

Background: KRIT1 FERM domain can bind Rap1 GTPase and HEG1 cytoplasmic tail.
Results: We solved the crystal structure of the KRIT1-Rap1-HEG1 ternary complex and show how KRIT1 F2 subdomain contributes to Rap1 binding specificity.
Conclusion: The structure rationalizes the capacity of KRIT1 to form this ternary complex.
Significance: We reveal the structure of Rap1 in complex with HEG1-bound KRIT1 and identify a novel determinant of Rap1 specificity.
Keywords: Cell Adhesion, Cell Junctions, Crystal Structure, Small GTPases, Vascular Biology
Abstract
Loss of function mutation in Krev interaction trapped 1 (KRIT1) causes autosomal dominant familial cerebral cavernous malformations and disrupts cardiovascular development. The biological function of KRIT1 requires that its FERM (band 4.1, ezrin, radixin, moesin) domain physically interact with both the small GTPase Rap1 and the cytoplasmic tail of the Heart of glass (HEG1) membrane anchor. In this study, we show that the KRIT1 FERM domain can bind both Rap1 and HEG1 simultaneously, and we solved the crystal structure of the KRIT1-Rap1-HEG1 ternary complex. Rap1 binds on the surface of the F1 and F2 subdomains, in an interaction that leaves its Switch II region accessible to other potential effectors. HEG1 binds in a hydrophobic pocket at the KRIT1 F1 and F3 interface, and there is no overlap with the Rap1-binding site. Indeed, the affinity of KRIT1 or the KRIT1-Rap1 complex for HEG1 is comparable (Kd = 1.2 and 0.96 μm, respectively) showing that there is no competition between the two sites. Furthermore, analysis of this structure revealed a specific ionic interaction between the F2 lobe of KRIT1 and Rap1 that could explain the remarkable Rap1 specificity of KRIT1. This structural insight enabled design of KRIT1(K570I), a mutant that binds Rap1 with 8-fold lower affinity and exhibits increased binding to HRas. These data show that HEG1 can recruit the Rap1-KRIT complex to the plasma membrane where Rap1's Switch II region remains accessible and reveals an important determinant of KRIT1's specificity for Rap1.
Introduction
Cell-cell and cell-extracellular matrix interactions are fundamental to the development of multicellular organisms, and genetic defects that affect cell adhesion can lead to disease. Cerebral cavernous malformations are relatively common, affecting 1 in 200 individuals, and those with the severe familial variety are afflicted by bleeding from weakened cerebral vessels resulting in chronic headaches, epilepsy, and hemorrhagic stroke. These cerebrovascular lesions, which can also arise outside the brain (1), are thought to be due to defective endothelial cell (EC)2 junctions (2, 3). CCMs occur in both sporadic and autosomal dominant inherited forms, and the latter are due to heterozygous mutations at three loci, CCM1 (KRIT1), CCM2 (OSM), and CCM3 (PDCD10) (4–7). KRIT1 protein is expressed in EC (8), and mice lacking KRIT1 die of vascular defects (9). Moreover, KRIT1 is localized in EC junctions, and this localization is mediated by Rap1 (10). EC monolayers depleted of KRIT1 exhibit increased permeability, and Rap1 is unable to reverse this effect; thus KRIT1 is required for the stabilizing effect of Rap1 on EC junctions.
KRIT1 protein contains three NPX(Y/F) motifs, four ankyrin repeats, and a C-terminal FERM domain. It was first identified as a Rap1-binding protein by yeast two-hybrid screen (11), and this interaction was shown to occur at the FERM domain (12). The interaction between Rap1 and its effector protein KRIT1 plays an important role in stabilization of EC junctions and regulation of cell-cell junction processes (10, 13). KRIT1 associates with microtubules (8), and Rap1 binding to the FERM domain frees KRIT1 from microtubules, enabling KRIT1 to localize to cell-cell junctions and to stabilize those junctions (14). The crystal structure of the KRIT1 FERM domain in complex with Rap1 shows that it binds the F1 lobe of KRIT1 in a GTPase-ubiquitin-like fold interaction but that it also binds the F2 lobe by a novel interaction (15). However, KRIT1 binds with 10-fold higher affinity to Rap1 versus HRas (16), but how it achieves its remarkable Rap1-specificity is not understood.
In zebrafish, loss of krit1 or heg (heart of glass) function leads to aberrant vascular morphogenesis and cardiac dilatation (17, 18). In mice, homozygous null mutation of HEG1 or KRIT1 leads to a similar vascular phenotype (19). These data suggest that KRIT1 and HEG1 interact genetically and play major roles in regulating vascular development and integrity. Indeed, KRIT1 binds the HEG1 cytoplasmic tail with Kd = 1.2 μm (20), and the structure of this assembly shows binding of the HEG1(Tyr1380–Phe1381) in a hydrophobic groove at the KRIT1 F1 and F3 subdomains interface. The KRIT1-HEG1 interaction is necessary for KRIT1 targeting to EC junctions and for cardiovascular development in zebrafish, establishing HEG1 as a membrane anchor of the CCM complex.
KRIT1 has a central role organizing and localizing the CCM protein complex, which has an essential role in the regulation of endothelial cell-cell junctions. Here, we show that KRIT1 can bind to both Rap1 and HEG1 simultaneously as follows: (i) the affinity of KRIT1 and KRIT1-Rap1 for HEG1 is very similar, and (ii) the KRIT1-Rap1 complex is not affected by the presence of HEG1 in size exclusion chromatography (SEC). Therefore, Rap1 and HEG1 neither compete nor bind cooperatively to KRIT1 FERM domain. We report the crystal structure of the KRIT1-Rap1-HEG1 ternary complex, and both Rap1 and HEG1 interact at nonoverlapping sites on the F1 lobe of the KRIT1 FERM domain. No major conformational changes that alter the others' binding site are observed after comparison of the KRIT1-Rap1-HEG1 ternary complex with the KRIT1-Rap1 and KRIT1-HEG1 structures, thus explaining the lack of competition or interaction between the two FERM domain ligands. Furthermore, KRIT1 is a Rap1-specific effector protein, and structure-based mutagenesis confirmed that the F2 lobe of the FERM domain contributes to the Rap1 specificity over HRas. More specifically, a salt bridge between KRIT1 Lys570 and Rap1 Glu45 confers specificity, and the less favorable binding to HRas Val45 can be increased by KRIT1(K570I) mutation. These data reveal the structural basis of a KRIT1-Rap1-HEG1 ternary complex where both ligands interact on different surfaces of the F1 subdomain lobe without affecting each others' binding interface and how the F2 subdomain contributes to the Ras GTPase recognition specificity.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human KRIT1 FERM domain, residues 417–736, wild-type, K570I, and K570E were expressed and purified as described previously (14). Briefly, KRIT1 was cloned into the expression vector pLEICS-07 (Protex, Leicester, UK) and expressed in Escherichia coli BL21 Star (DE3) (Invitrogen). Recombinant His-tagged KRIT1 was purified by nickel-affinity chromatography; the His tag was removed by cleavage with tobacco etch virus protease overnight, and the protein was further purified by cation exchange chromatography. The protein concentration was assessed using the A280 extinction coefficient of 45,090 m−1.
Glutathione S-transferase (GST)-KRIT1 FERM domains wild-type, K570I, and R452E were cloned into the expression vector pGEX-4T-1. The recombinant proteins were purified on glutathione-Sepharose 4 Fast Flow beads according to the manufacturer's instructions (GE Healthcare).
