

Background: IL-32β promotes IL-10 production in myeloid cells.
Results: IL-32β-mediated C/EBPα serine 21 phosphorylation by PKCδ induced the dissociation of C/EBPα from IL-10 promoter, thereby promoting IL-10 production.
Conclusion: IL-32β suppressed the inhibitory effect of C/EBPα on IL-10 production by mediating C/EBPα serine 21 phosphorylation by PKCδ.
Significance: Our data suggest that IL-32β functions as an intracellular regulator of IL-10 production.
Keywords: C/EBP Transcription Factor, Chromatin Immunoprecipitation (ChiP), Gene Regulation, Gene Silencing, Protein Kinase C (PKC), U937
Abstract
We previously reported that IL-32β promotes IL-10 production in myeloid cells. However, the underlying mechanism remains elusive. In this study, we demonstrated that IL-32β abrogated the inhibitory effect of CCAAT/enhancer-binding protein α (C/EBPα) on IL-10 expression in U937 cells. We observed that the phosphorylation of C/EBPα Ser-21 was inhibited by a PKCδ-specific inhibitor, rottlerin, or IL-32β knockdown by siRNA and that IL-32β shifted to the membrane from the cytosol upon phorbol 12-myristate 13-acetate treatment. We revealed that IL-32β suppressed the binding of C/EBPα to IL-10 promoter by using ChIP assay. These data suggest that PKCδ and IL-32β may modulate the effect of C/EBPα on IL-10 expression. We next demonstrated by immunoprecipitation that IL-32β interacted with PKCδ and C/EBPα, thereby mediating C/EBPα Ser-21 phosphorylation by PKCδ. We showed that IL-32β suppressed the inhibitory effect of C/EBPα on IL-10 promoter activity. However, the IL-10 promoter activity was reduced to the basal level by rottlerin treatment. When C/EBPα serine 21 was mutated to glycine (S21G), the inhibitory effect of C/EBPα S21G on IL-10 promoter activity was not modulated by IL-32β. Taken together, our results show that IL-32β-mediated C/EBPα Ser-21 phosphorylation by PKCδ suppressed C/EBPα binding to IL-10 promoter, which promoted IL-10 production in U937 cells.
Introduction
Interleukin 32 (IL-32) was first reported as a proinflammatory cytokine that induces IL-1β, IL-6, tumor necrosis factor-α (TNF-α), and IL-8 (1). Multiple studies have shown the association of IL-32 with various inflammatory diseases, such as rheumatoid arthritis, inflammatory bowel disease, and chronic obstructive pulmonary disease (2, 3). In addition to those inflammatory diseases, viral and bacterial infections and even cancers induce IL-32 expression. However, the majority of protein is detected in cellular lysates rather than in the supernatants (2, 4–6). Recently, the interactions of IL-32 with intracellular proteins, such as focal adhesion kinase 1 (FAK 1), paxillin, integrins, protein kinase Cδ (PKCδ), protein kinase Cϵ (PKCϵ), and STAT3, have been demonstrated, which implies that IL-32 may participate in inflammatory responses as an intracellular mediator (7–9).
The CCAAT/enhancer-binding protein (C/EBP)3 family of transcription factors is known to play important roles in cell proliferation and differentiation. The C/EBP family comprises six members: C/EBPα, C/EBPβ, C/EBPγ, C/EBPδ, C/EBPϵ, and C/EBPζ. Each member, except C/EBPζ, which lacks a canonical basic region, contains a similar basic region and leucine zipper sequence at its C terminus that mediate DNA binding and dimerization, respectively (10, 11). C/EBPα dimerizes with other members of the C/EBP family or with itself (12) and interacts with other proteins such as transcription factor II B (TFIIB), TATA-binding protein (TBP), retinoblastoma protein (Rb), p300/CREB-binding protein (CBP), p21, and members of the SWI/SNF complex (13–17). C/EBPα is known to up-regulate the expression of granulocytic lineage-specific genes (18–21), both directly and synergistically with other key genes such as core-binding factor (CBF) complex genes and PU.1 in myeloid development. C/EBPα also represses E2F-regulated genes by direct interaction with E2F, which induces cell cycle arrest (19, 22).
It has also been shown that C/EBPα is regulated by phosphorylation and SUMOylation (SUMO = small ubiquitin-like modifier) (23–25). Phosphorylation of Thr-222 and Thr-226 on C/EBPα by glycogen synthase kinase 3 (GSK3) causes profound conformational changes, which induce adipogenesis (25). C/EBPα phosphorylation on Ser-21 by extracellular signal-regulated kinase 1/2 (ERK1/2) inhibits granulopoiesis. Phosphorylation of C/EBPα Ser-21 induces conformational changes (26). An oncogenic FLT3 kinase also inhibits the transcriptional activity of C/EBPα by phosphorylating Ser-21 (27).
We previously reported that IL-32β promotes IL-10 production in myeloid cells (28). In this study, we further verified our results and demonstrated an intracellular mediatory role of IL-32β for IL-10 production by showing that IL-32β promoted IL-10 production in a PKCδ-dependent way, where it mediated the phosphorylation of C/EBPα Ser-21 by PKCδ. The phosphorylation of C/EBPα Ser-21 suppressed its inhibitory role in IL-10 induction. Our data suggest that IL-32β may function as an intracellular regulator of inflammatory response as well as an inducer of inflammation.
