

Abstract
Background
The adenosine triphosphate-sensitive potassium (KATP) channel opener, diazoxide, preserves myocyte volume homeostasis and contractility during stress via an unknown mechanism. Pharmacologic overlap has been suggested between succinate dehydrogenase (SDH) activity and KATP channel modulators. Diazoxide may be cardioprotective due to the inhibition of SDH which may form a portion of the mitochondrial KATP channel. To determine the role of inhibition of SDH in diazoxide's cardioprotection, this study utilized glutathione to prevent the inhibition of SDH.
Methods
SDH activity was measured in isolated mitochondria exposed to succinate (control), malonate (inhibitor of succinate dehydrogenase), diazoxide, and varying concentrations of glutathione alone or in combination with diazoxide. Enzyme activity was measured by spectrophotometric analysis.
To evaluate myocyte volume and contractility, cardiac myocytes were superfused with Tyrode's physiologic solution (20 minutes), followed by test solution (20 minutes) including: Tyrode's, hyperkalemic cardioplegia (stress), cardioplegia + diazoxide, cardioplegia + diazoxide + glutathione, or glutathione alone; followed by Tyrode's (20 minutes). Myocyte volume and contractility were recorded using image grabbing software.
Results
Both malonate and diazoxide inhibited succinate dehydrogenase. Glutathione prevented the inhibition of succinate dehydrogenase by diazoxide in a dose dependent manner.
The addition of diazoxide prevented the detrimental myocyte swelling due to cardioplegia alone and this benefit was lost with the addition of glutathione. However, glutathione elicited an independent cardioprotective effect on myocyte contractility.
Conclusion
The ability of diazoxide to provide beneficial myocyte homeostasis during stress involves the inhibition of succinate dehydrogenase, which may also involve the opening of a purported mitochondrial KATP channel.
Keywords: Myocardial Protection, Cardioplegia
Introduction
Diazoxide (DZX), an adenosine triphosphate sensitive potassium (KATP) channel opener, is cardioprotective by maintaining myocyte volume homeostasis and contractility during stress (1-5). DZX, unlike nonspecific potassium channel openers, is 2000 times more specific for the mKATP channel and it is widely believed that DZX works not through a sarcolemmal KATP (sKATP) channel but through a purported mitochondrial KATP (mKATP) channel (6). Additional evidence that the action of DZX is at a non-sKATP location is provided by the failure of DZX to generate a potassium current via the sKATP channel and by the abolition of the cardioprotective effect of DZX on myocyte volume in myocytes from mice lacking the sulfonylurea subunit type 1 receptor (SUR1) which is not present in the ventricular sKATP channel (composed of SUR2A and Kir 6.2) (7,8).
Several hypotheses have been proposed for a mitochondrial-based mechanism of action for DZX including: mKATP channel regulation of mitochondrial matrix volume to either activate the respiratory chain and provide more ATP or to alter the outer mitochondrial membrane permeability to adenosine diphosphate leading to preservation of segregated adenine nucleotides, mKATP channel reduction in calcium overload during stress leading to decrease in cellular injury, or via mKATP maintenance of the inner mitochondrial membrane potential allowing potassium influx into the mitochondrial matrix and hydrogen efflux (9-12).
Alternatively, one KATP channel-independent mechanism that has been proposed is the inhibition of succinate dehydrogenase (SDH) by DZX. SDH, a multi-protein complex also known as Respiratory Complex II, is found in the inner mitochondrial membrane and participates in the respiratory chain and the generation of electrons for the synthesis of ATP, among other mitochondrial functions. Succinate oxidation, via SDH results in generation of electrons as succinate is converted to fumarate. These electrons, in conjunction with protons generated from the other complexes within the respiratory chain combine to generate ATP. SDH, along with coenzyme Q, also generates reactive oxygen species which can be lethal to cells at high concentrations (13-14).
Diazoxide has been shown to partially inhibit SDH activity (14-17). Interestingly, another inhibitor of SDH, 3-nitropropionic acid (3-NPA), has also been shown to be cardioprotective and associated with decreased oxygen radical production (18,19). Additional evidence supporting the pharmacologic overlap between SDH and the mKATP channel involves malonate, a known inhibitory of SDH. Malonate has been shown to activate mKATP channels leading to mitochondrial matrix swelling (20). This observed effect was inhibited by KATP channel inhibitor 5-hydroxydeconoate (5-HD). Others have suggested that SDH and mKATP are inversely related (inhibition of SDH is correlated with channel opening) (20).
SDH has also been suggested to be part of a purported mKATP channel. Four mitochondrial proteins, mitochondrial ATP-binding cassette-1, phosphate carrier, adenine nucleotide translocator, and ATP Synthase, were identified to associate with SDH to form a supercomplex in the inner mitochondrial membrane (21). This multiprotein complex exhibited mKATP channel activity, generated a potassium current, and increased potassium influx in the presence of DZX. This was reversed by the addition of ATP, 5-HD, and glibenclamide, all known mKATP channel inhibitors.
We hypothesize that the cardioprotective mechanism of action of DZX involves the inhibition of SDH. Definitive evaluation of the mechanism of action of DZX at a purported mKATP channel still awaits molecular identification of this entity. Glutathione has been shown to prevent the inhibition of SDH (22). Glutathione was utilized in the present study to evaluate the contribution of SDH inhibition in DZX's cardioprotective mechanism (Figure 1).