Human Rap1 isoform Rap1b (residues 1–167) and HRas (residues 1–189) cloned into pTAC vector in the E. coli strain CK600K was the generous gift of Professor Alfred Wittinghofer (Max Planck Institute of Molecular Physiology, Germany). The Rap1 G12V, E45K, F64A, and M67A mutants were generated by site-directed mutagenesis. Cultures were grown at 37 °C until they reached an A600 of 0.8, transferred to an 18 °C shaker for an hour, and then induced with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside overnight. Untagged GTPases were purified by ion exchange, followed by Superdex-75 (26/60) gel filtration (GE Healthcare). The column was pre-equilibrated and run with 20 mm Tris, 50 mm NaCl, 3 mm MgCl2, and 2 mm DTT, pH 8, in a 6 °C cabinet. Nucleotide exchange for GMP-PNP was achieved as described by Gorzalczany et al. (21), and the final sample was dialyzed into isothermal titration calorimetry (ITC) buffer (20 mm sodium phosphate, 50 mm NaCl, 3 mm MgCl2, pH 6.5, containing 0.2 mm GMP-PNP). The protein concentration was assessed using the CB protein assay (Calbiochem). Synthetic human HEG1 C26 peptide (residues 1356–1381) was purchased from GenScript.
KRIT1 Protein Interaction Assays for Rap1 and HRas
Equal amounts of glutathione S-transferase (GST)-KRIT1 wild-type, K570I, and R452E mutant immobilized on glutathione-Sepharose beads were incubated with 10 μm GMP-PNP-loaded Rap1 and HRas. The buffer used was 20 mm sodium phosphate, 50 mm NaCl, 3 mm MgCl2, pH 6.5, containing 0.2 mm GMP-PNP and 0.2% Triton X-100. The mixture was incubated for 1 h on a rotary shaker in the cold room, and the beads were washed three times with 0.3 ml of ice-cold buffer and finally resuspended in sample buffer for SDS-PAGE analysis. The gels were stained with Coomassie Blue, which is a strong infrared fluorophore, and scanned at 700 nm using a fluorescence imaging system (Odyssey system, LiCoR), and the band intensity was quantified.
KRIT1 Binding to Rap1 and HRas by Size Exclusion Chromatography
SEC of recombinant KRIT1 FERM domain with Rap1 and HRas was performed using a Superdex-75 (10/300) GL (GE Healthcare) at room temperature. The proteins, 50 μm each, were mixed in a volume of 100 μl and incubated for 30 min at room temperature before loading onto the column, which was pre-equilibrated with and run with 20 mm Tris, 150 mm NaCl, 3 mm MgCl2, pH 7.4, 2 mm DTT (TBS).
For HEG1 competition assay, TBS containing a mixture of 50 μm of KRIT1 and 50 μm of Rap1 was incubated in the presence or absence of 200 μm HEG1 peptide for 30 min at room temperature before loading onto the Superdex-75 column.
Purification of the KRIT1-Rap1 Complex
Equal molar concentrations of KRIT1 FERM domain and GMP-PNP-loaded Rap1 were mixed and loaded on a Superdex-75 (26/60). The column was pre-equilibrated and run with 20 mm Tris, 50 mm NaCl, 3 mm MgCl2, and 2 mm DTT, pH 8, in a 6 °C cabinet. The final complex was concentrated to 9.5 mg/ml as determined by the CB protein assay (Calbiochem).
Isothermal Titration Calorimetry
ITC data were collected using a VP-ITC microcalorimeter (MicroCal Ltd., Northampton, MA) at 25 °C and analyzed by fitting to a single-site binding equation using MicroCal Origin software.
For HEG1 binding, 0.4 mm of synthetic human HEG1 C26 peptide was titrated from the syringe into the sample cell containing 40 μm KRIT1 FERM domain or KRIT1-Rap1 complex. Proteins and peptides were dialyzed into 20 mm sodium phosphate, 150 mm NaCl, 3 mm MgCl2, pH 6.5, before performing the experiment.
For Rap1 binding, 0.8 mm GMP-PNP-bound Rap1 was titrated from the syringe into the sample cell containing 67 μm KRIT1 FERM domain wild-type or K570I mutant. All proteins were dialyzed into 20 mm sodium phosphate, 50 mm NaCl, 3 mm MgCl2, pH 6.5, containing 0.2 mm GMP-PNP before performing the experiment.
Crystallization of the KRIT1-Rap1 and KRIT1-Rap1-HEG1 Complex
The purified KRIT1-Rap1 complex was used for crystallization in the absence or presence of equimolar concentrations of HEG1 peptide (3 mm stock in water). Crystals were grown at 5 °C using the sitting-drop method by mixing equal volumes of protein complex and reservoir solution (2 + 2 μl). The reservoir solution contained 15% PEG 2,000 MME, 100 mm Tris, 100 mm KCl, pH 8.5. The crystals were briefly transferred to reservoir solution containing 20% glycerol before freezing in liquid nitrogen.
Structure Determination
Diffraction data for the KRIT1-Rap1 and KRIT1-Rap1-HEG1 complexes were collected at Diamond Light Source beamlines I02 and I24, respectively. The data were processed with XDS (22).
For the KRIT1-Rap1 complex, initial phases were determined using the structure of human Rap1A excluding the nucleotide (Protein Data Bank (PDB) code 1c1y) with Phaser, and all the following software used are part of the CCP4 software suite (23). We then docked GTP into the model and ran cycles of maximum likelihood refinement in Refmac5. It became very apparent from that point that the Rap1 bound to GMP-PNP was present, and the quality of the map improved dramatically. Three cycles of buccaneer built parts of KRIT1, and this model was then input into ARP/wARP (49 cycles = Rwork of 29.3 and Rfree of 33.7). The model was then optimized using cycles of manual refinement with Coot and maximum likelihood refinement in Refmac5. The final Ramachandran plot shows 98.5% of residues in favored regions, 1.3% in allowed regions, and 0.2% in outlier region, as defined by PROCHECK. The crystal structure has been submitted to the Protein Data Bank with the accession number 4hbo.
The structure of the KRIT1-Rap1-HEG1 was solved by molecular replacement using Phaser with the structure of KRIT1-Rap1 complex. The HEG1 sequence was built into the density using Coot and the model refined as described above. The final Ramachandran plot shows 95.6% of residues in favored regions, 3% in allowed regions, and 1.5% in outlier regions, as defined by PROCHECK. The crystal structure has been submitted to the Protein Data Bank with the accession number 4hbq.
Differential Scanning Calorimetry
Differential scanning calorimetry experiments were performed at a scanning rate of 1 K/min under 3.0 atm of pressure using N-DSC II differential scanning calorimeter (Calorimetry Sciences Corp, Provo, UT). Differential scanning calorimetry samples contained phosphate-buffered saline (PBS), pH 7.4, and 1.0 mg/ml KRIT1 wild-type or K570I FERM domain.
RESULTS
KRIT1 Forms a Ternary Complex with Rap1 and HEG1
Using SEC, we found that the KRIT1 FERM domain formed a complex with GMP-PNP (a GTP analog)-bound Rap1 but not with GDP-bound Rap1 (Fig. 1A). Moreover, we found that the complex also formed in the presence of a 4-fold molar excess of HEG1 cytoplasmic tail peptide (Fig. 1B), suggesting that HEG1 does not compete for Rap1 binding to KRIT1. We purified the KRIT1/Rap1·GMP-PNP complex using SEC and quantified the binding affinity of the complex for the C terminus of the HEG1 cytoplasmic tail by ITC. The KRIT1 FERM domain in complex with Rap1 bound HEG1 tightly (Kd = 0.96 ± 0.14 μm) (Fig. 1C), very similar to the affinity of HEG1 for the isolated KRIT1 FERM domain (Kd = 1.2 ± 0.14 μm) (Fig. 1D). Thus, Rap1 has little effect on the affinity of the KRIT1 FERM domain for HEG1. Therefore, the three proteins can form a ternary complex.
