

Abstract
On account of the reported anticancer of pyrimidine and condensed pyrimidine, a new pyrimido [3,2-b]-1,2,4,5-tetrazine 3a,b, 5c,d, 6, 9, pyrimido [3,2-b]-1,2,4-triazine 10, 11, pyrimido [3,2-b]-1,2,4-triazole 12 and pyrimidine derivatives 1,2a,b, 4c,d, 8, 13, 14, 15 and 16 were synthesized through different chemical reactions. Structures of all synthesized compounds were supported by spectral and elemental analyses. The obtained compounds were evaluated for their in vitro antitumor activity against human liver cancer cell line (HEPG2).
Keywords: Pyrimido[3,2-b]-1,2,4,5-tetrazine; Pyrimido[3,2-b]1,2,4-triazine; Pyrimido[3,2-b]-1,2,4-triazole; Pyrimidine derivatives; Anticancer
1. Introduction
Pyrimidine and condensed pyrimidine are important classes of heterocyclic compounds that exhibit a broad spectrum of biological activities such as anticancer (Araguchi et al., 2004, Breault et al., 2000, Kim et al., 2003, Deng et al., 2008, Abou El Ella et al., 2008, Nguyen, 2008), antiviral (Depecker et al., 2004, Renau et al., 1996), antibacterial (Kuyper et al., 1996, Gregory et al., 2007), antioxidant (Andrus et al., 1997, Bundy et al., 1995), anxiolytic (Meada et al., 1998) and antidepressant activities (Hogenkamp et al., 2001).
Among its fused bicyclics, the pyrimido [1,2-b]-1,2,4,5-tetrazin-6-ones have inhibitory activity against the human cytomegalovirus (HCMV) protease (Martin et al., 2003).
Pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione derivatives confer significant cytoprotective effects from rotenone toxicity in a cellular rotenone stress assay and Parkinson’s disease (Zhou et al., 2009). Also, pyrimido[5,4-3]-1,2,4-triazine-5,7-diamine based—hypoglycemic agents with protein tyrosine phosphatases inhibitory activity (Kevin et al., 2003).
Fused azoloazine systems have attracted attention due primarily to the fact that they are widespread among natural biologically active compounds (Sidorenko et al., 2010).
Among the nitrogen containing heterocycles triazolopyrimidines represent a pharmaceutically important class of compounds because of their diverse range of biological activities such as antitumor (Navarro et al., 1998), cytotoxicity (Magan et al., 2004), therapeutic potentiality (Magan et al., 2005). Potent and selective ATP site directed inhibition of the EGF receptor protein tyrosine kinase (Traxler et al., 1996) and cardiovascular (Rusinov et al., 1986) activities. In addition, they have been found in DNA-interactive drugs (Lauria et al., 2002). Pyrimidine derivatives are attractive due to their pharmacological activity (Foroughifar et al., 2002, Tsuji and Ishikawa, 1994) and possess antiviral (Varma, 1999) antitumor (Kappe et al., 1997), antibacterial (Xie et al., 1999, Kappe, 1993), antihypertensive (Atwal et al., 1991), and antituberculous properties (Patel et al., 2006) and effective against wild type and various mutant strains of HIV-1 (Ludovici and Janseen, 2004). In light of the aforementioned facts.y I envisioned our approach toward the synthesis of novel pyrimido[2,3-b]-1,2,4,5-tetrazine, pyrimido[2,3-b]-1,2,4-triazine, pyrimido[2,3-b]-1,2,4-triazole and pyrimidine derivatives to evaluate their potential anticancer activities.
2. Results and discussion
The starting material 1-amino-4-methyl-6-phenyl pyrimidin-2-thione was prepared in good yields by the reaction of benzoylacetone and thiosemicarbazide in refluxing ethanol containing catalytic amounts of piperidine afforded a pale yellow product 1 (Scheme 1). The structure of compound 1 was established on the basis of its elemental analyses and spectral data. Thus structure of 1 is supported by its mass spectrum which showed a molecular ion corresponding to the formula C11H11N3S (M+ 217). Its IR spectrum revealed absorption bands due to NH2, C N and C S groups near 3361, 3308, 1644 and 1216 cm−1, respectively. The 1H NMR spectrum revealed (CH3) protons as a singlet signal around δ 2.35 ppm, singlet at δ 5.70 ppm assigned to the NH2 protons, singlet at δ 8.65 ppm assigned to the H-5 pyrimidine ring and multiplet at δ 7.30–8.20 ppm assigned to the aromatic protons. Thenoyl isothiocyanate (Coppo and Fawzi, 1997) or phenyl isothiocyanate reacted with compound 1 to give N-thenoyl or (N-phenyl)-N′-1-[(4-methyl-6-phenyl-2-thioxo pyrimidinyl] thiourea 2a,b. The IR spectrum of compound 2a showed the absence of an absorption band corresponding to the amino group and the appearance of new bands corresponding to (2NH) at 3242, 3135 cm−1 and 1H NMR showed signals at 12.79 and 10.00 ppm for (2NH).
Scheme 1.
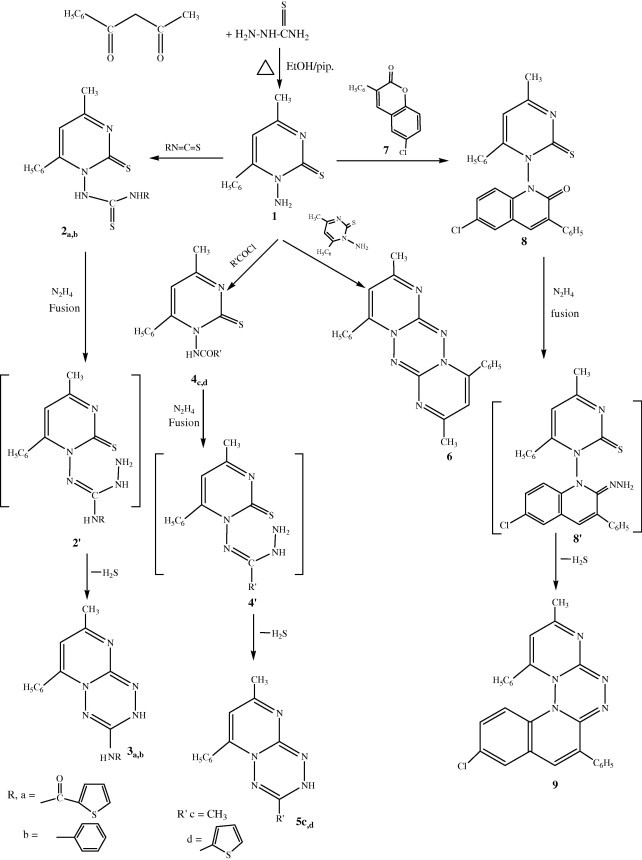
Treatment of 2a or b with hydrazine hydrate afforded 9-methyl-7-phenyl-4-(2-thenoyl amino (or) phenyl amino)-(3H) pyrimido[3,2-b]-1,2,4,5-tetrazine 3a,b. This reaction proceeds through the initial formation of the intermediate 2′, followed by interamolecular cyclization and loss of hydrogen sulfide (lead acetate paper). The structure of compounds 3a,b was confirmed by elemental analysis and their spectral data. The IR spectra of compound 3a showed strong absorption bands in the region 3305, 3259 cm−1 to NH tetrazine ring and NH of secondary amine.