Figure 1. Representation of the adenosine triphosphate – sensitive potassium channel (KATP) and the succinate dehydrogenase (SDH) enzyme complex on the inner mitochondrial membrane.

Adenosine triphosphate – sensitive potassium channel opener diazoxide provides myocyte volume homeostasis and preserves contractility during stress via an unknown mechanism. Diazoxide also inhibits SDH. The relationship between the KATP channel activity and SDH during stress is unknown. Glutathione was utilized to determine the role of inhibition of succinate dehydrogenase in diazoxide's cardioprotection. Mitochondria from our laboratory visualized via electron microscopy (mag 3000×). (KATP channel figure partially reproduced with permission from Sellitto et al., Diazoxide maintenance of myocyte volume and contractility during stress: Evidence for a non-sarcolemmal KATP channel location. J Thorac Cardiovasc Surg 2010; 140:1153 [8] and succinate dehydrogenase image from en.wikipedia.org).
Material and Methods
All animal procedures were approved by the Animal Studies Committee at Washington University School of Medicine and all animals received humane care in compliance with the National Institute of Health's Guide to Care and Use of Laboratory Animals (23).
Mitochondrial Succinate Dehydrogenase Activity
Mitochondria were isolated from hearts of C57BL mice. Mice (either sex, 6-24 weeks old, average 24 grams) were anesthetized with 3% Avertin (0.3 grams 2,2,2-tribromoethanol, 1.86μL 2-methyl-2-butanol, 9.841mL sterile water) intraperitoneally and rapid cardiectomy was performed. Ventricular tissue was rapidly minced and homogenized with a 7mL Dounce homogenizer containing cold buffer (in mM/L: 10 HEPES (N-[2-hydroxyethyl]piperazine-N-[4-butanesulfonic acid]), 1 EDTA-K2 (ethylene diamine tetraacetic acid potassium), 250 Sucrose, adjusted to a pH of 7.1 with 20% potassium hydroxide. The homogenate was transferred to microcentrifuge tubes and centrifuged at 900 × g for 10 minutes at 4°C. Supernatant was then centrifuged at 5000 × g for 15 minutes. Supernatant was discarded and 300μL homogenization buffer was added to each pellet. A Bradford protein assay (Thermo Scientific; Rockford, IL) was utilized to determine and normalize total protein per each pellet. Mitochondria were stored in -20°C freezer and thawed on ice just before use and kept on ice throughout each assay. All other solutions were kept at room temperature.
Mitochondria, at a concentration of 1.8μg, were exposed to one of the following solutions (in a 1ml reaction): 20mM succinate (control) (Sigma, St. Louis), 8mM malonate (competitive inhibitor of SDH) (Sigma; St. Louis, MO), 100μM DZX (KATP channel opener) (Sigma; St. Louis, MO), varying concentrations of Glutathione alone (10μM, 25μM, 100μM) (blocker of SDH inhibition, GLU) (Sigma; St. Louis, MO), or 100μM DZX with varying concentrations of GLU (10μM, 25μM, 100μM). Each reaction additionally contained 20mM succinate, 2mM potassium cyanide (KCN), 50μM 2,6-Dicholoroindophenol (DCIP), 1.625mM phenazine methosulfate (PMS) and was brought to 1M volume with potassium phosphate buffer and titrated to a pH of 7.4. Reactions were prepared in disposable cuvettes covered with parafilm and allowed to activate for 20 minutes at room temperature before the final addition of KCN, DCIP, and PMS. Succinate and malonate controls were included in each mitochondrial isolation to confirm a functional assay.
SDH activity was measured by spectrophotometric (UV-1700 Spectrophotometer, Shimadzu Scientific Instruments; Columbia, MD) analysis of 2,6-DCIP (Sigma; St. Louis, MO) reduction at 600 nm for 20 minutes. Measurements were collected at 5 minute intervals and normalized to protein content.
Myocyte Isolation
Ventricular myocytes were isolated from C57BL mice (either sex, 6-15 weeks old, 20-30 gram body weight). Mice were anesthetized with 3.0% Avertin intraperitoneally. Rapid cardiectomy was performed and the aorta was cannulated using a 28-gauge needle and attached to a Langendorff apparatus. The aorta was perfused with various solutions for extracellular tissue digestion and cellular isolation (Solution A for 5 minutes followed by Solution B until a drip rate of 1 drop per second over a 10-second period was obtained). The left ventricle was then removed and transferred into Solution C, where it was gently dispersed by glass pipette. The isolated myocytes were allowed to centrifuge by gravity, and serial washings were performed every 10 minutes for 30 minutes. Myocytes (2-4 per mouse) were used within 5 hours and randomly selected. No more than 2 myocytes were used per test solution in any experiment.