FIGURE 1.
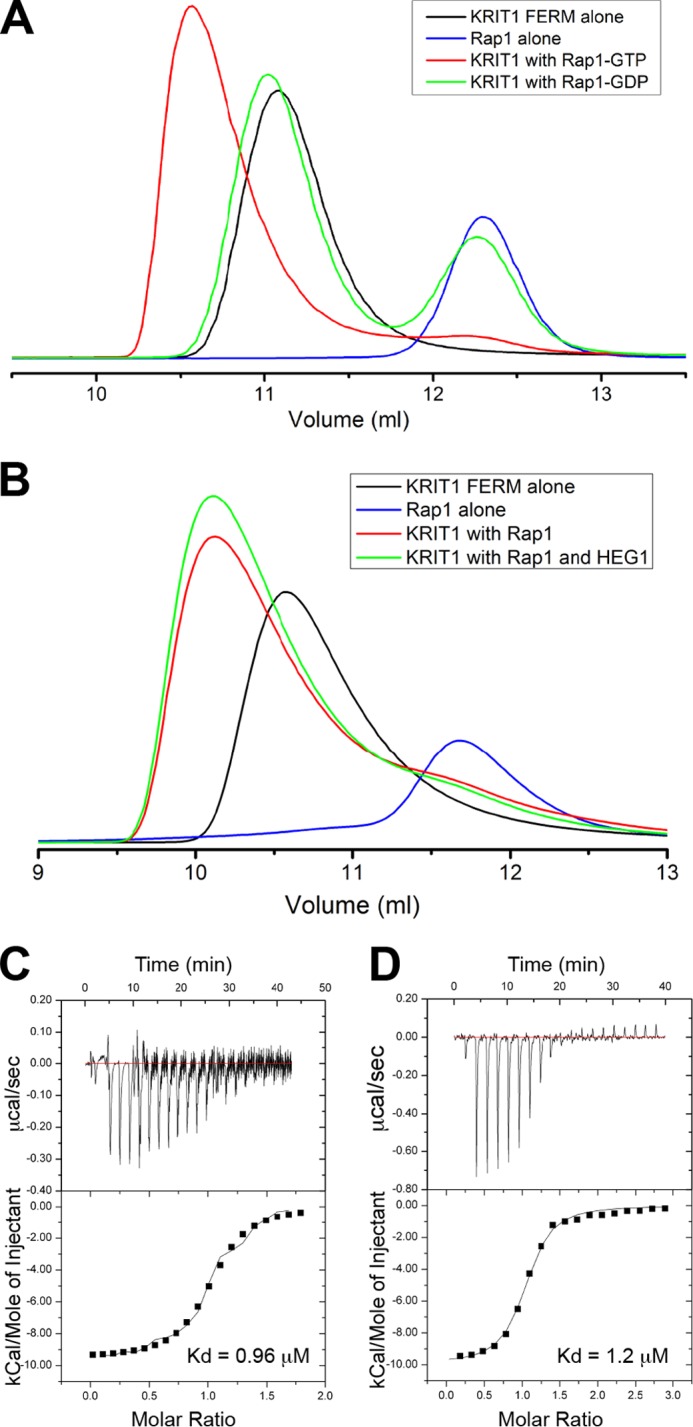
KRIT1-Rap1 binds HEG1, and they form a ternary complex. A and B, binding of the KRIT1 FERM domain to Rap1 was analyzed on a Superdex-75 (10/300) GL gel filtration column at room temperature. A, incubation of KRIT1 with Rap1 bound to GDP resulted in no complex formation, and most of the KRIT1 and Rap1 polypeptides remained in the free form. However, preincubation with Rap1 bound to GMP-PNP resulted in formation of a KRIT1-Rap1 complex. B, incubation of KRIT1 and Rap1-GTP (50 μm each) in the presence or absence 200 μm synthetic HEG1 cytoplasmic tail peptide. There is no competition between Rap1 and HEG1 for KRIT1 binding. C and D, calorimetric titration of HEG1 peptide out of the syringe into the following: C, KRIT1-Rap1 (Kd = 0.96 μm), and D, KRIT1 alone in the sample cell (Kd = 1.2 μm, re-plotted for comparison purposes (14)). For the titration of HEG1 into KRIT1-Rap1 the base line is noisy. We performed this experiment three times and obtained similar results.
Structure of the KRIT1-Rap1-HEG1 Ternary Complex and KRIT1-Rap1 Binary Complex
The human KRIT1 FERM domain (residues 417–736) in complex with human Rap1 (residues 1–167) was purified by SEC and crystallized in the presence or absence of a synthetic peptide containing the C-terminal 26 residues of the human HEG1 cytoplasmic tail. We determined the complex structures for KRIT1-Rap1 and KRIT1-Rap1-HEG1 to 1.67 and 1.95 Å resolution, respectively (Table 1). Both crystals have the same space group, almost identical cell dimensions, and the asymmetric unit contains one complex. In both high resolution structures we did not observe good electron density for the Rap1 residues 63–65 and higher than average B-factor for residues 60–69, or so-called Switch II.
TABLE 1.
Data collection and refinement statistics for the KRIT1 FERM domain in complex with (i) Rap1b and (ii) Rap1b and the HEG1 cytoplasmic tail
| Rap1b (4hdo) | Rap1b and HEG1 (4hdq) | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c | 57.9, 77.8, 58.9 Å | 57.3, 77.4, 58.6 Å |
| α, β, γ | 90.0, 91.2, 90.0° | 90.0, 95.6, 90.0° |
| Resolution | 50.0 to 1.67 Å (1.77 to 1.67 Å)a | 30 to 1.95 Å (2.07 to 1.95 Å)a |
| Rmerge | 4.6 (35.3) | 6.6 (34.7) |
| I/σI | 31.3 (4.4) | 21.0 (3.9) |
| Completeness | 96.0% (80.0%) | 99.1% (96.8%) |
| Redundancy | 3.6 (2.8) | 3.3 (2.9) |
| Refinement | ||
| Resolution | 32.4 to 1.67 Å | 29.2 to 1.95 Å |
| No. of reflections | 55,017 | 35,112 |
| Rwork/Rfree | 21.3/23.1 | 20.4/26.2 |
| No. of atoms | 4167 | 4014 |
| Protein | 3860 | 3883 |
| Ligand/ion | 39 | 39 |
| Water | 268 | 92 |
| B-factors | 23.4 | 38.0 |
| Protein | 23.0 | 38.1 |
| Ligand/ion | 20.4 | 34.7 |
| Water | 28.7 | 34.5 |
| Root mean square deviations | ||
| Bond lengths | 0.005 Å | 0.02 Å |
| Bond angles | 1.006° | 1.943° |
a The highest resolution shell is shown in parentheses.
As reported previously, the FERM domain of KRIT1 is arranged as a cloverleaf as observed for other ERM proteins (15, 20), and Rap1 binds at the F1 and F2 interface of KRIT1 (Fig. 2A) (15). The F1 region of KRIT1 has a ubiquitin fold similar to other Ras-binding domains (RBD). The major binding determinant is mediated by main-chain and side-chain interactions across a newly formed anti-parallel β-sheet formed by strand β2 from Rap1, also called Switch I, and strand β2A from the KRIT1 RBD (Fig. 2B). This region was previously shown to be important by mutagenesis (14, 15).