The absorption bands of the aroyl carbonyl for compound 3a were found at, 1665 cm−1 region. In the 1H NMR spectrum, the signal of NH proton of tetrazine ring recorded at 9.85 ppm (Hany and Hazem, 2008). The 13C NMR spectrum showed the expected resonance signal of C-5 of tetrazine ring around 111.00 ppm. This assignment is in good agreement with literature data for carbon flanked by two nitrogens and azomethine carbon in six-membered heterocycles (El-Abadelah et al., 1988). The mass spectrum of 3a showed a molecular ion peak m/z at 350 (M+, 2.72%).
Treatment of compound 1 with acetyl chloride or thenoyl chloride in pyridine resulted in the formation of the 1-(carboxamide (or) 2-thenoylamino)-4-methyl-6-phenyl-pyrimidine-2-thion 4c–d. The IR spectrum of compound 4c showed a strong band in the region of 1695 cm−1 characteristic of C O of secondary amide. The two bands in the region of 3190, 3320 cm−1 are the stretching modes of HNCO and N C–OH groups. The 1H NMR of 4c showed a singlet at δ 9.98, 10.76 ppm (1H, HNC O or N C–OH) and a singlet at δ 11.33 ppm (1H, HNCOCH3(. Compounds 5c or d were prepared, under fusion condition of compounds 4c or d with hydrazine hydrate to furnish the 4,9-dimethyl-7-phenyl-(3H) pyrimido[3,2-b]-1,2,4,5-tetrazine 5c and 9-methyl-7-phenyl-4-(2-thienyl)(3H) pyrimido[3,2-b]-1,2,4,5-tetrazine 5d. This reaction proceeded through the initial formation of the intermediate 4′ via loss of water, followed by intramolecular cyclization and loss of hydrogen sulfide (lead acetate paper). Structure of compound 5c was supported on the basis of the elemental analysis (sulfur free, IR, 1H NMR and mass spectral data). The IR spectrum exhibited the absence of (C O) band and the presence of NH band at 3329 cm−1. 1H NMR showed signal at δ 12.38 ppm for NH tetrazine ring. Mass spectrum of 5c revealed a molecular ion peak m/z at 239 (M+, 8.03%) with a base peak m/z at 77 (100%).
6,12-Dimethyl-4,10-diphenyl pyrimido[3,2-b]-1,2,4-5-tetrazino[3,2-b] pyrimidine 6 was prepared, via refluxing of compound 1 in pyridine for a long time. The structure of compound 6 was confirmed by elemental analysis (sulfur free), IR spectrum which showed the absence of (NH2) and (C S) band, also, 1H NMR revealed signals at δ 7.07–8.01 ppm for aromatic protons. The mass spectrum showed a molecular ion peak m/z at 366 (M+, 3.11%).
On repeating the same reaction in xylene the mono thion derivative 1 was obtained based on the IR data which showed the presence of (NH2) and (C S) bands. 1H NMR revealed signal at δ 5.20 ppm for NH2, 13C NMR signal at values of 205 ppm for (C S) functionality.
In addition, phenyl acetonitrile reacted with 5-chlorosalicyladehyde in ethanol piperidine (Abd El-Hamid, 1994) to give the corresponding coumarin derivative 7. The structure of compound 7 was established by IR, 1H NMR, and mass spectrometry. The interaction of compound 7 with compound 1 in boiling ethanol afforded the corresponding N-quinolinone derivative 8 (Scheme 1). Cyclization of 8 under fusion condition with hydrazine hydrate afforded the 4,12-diphenyl-14-methyl-pyrimido[3,2-b]-1,2,4,5-tetrazino[4,3-a] quinoline 9.
The structures of 8 and 9 were established on the basis of their elemental analysis and spectral data. Their IR and 1H NMR spectra proved disappearance of amino group (see experimental section) in addition to the mass spectrum of compound 9 which revealed molecular ion peaks m/z at 435.5 (M+, 1.12), 437.5 (M+, 3.22) (See scheme 2).
Scheme 2.
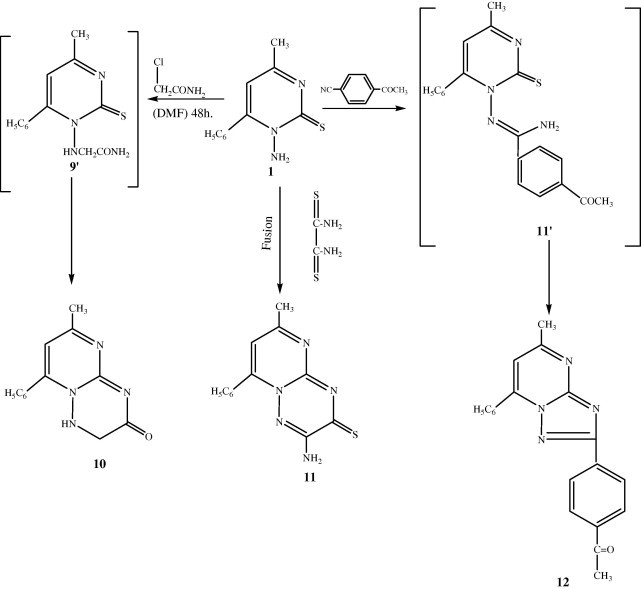
Interaction of 1 with chloroacetamide in dry DMF led to cyclo-condensation with elimination of HCl and H2S afforded pyrimido[3,2-b]-1,2,4-triazin-3-one derivative 10. The IR spectrum showed absorption bands at 3375 cm−1 and 1717 cm−1 corresponding to NH and C O groups (Aly et al., 1995) respectively, its 1H NMR spectrum showed signals at δ 4.00 and 9.20 ppm assigned to (CH2CO) and (NH) groups. Both elemental analysis (sulfur free) and spectral data of 10 were consistent with the assigned structure also, interaction of 1 with dithiooxamide, under fusion conditions (Abd El-hamid, 1994). Structure of compound 11 was supported on the basis of elemental analysis, IR, 1H NMR and mass spectral data. Its IR spectrum showed absorption bands at 3383–3319 cm−1 for the NH2 group. The 1H NMR showed amino protons at δ 4.78 ppm, in addition to the mass spectrum which revealed a molecular ion peak m/z at 269 (M+, 6.84%), 77 (M+, 100%).
When treating compound 1 with excess of potassium t-butoxide and one equivalent of p-cyano acetophenone in t-butanol under reflux for 48 h. (Molina et al., 1985), the 1-amino heterocycle 1 is directly converted into the corresponding 3-(4-acetophenone)-8-methyl-6-phenyl-pyrimido[3,2-b]-1,2,4-triazole 12 in moderate yield (50%). The reaction appears to be quite general for aromatic nitriles; however attempts to apply the method to aliphatic nitriles were unsuccessful. Structural elucidation of 12 is accomplished on the basis of spectral and elemental analysis (sulfur free) data. The 1H NMR spectrum showed two singlet signals at δ 2.42 and 3.50 ppm assigned to CH3 and CH3CO protons, also, its mass spectrum showed an intense peak at m/z = 328 (M+, 3.11%) corresponding to the molecular ion. Complete information about the 1H NMR, IR, 13 C NMR and mass spectra is presented in the experimental section.