Solution A consisted of (in mM/L, except as noted): 116 NaCl; 5.36 KCl; 0.97 Na2HPO4; 1.47 KH2PO4; 21.10 HEPES (N-[2-hydroxyethyl]piperazine-N′-[4-butanesulfonic acid]); 11.65 glucose; 26.50 μmol/L phenol red (Sigma; St. Louis, MO); 3.72 MgCl2; 4.40 NaHCO3; essential vitamins (100×, 10 mL, GIBCO; Grand Island, NY); and amino acids (50×, 20 mL, GIBCO; Grand Island, NY). Solution B consisted of solution A plus 10μM CaCl2 and 1.2 mg/mL collagenase (Type 2, Worthington Biochemical Corporation; Freehold, NJ). Solution C consisted of solution A plus 5 mg/mL bovine serum albumin (Sigma; St. Louis, MO), 1.25 mg/mL taurine, and l50μM CaCl2.
Experimental Protocol
Ventricular myocytes were superfused with control 37°C Tyrode's solution (in mM/L: 130 NaCl; 5 KCl; 2.5 CaCl2; 1.2 MgSO4; 24 NaHCO3; 1.75 Na2HP04; and 10 glucose) buffered to a pH of 7.4 using 95% O2–5% CO2 for 20 minutes for baseline measurements (average of the first 4 data points). Any baseline changes in cell volume secondary to the isolation or imaging protocol would be evident during this period. Myocytes were then superfused for 20 minutes with one of the following test solutions: control 37°C physiologic Tyrode's solution, 9°C hyperkalemic cardioplegic solution (CPG), 9°C CPG+100μM DZX, 9°C CPG+100μM DZX+100μM GLU, or 37°C Tyrode's+100μM GLU. Hyperkalemic cardioplegic solution is composed of Plegisol (Abbott Laboratories; North Chicago, IL) which contains (in mM/L): 110 NaCl; 16 KCl; 16 MgCl2; and 1.2 CaCl2 equilibrated with 95% O2–5% CO2 and titrated to pH 7.4 with 10% NaHCO3 solution alone. Cells were then re-exposed to 37°C physiologic Tyrode's solution for 20 minutes.
Myocyte Volume Measurement
Myocyte volume was measured as previously described (24). An aliquot of isolated myocytes was placed on a slide on a glass-bottomed chamber on an Olympus IX51 inverted microscope stage (Olympus Corporation, Tokyo Japan). After a 5 minute stabilization period, the chamber was superfused at a rate of 3 ml/min with 37°C Tyrode's. Chamber temperature was controlled by a waterbath system (Thermo Haake, Karlsruhe, Germany). Cell images were displayed on a video monitor using a charge-coupled device camera (KPM1U; Hitachi Denshi, Tokyo, Japan). Digital images of viable cells were captured using a video-frame grabber (Scion Corporation, Frederick, MD) every 5 minutes. Relative cell volume change was determined as previously described with the following formula (25):
where t represents test and c represents control, respectively.
Myocyte Contractility Measurement
Ventricular myocyte contractility was measured using a video-based edge detection system (IonOptix; Milton, MA) as previously described (4). Cells were paced using a field stimulator (MyoPacer, IonOptix; Milton, MA) with alternating 15 millivolt pulses, 5 milliseconds in duration at a rate of 1Hz. Data were recorded for 30 seconds at baseline and at 10 minutes and 20 minutes following re-exposure to Tyrode's. Variables were computed using IonWizard edge-detection software (IonOptix) (percentage shortening (PS), peak velocity of shortening (VS), and peak velocity of re-lengthening (VR)). Myocyte shortening was indicated as the percent difference between maximum and minimum cell length for each contraction. Peak velocity of shortening and re-lengthening was indicated as normalized maximal derivative of the downward and the upward portions of the digitized contraction curve to myocyte length, respectively.
Statistical Analysis
Data were analyzed using SYSTAT 13 (SYSTAT Software Inc., Chicago, IL). All data are presented as mean value ± standard error of the mean, with n equal to the number of experiments in each group. A repeated-measures analysis of variance was used for sequential time-based measurements for each test solution against its own baseline value in both mitochondria and myocyte experiments. For mitochondrial SDH analysis, group comparisons were made based on percent change in absorbance. For myocyte experiments, Tukey's HSD test was used for post hoc multiple comparisons between different test groups. A Shapiro-Wilks test was used to test for normality. If the data failed the normality test, the data were transformed or a nonparametric test was used. Probability values <0.05 were considered significant.
Results
Glutathione Prevented Diazoxide's Inhibition of Mitochondrial Succinate Dehydrogenase Activity
Succinate dehydrogenase activity is represented as change in absorbance over time (Figures 2-5). SDH activity is inversely related to absorbance. There was no difference in absorbance at baseline between groups.
Figure 2. Diazoxide inhibits SDH activity.

Isolated mitochondria were exposed to test solutions for 20 minutes. SDH activity is represented as percent change in absorbance over time. SDH activity is inversely proportional to absorbance. 10μM glutathione did not prevent inhibition of SDH by DZX.
Figure 5. Glutathione's ability to prevent the inhibition of SDH by DZX is dose dependent.