FIGURE 2.

Crystal structure of the ternary complex between the KRIT1 FERM domain, Rap1, and the HEG1 cytoplasmic tail. A, schematic representation of the KRIT1 FERM domain bound to Rap1 and the HEG1 cytoplasmic tail. The KRIT1 FERM domain contains three subdomains; F1 (green), F2 (red), and F3 (red). The F1 helix α2A is not seen in the other ubiquitin fold colored in blue. The HEG1 cytoplasmic tail (yellow) binds at the F1-F3 interface. Rap1 (magenta) bound to GMP-PNP binds at the surface of F1-F2. B, KRIT1 and Rap1 complex shows an extended binding interface involving both F1 and F2 regions of KRIT1. The KRIT1 β2A strand stacks against the β2 strand of Rap1 to form an extended β-sheet. The KRIT1 region surrounding Arg452 makes multiple water-mediated hydrogen bonds with Rap1. The KRIT1 residues Leu526 and Pro525 at the bottom of helix α1B form a hydrophobic surface that stacks with Ile27 of Rap1. A distal salt bridge is found between KRIT1 Lys570 and Glu45 of Rap1.
The crystal structure of the KRIT1-Rap1-HEG1 ternary complex showed binding of the C-terminal HEG1 sequence in a hydrophobic pocket at the F1 and F3 interface of the FERM domain (Fig. 2A). In brief, HEG1 contacts three regions on the FERM domain as follows: (i) polar interactions with the F1 lobe; (ii) hydrophobic area on the F3 lobe involving helix α1C, and (iii) the helix α2A closing the interface between F1 and F3 (Fig. 2A). As reported previously for the structure of the FERM domain in the absence of Rap1 (PDB = 3u7d) (20), the last three residues of HEG1 (1379DYF1381) show the best electron density, and the N-terminal region exits the pocket making very few contacts with KRIT1. In the high resolution structure reported here, a poor electron density was observed for the side chains of Arg1378 and Arg1377 and no density for the first 21 residues, suggesting that they remain unstructured in the complex. Thus, as in the structure of the Rap1-free FERM domain, the C-terminal HEG1 Tyr-Phe dipeptide is critical for HEG1 binding to KRIT1.
Rap1 Binding to KRIT1 FERM Domain Does Not Induce a Major Conformational Change in the HEG1 Binding Pocket
Because we had previously solved the structure of the KRIT1 FERM domain in complex with HEG1, we had the opportunity to examine the effect of Rap1 binding on the structure of the FERM domain and on the structure of the HEG1 binding pocket. Overall, the structure of the KRIT1 FERM domain bound to Rap1 and HEG1 reported here is very similar to the KRIT1-HEG1 structure reported previously (root mean square deviation = 0.79 Å for all atoms with PDB 3u7d) (Fig. 3A). The differences are small and clustered at the F1 and F2 surface where Rap1 binds, but the HEG1 binding pocket is essentially the same whether Rap1 is bound or not (Fig. 3B). These data show that Rap1 binding to KRIT1 does not induce large conformational changes in the FERM domain nor cause steric clashes that compromise HEG1 binding (Fig. 3, A and B), and both Rap1 and HEG1 can bind simultaneously to KRIT1. The similarity of the interactions of HEG1 with the Rap1 bound and free FERM domain explained the lack of effect of Rap1 on the affinity of HEG1 binding to KRIT1 (Fig. 1, C and D).
FIGURE 3.

Rap1 or HEG1 binding to the KRIT1 FERM domain does not induce conformational changes affecting binding at the other site. A, comparison of the KRIT1-HEG1 complex with the ternary complex structure. Only the FERM domain is shown to highlight that both structures are very similar. B, same superimposition as A with a focus on the HEG1 binding pocket located at the KRIT1 F1 and F3 interface. Overall the binding pocket is not affected by Rap1 binding. C, comparison of the KRIT1-Rap1 complex with the ternary complex structure. Only the FERM domain is shown to highlight that both structures are almost identical. D, same superimposition as C with a focus on the Rap1 binding interface at the KRIT1 F1/RBD and F2 surface. Overall the binding surface on KRIT1 is not affected by HEG1 binding.
HEG1 Binding to KRIT1 FERM Domain Does Not Induce a Conformational Change in the RBD Region
Here, we report the crystal structure of the KRIT1 FERM domain bound to Rap1 in the absence or presence of the HEG1 cytoplasmic tail. Both structures were almost identical, root mean square deviation = 0.48 Å for all atoms, suggesting that HEG1 binding does not induce large conformational changes in the FERM domain (Fig. 3C). Moreover, the residues at the binding interface between Rap1 and KRIT1 were in almost identical positions in both structures (Fig. 3D). These data show that both HEG1 and Rap1 can bind simultaneously and that HEG1 binding does not induce large conformational changes in the Rap1-occupied FERM domain.
KRIT1 F2 Lobe and Rap1 Specificity
KRIT1 binds Rap1 more tightly than its paralog HRas (10–12, 16), and Lys31 of Rap1 is responsible for specificity of interaction with the RBD of RalGDS (24). In contrast, Glu31 in HRas confers specificity for c-Raf, and mutagenesis of either Rap1(K31E) or HRas(E31K) can reverse their specificity (25, 26). In the present structure, Lys31 of Rap1 does not make contact with KRIT1, and we hypothesized that the specificity for Rap1 comes from the F2 lobe contacts (Fig. 2B). We were not able to express the isolated KRIT1 F1 subdomain, and we therefore sought to test the idea that the F2 subdomain contributes to Rap1 specificity.
Helix α1B, which is located in the F2 lobe of KRIT1, also makes contact with Rap1 in the Switch I region. Both Pro525 and Leu526 at the bottom of helix α1B stack against Rap1 Ile27 accounting for an extra hydrophobic interaction (Fig. 2B). Furthermore, the side chain of KRIT1 Lys570 makes a hydrogen bond with Rap1 Glu45. Those two F2-mediated interactions and the large buried surface area upon complex formation could explain the high affinity of KRIT1 for Rap1 but not how the specificity for Rap1 versus HRas was determined.
The structure described in this study created the opportunity to examine the KRIT1 FERM domain specificity for Rap1 versus HRas; the amino acid sequences of the two GTPases are very similar exhibiting 57.5% identity for residues 1–167. A first look at the surface charge of both Rap1 and HRas as seen from the KRIT1 binding interface (Fig. 4B) shows very little difference. Analysis of the KRIT1-Rap1 interface using PDBePISA identifies many hydrogen bonds between the two proteins, and many residues are buried by formation of the complex as summarized in Fig. 4A. The difference between Rap1 (Fig. 4C) and HRas (Fig. 4D) within the KRIT1 binding interface is limited to four residues as highlighted in Fig. 4A. We noted that KRIT1 Lys570, which is located in the F2 lobe, makes a hydrogen bond with Rap1 Glu45 and that the equivalent residue in HRas is Val45. Therefore, we hypothesized that substituting KRIT1 Lys570 for a hydrophobic residue would make HRas binding more favorable by stacking with Val45 and Rap1 binding less favorable by loss of a hydrogen bond with Glu45.
FIGURE 4.