The synthetic approach demonstrated here was extended to enable the synthesis of other functionally substituted pyrimidine for their biological evaluation. Thus, when compound 1 was reacted with ethylamine in refluxing ethanol, the 1-amino-2-ethylamino-4-methyl-6-phenyl pyrimidine 13 was obtained. The structure of 13 was established on the basis of their elemental analysis and spectral data. Refluxing of compound 13 in acetic anhydride afforded 1-carboxyamide-2-ethylamino-4-methyl-6-phenyl pyrimidine 14 (Scheme 3). Its IR spectrum showed disappearance of NH2 and appearance of a new band at 1668 cm−1 for (C O) group. Moreover, its 1H NMR spectrum revealed two characteristic singlet signals at 2.50 and 11.19 ppm due to methyl protons at C-4 of pyrimidine moiety and NH proton, respectively. 1-(1H-pyrrol-1-yl)-4-methyl-6-phenyl-pyrimidine-2-thione 15 was prepared, when compound 1 was condensed with 2,5-dimethoxy tetrahydrofuran in glacial acetic acid. The structure of compound 15 was confirmed on the basis of elemental analysis and spectral data. The IR spectrum of 15 revealed the disappearance of the NH2 bands. Additionally, mass spectrum of 15 revealed molecular ion peak 267 (1.02%).
Scheme 3.
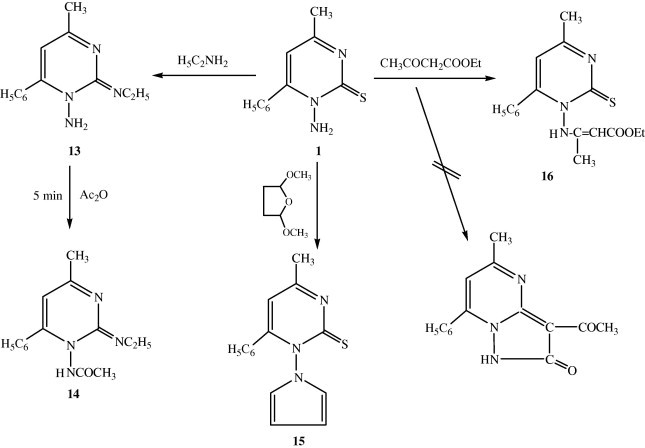
Compound 1 reacted with ethyl acetoacetate to afford ethyl-3-(4-methyl-6-phenylpyrimidine-2-thione-1-yl amino)but-2-enoate 16. The 1H NMR of 16, olefinic proton in the side-chain resonated as a singlet at δ 4.75 ppm and NH signal was located in the region δ 10.97 ppm.
3. Conclusion
In the present work, the synthesis of pyrimido[3,2-b]-1,2,4,5-tetrazine 3a,b,5c,d,6,9,pyrimido[3,2-b]-1,2,4-triazine 10,11,pyrimido[3,2-b]-1,2,4-triazole 12 and pyrimidine derivatives 1,2a,b,4c,d,8,13,14,15,16 is reported. All spectroscopic analyses confirmed the proposed structures of these compounds. Antitumor activity data have shown that the synthesized compounds have a significant antitumor activity against (HEPG2) cancer cell line.
4. Experimental
Melting points are uncorrected and were determined on a Stuart melting point apparatus. Elemental analyses (C,H,N) were performed on Perkin–Elmer 2400 analyzer. The IR spectra (KBr) were measured on (Pye unicam SP 1000 IR spectrophotometer). 1H NMR spectra were obtained on a Bruker proton NMR–Avance 300 (300 MHz) using tetramethyl silane (TMS) as internal standard. Mass spectra were run on Varian MAT 311-A 70ev. The synthesized compounds were screened in vitro antitumor activity at the regional center for mycology and biotechnology, Al-AZHAR UNIVERSITY.
4.1. 1-Amino-4-methyl-6-phenyl pyrimidin-2-thione 1
A mixture of benzoyl acetone (0.01 mol) and thiosemicarbazide (0.01 mol) in ethanol (50 ml) containing 3 drops of piperidine was refluxed for 5 h. The reaction mixture was concentrated and allowed to cool and the solid obtained was recrystallized from ethanol. Yield 79%; m.p. 160–162 °C; IR (KBr, cm−1) 3361, 3308 (NH2), 3050 (CH arom.), 2922 (CH aliph.), 1644 (C N), 1216 (C S). 1H NMR (DMSO-d6) δ 2.35 (s, 3H, CH3), 5.70 (s, 2H, NH2), 7.30–8.20 (m, 5H, Ar-H), 8.65 (s, 1H, (H-5) pyrimidine); 13C NMR: δ 16.10 (CH3), 111.18–145.10 (Caromatic), 205.00 (C S); MS, m/z (%): 217 [M+, 3.37], 77 [100]. Anal. Calcd. For: C11H11N3S: C, 60.82; H, 5.06; N, 19.35; S, 14.74. Found: C, 60.55; H, 5.00; N, 19.50; S, 14.80.
4.2. N-Thenoyl-N′-1-[(4-methyl-6-phenyl-2-thioxo)pyrimidinyl]thiourea 2a, N-phenyl-N′-1-[(4-methyl-6-phenyl-2-thioxo)pyrimidinyl]thiourea 2b
Thenoyl isothiocyanate or phenyl isothiocyanate (0.005 mol) was added to a solution of compound 1 (0.005 mol) in 20 ml of acetonitrile. The resulting solution was stirred and heated under reflux for 18 h. (A solid formed during the course of the reaction). The reaction mixture was cooled and the solid was removed by filtration, dried and recrystallized from ethanol to give, 2a; yield 64%; m.p. 190–192 °C; IR (KBr, cm−1): 3242, 3135 (2NH), 3020 (CH arom.), 2974 (CH aliph.) 1660 (C O), 1238 (C S); 1H NMR (DMSO-d6) δ 2.50(s,3H,CH3), 10.00 (br. s, 1H, NH), 12.79 (br. s, 1H, NH), 8.08–7.27 (m, 9H, Ar-H; pyrimidine H-5); Anal. Calcd. For C17H14ON4S3: C, 52.84; H, 3.62; N, 14.50; S, 24.87; Found: C, 52.55, H, 3.33; N, 14.80; S, 25.00.
2b, yield, 60%; m.p. 220–222 °C; IR(KBr, cm−1): 3280, 3200 (2NH), 3100 (CH arom.), 2920 (CH aliph), 1220 (C S); 1H NMR (DMSO-d6) δ 10.11 (br. s,2H, 2NH); 7.78–7.50 (m, 11H, Ar-H, pyrimidin H-5); Anal. Calcd. For C18H16N4S2: C, 61.36; H, 4.54; N, 15.90; S, 18.18. Found: C, 61.30; H, 4.50; N, 16.00 S, 18.50.
4.3. 9-Methyl-7-phenyl-4-(2-thenoyl amino) (3H) pyrimido[3,2-b]-1,2,4,5-tetrazine 3a, 9-methyl-7-phenyl-4-phenylamino (3H) pyrimido[3,2-b]-1,2,4,5-tetrazine 3b
A mixture of compound 2a or b (0.01 mol) and hydrazine hydrate (0.01 mol) was fused on an oil bath at 165 °C for 4 h., the solid obtained was recrystallized from ethanol, 3a; yield 55%; m.p. 140–142 °C; IR (KBr, cm−1):3463(OH), 3305, 3259 (2NH), 3020 (CH arom.), 2888 (CH aliph.), 1665 (C O), 1640 (C N); 1H NMR (DMSO-d6) δ: 2.50 (s, 3H, CH3), 7.33–8.08 (m, 8H, Ar-H), CH-pyrimidine), 9.85 (s, 1H, NH), 12.79 (s, 1H, NH); 13C NMR (DMSO-d6) δ 20.28 (CH3), 111.00–150.03 (Caromatic), 180.22 (C O); Ms, m/z (%): 350 [M+] (2.72), 77 (100), Anal. Calcd. For C17H14ON6S: C, 58.28; H, 4.00; N, 24.00; S, 9.14; Found: C, 58.50; H, 4.11; N, 24.28, S, 9.50.