EXPERIMENTAL PROCEDURES
Reagents and Cell Culture
The human promyelomonocytic U937 cell line was grown in RPMI 1640 (WelGENE, Daegu, Korea) supplemented with 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (HyClone, Logan, UT). Human embryonic kidney 293 (HEK293) and lung carcinoma A549 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (10%) and antibiotics. Phorbol 12-myristate 13-acetate (PMA), lipopolysaccharide (LPS), and polyinosinic:polycytidylic acid (poly(I:C)) were purchased from Sigma. MAPK inhibitor (PD98059), and PKCδ inhibitor (rottlerin) and classical PKC inhibitor (Gö6976) were purchased from Calbiochem.
Measurement of IL-10 Expression Levels and Enzyme-linked Immunosorbent Assay (ELISA)
IL-10 mRNA expression was detected by reverse transcription-polymerase chain reaction (RT-PCR) of total RNAs extracted from U937 cells after the various treatments with 10 nm PMA, 1 μg/ml LPS, or 10 μg/ml poly(I:C) for 24 h. U937 cells were also treated with increasing amounts of the PKCδ-specific inhibitor rottlerin for 24 h. The culture media were collected for ELISA. The IL-10 primer set used for PCR was as follows: sense 5′-AACCTGCCTAACATGCTTCGA-3′; antisense 5′-CTCATGGCTTTGTAGATGCCT-3′. Human IL-10, TNFα, and IL-8 ELISAs were performed using ELISA kits from R&D systems ( Minneapolis, MN).
Immunofluorescence Analyses
For immunofluorescence staining, U937 cells were treated with 50 nm PMA or 1 μg/ml LPS for 40 min; thereafter, 1 × 105 cells were transferred into a 1.5-ml microcentrifuge tube. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 20 min. After blocking with 1% bovine serum albumin (BSA) for 30 min, KU32-52 antibody to IL-32 (1:200) was incubated for 1 h. Fluorescein isothiocyanate (FITC)-conjugated anti-mouse secondary antibody (1:500) was used for staining. 4′,6-Diamidino-2-phenylindole (DAPI) was used for nuclei staining. Fluorescence images were obtained using a Zeiss LSM5 confocal microscope with Plan-Apochromat 63×/1.4 oil differential interference contrast objectives.
Construction of Expression Vectors
IL-32β cDNA was subcloned into pcDNA3.1 + 6×Myc vector by using EcoRI and XhoI. cDNAs for PKCα, PKCδ, PKCϵ, and PKCθ were subcloned into the pcDNA3.1 + 5×FLAG vector by using EcoRI and XhoI (7). Human intronless C/EBPα gene was PCR-amplified from THP-1 genomic DNA by using two overlapping primer sets to obtain the entire cDNA; PCR was then conducted again using the two-PCR fragment as templates. The primers sets were as follows: forward (F) 5′-GAATTCATGGAGTCGGCCGACTTCTAC-3′; reverse (R) 5′-CTCGAGTCACGCGCAGTT-GCCCATGGC-3′; forward 2 (F2) 5′-AGATCTCGCACTGCGGCCAGACCACCA-3′; reverse 2 (R2) 5′-AGATCTGGAACTGCAGGTGCGGGGCGG-3′. The entire amplified gene was sequenced and cloned into pCS3MT 6×Myc vector using EcoRI and XhoI. A mutant C/EBPα at serine 21 to glycine (S21G) was generated by site-directed mutagenesis. The primer set for mutagenesis was as follows: forward 5′-AGCAGCCACCTGCAGGGCCCCCCGCAC-3′; reverse 5′-GGGCGCGTGCGGGGGGCCCTGCAGGTG-3′. C/EBPα S21G was subcloned into 6×Myc pCS3MT vector and confirmed by sequencing. The deletion mutants of C/EBPα and IL-32β were generated by PCR, and the PCR products were cloned into the pCS3MT 6×Myc vector.
Electroporation
U937 promyelomonocytic cells were transfected with 1.5 μg of IL-32 siRNA or nontargeting siRNA using a NeonTM transfection system (Invitrogen). PKCδ siRNA was transfected in the same way. PKCδ and IL-32 siRNA and nontargeting siRNA were purchased from Dharmacon (Lafayette, CO). The transfected cells were incubated overnight, and 10 nm PMA was then applied for the indicated time, after which cell lysates were prepared for Western blotting.
Western Blotting and Immunoprecipitation
HEK293 cells were co-transfected with pcDNA3.1 + 6×Myc-IL-32β and pcDNA3.1 + 5×FLAG-PKCs (PKCα, PKCδ, PKCϵ, and PKCθ). The different combinations of IL-32β, PKCδ, C/EBPα, and mutant C/EBPα S21G were co-transfected into HEK293. After overnight incubation, cells were treated with 10 μm rottlerin or 10 μm PD98059 for 1 h for the inhibitor-treated samples. 20 nm PMA was treated for a further 90 min and then lysed in 50 mm HEPES, pH 7.5; 150 mm NaCl; 5% glycerol; 20 mm β-glycerophosphate; 1% Nonidet P-40; 0.5% Triton X-100; and 1 mm EDTA. Western blotting was performed using the Myc tag antibody (Millipore-Upstate, Bedford, MA), pC/EBPα (serine 21) antibody (Cell Signaling, Danvers, MA), and C/EBPα antibody (Santa Cruz Biotechnology). U937 cells were lysed after PMA treatment for the indicated time. Treatment with inhibitors was performed 1 h before PMA treatment. The optical density of the bands was measured by using Image Studio Lite Software after scanning the blots (LI-COR Biosciences, Lincoln, NE). For immunoprecipitation, cell lysates were mixed with 1 μg of Myc antibody, 3 μg of C/EBPα antibody, or 3 μg of goat polyclonal IL-32 antibody (R&D systems) for 1 h, respectively, and then pulled down using 35 μl of protein G-agarose bead (Kirkegaard & Perry Laboratories, Gaithersburg, MD).