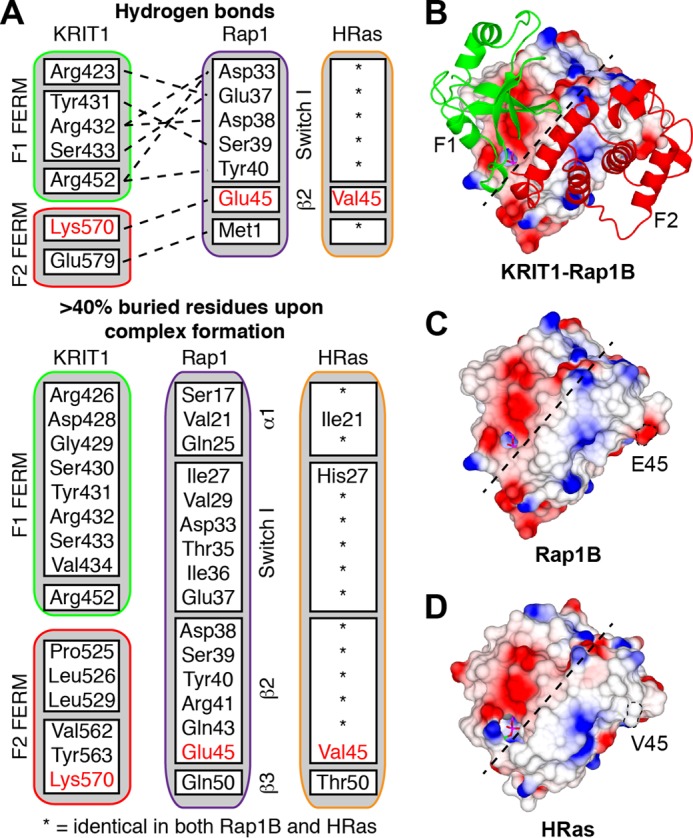
Rap1 binding interface for KRIT1 is highly homologous to HRas. A, summary of the KRIT1-Rap1 interactions as identified using PDBePISA. The upper section shows the hydrogen bonds between the two proteins, and the lower section shows the residues buried by formation of the complex. HRas is very similar to Rap1, and the residues that are different are highlighted. B, view of the KRIT1 F1 and F2 subdomains on the surface of Rap1. Both the F1 and F2 subdomains make contact with Rap1. C and D, surface electrostatic potential as follows: C, Rap1; and D, HRas (PDB 1lfd). The dotted line separates the binding interface for KRIT1 F1 and F2 subdomains as shown in B.
To analyze the specificity of the KRIT1 FERM domain for Rap1 versus HRas, we created a KRIT1(K570I) mutant and tested its interaction with both GTPases. KRIT1(K570I) was well folded as judged by its sharp melting temperature of 58 °C in differential scanning calorimetry (Fig. 5A). We first quantified the binding affinity of the FERM domain for Rap1 by ITC. The KRIT1 FERM domain wild-type bound Rap1 with a Kd = 0.36 ± 0.11 μm (Fig. 5B); however, the KRIT1(K570I) FERM bound with a Kd = 2.93 ± 0.77 μm, an ∼8-fold lower affinity (Fig. 5C).
FIGURE 5.

KRIT1(K570I) has reduced binding affinity for Rap1. A, KRIT1(K570I) FERM mutant does not disrupt protein folding. Differential scanning calorimetry results of FERM WT and K570I proteins exhibit narrowly defined melting points, indicating that they are well folded. B and C, calorimetric titration of Rap1, out of the syringe into the following: A, KRIT1 wild-type, and B, KRIT1(K570I) mutant in the sample cell (Kd = 0.36 and 2.93 μm, respectively). The binding affinity is reduced by 8-fold.
We then used SEC to evaluate the effect of the KRIT1(K570I) mutation on the interaction with Rap1. Both proteins formed a complex with Rap1 with a shift toward an increased volume of elution (Ve) with the KRIT1(K570I) mutant (Fig. 6A). Persistence of a complex through the SEC column implies that the proteins physically interact and equilibrate quickly enough to associate on a scale of minutes to hours. SEC is sensitive to both on- and off-rates; a differentiated early peak corresponds to high nanomolar binding, and an early shoulder usually corresponds to low-to-mid-micromolar affinity of binding (27). The SEC data are in agreement with the ITC data showing that KRIT1(K570I) binds Rap1 with lower affinity than wild type. Interestingly, the same experiment with HRas shows the opposite result (Fig. 6B). The wild-type FERM binds HRas, but the complex with the KRIT1(K570I) mutant shifts toward reduced Ve suggesting that its affinity for HRas is increased.
FIGURE 6.

KRIT1 is a Rap1-specific effector, and KRIT1 Lys570 is important for high affinity and specificity binding toward Rap1. A and B, binding of the KRIT1 FERM domain wild-type and FERM(K570I) to the following: A, Rap1, and B, HRas as analyzed on a Superdex-75 (10/300) GL gel filtration column at room temperature. A, incubation of KRIT1(K570I) with Rap1 results in complex formation with a shift toward increased Ve compared with wild-type KRIT1, suggesting a reduced affinity. B, in contrast, incubation of KRIT1(K570I) with HRas results in complex formation with a shift toward lower Ve compared with wild-type KRIT1, suggesting an increase in affinity. C, Coomassie stain of SDS-PAGE after KRIT1 pulldown. GST-KRIT1 wild-type pulls down Rap1 almost exclusively, whereas the KRIT1(K570I) pulls down both Rap1 and HRas. The KRIT1(R452E) does not bind either Rap1 or HRas. KRIT1 Lys570 is important for Rap1 binding specificity. D, quantification of Rap1 and HRas binding from three independent experiments such as shown in C. The KRIT1(K570I) mutation caused a 4-fold increase in HRas binding and a 25% reduction in Rap1 binding.
To learn more about the relative affinity of KRIT1 for both GTPases, we performed competition assays using affinity chromatography. Rap1, but not HRas, bound strongly to the wild-type KRIT1 FERM domain confirming that it is indeed a Rap1-specific effector (Fig. 6C). The KRIT1(K570I) exhibited binding to both GTPases with a 25% reduction in binding to Rap1 and a 4-fold increase in binding to HRas (Fig. 6D). As expected, the previously described KRIT1(R452E) mutant compromised binding to both GTPases (Rap1 binding Kd = 1.8 ± 0.25 and 67 ± 18.7 μm for KRIT1 wild-type and mutant respectively (14)). Our data show that indeed the F2 lobe of KRIT1 contributes to determining KRIT1 specificity for Rap1 versus HRas.
Rap1 Glu45 Determines KRIT1 Specificity
To further test the role of Rap1 Glu45 in KRIT1 specificity, we generated a Rap1 mutant where we reversed the charge of Glu45 to a lysine (Fig. 7A). We hypothesized that Rap1(E45K) would have reduced affinity for KRIT1 as it will be repulsed by KRIT1 Lys570. Using SEC, we observed formation of a complex with a large shift toward an increased Ve with the Rap1(E45K) (Fig. 7C) in comparison with wild-type Rap1 (Fig. 7B). We then generated a KRIT1(K570E) mutant with the aim to rescue binding. Indeed, using SEC with the KRIT1(K570E) and Rap1(E45K), we observed formation of a complex at approximately the same Ve (Fig. 7D) as the wild-type complex (Fig. 7B). Our data show that the ionic interaction between KRIT1 Lys570 and Rap1 Glu45 is important for high affinity binding and that charge reversal rescues binding.
FIGURE 7.