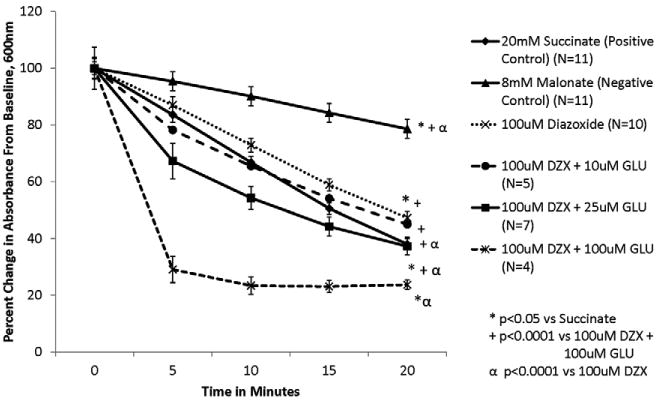
Isolated mitochondria were exposed to test solutions for 20 minutes. SDH activity is represented as percent change in absorbance over time. SDH activity is inversely proportional to absorbance.
Succinate, the substrate for succinate dehydrogenase, served as control for SDH activity (Figures 2-5). Exposure of mitochondria to malonate, a known inhibitor of SDH, resulted in a significant decline in SDH activity (Figures 2-5, p<0.0001 vs succinate alone). Exposure of mitochondria to 100 μM DZX also demonstrated a statistically significant decrease in SDH activity compared to succinate alone (p=0.029 vs succinate) (Figures 2-5), although less dramatic than that seen with malonate.
Glutathione reversed DZX's inhibition of SDH in a dose dependent fashion (Figure 5). At glutathione dose of 10μM had no effect on DZX's inhibition of SDH (p=0.362 vs 100μM DZX) (Figure 2). GLU (25μM) prevented DZX's inhibition of SDH activity (p<0.0001) (Figure 3). Similar but stronger effects were observed with 100μM GLU (p<0.0001) as shown in Figure 4.
Figure 3. Glutathione (25μM) prevents DZX's inhibition of SDH.
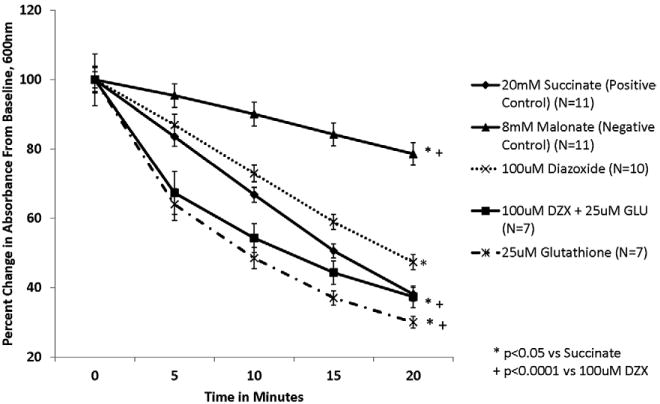
Isolated mitochondria were exposed to test solutions for 20 minutes. SDH activity is represented as percent change in absorbance over time. SDH activity is inversely proportional to absorbance. The addition of 25μM Glutathione prevented the inhibition of SDH by DZX.
Figure 4. Glutathione (100μM) prevents DZX's inhibition of SDH.
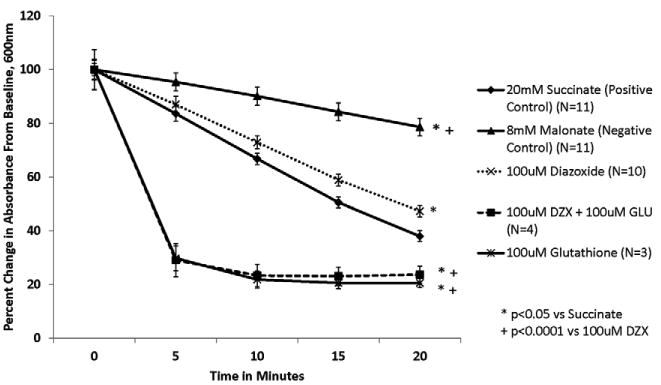
Isolated mitochondria were exposed to test solutions for 20 minutes. SDH activity is represented as percent change in absorbance over time. SDH activity is inversely proportional to absorbance. The addition of 100μM glutathione prevented the inhibition of SDH activity by DZX.
Glutathione alone was also associated with a dose dependent increase in SDH activity that was significantly greater than succinate at higher glutathione doses (p<0.001 for doses of 25μM and 100μM glutathione vs succinate) (Figures 3,4).
Glutathione Reversed DZX Prevention of Myocyte Swelling due to Cardioplegia
There were no statistically significant differences in cell volume during the initial baseline exposure (time 0-20 minutes) or during re-exposure to Tyrode's solution (time 40-60 minutes) between groups (Figure 6).
Figure 6. Glutathione prevented volume homeostasis provided by DZX during stress.
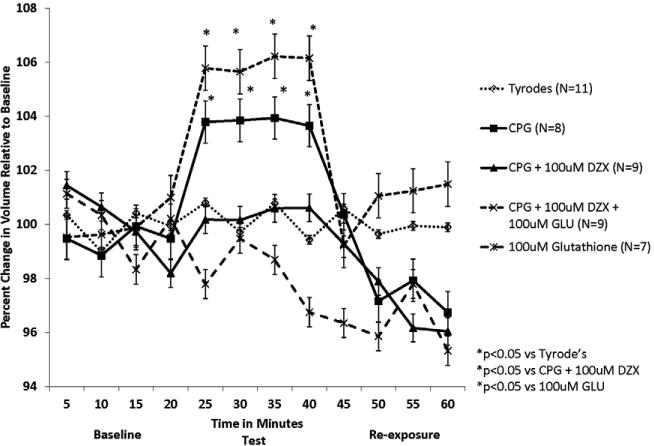
Isolated myocytes were exposed to Tyrode's during time 0-20 minutes, test solutions during time 20-40 minutes, and Tyrode's during time 40-60 minutes. Stress in the form of hyperkalemic cardioplegia (CPG) resulted in significant myocyte swelling that was prevented by diazoxide. Glutathione prevented the beneficial effect noted with diazoxide during myocyte stress.