3b, yield 50%; m.p. 170–172 °C; IR (KBr, cm−1): 3336, 3266 (2NH), 3050 (CH arom.), 2992 (CH aliph.), 1618 (C N); 1H NMR (DMSO-d6) δ: 2.38 (s, 3H, CH3), 7.18–7.90 (m, 10H, Ar-H), 8.05 (s, 1H, CH-pyrimidine), 11.18 (s, 1H, NH), 13.90 (s, 1H, NH); MS, m/z (%): 316 (1.11), 77 (100); Anal. Calcd. For C18H16N6: C, 68.35; H, 5.06, N, 26.58; Found: C, 68.30; H, 5.00, N, 26.60.
4.4. 1-Carboxamide-4-methyl-6-phenyl-pyrimidine-2-thione 4c; 1-(2-thenoyl amino)-4-methyl-6-phenyl-pyrimidine-2-thione 4d
A solution of 1 (0.01 mol) in acetyl chloride or thenoyl chloride (10 ml) was refluxed for 4 h. Decomposition over ice-cold water gave a solid that recrystallized from ethanol 4c: yield, 55%; m.p. 260–262 °C; IR (KBr, cm−1) 3320 (OH), 3190 (NH), 3005 (CH arom), 2999 (CH aliph.), 1695 (C O), 1644 (C N), 1228 (C S); 1H NMR (DMSO-d6) δ 2.28 (s, 3H, CH3), 3.80 (s, 3H, CH3), 7.13–8.80 (m, 6H, Ar-H), 9.98, 10.76 (2s, 1H, HNC O and N C–OH), 11.33 (s, 1H, HNCOCH3); Anal. Calcd. For: C13H13ON3S: C, 60.23; H, 5.01; N, 16.21; S, 12.35. Found: C, 60.30; H, 5.00, N, 16.28; S, 12.30.
4d, yield, 62%; m.p. 295–297 °C; IR (KBr, cm−1): 3320 (OH), 3190 (NH) 3100 (CH arom.), 2980 (CH aliph.), 1690 (C O), 1644 (C N), 1220 (C S) 1H NMR (DMSO-d6) δ 2.32 (s, 3H, CH3); 7.33–7.44 (m 9H, Ar-H); 9.81, 9.13 (2s, 1H, HNC O and N C–OH), 10.68 (s, 1H, HNCOR); Anal. Calcd. For: C16H13ON3S2: C, 58.71; H, 3.97; N, 12.84; S, 19.57. Found: C, 58.70; H, 4.00; N, 12.80, 5, 19.62.
4.5. 4,9-Dimethyl-7-phenyl (3H) pyrimido[3,2-b]-1,2,4,5-tetrazine 5c and 9-methyl-7-phenyl-4-(2-thienyl) (3H) pyrimido[3,2-b]-1,2,4,5-tetrazine 5d
A mixture of compound 4c or d (0.01 mol) and hydrazine hydrate (0.01 mol) was fused in an oil bath at 180 °C for 4 h. The solid obtained was recrystallized from ethanol;
5c yield 50%; m.p. 140–142 °C; IR (KBr, cm−1) 3329 (NH), 3095 (CH arom.), 2980–2888 (CH aliph.), 1640 (C N), 1H NMR (DMSO-d6) δ 2.30 (s, 3H, CH3), 2.50 (s, 3H, CH3), 7.85–8.11 (m, 6H, Ar-H), 12.38 (s, 1H, NH); MS, m/z (%): 239 [M+] (8.03), 77 [100]. Anal. Calcd. for: C13H13N: C, 66.27; H, 5.43; N, 29.28. Found: C, 66.20; H, 5.50; N, 29.80.
5d yield 57%; m.p. 240–242 °C; IR (KBr, cm−1): 3300 (NH), 3055 (CH arom.), 2988 (CH aliph.), 1648 (C N), 1H NMR (DMSO-d6) δ 2.33 (s, 3H, CH3), 7.32–8.11 (m, 8H, Ar-H), 9.52 (s, 1H, C–H pyrimidine), 12.99 (s, 1H, NH); MS, m/z (%): 307 [M+] (3.11), 77 [100]. Anal. Calcd. For: C16H13N5S: C, 62.54; H, 4.23; N, 22.80, S, 10.42. Found: C, 62.54; H, 4.23; N, 22.80; S, 10.42.
4.6. 6,12-Dimethyl-4,10-diphenyl pyrimido[3,2-b]-1,2,4,5-tetrazino[3,2-b] pyrimidin 6
A solution of 1 (0.01 mol) in pyridine (20 ml) was refluxed for 48 h. The reaction mixture was poured into ice cold water and the obtained solid was recrystallized from dioxan; yield 62%, m.p. >360 °C; IR (KBr, cm−1) 3055 (CH arom.), 2995–2870 (CH aliph.); 1H NMR (DMSO-d6) δ 2.42 (s, 6H, 2CH3), 7.07–8.01 (m, 12H, Ar-H); MS, m/z (%): 366 [M+] (3.11), 77 [100] Anal. Calcd. For: C22H18N6: C, 72.13; H, 4.91; N, 22.95. Found: C, 72.00; H, 5.00, N, 22.80.
4.7. 6-Chloro-3-phenyl coumarine 7
A mixture of the appropriate phenylacetonitrile (0.005 mol) and 5-chloro-salicyladehyde (0.005 mol) in ethanol (35 ml) containing piperidine (5 drops) was refluxed for 4 h. The reaction mixture was cooled and then acidified with dilute hydrochloric acid. The solid so formed was collected and recrystallized from ethanol, yield 80%; m.p. 200–202 °C; IR (KBr, cm−1) 3050 (CH arom.), 1680 (C O), 1600 (C C); 1H NMR (DMSO-d6) δ 7.00–8.22 (m, 9H, Ar-H); MS, m/z (%): 256.5 [M+] (3.11), 258.5 [M+] (11.22), 77 (100). Anal. Calcd. For: C15H9O2Cl: C, 70.17; H, 3.50; Cl, 13.84. Found: C, 70.30; H, 3.33; Cl, 14.00.
4.8. 6-Methy-4-phenyl-3[3-phenylquinoline-2-one] pyrimidin-2-thione 8
A mixture of 1 (0.01 mol) and 3-phenyl-6-chloro coumarin 7 (0.012 mol) was refluxed in ethanol (50 ml) for 12 h. the solid product was recrystallized from dioxane; yield 65%; m.p. 120–122 °C; IR (KBr, cm−1) 3055 (CH arom.), 2900–2880 (CH aliph.), 1680 (C O), 1222 (C S), 1648 (C N), 1600 (C C). 1H NMR (DMSO-d6) δ 2.33 (s, 3H, CH3), 7.29–8.41 (m, 15H, Ar-H). Anal. Calcd. For: C26H18ClON3S: C, 68.49; H, 3.95; N, 9.22; Cl, 7.79; S, 7.02 Found: C, 68.50, H, 3.70, N, 9.50, Cl, 7.80, S, 7.12.
