecA and Rad51 filaments may be due to convergent evolution because they have no homology in either sequence or structure. We can directly visualize that these lobes exist in different conformations in both Rad51 and RecA, and the conformation of these lobes depends on the nucleotide cofactor. Most importantly, the change in activity of both RecA and Rad51 appears to be correlated with the conformation of these lobes, and these domains may play a large regulatory role.
Acknowledgments
This research was supported by National Institutes of Health Grant GM35269 (to E.H.E.), Human Frontier Sciences Program RG335 (to E.H.E.), and the Imperial Cancer Research Fund (to S.C.W.).
Abbreviations
- dsDNA
double-stranded DNA
- ssDNA
single-stranded DNA
Footnotes
This paper results from the National Academy of Sciences colloquium, “Links Between Recombination and Replication: Vital Roles of Recombination,” held November 10–12, 2000, in Irvine, CA.
References
- 1.Kowalczykowski S C, Dixon D A, Eggleston A K, Lauder S D, Rehrauer W M. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roca A I, Cox M M. Prog Nucleic Acid Res Mol Biol. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- 3.Tashiro S, Walter J, Shinohara A, Kamada N, Cremer T. J Cell Biol. 2000;150:283–291. doi: 10.1083/jcb.150.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tashiro S, Kotomura N, Shinohara A, Tanaka K, Ueda K, Kamada N. Oncogene. 1996;12:2165–2170. [PubMed] [Google Scholar]
- 5.Kato M, Yano K, Matsuo F, Saito H, Katagiri T, Kurumizaka H, Yoshimoto M, Kasumi F, Akiyama F, Sakamoto G, et al. J Hum Genet. 2000;45:133–137. doi: 10.1007/s100380050199. [DOI] [PubMed] [Google Scholar]
- 6.Lim D S, Hasty P. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Proc Natl Acad Sci USA. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonoda E, Sasaki M S, Buerstedde J M, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinohara A, Ogawa H, Ogawa T. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 10.Shinohara A, Ogawa H, Matsuda Y, Ikeo K, Ushio N, Ogawa T. Nat Genet. 1993;4:239–243. doi: 10.1038/ng0793-239. [DOI] [PubMed] [Google Scholar]
- 11.Abrahams J P, Leslie A G, Lutter R, Walker J E. Nature (London) 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 12.Subramanya H S, Bird L E, Brannigan J A, Wigley D B. Nature (London) 1996;384:379–383. doi: 10.1038/384379a0. [DOI] [PubMed] [Google Scholar]
- 13.Korolev S, Hsieh J, Gauss G H, Lohman T M, Waksman G. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 14.Sawaya M R, Guo S, Tabor S, Richardson C C, Ellenberger T. Cell. 1999;99:167–177. doi: 10.1016/s0092-8674(00)81648-7. [DOI] [PubMed] [Google Scholar]
- 15.Singleton M R, Sawaya M R, Ellenberger T, Wigley D B. Cell. 2000;101:589–600. doi: 10.1016/s0092-8674(00)80871-5. [DOI] [PubMed] [Google Scholar]
- 16.Aihara H, Ito Y, Kurumizaka H, Terada T, Yokoyama S, Shibata T. J Mol Biol. 1997;274:213–221. doi: 10.1006/jmbi.1997.1403. [DOI] [PubMed] [Google Scholar]
- 17.Kurumizaka H, Aihara H, Ikawa S, Kashima T, Bazemore L R, Kawasaki K, Sarai A, Radding C M, Shibata T. J Biol Chem. 1996;271:33515–33524. doi: 10.1074/jbc.271.52.33515. [DOI] [PubMed] [Google Scholar]
- 18.Aihara H, Ito Y, Kurumizaka H, Yokoyama S, Shibata T. J Mol Biol. 1999;290:495–504. doi: 10.1006/jmbi.1999.2904. [DOI] [PubMed] [Google Scholar]
- 19.Stasiak A, DiCapua E. Nature (London) 1982;229:185–186. [Google Scholar]
- 20.Stasiak A, DiCapua E, Koller T. J Mol Biol. 1981;151:557–564. doi: 10.1016/0022-2836(81)90010-3. [DOI] [PubMed] [Google Scholar]
- 21.Yu X, Egelman E H. J Mol Biol. 1993;232:1–4. doi: 10.1006/jmbi.1993.1363. [DOI] [PubMed] [Google Scholar]