Myocyte volume remained at baseline during the one-hour exposure to physiologic Tyrode's solution throughout the experiments. Exposure to hyperkalemic CPG resulted in significant cell swelling compared to Tyrode's (p=0.003). The addition of 100μM DZX to CPG prevented this detrimental effect (p=0.0045 vs CPG alone; p=0.879 vs Tyrode's). The addition of 100μM GLU reversed DZX's prevention of cell swelling due to CPG (p=0.002 vs CPG + DZX). Tyrode's solution with 100μM GLU alone produced no significant change in myocyte volume (p=0.053 vs Tyrode's).
Myocyte Contractility
After 10 and 20 minutes of re-exposure to Tyrode's, contractile function of cells exposed to Tyrode's was between 30-40% of baseline (Figures 7A, 7B). Cells exposed to CPG demonstrated a decline in contractile function at 10 minutes after re-exposure to Tyrode's (vs Tyrode's, VS p=0.043, PS=0.044, VR p=0.202) and at 20 minutes following re-exposure (vs Tyrode's, VS p=0.055, PS p=0.065, VR p=0.122). The addition of 100μM DZX to CPG demonstrated improvement in contractility at 10 minutes (vs CPG, VS p=0.870, PS p=1.0, VR p=0.994) and at 20 minutes following re-exposure (vs CPG, VS p=0.807, PS p=0.708, VR p=0.471) although not statistically different. Similarly, the addition of 100μM GLU was associated with further improvement in contractile function at 10 minutes (vs CPG+DZX, VS p=0.129, PS p=0.127, VR p=0.746) and at 20 minutes after re-exposure (vs CPG+DZX, VS p=0.625, PS p=0.953, VR p=0.737) although not statistically different.
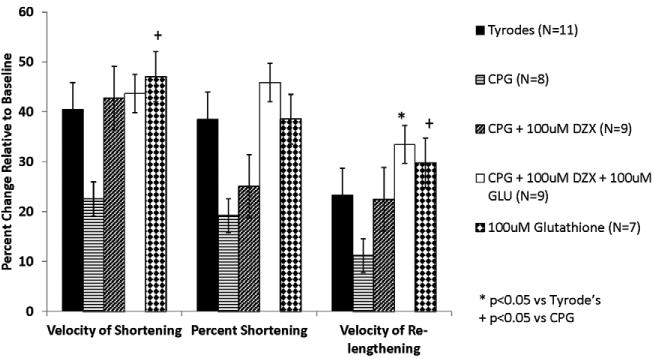
Figure 7. Glutathione improved myocyte contractility following re-exposure to Tyrode's.
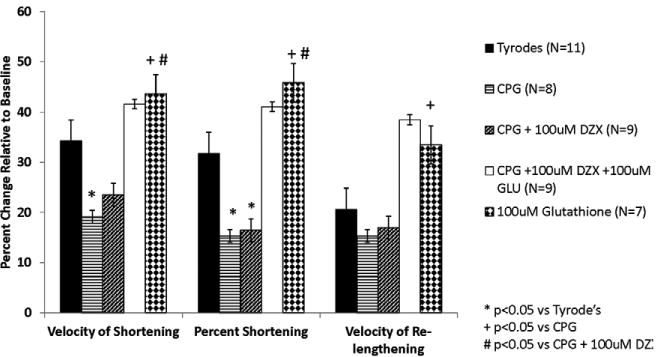
A: Myocyte contractility at 10 minutes following re-exposure to Tyrode's. Isolated myocytes were exposed to Tyrode's during time 0-20 minutes, test solutions during time 20-40 minutes, and Tyrode's during time 40-60 minutes. Glutathione was associated with an improvement in contractility with or without myocyte stress.
B: Myocyte contractility at 20 minutes following re-exposure to Tyrode's. Isolated myocytes were exposed to Tyrode's during time 0-20 minutes, test solutions during time 20-40 minutes, and Tyrode's during time 40-60 minutes. Glutathione was associated with an improvement in contractility with or without myocyte stress.
Exposure of cells to GLU alone resulted in contractile function similar to cells exposed to Tyrode's alone at 10 minutes (vs Tyrode's, VS p=0.679, PS p=0.792, VR p=0.734) and 20 minutes after re-exposure (VS p=0.896, PS p=0.957, VR p=0.848). Contractile function in the GLU alone group was significantly improved compared to cells exposed to CPG alone at 10 minutes (VS p=0.004, PS p= 0.007, VR p=0.026) and 20 minutes following re-exposure (VS p=0.026, PS p= 0.052, VR p=0.039).