IL-10 Reporter Plasmid and Luciferase Assay
IL-10 promoter region (from −1338 to +24) was subcloned into pGL3 Basic using XhoI and HindIII sites. The primer set was as follows: sense 5′-CTCGAGTGTGGAAGGGGAAGGTGAAGG-3′; antisense 5′-AAGCTTACAGAGCAGTGCTGAGCTGTG-3′. HEK293 cells were co-transfected with the different combinations of pGL3-IL-10 promoter, C/EBPα, mutant C/EBPα S21G, and IL-32β by using Lipofectamine® 2000 reagent (Invitrogen). After overnight incubation, the samples were treated with 10 μm rottlerin or 10 μm PD98059, after which they were treated with 20 nm PMA for a further 24 h. Luciferase assays were performed using Dual-Luciferase® reporter assay system (Promega, Madison, WI).
Chromatin Immunoprecipitation (ChIP) Assay
We used the commercially available ChIP assay kit (Millipore) according to the manufacturer's instructions. Briefly, U937 cells were electroporated with IL-32 siRNA or nontargeting RNA. After overnight incubation, cells were treated with PMA for 90 min. The cells were fixed with 1% formaldehyde, lysed in kit lysis buffer, and sonicated with 5 pulses for 5 s each. After centrifugation at 13,000 rpm for 20 min, the supernatants were precleared with 45 μl of protein A-agarose/salmon sperm DNA (50% slurry) for 60 min. After a brief centrifugation, the supernatants were mixed with 3 μg of C/EBPα antibody and maintained overnight. Sixty microliters of protein A-agarose/salmon sperm DNA (50% slurry) was added to each sample, and the pulled-down DNA fragments were eluted. PCR amplification using the eluted DNA as the template was performed for 35 cycles at an annealing temperature of 61 °C. The primers for PCR amplification of the IL-10 promoter were as follows: sense (from −577 to −557) 5′-CTTTGAGGATATTTAGCC-CAC-3′; antisense (from −296 to −276) 5′-GGGCTACCTCTCTTAGAATAA-3′. Primers for Bcl-2 and lactoferrin ChIP were: Bcl-2 sense (from −395 to −375) 5′-CATTCTTTTTAGCCGTGTTAC-3′; antisense (from −238 to −218) 5′-GATCTTTATTTCATGAGGCAC-3′; lactoferrin sense (from −211 to −192) 5′-CAGCCAGGCAGAACCAGGAC-3′; antisense (from −23 to −4) 5′-GCCACTTGCGCCTGCCCTGC-3′. We measured the optical intensity of the bands by using GelQuant.NET software provided by BiochemLabSolutions. The ChIP products were also analyzed by quantitative real-time PCR by using iQTM SYBR® Green supermix (Bio-Rad).
Statistical Analysis
Statistical analysis was performed using the unpaired two-tailed Student's t test. Differences were considered statistically significant at p < 0.05.
RESULTS
IL-32β Promotes IL-10 Production in U937 Cells upon PMA Stimulation
We demonstrated previously that IL-32β promoted the production of the anti-inflammatory cytokine IL-10 in myeloid cells (28). To verify that IL-32β is involved in IL-10 production, we knocked down IL-32β by using siRNA in U937 cells (Fig. 1A). Only the production of IL-10 was decreased upon PMA treatment after IL-32β silencing, whereas the levels of TNF-α and IL-8 were not changed (Fig. 1). The ectopic introduction of IL-32β into U937 cells further increased IL-10 production (data not shown). Therefore, these data confirmed that IL-32β up-regulated the production of IL-10 by PMA treatment in U937 cells.
FIGURE 1.

The involvement of IL-32β in IL-10 production in U937 cells. A, IL-32β was knocked down in U937 cells by transfection with 1.5 μg of IL-32 siRNA or nontargeting siRNA (NT), and the cells were then treated with 10 nm PMA for 24 h. The culture media were collected for ELISA, and cell lysates were prepared for Western blotting. con, control. The silencing of IL-32β was confirmed by Western blotting. B–D, ELISAs for IL-10 (B), TNF-α (C), and IL-8 (D) were performed. All values are mean ± S.E. (n = 3). *, p < 0.03 (B). siIL-32, IL-32 siRNA; siCtrl, siRNA control.
IL-32β Associates with PMA-activated PKCδ
We localized IL-32β in U937 cells by using confocal microscopy. Under normal conditions, IL-32β was distributed in the cytosol, but it shifted to the membrane upon PMA stimulation. LPS treatment did not result in the movement of IL-32β in U937 cells (Fig. 2). This finding suggests that IL-32β played an indirect mediatory role rather than a role as a direct transcriptional activator for IL-10 expression. Because the effect of IL-32β on IL-10 production was PMA-dependent, and its membrane localization upon PMA stimulation resembled the movement of PKC, we screened the relevant PKCs by immunoprecipitation, as we had previously performed with IL-32α (7). IL-32β specifically interacted with PMA-activated PKCδ because the interaction was inhibited by a PKCδ-specific inhibitor, rottlerin, but was not inhibited by a classical PKC inhibitor, Gö6976 (Fig. 3, A and B). Meanwhile, several studies demonstrated that PKCδ is involved in IL-10 production (29–31). Thus, we examined whether PKCδ was involved in IL-10 production in U937 cells as well. When U937 cells were treated with increasing doses of rottlerin, the production of IL-10 was decreased in a rottlerin dose-dependent manner (Fig. 3C). The TNF-α level was also affected by rottlerin, but IL-8 was not (Fig. 3, D and E). These data suggest that IL-32β may be inter-related with PKCδ for IL-10 production.
FIGURE 2.