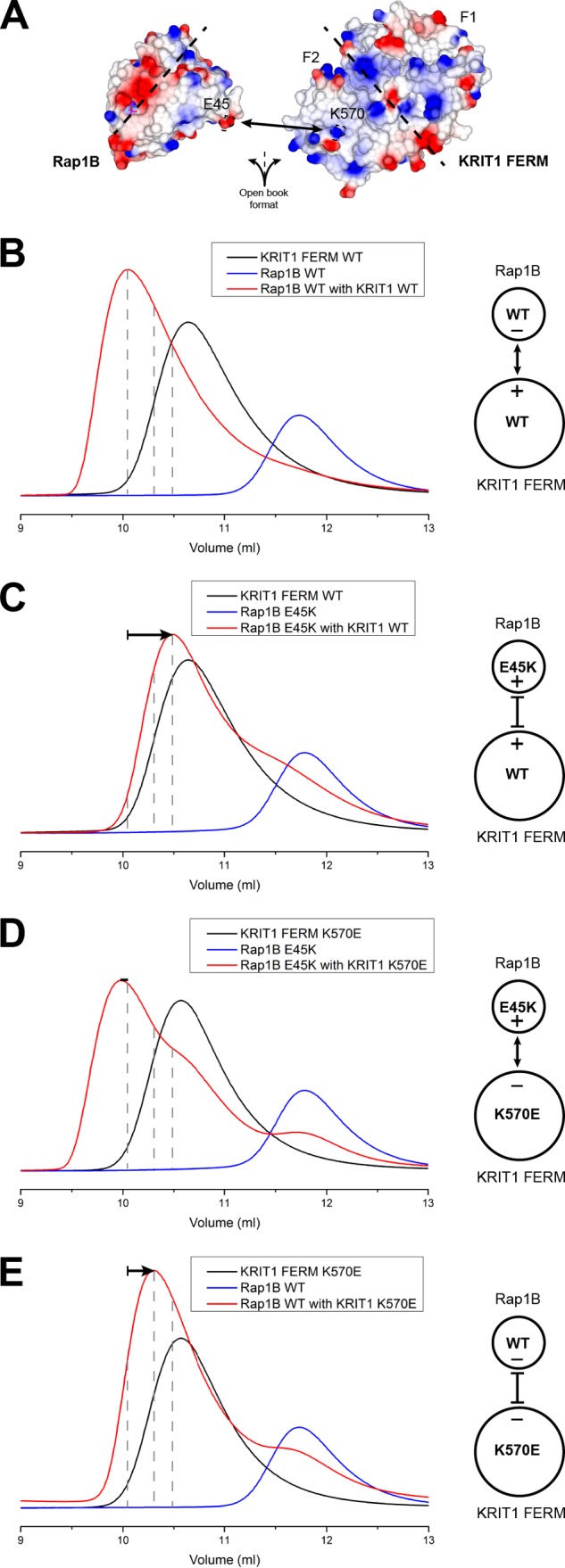
Ionic interaction between Rap1 Glu45 and KRIT1 Lys570 is important for high affinity binding. A, surface electrostatic potential of the Rap1 (left) and KRIT1 FERM (right) binding interface as open book view. The ionic interaction between Rap1 Glu45 and KRIT1 Lys570 is highlighted. The dotted line separates the binding interface for KRIT1 F1 and F2 subdomains. B–E, binding of the KRIT1 FERM domain to Rap1 as analyzed on a Superdex-75 (10/300) GL gel filtration column at room temperature. B, incubation of KRIT1 WT with Rap1 WT results in complex formation. C, incubation of KRIT1 WT with Rap1(E45K) results in complex formation with a shift toward increased Ve compared with wild-type KRIT1, suggesting a reduced affinity. D, incubation of KRIT1(K570E) with Rap(E45K) results in complex formation with a shift comparable with wild-type complex, suggesting binding has been recovered. E, incubation of KRIT1(K570E) with Rap1 WT results in complex formation with a shift toward increased Ve compared with wild-type KRIT1, suggesting a reduced affinity. The shift is less significant than C.
We also performed a SEC experiment using Rap1 wild type with the KRIT1(K570E) and also observed a shifted peak for the complex toward an increased Ve (Fig. 7E); however, the peak is less shifted in comparison with the complex of Rap1(E45K) with KRIT1 wild type (Fig. 7C). These data show that mutation of Rap1 Glu45 has a more drastic effect than mutation of KRIT1 Lys570 on the interaction, suggesting that Rap1 Glu45 plays an important role in KRIT1 specificity versus HRas Val45.
Rap1 Switch II Does Not Interact with KRIT1
In the both our crystal structures of KRIT1 in complex with Rap1, we did not observe good electron density for the Rap1 Switch II, suggesting that Switch II is flexible and not interacting with KRIT1 FERM domain (Fig. 8A). In the reported structure of KRIT1 in complex with Rap1(G12V) (15), hydrophobic interactions were observed between Rap1 Met67 and Phe64 with the KRIT1 F1 subdomain, similar to analogous Tyr64 in HRas complexes to other effectors like Nore1, PI3K, and PLC (28–30). Analysis of those HRas crystal structures showed clear electron density for Switch II in contrast with our structure. We superimposed other Rap1 structures with ours and modeled the missing residues in Switch II (Fig. 8A). We noticed that KRIT1 Tyr419 hydroxyl group points toward the usual positioning of Rap1 Phe64, possibly destabilizing Switch II. To test the role of Rap1 Switch II in the interaction and to be certain that crystal packing is not preventing normal positioning of Switch II, we created two alanine substitutions in this region, M67A and F64A, for comparison. Using SEC, we observed no clear shift in the Ve of the complex formed with the Rap1(M67A) mutant (Fig. 8B, green trace), suggesting that it does not play an important role in the interaction. Interestingly, the complex formed by the Rap1(F64A) mutant shows a very small shift toward higher Ve (Fig. 8B, pink trace), and this shift is very similar to the one observed for Rap1(G12V) (Fig. 8B, gray trace). We therefore concluded that the role of Switch II of Rap1 in the interaction is minor in comparison with the portion of Rap1 that interacts with the KRIT1 F2 lobe.
FIGURE 8.
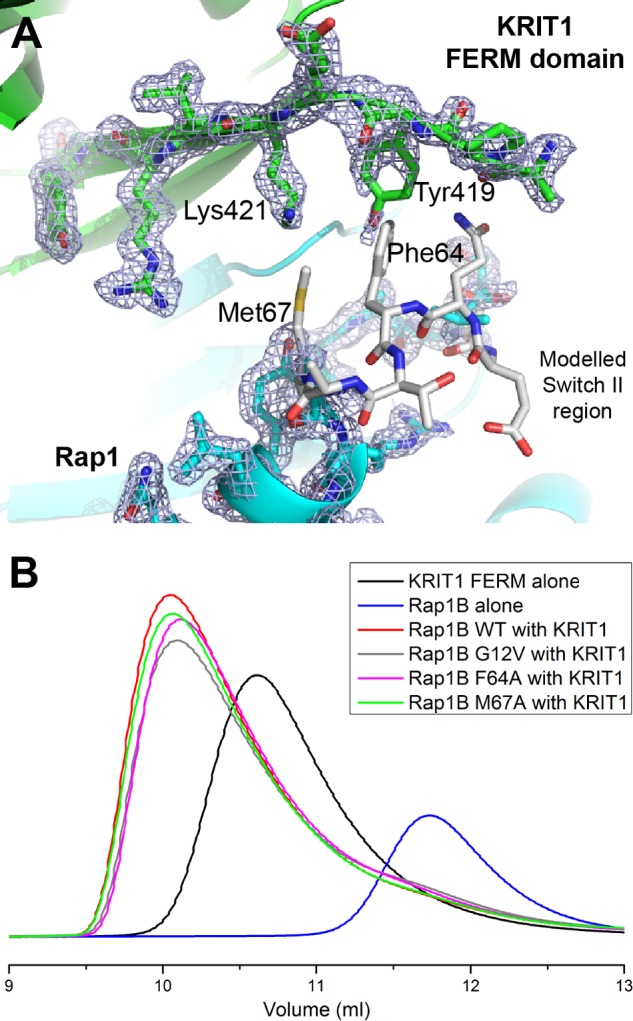
Rap1 Switch II is disordered in the crystal structure and does not play an important role in KRIT1 binding. A, close-up view of the KRIT1-Rap1 interaction in the vicinity of Rap1 Switch II region. The electron density is shown to highlight the absence of density for Rap1 residues 63–66 (2Fo − Fc map contoured at 1.0σ). KRIT1 is shown in green, Rap1 in blue, and the modeled region of Rap1 Switch II in white. B, binding of the KRIT1 FERM domain to Rap1 as analyzed on a Superdex-75 (10/300) GL gel filtration column at room temperature. Incubation of KRIT1 WT with various Rap1 mutants results in complex formation with a Ve comparable with wild-type complex, suggesting that binding is not affected.