- 22.Ogawa T, Yu X, Shinohara A, Egelman E H. Science. 1993;259:1896–1899. doi: 10.1126/science.8456314. [DOI] [PubMed] [Google Scholar]
- 23.Benson F E, Stasiak A, West S C. EMBO J. 1994;13:5764–5771. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X, Egelman E H. J Struct Biol. 1991;106:243–254. doi: 10.1016/1047-8477(91)90074-7. [DOI] [PubMed] [Google Scholar]
- 25.Yu X, Egelman E H. J Mol Biol. 1992;227:334–346. doi: 10.1016/0022-2836(92)90702-l. [DOI] [PubMed] [Google Scholar]
- 26.Baumann P, Benson F E, Hajibagheri N, West S C. Mutat Res. 1997;384:65–72. doi: 10.1016/s0921-8777(97)00028-1. [DOI] [PubMed] [Google Scholar]
- 27.Egelman E H. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 28.Yonesaki T, Minagawa T. EMBO J. 1985;4:3321–3327. doi: 10.1002/j.1460-2075.1985.tb04083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu X, Egelman E H. J Mol Biol. 1992;225:193–216. doi: 10.1016/0022-2836(92)91036-o. [DOI] [PubMed] [Google Scholar]
- 30.Paulus B F, Bryant F R. Biochemistry. 1997;36:7832–7838. doi: 10.1021/bi970576+. [DOI] [PubMed] [Google Scholar]
- 31.Fujisawa H, Yonesaki T, Minagawa T. Nucleic Acids Res. 1985;13:7473–7481. doi: 10.1093/nar/13.20.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stasiak A, Egelman E H. Experientia. 1994;50:192–203. doi: 10.1007/BF01924002. [DOI] [PubMed] [Google Scholar]
- 33.Menetski J P, Bear D G, Kowalczykowski S C. Proc Natl Acad Sci USA. 1990;87:21–25. doi: 10.1073/pnas.87.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Namsaraev E A, Berg P. (1998) Biochemistry. 1939;37:11932–11939. doi: 10.1021/bi9810297. [DOI] [PubMed] [Google Scholar]
- 35.Namsaraev E A, Berg P. J Biol Chem. 1998;273:6177–6182. doi: 10.1074/jbc.273.11.6177. [DOI] [PubMed] [Google Scholar]
- 36.Yu X, Egelman E H. J Mol Biol. 1993;231:29–40. doi: 10.1006/jmbi.1993.1254. [DOI] [PubMed] [Google Scholar]
- 37.Story R M, Weber I T, Steitz T A. Nature (London) 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 38.Yu X, Shibata T, Egelman E H. J Mol Biol. 1998;283:985–992. doi: 10.1006/jmbi.1998.2141. [DOI] [PubMed] [Google Scholar]
- 39.Ruigrok R W, Bohrmann B, Hewat E, Engel A, Kellenberger E, DiCapua E. EMBO J. 1993;12:9–16. doi: 10.1002/j.1460-2075.1993.tb05626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruigrok R W, DiCapua E. Biochimie. 1991;73:191–198. doi: 10.1016/0300-9084(91)90202-c. [DOI] [PubMed] [Google Scholar]
- 41.DiCapua E, Cuillel M, Hewat E, Schnarr M, Timmins P A, Ruigrok R W H. J Mol Biol. 1992;226:707–719. doi: 10.1016/0022-2836(92)90627-v. [DOI] [PubMed] [Google Scholar]
- 42.Hewat E A, Ruigrok R W, DiCapua E. EMBO J. 1991;10:2695–2698. doi: 10.1002/j.1460-2075.1991.tb07813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egelman E H, Stasiak A. Micron. 1993;24:309–324. [Google Scholar]
- 44.Chang C F, Rankert D A, Jeng T W, Morgan D G, Schmid M F, Chiu W. J Ultrastruct Mol Struct Res. 1988;100:166–172. doi: 10.1016/0889-1605(88)90023-7. [DOI] [PubMed] [Google Scholar]
- 45.Hewat E A, Ruigrok R W H, DiCapua E. EMBO J. 1991;10:2695–2698. doi: 10.1002/j.1460-2075.1991.tb07813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levin-Zaidman S, Frenkiel-Krispin D, Shimoni E, Sabanay I, Wolf S G, Minsky A. Proc Natl Acad Sci USA. 2000;97:6791–6796. doi: 10.1073/pnas.090532397. . (First Published May 30, 2000; 10.1073/pnas.090532397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bird L E, Subramanya H S, Wigley D B. Curr Opin Struct Biol. 1998;8:14–18. doi: 10.1016/s0959-440x(98)80004-3. [DOI] [PubMed] [Google Scholar]