4.9. 4,12-Diphenyl-14-methyl pyrimido[3,2-b]-1,2,4,5-tetrazino[4,3-a] quinoline 9
A mixture of compound 8 (0.01 mol) and hydrazine hydrate (0.01 mol) was refluxed for 4 h. The reaction mixture was cooled and the solid obtained was recrystallized from dioxane, yield 79%; m.p. >360 °C, IR (KBr, cm−1) 3105 (CH arom.), 2989 (CH aliph.); 1H NMR (DMSO-d6) δ 2.33 (s, 3H, CH3), 7.29–8.38 (m, 16H, Ar-H); M, m/z (%): 435.5 [M+] (1.12), 437.5 [M+] (3.22), 77 (100). Anal. Calcd. For: C26H18N5Cl: C, 71.64; H, 4.13; N, 16.07, Cl, 8.15. Found: C, 71.80; H, 4.00; N, 16.00; Cl, 8.30.
4.10. 9-Methyl-7-pheny-(5H) pyrimido[3,2-b]-1,2,4-triazin-3-one 10
A mixture of compound 1 (0.005 mol) and chloroacetamide (0.005 mol) in dry DMF (30 ml) was refluxed for 48 h. The reaction mixture was cooled, the solid was filtered off and recrystallized from DMF. Yield 53%; m.p. 320–322 °C. IR (KBr cm−1) 3375 (NH), 3055 (CH arom.), 2988 (CH aliph.), 1717 (C O), 1640 (C N) 1H NMR (DMSO-d6) δ 2.49 (s, 3H, CH3) 4.00 (s, 2H, CH2CO), 7.49–7.99 (m, 5H, Ar-H), 8.00 (s, 1H, CH-pyrimidine), 9.20 (s, 1H, NH); 13C NMR: δ 11.4 (CH3), 61.30 (CH2), 101.00, 110.00, 118.20, 119.80, 142.70, 148.20, 149.20, 150.11, 153.40, 155.50, (Caromatic), 180.60 (C O). MS (m/z) (%): 240 (M+), 6.09), 77 (100).
Anal. Calcd. For: C13H12N4O: C, 65.00. H, 5.00; N, 23.33. Found: C, 65.11; H, 5.28; N, 23.50.
4.11. 4-Amino-9-methyl-7-phenyl pyrimido[3,2-b]-1,2,4-triazin-3-thione 11
A mixture of compound 1 (0.01 mol) and dithiooxamide (0.01 mol) was fused on an oil bath at 175 °C for 4 h., the solid product was recrystallized from ethanol; yield, 70%; m.p. >360; IR (KBr, cm−1) 3383–3319 (NH2), 3005 (CH arom.), 2900 (CH aliph), 1644 (C N), 1222 (C S). 1H NMR (DMSO-d6) δ 2.33 (s, 3H, CH3), 4.78 (s, 2H, NH2), 7.11–7.71 (m, 6H, Ar-H), 13C NMR: δ 11.40 (CH3), 112.20–142.11, Caromatic; 200.00 (C S); MS, m/z (%): 269 [M+] (6.84), 77 [100]. Anal. Calcd. For: C13H11N5S: C, 57.99; H, 4.08; N, 26.02; S, 11.89; Found: C, 57.30; H, 4.00; N, 26.38.
4.12. 3-(4-Acetophenone)-8-methyl-6-phenyl pyrimido[3,2-b]1,2,4-triazole 12
A mixture of compound 1 (10 mmol), p-cyanoacetophenone (10 mmol), and Potassium t-butoxide (20 mmol) in t-butanol (50 ml) was refluxed for 48 h. After cooling, the solvent was evaporated under reduced pressure, the crude product was washed with cold water (50 ml), separated by filtrations and recrystallized from ethanol to give 12, yield 50%; m.p. 260–262 °C. IR (KBr cm−1) 3055 (CH arom), 2999–3880 (CH aliph), 1648 (C N); 1H NMR (DMSO-d6) δ 2.42, 3.50 (2s, 6H, (CH3) and (CH3CO) 7.52–8.80 (m, 10H, Ar–H), 13C NMR: δ 10.13 (CH3), 30.13 (CH3CO), 114.11–139.01 Caromatic.; MS (m/z): 328 [M+] (3.11), 76 (100). Anal. Calcd. For: C20H16N4O: C, 73.17; H, 4.87; N, 17.07 Found: C, 73.00; H, 4.80; N, 17.31.
4.13. 1-Amino-2-ethylamino-4-methyl-6-phenyl-pyrimidine 13
A mixture of compound 1 (0.01 mol) and ethyl amine (0.01 mol) in ethanol (30 ml) was heated under reflux for 2 h. After cooling, the solid formed was collected and recrystallized from ethanol yield 72% m.p. 190–192 °C; IR (KBr cm−1) 3400, 3300 (NH2), 3050 (CH arom.), 2900–2880 (CH aliph.) 1640 (C N); 1H NMR (DMSO-d6) δ 2.33 (s, 3H, CH3) 1.33 (t, 3H, CH2CH3, J = 9.22 Hz), 4.23 (q, 2H, CH2CH3, J = 11.82 Hz), 7.22–7.82 (m, 5H, Ar-H), 8.81 (s, 1H, CH-pyrimidine); Anal. Calcd. For C13H16N4: C, 68.42; H, 7.01; N, 24.56; Found: C, 68.60, H, 7.18; N, 24.60.
4.14. 1-Carboxyamid-2-ethylamino-4-methyl-6-phenyl-pyrimidine 14
A solution of compound 13 (0.005 mol) in acetic anhydride (20 ml) was heated for 10 min. The solvent was evaporated under reduced pressure, then the reaction mixture was poured into ice-water (40 ml) to give a solid precipitate which was filtered off and recrystallized from petroleum ether 60/80 to give 14.; yield 65%; m.p. 300–302 °C. IR (KBr cm−1) 3183 (NH), 3057 (CH arom.), 2900–2882 (CH aliph.), 1637 (C N), 1668 (C O). 1H NMR (DMSO-d6) 2.50 (s, 3H, CH3), 2.64 (s, 3H, CH3CO), 1.14 (t, 3H, CH2CH3, J = 8.22 Hz), 4.25 (q, 2H, CH2CH3), J = 11.30 Hz), 7.06–7.95 (m, 5H, Ar-H),8.32 (s, 1H, CH-pyrimidine), 11.19 (s, 1H, NH). Anal. Calcd. For C15H18N4O: C, 66.66, H, 6.66, N,20.74. Found: C, 66.50; H, 6.51; N, 20.70.
4.15. 1-(1H-Pyrrol-1-yl)-4-methyl-6-phenyl-pyrimidin-2-thione 15
A solution of compound 1 (0.09 mol), and 2,5-dimethoxytetra-hydrofuran (0.13 mol) in glacial acetic acid (25 ml) was refluxed for 6 h. The reaction mixture was concentrated and the obtained solid was recrystallized from (ethanol-dioxane; 1:1); yield 72%; m.p. >360 °C; IR (KBr cm−1) 3090 (CH arom.), 2990 (CH aliph.) 1646 (C N), 1222 (C S); 1H NMR (DMSO-d6) δ 2.50 (s, 3H, CH3), 7.19–7.95 (m, 9H, Ar-H and CH-pyrrol) 8.32 (s, 1H, CH-pyrimidine); MS: m/z (%): 267 [M+] (1.02), 77 (100). Anal. Calcd. For: C15H13N3S: C, 67.41; H, 4.86; N, 15.73; S, 11.98. Found: C, 67.50; H, 4.90; N, 16.00; S, 11.90.