DISCUSSION
The capacity of KRIT1 to bind to both HEG1 (20) and Rap1 (14) is essential for cardiovascular development. Here, we report that KRIT1 can simultaneously bind both GTP-bound Rap1 and HEG1 to assemble a ternary complex. Furthermore, we report that even though Rap1 and HEG1 interact with the F1 lobe of the KRIT1 FERM domain that KRIT1 and KRIT1-Rap1 bind HEG1 with similar affinities, suggesting that HEG1 and Rap1 neither compete nor bind cooperatively to the KRIT1 FERM domain. We report the structure of the HEG1-Rap1-KRIT1 ternary complex, and comparison with the Rap1-KRIT1 and HEG1-KRIT structures show that interactions of either HEG1 or Rap1 with the KRIT1 FERM domain do not induce major conformational changes that alter the others' binding interface, thus explaining the lack of interactions between the two ligands. The structure of the KRIT1-Rap1 complex also revealed a contact with the KRIT1 F2 subdomain that contributes to the specificity of Rap1 over HRas binding to KRIT1. These data define the structure of a ternary complex that regulates vascular integrity and development and identify a novel mechanism for establishing effector specificity in the Ras family of small GTPases.
KRIT1, Rap1, and HEG1 can form a ternary complex, and Rap1 neither competes with HEG1 nor binds cooperatively to the KRIT1 FERM domain. By ITC measurements, we found that the presence of bound Rap1 had a negligible effect on the affinity of KRIT1 for the HEG1 C terminus, and conversely, HEG1 had little effect on the affinity of KRIT1 for Rap1 as judged by SEC. Thus, although both ligands interact with the KRIT1 F1 domain (14, 20) they do not appear to appreciably influence each others' binding. Indeed, they formed a ternary complex, whose structure explained why neither ligand can sterically or allosterically hinder the binding of the other. Specifically, the HEG1 binds at the F1-F3 interface, whereas Rap1 binds another face of the F1 subdomain. Furthermore, neither ligand induces significant conformational changes in the FERM domain. KRIT1 functions at endothelial cell-cell junctions (10), and its interaction with both Rap1 (14) and HEG1 (20) is required for localization. The ternary complex reported here indicates that Rap1 can remain bound to KRIT1 when HEG1 captures KRIT1 at cell-cell junctions. Thus, the prenylated Rap1 C terminus can provide an additional membrane anchor for KRIT1.
Interestingly, in both our high resolution structures of the KRIT1-Rap1 complex, we do not observe good electron density for the Rap1 Switch II, suggesting that it is flexible and not involved in KRIT1 FERM binding. In agreement with this observation, we also find that Rap1(F64A) and Rap1(M67A) have a minimal effect on complex formation. The previously described KRIT1-Rap1 structure showed a hydrophobic interaction between KRIT1 Phe419 and Rap1 Phe64 (15); however, KRIT1 Phe419 was a mutation at Tyr419 introduced during cloning. This mutation could have led to a difference in crystal packing of Switch II in comparison with our studies that used the wild-type sequence. Our data show that Rap1 Switch II has a minimal role in KRIT1 FERM domain binding. Furthermore, because Switch II remains accessible after KRIT1 binding, other proteins can potentially bind to Switch II. That said, because Rap1 Switch I is engaged by KRIT1, the KRIT1-Rap1 complex is unlikely to bind strongly to other canonical Rap effectors.
The structure of the KRIT1-Rap1 complex reveals a new mechanism governing the recognition specificity of Ras family GTPases by a FERM domain-containing protein. KRIT1 binds Rap1 with higher affinity than HRas (Kd = 4.7 and 33 μm, respectively) (16). The KRIT1(R452E) mutant in the F1 subdomain exhibits ∼40-fold reduced binding affinity for Rap1 (14), and the crystal structure of the KRIT1-Rap1 complex shows that, as expected, KRIT1 recognizes Rap1 through the interaction of the Switch I region of Rap1 with the F1/RBD domain of KRIT1 (15). Our structure of KRIT1 in complex with Rap1 also shows a large binding interface that involves both the F1 and F2 subdomain of the FERM domain, and we speculated that F2 contributes to the specificity for Rap1 over HRas. In particular, we noted that the F2 subdomain forms a salt bridge with Rap1 Glu45, which is a leucine in HRas. We found that KRIT1(K570I) reduced KRIT1 affinity for Rap1 by ∼8-fold and increased binding to HRas. Furthermore, KRIT1(K570E) binds Rap1 weakly in SEC, and Rap1(E45K), which binds weakly to wild-type KRIT1, shows increased binding to KRIT1(K570E), verifying the importance of this salt bridge. Comparison of the SEC elution profile between KRIT1-Rap1(E45K) and KRIT1(K570E)-Rap1 complexes also suggests that Rap1 Glu45 is more important than KRIT1 Lys570 for the interaction, supporting our hypothesis that the difference between Rap1 Glu45 and HRas Val45 plays an important role in KRIT1 specificity. Thus, these data show how the F2 subdomain can contribute to the GTPase recognition specificity of this Ras family effector. Other FERM domain-containing proteins interactions with GTPases have been observed, such as SNX17 and Ezrin (31, 32), and it would be interesting to see if, like KRIT1, they also use the F2 subdomain to achieve specificity for their respective GTPase.
Acknowledgments
We thank I. L. Barsukov and A. A. Bobkov for help with ITC. We also thank the beamline scientists at the ESRF and Diamond Light Source and the United Kingdom Midlands Block Allocation Groups mx310 for DLS and mx1218 for ESRF.
This work was supported, in whole or in part, by National Institutes of Health Grants HL106489 and GM094663 (to M. H. G.). This work was also supported by American Heart Association Scientist Development Grant 12SDG11610043 (to A. R. G.).
The atomic coordinates and structure factors (codes 4HDO and 4HDQ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- EC
- endothelial cell
- CCM
- cerebral cavernous malformation
- KRIT1
- Krev interaction trapped 1
- HEG1
- heart of glass
- SEC
- size exclusion chromatography
- FERM
- band 4.1, ezrin, radixin, moesin
- ITC
- isothermal titration calorimetry
- GMP-PNP
- guanosine 5′-[β,γ-imido]triphosphate
- PDB
- Protein Data Bank
- RBD
- Ras-binding domain.