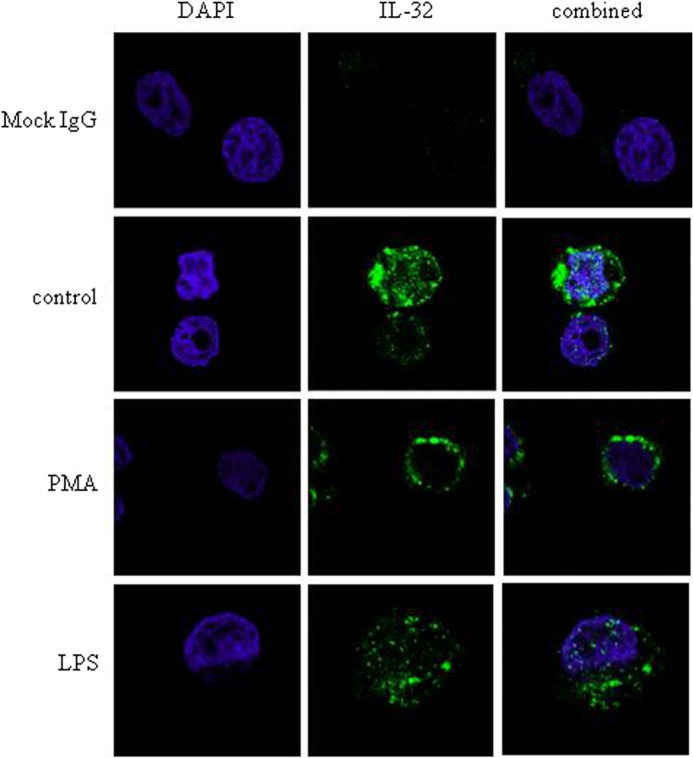
PMA-dependent shift of IL-32β localization in U937 cells. Immunofluorescence staining was performed to localize the endogenous IL-32β in U937 cells. Cells were treated with 50 nm PMA or 1 μg/ml LPS for 40 min. Cells were fixed, permeabilized, and labeled for nuclei with DAPI and endogenous IL-32 with FITC. Fluorescence signals were analyzed by confocal microscopy. The same amount of mock IgG (1 μg) as KU32-52 antibody was used for control.
FIGURE 3.

The interaction of PKCδ with IL-32β and its involvement in IL-10 production. A, IL-32β-interacting PKC isoforms were screened by immunoprecipitation. HEK293 cells were co-transfected with 6×Myc-tagged IL-32β and each of 5×FLAG-tagged PKC isoforms (α, δ, ϵ, and θ). After overnight incubation, PMA (20 nm) treatment was performed for 90 min. Immunoprecipitation (IP) was carried out with 1 μg of Myc tag antibody. The expression level of each gene was determined by Western blotting (WB) with 30 μg of whole cell lysates (WCL). B, after co-transfection of HEK293 cells with IL-32β and PKCδ, cells were treated with 10 μm rottlerin (Rott) or Gö6976 (6976), a classical PKC inhibitor, for 1 h before PMA (20 nm) treatment. Immunoprecipitation was performed with 3 μg of PKCδ antibody. U937 cells were treated with increasing doses of rottlerin 1 h before the PMA (10 nm) treatment; thereafter, the cells were incubated for 24 h. C–E, culture media were collected for IL-10 (C), TNF-α (D), and IL-8 (E) ELISA. Nontreated cells were included as a control (con). All values are mean ± S.E. (n = 3). *, p < 0.05; 0 versus 1 μm rottlerin, **, p < 0.02; 1 versus 10 μm rottlerin (B), ***, p < 0.01; 0 versus 10 μm rottlerin (C).
Serine 21 of C/EBPα Is Phosphorylated by PKCδ in an IL-32β-involved Way in U937 cells
The human promyelomonocytic U937 cells undergo monocytic differentiation by PMA stimulation. A tissue-specific transcription factor, C/EBPα, blocks 12-O-tetradecanoylphorbol-13-acetate-induced monocytic differentiation of bipotential myeloid cells when ectopically expressed (32) and induces growth arrest by repressing proliferation genes (10). Hence, we investigated the status of C/EBPα in U937 cells. C/EBPα was expressed in U937 cells and was phosphorylated at serine 21 by PMA stimulation (Fig. 4A). The phosphorylation of C/EBPα Ser-21 was inhibited by the treatment of rottlerin, a PKCδ-specific inhibitor, which means that C/EBPα Ser-21 could be phosphorylated by PKCδ as well. C/EBPα Ser-21 phosphorylation was also inhibited by PD98059, an ERK1/2 inhibitor, to a lesser extent than rottlerin, as consistent with the findings of a previous study (26) (Fig. 4B). To further verify that PKCδ is involved in C/EBPα Ser-21 phosphorylation, PKCδ was knocked down with 80% efficiency compared with GAPDH by using PKCδ-specific siRNA. As shown in Fig. 4C, phospho-C/EBPα at Ser-21 was significantly decreased by PKCδ silencing, which means that PKCδ is also involved in C/EBPα Ser-21 phosphorylation. We then examined whether IL-32β might affect the phosphorylation of C/EBPα. When IL-32β was knocked down by siRNA, the phosphorylation of Ser-21 of C/EBPα was not induced upon PMA treatment (Fig. 4D), which suggests that IL-32β involved is in C/EBPα Ser-21 phosphorylation. To analyze the relevance of C/EBPα to IL-10 expression, we performed ChIP for the putative C/EBPα-binding site on IL-10 promoter after silencing of IL-32β. We observed that the binding of C/EBPα to IL-10 promoter was increased by IL-32β knockdown. However, C/EBPα binding to Bcl-2 or lactoferrin promoter was not affected by IL-32β silencing. We further verified the ChIP results by quantitative real-time PCR (Fig. 5). This finding implies that IL-32β suppressed C/EBPα binding to IL-10 promoter, thereby relieving the IL-10 promoter from C/EBPα repression. ERK1/2 is known to directly phosphorylate C/EBPα Ser-21 through an FXFP docking site. However, we speculated that PKCδ might phosphorylate C/EBPα Ser-21 in a IL-32β-mediated way, which inhibited the binding of C/EBPα to IL-10 promoter.