4.16. Ethyl-3-(4-methyl-6-phenyl-pyrimidine-2-thione-1-yl-amino) but-2-enoate 16
A mixture of 1(5.62 mmol) and (8 ml) of ethylacetoacetate was heated for 8 h. Excess ethylacetoacetate was distilled off under reduced pressure and the, residue was purified by recrystallization from ethanol; yield 50%; m.p. >360 °C; IR (KBr cm−1): 3220 (NH), 3070 (CH arom.), 2900–2780 (CH aliph.), 1710 (C O), 1220 (C S), 1H NMR (DMSO-d6) δ 1.19 (t, 3H, OCH2CH3, J = 8.11 Hz) 2.50 (s, 3H, CH3), 2.73 (s, 3H, CH3), 4.35 (q, 2H, OCH2, J = 11.15 Hz), 4.75 (s, 1H, CH), 7.22–7.78 (m, 5H, Ar-H), 8.32 (s, 1H, CH-pyrimidine); 10.97 (s, 1H, NH). Anal. Calcd. For: C17H19N3O2S: C, 62.00; H, 5.77; N, 12.76; S, 9.72. Found: C, 62.18; H, 5.50; N, 12.70; S, 10.00.
5. Biological testing
5.1. Materials
Doxorubicin, the reference drug used in this study, is one of the most effective antitumor agents.
HEPG2 cells (human cell line of a well differentiated hepatocellular carcinoma isolated from a liver biopsy of male Caucasian aged 15 years) were obtained from the American type culture collection (ATCC). All other chemicals were obtained from sigma chemical company (st. Louis, Mo., USA).
5.2. Evaluation of celllular cytotoxicity
The cytotoxic activity of the target compounds against HEPG2 cells was determined using cytotoxicity assay. In brief, the cells were seeded in a 96-well plate at a cell concentration of 1 × 104 cell per well in 100 μl of growth medium and fresh medium containing different concentrations of the test sample was added after 24 h of seeding. Serial twofold dilution of the tested chemical compound was added to confluent cell monolayers dispensed into 96-well, flat-bottomed microtiter plates (Falcon, NJ, USA) using a multichannel pipette. The microtiter plates were incubated at 37 °C in a humidified incubator with 5% CO2 for a period of 48 h. Three wells were used for each concentration of the test sample. Control cells were incubated without test sample and with or without DMSO. The low percentage of DMSO present in the wells (maximal 0.1%) was found not to affect the experiment. After incubation of the cells for 24 h at 37 °C, various concentrations of sample (50, 25, 12.5, 6.25, 3.125 & 1.56 μg) were added, and the incubation was continued for 48 h and viable cell yield was determined by a colorimetric method. In brief, after the end of the incubation period, media were aspirated and the crystal violet solution (1%) was added to each well for at least 30 min. The stain was removed and the plates were rinsed using tap water until all excess stain was removed. Glacial acetic acid (30%) was then added to all wells and mixed thoroughly, and then the absorbances of the plates were measured after gently shaking on microplate reader, using a test wavelength of 490 nm. All results were corrected for background absorbance detected in wells without added stain. Treated samples were compared with the cell control in the absence of the tested compounds. All experiments were carried out in triplicate. The cell cytotoxic effect of each tested compound was calculated (Mosmann, 1983, Vijayan et al., 2004) (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10, Figure 11, Figure 12, Figure 13, Figure 14, Figure 15, Figure 16, Figure 17, Figure 18, Figure 19, Figure 20).
Figure 1.
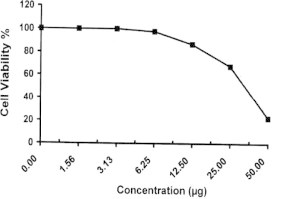
Sample No. 1. The inhibitory effect of compound 1 concentration on HEPG2 cells activity.
Figure 2.
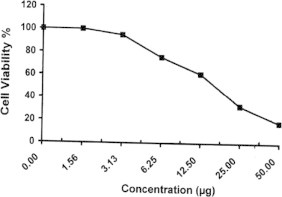
Sample No. 2a. The inhibitory effect of compound 2a concentration on HEPG2 cells activity.
Figure 3.
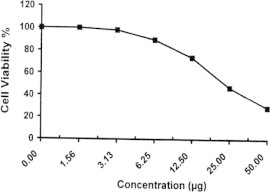
Sample No. 2b. The inhibitory effect of compound 2b concentration on HEPG2 cells activity.
Figure 4.
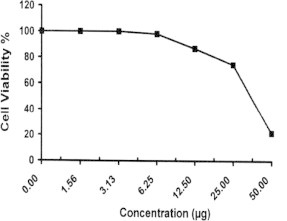
Sample No. 3a. The inhibitory effect of compound 3a concentration on HEPG2 cells activity.
Figure 5.
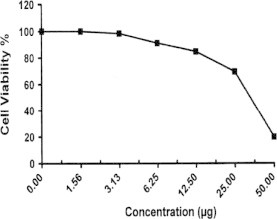
Sample No. 3b. The inhibitory effect of compound 3b concentration on HEPG2 cells activity.
Figure 6.
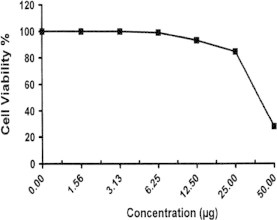
Sample No. 4c. The inhibitory effect of compound 4c concentration on HEPG2 cells activity.
Figure 7.
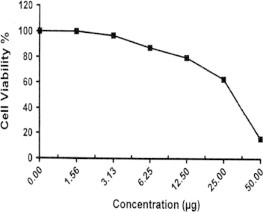
Sample No. 4d. The inhibitory effect of compound 4d concentration on HEPG2 cells activity.
Figure 8.
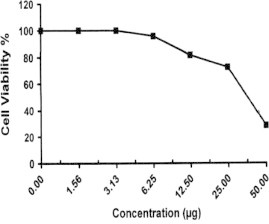
Sample No. 5c. The inhibitory effect of compound 5c concentration on HEPG2 cells activity.
Figure 9.
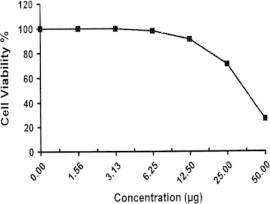
Sample No. 5d. The inhibitory effect of compound 5d concentration on HEPG2 cells activity.
Figure 10.
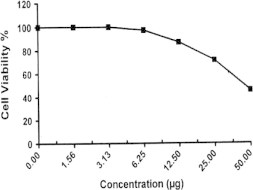
Sample No. 6. The inhibitory effect of compound 6 concentration on HEPG2 cells activity.
Figure 11.
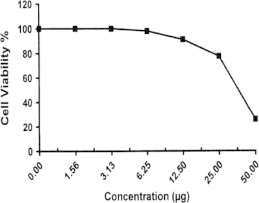
Sample No. 8. The inhibitory effect of compound 8 concentration on HEPG2 cells activity.