REFERENCES
- 1. Eerola I., Plate K. H., Spiegel R., Boon L. M., Mulliken J. B., Vikkula M. (2000) KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum. Mol. Genet. 9, 1351–1355 [DOI] [PubMed] [Google Scholar]
- 2. Wong J. H., Awad I. A., Kim J. H. (2000) Ultrastructural pathological features of cerebrovascular malformations: a preliminary report. Neurosurgery 46, 1454–1459 [DOI] [PubMed] [Google Scholar]
- 3. Clatterbuck R. E., Eberhart C. G., Crain B. J., Rigamonti D. (2001) Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatry 71, 188–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dubovsky J., Zabramski J. M., Kurth J., Spetzler R. F., Rich S. S., Orr H. T., Weber J. L. (1995) A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum. Mol. Genet. 4, 453–458 [DOI] [PubMed] [Google Scholar]
- 5. Craig H. D., Günel M., Cepeda O., Johnson E. W., Ptacek L., Steinberg G. K., Ogilvy C. S., Berg M. J., Crawford S. C., Scott R. M., Steichen-Gersdorf E., Sabroe R., Kennedy C. T., Mettler G., Beis M. J., Fryer A., Awad I. A., Lifton R. P. (1998) Multilocus linkage identifies two new loci for a Mendelian form of stroke, cerebral cavernous malformation, at 7p15–13 and 3q25.2–27. Hum. Mol. Genet. 7, 1851–1858 [DOI] [PubMed] [Google Scholar]
- 6. Labauge P., Denier C., Bergametti F., Tournier-Lasserve E. (2007) Genetics of cavernous angiomas. Lancet Neurol. 6, 237–244 [DOI] [PubMed] [Google Scholar]
- 7. Liquori C. L., Berg M. J., Squitieri F., Leedom T. P., Ptacek L., Johnson E. W., Marchuk D. A. (2007) Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am. J. Hum. Genet. 80, 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gunel M., Laurans M. S., Shin D., DiLuna M. L., Voorhees J., Choate K., Nelson-Williams C., Lifton R. P. (2002) KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc. Natl. Acad. Sci. U.S.A. 99, 10677–10682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whitehead K. J., Plummer N. W., Adams J. A., Marchuk D. A., Li D. Y. (2004) Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 131, 1437–1448 [DOI] [PubMed] [Google Scholar]
- 10. Glading A., Han J., Stockton R. A., Ginsberg M. H. (2007) KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell-cell junctions. J. Cell Biol. 179, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Serebriiskii I., Estojak J., Sonoda G., Testa J. R., Golemis E. A. (1997) Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene 15, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 12. Béraud-Dufour S., Gautier R., Albiges-Rizo C., Chardin P., Faurobert E. (2007) Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. FEBS J. 274, 5518–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Glading A. J., Ginsberg M. H. (2010) Rap1 and its effector KRIT1/CCM1 regulate β-catenin signaling. Dis. Model. Mech. 3, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu J. J., Stockton R. A., Gingras A. R., Ablooglu A. J., Han J., Bobkov A. A., Ginsberg M. H. (2011) A mechanism of Rap1-induced stabilization of endothelial cell-cell junctions. Mol. Biol. Cell 22, 2509–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li X., Zhang R., Draheim K. M., Liu W., Calderwood D. A., Boggon T. J. (2012) Structural basis for small G protein effector interaction of ras-related protein 1 (Rap1) and adaptor protein Krev interaction trapped 1 (KRIT1). J. Biol. Chem. 287, 22317–22327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wohlgemuth S., Kiel C., Krämer A., Serrano L., Wittinghofer F., Herrmann C. (2005) Recognizing and defining true Ras binding domains I: biochemical analysis. J. Mol. Biol. 348, 741–758 [DOI] [PubMed] [Google Scholar]
- 17. Mably J. D., Mohideen M. A., Burns C. G., Chen J. N., Fishman M. C. (2003) Heart of glass regulates the concentric growth of the heart in zebrafish. Curr. Biol. 13, 2138–2147 [DOI] [PubMed] [Google Scholar]
- 18. Mably J. D., Chuang L. P., Serluca F. C., Mohideen M. A., Chen J. N., Fishman M. C. (2006) Santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development 133, 3139–3146 [DOI] [PubMed] [Google Scholar]
- 19. Kleaveland B., Zheng X., Liu J. J., Blum Y., Tung J. J., Zou Z., Sweeney S. M., Chen M., Guo L., Lu M. M., Zhou D., Kitajewski J., Affolter M., Ginsberg M. H., Kahn M. L. (2009) Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat. Med. 15, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gingras A. R., Liu J. J., Ginsberg M. H. (2012) Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J. Cell Biol. 199, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gorzalczany Y., Sigal N., Itan M., Lotan O., Pick E. (2000) Targeting of Rac1 to the phagocyte membrane is sufficient for the induction of NADPH oxidase assembly. J. Biol. Chem. 275, 40073–40081 [DOI] [PubMed] [Google Scholar]
- 22. Kabsch W. (2010) Xds. J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 23. Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PubMed] [Google Scholar]
- 24. Nassar N., Horn G., Herrmann C., Block C., Janknecht R., Wittinghofer A. (1996) Ras/Rap effector specificity determined by charge reversal. Nat. Struct. Biol. 3, 723–729 [DOI] [PubMed] [Google Scholar]
- 25. Nassar N., Horn G., Herrmann C., Scherer A., McCormick F., Wittinghofer A. (1995) The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature 375, 554–560 [DOI] [PubMed] [Google Scholar]
- 26. Huang L., Weng X., Hofer F., Martin G. S., Kim S. H. (1997) Three-dimensional structure of the Ras-interacting domain of RalGDS. Nat. Struct. Biol. 4, 609–615 [DOI] [PubMed] [Google Scholar]
- 27. Mayer C. L., Snyder W. K., Swietlicka M. A., Vanschoiack A. D., Austin C. R., McFarland B. J. (2009) Size-exclusion chromatography can identify faster-associating protein complexes and evaluate design strategies. BMC Res. Notes 2, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pacold M. E., Suire S., Perisic O., Lara-Gonzalez S., Davis C. T., Walker E. H., Hawkins P. T., Stephens L., Eccleston J. F., Williams R. L. (2000) Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase γ. Cell 103, 931–943 [DOI] [PubMed] [Google Scholar]
- 29. Bunney T. D., Harris R., Gandarillas N. L., Josephs M. B., Roe S. M., Sorli S. C., Paterson H. F., Rodrigues-Lima F., Esposito D., Ponting C. P., Gierschik P., Pearl L. H., Driscoll P. C., Katan M. (2006) Structural and mechanistic insights into Ras association domains of phospholipase Cϵ. Mol. Cell 21, 495–507 [DOI] [PubMed] [Google Scholar]
- 30. Stieglitz B., Bee C., Schwarz D., Yildiz O., Moshnikova A., Khokhlatchev A., Herrmann C. (2008) Novel type of Ras effector interaction established between tumour suppressor NORE1A and Ras switch II. EMBO J. 27, 1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hatzoglou A., Ader I., Splingard A., Flanders J., Saade E., Leroy I., Traver S., Aresta S., de Gunzburg J. (2007) Gem associates with Ezrin and acts via the Rho-GAP protein Gmip to down-regulate the Rho pathway. Mol. Biol. Cell 18, 1242–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ghai R., Mobli M., Norwood S. J., Bugarcic A., Teasdale R. D., King G. F., Collins B. M. (2011) Phox homology band 4.1/ezrin/radixin/moesin-like proteins function as molecular scaffolds that interact with cargo receptors and Ras GTPases. Proc. Natl. Acad. Sci. U.S.A. 108, 7763–7768 [DOI] [PMC free article] [PubMed] [Google Scholar]