FIGURE 4.

The involvement of IL-32β in C/EBPα phosphorylation by PKCδ. A, U937 cells were treated with 10 nm PMA for the indicated time. The phosphorylation of C/EBPα at serine 21 was detected by phospho-C/EBPα (S21)-specific antibody. Quantitation is shown below the blots, normalized to 0 min (for 10, 30, and 60 min). B, U937 cells were treated with 10 μm rottlerin (Rott) or 10 μm PD98059 (PD) for 1 h; thereafter, they were treated with 10 nm PMA for a further 60 min. Quantitation is shown below the blots, normalized to nontreated control for the others. C, U937 cells were transfected with 2 μg of PKCδ siRNA and nontargeting siRNA (NT). Cells were treated with 10 nm PMA for 30 min after overnight incubation, and then Western blotting was performed. D, 1.5 μg of IL-32 siRNA and nontargeting siRNA (NT) was introduced into U937 cells. After overnight incubation, cells were treated with 10 nm PMA for the indicated time. IL-32β band (25 kDa) was detected with KU32-52 antibody.
FIGURE 5.

The suppression of C/EBPα binding to IL-10 promoter by IL-32β. ChIP was performed with U937 cells after silencing of IL-32β. U937 cells were transfected with 1.5 μg of IL-32 siRNA and nontargeting siRNA (NT). After an overnight incubation following transfection, 10 nm PMA was treated for 90 min. ChIP was carried out with 3 μg of C/EBPα antibody. Quantitation is shown below the PCR data, normalized to control (con) nontargeting siRNA for the others. The consensus binding sequence of C/EBP and a putative C/EBPα-binding site and its location on IL-10 promoter are presented. ChIP results from Bcl-2 and lactoferrin promoters are presented as controls. IL-10 ChIP products were also measured by quantitative real-time PCR. The results are given as -fold change of the IL-10/lactoferrin ChIP ratio as compared with the value of nontreated nontargeting siRNA. All values are mean ± S.E. (n = 3).
IL-32β Interacts with PKCδ and C/EBPα and Mediates C/EBPα Ser-21 Phosphorylation
We next investigated how IL-32β mediated C/EBPα Ser-21 phosphorylation by PKCδ. IL-32β association with PMA-activated PKCδ was shown in Fig. 3, A and B. We then examined whether C/EBPα interacted with these molecules together. We cloned human C/EBPα cDNA into 6×Myc-tagged expressing vector and then performed immunoprecipitations after co-transfection of HEK293 with different combinations of IL-32β, C/EBPα, and PKCδ. IL-32β co-immunoprecipitated with C/EBPα and PKCδ, and this interaction was inhibited by rottlerin, but not by PD98059, an ERK1/2 inhibitor (Fig. 6, A and B). The interaction of C/EBPα with IL-32β was also confirmed in U937 cells. The interaction was inhibited by the treatment of rottlerin, but not by PD98059 (Fig. 6C). We then proved that IL-32β mediated the phosphorylation of C/EBPα Ser-21 by PKCδ. We transfected HEK293 cells with 6×Myc-tagged C/EBPα with or without IL-32β and then immunoprecipitated C/EBPα with Myc antibody. C/EBPα Ser-21 was not phosphorylated in the absence of IL-32β. The phosphorylation of Ser-21 was inhibited by rottlerin, but the phospho-C/EBPα Ser-21 was still detected even in the presence of a ERK1/2 inhibitor, PD98059 (Fig. 6D), which means that PKCδ also phosphorylated C/EBPα Ser-21 in a IL-32β-mediated way. When C/EBPα serine 21 was mutated to glycine, it was no longer phosphorylated (Fig. 6E). However, the interaction of C/EBPα with PKCδ and IL-32β was not inhibited by the mutation of serine 21 to glycine (S21G). We reconfirmed the interaction of IL-32β with mutant C/EBPα S21G in A549 lung carcinoma cells, which means that the phosphorylation of C/EBPα Ser-21 did not influence its association with IL-32β and PKCδ (Fig. 6, F and G). We also characterized the interaction domains of C/EBPα and IL-32β. IL-32β interacted with the full-length C/EBPα (FL) as well as F2 and F4 (Fig. 7A). F2 contains the DNA-binding domain and the basic leucine zipper domain, and F4 contains only the basic leucine zipper domain. Therefore, these data mean that IL-32β binds to the basic leucine zipper region of C/EBPα. C/EBPα bound to the N-terminal region of IL-32β (Fig. 7B). Most isoforms of IL-32 contain the same C terminus (D2), but the N terminus (D1) is diverse because of alternative splicing. Meanwhile, the two mutants of IL-32β had no effect on IL-10 production, which means that only the intact form of IL-32β is functional (data not shown).
FIGURE 6.