Figure 12.
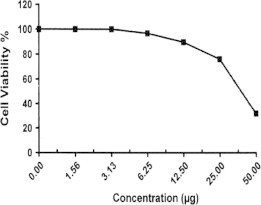
Sample No. 9. The inhibitory effect of compound 9 concentration on HEPG2 cells activity.
Figure 13.
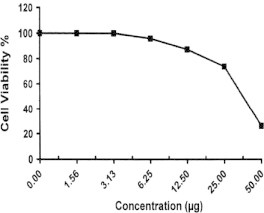
Sample No. 10. The inhibitory effect of compound 10 concentration on HEPG2 cells activity.
Figure 14.
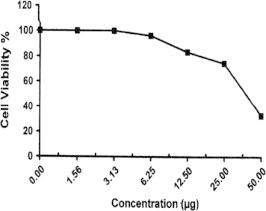
Sample No. 11. The inhibitory effect of compound 11 concentration on HEPG2 cells activity.
Figure 15.
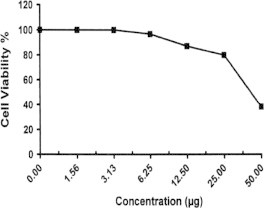
Sample No. 12. The inhibitory effect of compound 12 concentration on HEPG2 cells activity.
Figure 16.
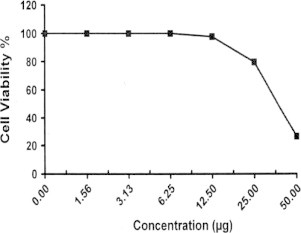
Sample No.13. The inhibitory effect of compound 13 concentration on HEPG2 cells activity.
Figure 17.
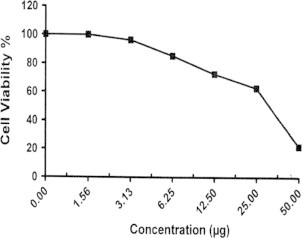
Sample No.14. The inhibitory effect of compound 14 concentration on HEPG2 cells activity.
Figure 18.
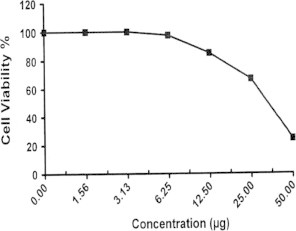
Sample No.15. The inhibitory effect of compound 15 concentration on HEPG2 cells activity.
Figure 19.
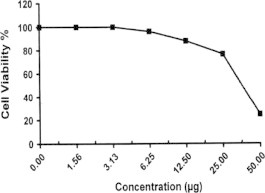
Sample No. 16. The inhibitory effect of compound 16 concentration on HEPG2 cells activity.
Figure 20.
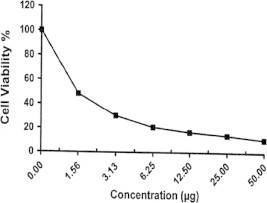
Doxorubicin.
5.3. Evaluation of cytotoxicity activity against the human liver cancer (HEPG2) cell line
The antitumor activities of compounds were assessed against HEPG2 cancer cell line in comparison to the traditional anticancer drug (Doxorubicin) on the basis of monitoring the inhibition of the growth of human cancer cells, a series of synthesized compounds possessing a broader spectrum of antitumor activity. Ninety tested compounds (1; 2a,b; 3a,b; 4c,d; 5c,d; 6;8;9;10;11;12;13;14;15 and 16) were subjected to a screening system for investigation of their antitumor potency against liver (HEPG2) cell line. The antitumor activity results indicated that most of the compounds showed inhibition activity against the tested cell line but varying intensity extents in comparison to the known anticancer drug (Doxorubicin) (See Table 1).
Table 1.
In vitro antitumor activity of the synthesized compounds.
| Comp. No | Viability % | IC50(μg/ml)a HEPG2b | |||||
|---|---|---|---|---|---|---|---|
| Sample concentration (μg m−1) | |||||||
| 50 | 25 | 12.5 | 6.25 | 3.12 | 1.56 | ||
| 1 | 22.40 | 67.74 | 86.99 | 97.83 | 100.0 | 100.0 | 34.8 |
| 2a | 27.81 | 84.66 | 93.29 | 99.12 | 100.0 | 100.0 | 17.4 |
| 2b | 28.72 | 46.99 | 73.91 | 89.63 | 98.12 | 100.0 | 23.6 |
| 3a | 26.60 | 71.09 | 91.41 | 98.18 | 100.0 | 100.0 | 36.5 |
| 3b | 19.87 | 69.49 | 84.74 | 91.09 | 98.46 | 100.0 | 34.8 |
| 4c | 27.81 | 84.66 | 93.29 | 99.12 | 100.0 | 100.0 | 40.2 |
| 4d | 15.83 | 62.88 | 79.62 | 87.44 | 96.92 | 100.0 | 31.8 |
| 5c | 28.33 | 72.31 | 81.54 | 95.86 | 100.0 | 100.0 | 37.7 |
| 5d | 26.60 | 71.09 | 91.41 | 98.18 | 100.0 | 100.0 | 36.9 |
| 6 | 45.77 | 71.28 | 86.92 | 97.24 | 100.0 | 100.0 | 45.9 |
| 8 | 28.82 | 77.60 | 91.30 | 98.11 | 100.0 | 100.0 | 38.30 |
| 9 | 31.85 | 76.16 | 89.79 | 96.84 | 100.0 | 100.0 | 39.8 |
| 10 | 26.43 | 73.86 | 87.43 | 96.07 | 100.0 | 100.0 | 37.6 |
| 11 | 33.08 | 74.36 | 83.44 | 96.18 | 100.0 | 100.0 | 39.8 |
| 12 | 38.33 | 79.94 | 87.12 | 96.86 | 100.0 | 100.0 | 43.0 |
| 13 | 26.44 | 79.59 | 97.67 | 100.0 | 100.0 | 100.0 | 38.9 |
| 14 | 21.71 | 62.81 | 72.67 | 85.44 | 96.39 | 100.0 | 32.8 |
| 15 | 24.42 | 66.67 | 85.06 | 97.48 | 100.0 | 100.0 | 34.9 |
| 16 | 24.62 | 76.35 | 87.83 | 96.14 | 100.0 | 100.0 | 37.7 |
| Doxorubicinc | 10.95 | 14.29 | 16.90 | 21.03 | 30.32 | 100.0 | 1.2 |
“IC50, compound concentration required to inhibit tumor cell proliferation by 50%”.
Human liver cell line (HEPG2).
Positive control.
Compounds 2a,b showed significant in vitro antitumor activity (IC50, 17.4 μg/ml, 23.6 μg/ml) and possess thiouredo moiety which is known to have antitumor activity (Abou El Ella et al., 2008, Agrawal et al., 2002). Also, fusion of pyrimido[3,2-b]-1,2,4,5-tetrazine in tri(or) tetracyclic structure namely pyrimido tetrazinopyrimidine (6) (IC50, 45.9 μg/ml) or pyrimidotetrazino quinoline (9) (IC50, 39.8 μg/ml) enhances the antitumor activity.
Footnotes
Peer review under responsibility of King Saud University.