- 48.Shiratori A, Shibata T, Arisawa M, Hanaoka F, Murakami Y, Eki T. Yeast. 1999;15:219–253. doi: 10.1002/(SICI)1097-0061(199902)15:3<219::AID-YEA349>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 49.Bishop D K, Park D, Xu L, Kleckner N. Cell. 1992;69:439–456. doi: 10.1016/0092-8674(92)90446-j. [DOI] [PubMed] [Google Scholar]
- 50.Fass D, Bogden C E, Berger J M. Structure Fold Des. 1999;7:691–698. doi: 10.1016/s0969-2126(99)80090-2. [DOI] [PubMed] [Google Scholar]
- 51.Allison T J, Wood T C, Briercheck D M, Rastinejad F, Richardson J P, Rule G S. Nat Struct Biol. 1998;5:352–356. doi: 10.1038/nsb0598-352. [DOI] [PubMed] [Google Scholar]
- 52.Briercheck D M, Wood T C, Allison T J, Richardson J P, Rule G S. Nat Struct Biol. 1998;5:393–399. doi: 10.1038/nsb0598-393. [DOI] [PubMed] [Google Scholar]
- 53.Machius M, Henry L, Palnitkar M, Deisenhofer J. Proc Natl Acad Sci USA. 1999;96:11717–11722. doi: 10.1073/pnas.96.21.11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakagawa N, Sugahara M, Masui R, Kato R, Fukuyama K, Kuramitsu S. J Biochem (Tokyo) 1999;126:986–990. doi: 10.1093/oxfordjournals.jbchem.a022566. [DOI] [PubMed] [Google Scholar]
- 55.Sohi M, Alexandrovich A, Moolenaar G, Visse R, Goosen N, Vernede X, Fontecilla-Camps J C, Champness J, Sanderson M R. FEBS Lett. 2000;465:161–164. doi: 10.1016/s0014-5793(99)01690-7. [DOI] [PubMed] [Google Scholar]
- 56.Theis K, Chen P J, Skorvaga M, Van Houten B, Kisker C. EMBO J. 1999;18:6899–6907. doi: 10.1093/emboj/18.24.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.San Martin M C, Stamford N P J, Dammerova N, Dixon N E, Carazo J M. J Struct Biol. 1995;114:167–176. doi: 10.1006/jsbi.1995.1016. [DOI] [PubMed] [Google Scholar]
- 58.Yu X, Jezewska M J, Bujalowski W, Egelman E H. J Mol Biol. 1996;259:7–14. doi: 10.1006/jmbi.1996.0297. [DOI] [PubMed] [Google Scholar]
- 59.Rusche J R, Konigsberg W, Howard-Flanders P. J Biol Chem. 1985;260:949–955. [PubMed] [Google Scholar]
- 60.Benedict R C, Kowalczykowski S C. J Biol Chem. 1988;263:15513–15520. [PubMed] [Google Scholar]
- 61.Tateishi S, Horii T, Ogawa T, Ogawa H. J Mol Biol. 1992;223:115–129. doi: 10.1016/0022-2836(92)90720-5. [DOI] [PubMed] [Google Scholar]
- 62.Higashitani N, Higashitani A, Roth A, Horiuchi K. J Bacteriol. 1992;174:1612–1618. doi: 10.1128/jb.174.5.1612-1618.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sassanfar M, Roberts J W. J Mol Biol. 1990;212:79–96. doi: 10.1016/0022-2836(90)90306-7. [DOI] [PubMed] [Google Scholar]
- 64.Liu S K, Eisen J A, Hanawalt P C, Tessman I. J Bacteriol. 1993;175:6518–6529. doi: 10.1128/jb.175.20.6518-6529.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horii T, Ozawa N, Ogawa T, Ogawa H. J Mol Biol. 1992;223:105–114. doi: 10.1016/0022-2836(92)90719-z. [DOI] [PubMed] [Google Scholar]
- 66.Menetski J P, Varghese A, Kowalczykowski S C. Biochemistry. 1988;27:1205–1212. doi: 10.1021/bi00404a021. [DOI] [PubMed] [Google Scholar]
- 67.Menetski J P, Kowalczykowski S C. J Mol Biol. 1985;181:281–295. doi: 10.1016/0022-2836(85)90092-0. [DOI] [PubMed] [Google Scholar]
- 68.Lee J W, Cox M M. Biochemistry. 1990;29:7677–7683. doi: 10.1021/bi00485a017. [DOI] [PubMed] [Google Scholar]
- 69.Lee J W, Cox M M. Biochemistry. 1990;29:7666–7676. doi: 10.1021/bi00485a016. [DOI] [PubMed] [Google Scholar]
- 70.Menetski J P, Kowalczykowski S C. J Biol Chem. 1987;262:2085–2092. [PubMed] [Google Scholar]
- 71.Menetski J P, Kowalczykowski S C. J Biol Chem. 1987;262:2093–2100. [PubMed] [Google Scholar]
- 72.Lindsley J E, Cox M M. J Mol Biol. 1989;205:695–711. doi: 10.1016/0022-2836(89)90315-x. [DOI] [PubMed] [Google Scholar]
- 73.Weinstock G M, McEntee K, Lehman I R. J Biol Chem. 1981;256:8850–8855. [PubMed] [Google Scholar]
- 74.Kowalczykowski S C. Biochemistry. 1986;25:5872–5881. doi: 10.1021/bi00368a006. [DOI] [PubMed] [Google Scholar]