IL-32β-mediated phosphorylation of C/EBPα Ser-21 by PKCδ. A and B, IL-32β was associated with C/EBPα and PKCδ, and then mediated the phosphorylation of C/EBPα serine 21 by PKCδ. HEK293 cells were co-transfected with 6×Myc-tagged IL-32β and C/EBPα and with 5×FLAG-tagged PKCδ. After overnight incubation, cells were treated with 10 μm rottlerin (Rott) or 10 μm PD98059 (PD) for 1 h for the inhibitor-treated samples. 20 nm PMA was treated for 90 min. Three micrograms of KU32-52 antibody (A) or C/EBPα antibody (B) was used. IP, immunoprecipitation; WCL, whole cell lysate. C, the expression levels of the transfected genes were determined by Western blotting with 30 μg of whole cell lysates (WCL). Immunoprecipitations (IP) were performed with U937 cells after 10 μm rottlerin or PD98059 treatment for 1 h and treatment with 20 nm PMA for a further 90 min. 3 μg of goat polyclonal IL-32 antibody was used. After co-transfection of HEK293 cells with 6×Myc-tagged IL-32β and C/EBPα, cells were treated with inhibitors and PMA in the same way as in A. D, cell lysates were immunoprecipitated with 1 μg of Myc tag antibody; the phosphorylated C/EBPα at serine 21 was detected using phospho-C/EBPα Ser-21-specific antibody. E and F, a mutant C/EBPα S21G was co-transfected with IL-32β and PKCδ into HEK293 cells, and then immunoprecipitations were performed with Myc tag antibody (E) or KU32-52 antibody (F). G, A549 lung carcinoma cells were co-transfected with a mutant C/EBPα S21G, IL-32β, and PKCδ. Each plasmid was also transfected as controls. Immunoprecipitation was carried out with Myc tag antibody. The expression levels of the introduced genes were determined by Western blotting with 30 μg of whole cell lysates (WCL).
FIGURE 7.

Interaction domain mapping of C/EBPα and IL-32β. A, functional domains of C/EBPα and its deletion mutants are schematically illustrated. TAD; transactivation domain, RD; regulatory domain, DBD; DNA-binding domain, bLZ; basic leucine zipper domain. HEK293 cells were co-transfected with 6×Myc-tagged C/EBPα mutants and 5×FLAG-tagged IL-32β. After overnight incubation, cells were treated with 20 nm PMA for 90 min. Immunoprecipitation was performed with 1 μg of FLAG antibody. B, a schematic illustration of IL-32β deletion mutants is presented. HEK293 cells were co-transfected with 5×FLAG-tagged C/EBPα and 6×Myc-tagged IL-32β mutants. Immunoprecipitation was performed as in A with 1 μg of FLAG antibody. The pulled down D1 is marked by arrow. The expression levels of the transfected genes were determined by Western blotting with 30 μg of whole cell lysates (WCL). Immunoglobulin G light chain (IgG L) is indicated by an arrow.
IL-32β Suppressed the Inhibitory Effect of C/EBPα on IL-10 Expression
We further investigated the effect of C/EBPα on IL-10 expression using IL-10 promoter reporter assay. We observed that IL-10 promoter was not activated under C/EBPα expression without IL-32β. However, in the presence of IL-32β, the promoter was activated (Fig. 8A). The promoter activity was inhibited by rottlerin, but not by PD98059, which means that ERK1/2 was not involved in IL-10 expression in this system. The activity of C/EBPα is repressed by the phosphorylation of Ser-21 (26); as is consistent with this study, a mutant C/EBPα S21G, which could not be phosphorylated by PKCδ, repressed IL-10 promoter activity even under co-expression with IL-32β (Fig. 8B). When the cells were treated with rottlerin under mutant C/EBPα S21G expression, the promoter activity was further reduced, but was not modulated by PD98059 (Fig. 8C). These data therefore suggest that IL-32β-mediated phosphorylation of C/EBPα Ser-21 by PKCδ suppressed the inhibitory effect of C/EBPα on IL-10 production.
FIGURE 8.

The release of C/EBPα block on IL-10 expression by IL-32β. A, HEK293 cells were co-transfected with pGL3-IL-10 promoter, IL-32β, or C/EBPα. After overnight incubation, cells were treated with 20 nm PMA for 24 h, and then firefly luciferase assay was performed. All values are mean ± S.E. *, **, p < 0.001. B, luciferase assay was performed in the same way as in A after co-transfection of HEK293 cells with pGL3-IL-10 promoter, IL-32β, C/EBPα, or mutant C/EBPα S21G. For inhibitor-treated samples, treatment with 10 μm rottlerin (Rott) and 10 μm PD98059 (PD) was performed 1 h before PMA (20 nm) treatment for 24 h. All values are mean ± S.E. *, **, p < 0.001. C, HEK293 cells were co-transfected with a mutant C/EBPα S21G, IL-32β, and pGL3-IL-10 promoter, and then luciferase assay was performed in the same way as in B. All values are mean ± S.E. (n = 3). *, p < 0.001.