Comment
The KATP channel opener, diazoxide, maintains myocyte volume homeostasis and contractility during stress (1-5). Although diazoxide is referred to as a KATP channel opener (very weak at the sarcolemmal channel and presumed activity at a mitochondrial channel), the components of the mitochondrial channel have not been defined. Recently, it has been suggested that the Kir (inward rectifying) subunit is a ROMK (renal outer medullary) potassium channel (26). Because the mitochondrial channel has not been cloned or defined and the measurement of ion flux across a mitochondrial membrane in myocardial tissue to confirm channel activity is not feasible, the investigation of the mechanism of action of diazoxide requires indirect methods.
Using genetic deletion, it has been determined that the cardioprotection provided by diazoxide requires the SUR1 subunit of the KATP channel and involves a non-sarcolemmal KATP channel location (8). Interestingly, DZX also inhibits SDH activity in mitochondria of SUR1 knockout mice suggesting that SDH and SUR1 are genetically distinct (27). An inverse relationship between SDH activity and KATP channel opening has also been proposed (20). The exact mechanism of cardioprotection by diazoxide has been proposed to involve the inhibition of SDH which may form a portion of the purported mKATP channel. This is consistent with the finding that the inhibition of SDH by other compounds (3-NPA, malonate) is cardioprotective (16, 18-21). Alternatively, SDH and SUR1 may be separate components of a purported mKATP channel or SDH may be a modulator of the mKATP channel.
Similar to findings of others, DZX inhibited SDH activity in the present study. Glutathione prevented the inhibition of SDH in a dose-dependent fashion and was utilized to examine the potential role of SDH inhibition in the cardioprotection provided by DZX during stress.
Diazoxide alone has been demonstrated to result in no change or a slight decrease in cellular volume; and DZX has been shown to prevent the detrimental myocyte swelling associated with CPG (2,3,8). In a detrimental fashion, glutathione prevented the cardioprotective volume homeostasis provided by DZX during stress (CPG) in the present study. Glutathione is a known reducing agent, an intrinsic anti-oxidant, and a potent scavenger of hydrogen peroxide (28, 29). It has been implicated in ischemic preconditioning and shown to provide cardioprotection (30-32). These beneficial properties would support a synergistic cardioprotection with that of diazoxide; however, this was not observed in the present study of myocyte volume response to stress. Alternatively, these findings suggest that glutathione was acting to prevent the beneficial inhibition of SDH by DZX.
In contrast to the volume observations, glutathione did not prevent the contractility benefit previously noted with DZX during the stress of CPG. Glutathione provided additional beneficial effects during the re-exposure to Tyrode's in the present study (when contractility was measured) that may be attributed to its antioxidant properties as reperfusion injury (or injury in the recovery period following stress) is known to involve reactive oxygen species.
Glutathione alone at 25μM and 100μM demonstrated greater SDH activity than both succinate alone and CPG+DZX+GLU. This may be related to a separate mechanism (the ability to reduce DCIP) of glutathione independent of its interaction with DZX. However, glutathione clearly had a detrimental effect on myocyte homeostasis when added to CPG + DZX and glutathione alone had no effect on myocyte volume suggesting that glutathione was acting via the prevention of the inhibition of SDH. In addition, a similar SDH assay (with DCIP as the terminal electron acceptor) has been utilized by others to document glutathione's ability to prevent the inhibition of SDH (22).
Exposure to CPG increased myocyte volume and decreased contractility that was prevented by DZX as reported previously (2,3,8). The findings that glutathione alone prevented the beneficial preservation of volume homeostasis by diazoxide (detrimental) and improved contractility following stress (beneficial), are in contrast to our previous work in which we found an inverse relationship between myocyte volume and contractility (1). This suggests that volume derangement represents only one mechanism of myocardial stunning.
The present study suggests that the inhibition of SDH is involved in the cardioprotection provided by DZX (particularly volume homeostasis) and supports a potential mitochondrial location of action of DZX. SDH may form a part of a purported mKATP channel or may be a modulator of mKATP activity. This is consistent with the work of others (20) who propose that mKATP channel opening is not a pre-requisite for SDH inhibition and by previous work from our laboratory that documented SDH inhibition by diazoxide in animals lacking the SUR1 KATP channel subunit (27).
Because the KATP channel uniquely provides cardioprotection during myocyte stress and is inhibited by ATP, it represents a tremendous potential target for pharmacologic therapy during myocyte stress. The present study further elucidates the mechanism of action of diazoxide which is vital prior to its adoption for clinical use as a cardioprotective agent during myocardial stress.
Study Limitations
Glutathione was utilized to prevent the inhibition of SDH by DZX, however, this potent antioxidant introduced another cardioprotective mechanism and complicated observed results.
It is important to note that cardiac myocytes exposed to physiologic Tyrode's solution demonstrated contractility values that were lower than previously published (75-90% of baseline), although the model has remained unchanged throughout these studies (1,2,4). Importantly, however, the same relationships in contractility were observed between control Tyrode's, stress (CPG), and stress with diazoxide as in previous work.
Acknowledgments
This study was supported by AHA Grant in Aid 09GRNT202045 (JSL), Thoracic Surgery Foundation for Research and Education Nina Starr Braunwald Career Award (JSL), NIH RO1HL098182-01A1 (JSL), and NIH T325T32HL007776 (MMA).