References
- Araguchi K.H., Kubota Y., Tanaka H. J. Org. Chem. 2004;69:1831. doi: 10.1021/jo030262u. [DOI] [PubMed] [Google Scholar]
- Abou El Ella D.A., Ghorab M.M., Noaman E., Heiba H.I., Khalil A.I. Bioorg. Med. Chem. Lett. 2008;16:2391–2402. doi: 10.1016/j.bmc.2007.11.072. [DOI] [PubMed] [Google Scholar]
- Andrus P.K., Fleck T.J., Oostveen J.A., Hall E.D. J. Neurosci. Res. 1997;47:650–654. doi: 10.1002/(sici)1097-4547(19970315)47:6<650::aid-jnr11>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Atwal K.S., Swanson B.N., Unger S.E. J. Med. Chem. 1991;34:806–811. doi: 10.1021/jm00106a048. [DOI] [PubMed] [Google Scholar]
- Abd El-Hamid A.O. Phosphorus Sulfur Silicon. 1994;88:217. [Google Scholar]
- Aly A.S., Fathy N.M., Swelam S.A., Abdel Megeid F.M.E. J. Pharm. Sci. 1995;36:1577–1586. [Google Scholar]
- Agrawal V.K., Sharma R., Khadikar V.P. Bioorg. Med. Chem. Lett. 2002;10:2993–2999. doi: 10.1016/s0968-0896(02)00123-2. [DOI] [PubMed] [Google Scholar]
- Breault, G.A., Pease, J.E., 2000. PCT Int. Appl. WO 2000012485.
- Bundy G.L., Ayer D.E., Banitt L.S., Belonga K.L., Mizsak S.A., Palmer J.R., Tustin J.M., Chin J.E., Hall E.D. J. Med. Chem. 1995;38:4161–4163. doi: 10.1021/jm00021a004. [DOI] [PubMed] [Google Scholar]
- Coppo F.T., Fawzi M.M. J. Heterocycl. Chem. 1997;34:1351. [Google Scholar]
- Deng,Y., Wang, Y., Cherian, C., Hou, Z., Buck, S.A., Matherly, L.H., Gangiee, A., 2008, 51, 5052–5063. [DOI] [PMC free article] [PubMed]
- Depecker G., Patino N., Giorgio C.D., Terreux R., Gobrol bass D., Bailly C., Aubertin A., Candom R. Org. Biomol. Chem. 2004;2:74. doi: 10.1039/b311775h. [DOI] [PubMed] [Google Scholar]
- El-Abadelah M.M., Hussein A.Q., Kamal M.R., Al-Adhami K.H. Heterocycles. 1988;27:917–924. [Google Scholar]
- Foroughifar N., Mobinikhaledi A.J., Shariatzadeh S.M. Asian J. Chem. 2002;14:782–790. [Google Scholar]
- Gregory, B., Haibong, N., Brian, S., Fei, Z., 2007. WO2007071965.
- Hogenkamp, D.J., Nguyen, P., Shao, B., 2001. WO 0168612.
- Hany M., Hazem M. Org. Commun. 2008;1(1):1–8. [Google Scholar]
- Kim D.C., Lee Y.R., Yang B.S., Shin K.J., Kim D.J., Chung B.Y., Yoo K.H. Eur. J. Med. Chem. 2003;38:525–532. doi: 10.1016/s0223-5234(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Kuyper L.F., Garvey J.M., Baccanari D.P., Champness J.N., Stammers D.K., Beddel C.R. Bioorg. Med. Chem. Lett. 1996;4:593–602. doi: 10.1016/0968-0896(96)00045-4. [DOI] [PubMed] [Google Scholar]
- Kevin R., Lina S., Lida Q., Rachel M., Brian W., Dymock S., Hilary O., Mathew T., Glyn W., Joseph A., Sergi K., Ying P., Marcia R., Rogely B., Fiorenza F., Ralph G., Andree R. Bioorg. Med. Chem. Lett. 2003;13:2895–2898. doi: 10.1016/s0960-894x(03)00623-1. [DOI] [PubMed] [Google Scholar]
- Kappe C.O., Fabian W.M.F., Semones M.A. Tetrahedron. 1997;53:2803–2816. [Google Scholar]
- Kappe, C.O. 1993. 49, 6937–6963.
- Lauria A., Diana P., Barraja P., Montalbano A., Cirrinicione G., Dattolo G., Almerico A.M. Tetrahedron. 2002;58:9723. [Google Scholar]
- Ludovici D., Janseen P. Bioorg. Med. Chem. Lett. 2004;11:2335–2339. [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival application to proliferation and cytotoxicity assays. J. Immuol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Magan R., Marin C., Salas J., Perez M., Rosales M. J. Men Instaswaldo Cruz Riode Janeiro. 2004;99(6):651. doi: 10.1590/s0074-02762004000600021. [DOI] [PubMed] [Google Scholar]
- Magan R., Marin C., Rosales M. J.M. Pharmacology. 2005;73:41. doi: 10.1159/000081073. [DOI] [PubMed] [Google Scholar]
- Molina, P., Alagarin, M., Devega, M., 1985, 23, 10.
- Martin J., Kevin J., Ellen Z., Geraldine B., George A., Wei-Dong D., Stanley A., Miriam R., Jonathan D. Bioorg. Med. Chem. Lett. 2003;13:3483–3486. [Google Scholar]
- Meada E.A., Sznaidman M., Pollard G.T., Beauchamp L.M., Howard J.L. Eur. J. Med. Chem. 1998;33:363–374. [Google Scholar]
- Nguyen T.L. Anticancer Agents Med. Chem. 2008;8:710–716. doi: 10.2174/187152008785914770. [DOI] [PubMed] [Google Scholar]
- Navarro J., Salas J., Romero M., Vilaplana R., Faure R. J. Med. Chem. 1998;41:332. doi: 10.1021/jm970358e. [DOI] [PubMed] [Google Scholar]
- Patel R.B., Desai K.R., Chikalia K.H. J. Ind. Chem. 2006;45B:773–779. [Google Scholar]
- Renau T.C., Kennedy C., Ptak R.G., Breitenbach J.M., Drach J.C., Townsend L.B. J. Med. Chem. 1996;39:3470–3476. doi: 10.1021/jm950835y. [DOI] [PubMed] [Google Scholar]
- Rusinov V., Yu A., Petrov T., Chupakhin O.N., Kovalev G., Komina E.R. Khimiko-Farmatsevticheskii Zh. 1986;20:178. [Google Scholar]
- Traxler P., Furet P., Mett H., Buchdunger E., Meyer T., Lydon N. J. Med. Chem. 1996;39:2285. doi: 10.1021/jm960118j. [DOI] [PubMed] [Google Scholar]
- Tsuji K., Ishikawa H. Bioorg. Med. Chem. Lett. 1994;4:1601–1611. [Google Scholar]
- Varma R.S. Green Chem. 1999;1:43–55. [Google Scholar]
- Vijayan P., Raghu C., Ashok G., Dhanaraj S.A., Suresh B. Antiviral activity of medicinal plants of Nilgiris. Indian J. Med. Res. 2004;120:24–29. [PubMed] [Google Scholar]
- Xie W., Jin Y., Wang P.G. Chemtech. 1999;2:23–29. [Google Scholar]
- Zhou Y., Wei L., Brady T., Reddy P., Nguyen T., Chen J., Au O., Yoon I., Yip G., Zhang B., Barber Bioorg. Med. Chem. Lett. 2009;19:4303. doi: 10.1016/j.bmcl.2009.05.073. [DOI] [PubMed] [Google Scholar]