DISCUSSION
The initiation of the proinflammatory response is a prerequisite for the proper immune responses upon viral or bacterial infections. Although the proinflammatory reaction is essential, it should be resolved to prevent damage to the host. IL-32 has been studied for its proinflammatory function because multiple studies have shown its association with inflammatory diseases. IL-32β is known to induce TNF-α, IL-8, IL-6, and macrophage inflammatory protein-1α (MIP-1α), all of which are proinflammatory mediators. However, we showed that IL-32β induced an anti-inflammatory cytokine IL-10 in myeloid cells. In this study, we questioned the mechanism of action of IL-32β because IL-32β protein was detected only with cellular lysates, not in the culture medium. In this study, we solved our previous question by revealing the mechanism by which IL-32β promotes IL-10 production. Recently, several studies have shown the intracellular mediatory function of IL-32 (7, 8, 33). We demonstrated that IL-32β mediated the phosphorylation of serine 21 on C/EBPα by PMA-activated PKCδ, which resulted in IL-10 production. Phosphorylation of C/EBPα at Ser-21 inhibits its transcriptional activity through induction of conformational changes. The involvement of PKCδ in IL-10 production is known, but there is little evidence of the effect of C/EBPα on IL-10 production. Although a previous study showed that C/EBPα induced IL-10 production (34), the effect was synergistic with cAMP-responsive element-binding protein/activating transcription factor (CREB/ATF). C/EBPα not only activates the granulopoiesis-related genes, but also represses cell cycle-related genes, which means that C/EBPα acts as a transcriptional activator as well as a transcriptional inhibitor. There are a couple of models explaining the inhibitory function of C/EBPα. C/EBPα may inhibit S-phage genes indirectly through an association with E2F, or by direct binding to E2F sites. Alternatively, C/EBPα may directly inhibit S-phage genes by binding to the C/EBP-binding site on promoters of those genes (10). On the grounds of these studies, we examined the effect of IL-32β on C/EBPα and showed that IL-32β inhibited C/EBPα binding to IL-10 promoter by mediating Ser-21 phosphorylation by PKCδ. However, whether the inhibition of the IL-10 gene by C/EBPα results from the binding of C/EBPα to IL-10 promoter or the interaction of other inhibitory proteins with C/EBPα located on the IL-10 promoter remains unclear. In any event, according to our results, IL-32β has an intracellular mediatory function in addition to its proinflammatory function.
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (2012R1A2A2A 02008751 and 2011-0028213).
- C/EBP
- CCAAT/enhancer-binding protein
- CREB
- cAMP-responsive element-binding protein
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Kim S. H., Han S. Y., Azam T., Yoon D. Y., Dinarello C. A. (2005) Interleukin-32: a cytokine and inducer of TNFα. Immunity 22, 131–142 [DOI] [PubMed] [Google Scholar]
- 2. Shioya M., Nishida A., Yagi Y., Ogawa A., Tsujikawa T., Kim-Mitsuyama S., Takayanagi A., Shimizu N., Fujiyama Y., Andoh A. (2007) Epithelial overexpression of interleukin-32α in inflammatory bowel disease. Clin. Exp. Immunol. 149, 480–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Joosten L. A., Netea M. G., Kim S. H., Yoon D. Y., Oppers-Walgreen B., Radstake T. R., Barrera P., van de Loo F. A., Dinarello C. A., van den Berg W. B. (2006) IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 103, 3298–3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nishida A., Andoh A., Shioya M., Kim-Mitsuyama S., Takayanagi A., Fujiyama Y. (2008) Phosphatidylinositol 3-kinase/Akt signaling mediates interleukin-32α induction in human pancreatic periacinar myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G831–G838 [DOI] [PubMed] [Google Scholar]
- 5. Netea M. G., Azam T., Lewis E. C., Joosten L. A., Wang M., Langenberg D., Meng X., Chan E. D., Yoon D. Y., Ottenhoff T., Kim S. H., Dinarello C. A. (2006) Mycobacterium tuberculosis induces interleukin-32 production through a caspase- 1/IL-18/interferon-γ-dependent mechanism. PLoS Med. 3, e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goda C., Kanaji T., Kanaji S., Tanaka G., Arima K., Ohno S., Izuhara K. (2006) Involvement of IL-32 in activation-induced cell death in T cells. Int. Immunol. 18, 233–240 [DOI] [PubMed] [Google Scholar]
- 7. Kang J. W., Park Y. S., Lee D. H., Kim J. H., Kim M. S., Bak Y., Hong J., Yoon D. Y. (2012) Intracellular interaction of interleukin (IL)-32α with protein kinase Cϵ (PKCϵ) and STAT3 protein augments IL-6 production in THP-1 promonocytic cells. J. Biol. Chem. 287, 35556–35564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heinhuis B., Koenders M. I., van den Berg W. B., Netea M. G., Dinarello C. A., Joosten L. A. (2012) Interleukin 32 (IL-32) contains a typical α-helix bundle structure that resembles focal adhesion targeting region of focal adhesion kinase-1. J. Biol. Chem. 287, 5733–5743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoon S., Woo S. U., Kang J. H., Kim K., Shin H. J., Gwak H. S., Park S., Chwae Y. J. (2012) NF-κB and STAT3 cooperatively induce IL-6 in starved cancer cells. Oncogene 31, 3467–3481 [DOI] [PubMed] [Google Scholar]
- 10. Johnson P. F. (2005) Molecular stop signs: regulation of cell-cycle arrest by C/EBP transcription factors. J. Cell Sci. 118, 2545–2555 [DOI] [PubMed] [Google Scholar]
- 11. Darlington G. J., Ross S. E., MacDougald O. A. (1998) The role of C/EBP genes in adipocyte differentiation. J. Biol. Chem. 273, 30057–30060 [DOI] [PubMed] [Google Scholar]
- 12. Lekstrom-Himes J., Xanthopoulos K. G. (1998) Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J. Biol. Chem. 273, 28545–28548 [DOI] [PubMed] [Google Scholar]