Abbreviations and Acronyms
- 3-NPA
3-Nitropropionic Acid
- 5-HD
5-Hydroxydecanoate
- CPG
cardioplegia
- DCIP
2,6-dichloroindophenol
- DZX
Diazoxide
- EDTA
K2 – ethylene diamine tetraacetic acid potassium
- GLU
Glutathione
- HEPES
N-[2-hydroxyethyl] piperazine-N-[4-butanesulfonic acid]
- KATP
Adenosine triphosphate - sensitive potassium
- KCN
potassium cyanide
- Kir 6.2
potassium inward rectifying subunit 6.2
- mKATP
mitochondrial KATP
- PMS
phenazine methosulfate
- PS
percentage shortening
- ROMK
renal outer medullary potassiums KATP- sarcolemmal KATP
- SDH
succinate dehydrogenaseSUR1- sulfonylurea receptor subunit 1
- VR
peak velocity of re-lengthening
- VS
peak velocity of shortening
Footnotes
Presented at Southern Thoracic Surgical Association 59th Annual Meeting, Naples, Florida, November 9, 2012.
References
- 1.Mizutani S, Prasad SM, Sellitto AD, Schuessler RB, Damiano RJ, Lawton JS. Myocyte volume and function in response to osmotic stress: observations in the presence of an adenosine triphosphate-sensitive potassium channel opener. Circulation. 2005;112:219–223. doi: 10.1161/CIRCULATIONAHA.104.523746. [DOI] [PubMed] [Google Scholar]
- 2.Mizutani S, Al-Dadah AS, Block JB, et al. Hyperkalemic cardioplegia-induced myocyte swelling and contractile dysfunction: prevention by diazoxide. Annals of Thoracic Surgery. 2006;81:154–159. doi: 10.1016/j.athoracsur.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 3.Prasad SM, Al-Dadah AS, Byrd GD, et al. Role of the sarcolemmal adenosine triphosphate sensitive potassium channel in hyperkalemic cardioplegia-induced myocyte swelling and reduced contractility. Annals of Thoracic Surgery. 2006;81:148–153. doi: 10.1016/j.athoracsur.2005.06.055. [DOI] [PubMed] [Google Scholar]
- 4.Al-Dadah AS, Voeller RK, Schuessler RB, Damiano RJ, Lawton JS. Maintenance of myocyte volume homeostasis during stress by diazoxide is cardioprotective. Annals of Thoracic Surgery. 2007;84:857–863. doi: 10.1016/j.athoracsur.2007.04.103. [DOI] [PubMed] [Google Scholar]
- 5.Maffit SK, Sellitto AD, Al-Dadah AS, Schuessler RB, Damiano R, Jr, Lawton JS. Diazoxide maintains human myocyte volume homeostasis during stress. Journal of the American Heart Association. 2012;1:1–8. doi: 10.1161/JAHA.112.000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garlid KD, Paucek P, Yarov-Yarovoy V, et al. Cardioprotective effect of diazoxide and its interactions with mitochondrial ATP-sensitive K+ channels: possible mechanism of cardioprotection. Circulatory Research. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 7.Flagg TP, Kurata HT, Masia R, et al. Differential structure of atrial and ventricular KATP: atrial KATP channels require SUR1. Circulation Research. 2008;103:1458–1465. doi: 10.1161/CIRCRESAHA.108.178186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sellitto AD, Maffit SK, Al-Dada AS, et al. Diazoxide maintenance of myocyte volume and contractility during stress: evidence for a non-sarcolemmal KATP channel location. Journal of Thoracic and Cardiovascular Surgery. 2010;140(5):1153–1159. doi: 10.1016/j.jtcvs.2010.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dos Santos P, Kowaltowski AJ, Laclau MN, et al. Mechanisms by which opening the mitochondrial ATP-Sensitive K+channel protects the ischemic heart. American Journal of Physiology Heart Circulation Physiology. 2001;283:H284–H295. doi: 10.1152/ajpheart.00034.2002. [DOI] [PubMed] [Google Scholar]
- 10.Das M, Parker JE, Halestrap AP. Matrix volume measurements challenge the existence of diazoxide/glibenclamide-sensitive KATP channels in rat mitochondria. Journal of Physiology. 2003;547(3):893–902. doi: 10.1113/jphysiol.2002.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McFalls EO, Liem D, Schoonderwoerd K, Lamers J, Sluiter W, Duncker D. Mitochondrial function: the heart of myocardial preservation. Journal of Laboratory and Clinical Medicine. 2003;142:141–149. doi: 10.1016/S0022-2143(03)00109-4. [DOI] [PubMed] [Google Scholar]
- 12.Rousou AJ, Ericsson M, Federman M, Levitsky S, McCully JD. Opening of mitochondrial KATP channels enhances cardioprotection through the modulation of mitochondrial matrix volume, calcium accumulation, and respiration. American Journal of Physiology Heart Circulation Physiology. 2004;287:H1967–H1976. doi: 10.1152/ajpheart.00338.2004. [DOI] [PubMed] [Google Scholar]
- 13.Van den Hoek TL, Shao Z, Li C, Schumacker PT, Becker LB. Mitochondrial electron transport can become a significant source of oxidative injury in cardiomyocytes. Journal of Molecular and Cellular Cardiology. 1997;29:2441–2450. doi: 10.1006/jmcc.1997.0481. [DOI] [PubMed] [Google Scholar]
- 14.Dzeja PP, Bast P, Ozcan C, et al. Targeting nucleotide-requiring enzymes: implications for diazoxide-induced cardioprotection. American Journal of Physiology Heart Circulation Physiology. 2003;254:H1048–H1056. doi: 10.1152/ajpheart.00847.2002. [DOI] [PubMed] [Google Scholar]
- 15.Shafer G, Portenhauser R, Trolp R. Inhibition of mitochondrial metabolism by the diabetogenic thiadiazine diazoxide. Action on succinate dehydrogenase and TCA-Cycle oxidations Biochemical Pharmacology. 1971;20:1271–1280. doi: 10.1016/0006-2952(71)90358-3. [DOI] [PubMed] [Google Scholar]
- 16.Hanley PJ, Mickel M, Loffler M, Brandt U, Daut J. KATP channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. Journal of Physiology. 2002;542(3):735–741. doi: 10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim KHH, Javadov SA, Das M, Clarke SJ, Suleiman MS, Halestrap AP. The effects of ischemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. Journal of Physiology. 2002;545(3):961–974. doi: 10.1113/jphysiol.2002.031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ockaili RA, Bhargave P, Kukreja RC. Chemical preconditioning with 3-nitropropionic acid in hearts: role of mitochondrial KATP channel. American Journal of Physiology Heart Circulation Physiology. 2001;280:H2406–H2411. doi: 10.1152/ajpheart.2001.280.5.H2406. [DOI] [PubMed] [Google Scholar]
- 19.Busija DW, Katakam P, Rajapakse NC, et al. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Research Bulletin. 2005;66:85–90. doi: 10.1016/j.brainresbull.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 20.Wojtovich AP, Brookes PS. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochimica et Biophysica Acta. 2008;1777:882–889. doi: 10.1016/j.bbabio.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ardehali H, Chen Z, Ko Y, Mejia-Alvarez R, Marban E. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. PNAS. 2004;101(32):11880–11885. doi: 10.1073/pnas.0401703101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muraoka S, Miura T. Inactivation of mitochondrial succinate dehydrogenase by adriamycin activated by horseradish peroxidase and hydrogen peroxide. Chemico-Biological Interactions. 2003;145:67–75. doi: 10.1016/s0009-2797(02)00239-9. [DOI] [PubMed] [Google Scholar]
- 23.Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Guide to Care and Use of Laboratory Animals. 8th. Institutes for Laboratory Animal Research, Division on Earth and Life Studies, National Research Council, National Institutes of Health; 2010. [Google Scholar]
- 24.Sellitto AD, Al-Dadah AS, Schuessler RB, Nichols CG, Lawton JS. An open sarcolemmal adenosine triphosphate-sensitive potassium channel is necessary for detrimental myocyte swelling secondary to stress. Circulation. 2011;124:S70–S74. doi: 10.1161/CIRCULATIONAHA.110.012039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drewnowska K, Clemo HF, Baumgarten CM. Prevention of myocardial intracellular edema induced by St. Thomas' Hospital cardioplegic solution. Journal of Molecular Cellular Cardiology. 1991;23:1215–1221. doi: 10.1016/0022-2828(91)90079-2. [DOI] [PubMed] [Google Scholar]
- 26.Foster DB, Ho AS, Rucker J, et al. Mitochondrial ROMK channel is a molecular component of mitoKATP. Circ Res. 2012;111:446–454. doi: 10.1161/CIRCRESAHA.112.266445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anastacio MM, Kanter EM, Keith AD, Schuessler RB, Nichols CG, Lawton JS. The inhibition of succinate dehydrogenase by diazoxide is independent of the KATP channel subunit SUR1 (abstract) Journal of the American College of Surgeons. 2012;215(3 Supplement):535–536. doi: 10.1016/j.jamcollsurg.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blaustein A, Deneke SM, Stolz RI, Baxter D, Healey N, Fanburg BL. Myocardial glutathione depletion impairs recovery after short periods of ischemia. Circulation. 1989;80:1449–1457. doi: 10.1161/01.cir.80.5.1449. [DOI] [PubMed] [Google Scholar]
- 29.Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of myocardial antioxidants in ischemiareperfusion injury. Cardiovascular Research. 2000;47:446–456. doi: 10.1016/s0008-6363(00)00078-x. [DOI] [PubMed] [Google Scholar]
- 30.Das DK, Engelman RM, Kimura Y. Molecular adaptation of cellular defenses following preconditioning of the heart by repeated ischemia. Cardiovascular Research. 1993;27:578–584. doi: 10.1093/cvr/27.4.578. [DOI] [PubMed] [Google Scholar]
- 31.Yoshida T, Watanabe M, Engelman DT, et al. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. Journal of Molecular and Cellular Cardiology. 1996;28:1759–1767. doi: 10.1006/jmcc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 32.Aldakkak M, Stowe DF, Heisner JS, Riess ML, Camara AKS. Adding ROS quenchers to cold K+ cardioplegia reduces superoxide emission during 2-hour global cold cardiac ischemia. Journal of Cardiovascular Pharmacology and Therapeutics. 2011;17(1):93–101. doi: 10.1177/1074248410389815. [DOI] [PMC free article] [PubMed] [Google Scholar]