- 13. Pedersen T. A., Kowenz-Leutz E., Leutz A., Nerlov C. (2001) Cooperation between C/EBPα TBP/TFIIB and SWI/SNF recruiting domains is required for adipocyte differentiation. Genes Dev. 15, 3208–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Erickson R. L., Hemati N., Ross S. E., MacDougald O. A. (2001) p300 coactivates the adipogenic transcription factor CCAAT/enhancer-binding protein α. J. Biol. Chem. 276, 16348–16355 [DOI] [PubMed] [Google Scholar]
- 15. Timchenko N. A., Harris T. E., Wilde M., Bilyeu T. A., Burgess-Beusse B. L., Finegold M. J., Darlington G. J. (1997) CCAAT/enhancer binding protein α regulates p21 protein and hepatocyte proliferation in newborn mice. Mol. Cell Biol. 17, 7353–7361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen P. L., Riley D. J., Chen Y., Lee W. H. (1996) Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 10, 2794–2804 [DOI] [PubMed] [Google Scholar]
- 17. Nerlov C., Ziff E. B. (1995) CCAAT/enhancer binding protein-α amino acid motifs with dual TBP and TFIIB binding ability co-operate to activate transcription in both yeast and mammalian cells. EMBO J. 14, 4318–4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reddy V. A., Iwama A., Iotzova G., Schulz M., Elsasser A., Vangala R. K., Tenen D. G., Hiddemann W., Behre G. (2002) Granulocyte inducer C/EBPα inactivates the myeloid master regulator PU. 1: possible role in lineage commitment decisions. Blood 100, 483–490 [DOI] [PubMed] [Google Scholar]
- 19. Johansen L. M., Iwama A., Lodie T. A., Sasaki K., Felsher D. W., Golub T. R., Tenen D. G. (2001) c-Myc is a critical target for c/EBPα in granulopoiesis. Mol. Cell Biol. 21, 3789–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klempt M., Melkonyan H., Hofmann H. A., Eue I., Sorg C. (1998) The transcription factors c-myb and C/EBPα regulate the monocytic/myeloic gene MRP14. Immunobiology 199, 148–151 [DOI] [PubMed] [Google Scholar]
- 21. Smith L. T., Hohaus S., Gonzalez D. A., Dziennis S. E., Tenen D. G. (1996) PU. 1 (Spi-1) and C/EBPα regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood 88, 1234–1247 [PubMed] [Google Scholar]
- 22. Slomiany B. A., D'Arigo K. L., Kelly M. M., Kurtz D. T. (2000) C/EBPα inhibits cell growth via direct repression of E2F-DP-mediated transcription. Mol. Cell Biol. 20, 5986–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Subramanian L., Benson M. D., Iñiguez-Lluhí J. A. (2003) A synergy control motif within the attenuator domain of CCAAT/enhancer-binding protein α inhibits transcriptional synergy through its PIASy-enhanced modification by SUMO-1 or SUMO-3. J. Biol. Chem. 278, 9134–9141 [DOI] [PubMed] [Google Scholar]
- 24. Kim J., Cantwell C. A., Johnson P. F., Pfarr C. M., Williams S. C. (2002) Transcriptional activity of CCAAT/enhancer-binding proteins is controlled by a conserved inhibitory domain that is a target for sumoylation. J. Biol. Chem. 277, 38037–38044 [DOI] [PubMed] [Google Scholar]
- 25. Ross S. E., Erickson R. L., Hemati N., MacDougald O. A. (1999) Glycogen synthase kinase 3 is an insulin-regulated C/EBPα kinase. Mol. Cell Biol. 19, 8433–8441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ross S. E., Radomska H. S., Wu B., Zhang P., Winnay J. N., Bajnok L., Wright W. S., Schaufele F., Tenen D. G., MacDougald O. A. (2004) Phosphorylation of C/EBPα inhibits granulopoiesis. Mol. Cell Biol. 24, 675–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Radomska H. S., Bassères D. S., Zheng R., Zhang P., Dayaram T., Yamamoto Y., Sternberg D. W., Lokker N., Giese N. A., Bohlander S. K., Schnittger S., Delmotte M. H., Davis R. J., Small D., Hiddemann W., Gilliland D. G., Tenen D. G. (2006) Block of C/EBPα function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J. Exp. Med. 203, 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kang J. W., Choi S. C., Cho M. C., Kim H. J., Kim J. H., Lim J. S., Kim S. H., Han J. Y., Yoon D. Y. (2009) A proinflammatory cytokine interleukin-32β promotes the production of an anti-inflammatory cytokine interleukin-10. Immunology 128, e532–e540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leghmari K., Bennasser Y., Tkaczuk J., Bahraoui E. (2008) HIV-1 Tat protein induces IL-10 production by an alternative TNF-α-independent pathway in monocytes: role of PKC-δ and p38 MAP kinase. Cell Immunol. 253, 45–53 [DOI] [PubMed] [Google Scholar]
- 30. Wilmanski J., Siddiqi M., Deitch E. A., Spolarics Z. (2005) Augmented IL-10 production and redox-dependent signaling pathways in glucose-6-phosphate dehydrogenase-deficient mouse peritoneal macrophages. J. Leukoc. Biol. 78, 85–94 [DOI] [PubMed] [Google Scholar]
- 31. Contreras X., Bennasser Y., Bahraoui E. (2004) IL-10 production induced by HIV-1 Tat stimulation of human monocytes is dependent on the activation of PKCβ(II) and δ isozymes. Microbes Infect. 6, 1182–1190 [DOI] [PubMed] [Google Scholar]
- 32. Rangatia J., Vangala R. K., Treiber N., Zhang P., Radomska H., Tenen D. G., Hiddemann W., Behre G. (2002) Downregulation of c-Jun expression by transcription factor C/EBPα is critical for granulocytic lineage commitment. Mol. Cell Biol. 22, 8681–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heinhuis B., Netea M. G., van den Berg W. B., Dinarello C. A., Joosten L. A. (2012) Interleukin-32: a predominantly intracellular proinflammatory mediator that controls cell activation and cell death. Cytokine 60, 321–327 [DOI] [PubMed] [Google Scholar]
- 34. Brenner S., Prösch S., Schenke-Layland K., Riese U., Gausmann U., Platzer C. (2003) cAMP-induced Interleukin-10 promoter activation depends on CCAAT/enhancer-binding protein expression and monocytic differentiation. J. Biol. Chem. 278, 5597–5604 [DOI] [PubMed] [Google Scholar]