

Background: The structural basis for interaction between a master regulator and DNA remains unclear.
Results: We solved the crystal structures of a broad regulator Ms6564 and its protein-operator complex.
Conclusion: Ms6564 binds DNA with strong affinity but makes flexible contacts with DNA.
Significance: Ms6564 might slide more easily along the genomic DNA and extensively regulate the expression of diverse genes.
Keywords: Crystal Structure, DNA-binding Protein, DNA Transcription, DNA-Protein Interaction, Repressor Protein, Mycobacterium, TetR Regulator
Abstract
Master regulators, which broadly affect expression of diverse genes, play critical roles in bacterial growth and environmental adaptation. However, the underlying mechanism by which such regulators interact with their cognate DNA remains to be elucidated. In this study, we solved the crystal structure of a broad regulator Ms6564 in Mycobacterium smegmatis and its protein-operator complex at resolutions of 1.9 and 2.5 Å, respectively. Similar to other typical TetR family regulators, two dimeric Ms6564 molecules were found to bind to opposite sides of target DNA. However, the recognition helix of Ms6564 inserted only slightly into the DNA major groove. Unexpectedly, 11 disordered water molecules bridged the interface of TetR family regulator DNA. Although the DNA was deformed upon Ms6564 binding, it still retained the conformation of B-form DNA. Within the DNA-binding domain of Ms6564, only two amino acids residues directly interacted with the bases of cognate DNA. Lys-47 was found to be essential for the specific DNA binding ability of Ms6564. These data indicate that Ms6564 can bind DNA with strong affinity but makes flexible contacts with DNA. Our study suggests that Ms6564 might slide more easily along the genomic DNA and extensively regulate the expression of diverse genes in M. smegmatis.
Introduction
Protein-DNA interactions play critically important roles in many biological processes (1, 2). This is particularly true with transcriptional regulation, because a regulator can function only when it successfully recognizes its target DNA. In recent years, the functions of some master regulators, which regulate expression of a large number of genes, have been characterized. The structural basis for such a broad regulation, however, remains largely unclear.
A protein's specificity and affinity for binding DNA are usually determined by the base readout mechanism (recognition of DNA bases) or the shape readout mechanism (recognition of DNA shape) (1, 3). The α-helix and β-strands are two common secondary structure elements used for the base readout mechanism (1, 2, 4). In contrast, helix-turn-helix (1, 2, 4–7) and helix-loop-helix motifs (8) (category mainly α) are frequently used to recognize the DNA major groove. Interestingly, some regulators utilize both DNA base and shape recognition mechanisms to interact with their target DNA. One example is the two-component regulator, NarL, which can control expression of many respiration-related operons (9–11). Structural analysis of the signal output domain of NarL (NarLC) in complex with DNA reveals that NarLC acts as a dimer. The recognition helices contact the floor of the major groove of DNA, which is bent and transformed into the A-form (9). In contrast, transcription activator-like effectors can bind almost any DNA or DNA-RNA hybrid sequence primarily through a DNA-based recognition of a central domain of tandem repeats (12–14).
The TetR family of transcriptional regulators (TFRs)4 comprises a large group of transcriptional regulators. Their prototype is an Escherichia coli TetR gene that regulates the expression of a tetracycline efflux pump in Gram-negative bacterium (15). TFRs often serve as repressors and regulate a variety of bacterial physiological processes (15). They usually act as homodimers in which each monomer consists of an N-terminal DNA-binding domain (DBD) and a C-terminal ligand-binding domain (15–17). For example, Staphylococcus aureus QacR regulates the expression of a multidrug transporter (18) by acting as a pair of dimers that bind a 28-bp operator DNA, and each half-site of the operator is recognized by the DBD of the QacR dimer on the opposite sides of the DNA (19). Similarly, a pair of Corynebacterium glutamicum CgmR dimers also docks on the opposite sides of its operator (20). Some master TFRs are reported to regulate the expression of a large number of genes. For example, SmcR controls at least 121 genes (21). KstR is directly involved in regulating the expression of 83 and 74 genes in Mycobacterium smegmatis and Mycobacterium tuberculosis, respectively (15, 22). More recently, Ms6564 is characterized as a master regulator that regulates the expression of 339 potential target genes in M. smegmatis (15). However, the mechanisms through which such master regulators recognize specific DNA motifs are poorly understood.
In the present study, we determined the crystal structure of a TetR master regulator, Ms6564, and the Ms6564-operator complex at resolutions of 1.9 and 2.5 Å, respectively. We report that two dimeric Ms6564 molecules bind to opposite sides of its operator, which is similar to the case of other TFR regulators such as QacR and CgmR (19, 20). However, Ms6564 demonstrates flexible contact with DNA base pairs, and strikingly, 11 water molecules are incorporated into the protein-DNA interface. In addition, only two residues in the DBDs of Ms6564, Lys-47 and Lys-48 directly interact with the cognate DNA. Therefore, Ms6564 can bind DNA with good affinity but makes flexible contacts with DNA, which allows Ms6564 to extensively regulate the expression of diverse genes in M. smegmatis.
EXPERIMENTAL PROCEDURES
Strains, Enzymes, Plasmids, and Chemicals
E. coli BL21(DE3) strains and the pET28a expression vector were purchased from Novagen. All enzymes including DNA polymerase, restriction enzymes and DNA ligase, deoxynucleoside triphosphates (dNTPs), and all antibiotics were purchased from TaKaRa Biotech. β-d-1-Thiogalactopyranoside, DTT, and all chemicals were purchased from Sigma. PCR primers were synthesized by Invitrogen.
Protein Expression and Purification
The gene encoding truncated Ms6564 (residues 9–189) were amplified from the genomic DNA of M. smegmatis mc2 155. The PCR products were cloned into a pET28a vector to produce recombinant vectors. After transformation with these recombinant plasmids, E. coli BL21(DE3) cells were grown in LB medium up to an A600 of 0.8 at 37 °C, and protein expression was induced with 0.1 mm β-d-1-thiogalactopyranoside at 16 °C. Selenomethionine-labeled (SeMet) Ms6564 was expressed in M9 medium. Both native and SeMet Ms6564 were purified on Ni2+ affinity columns as previously described (15). The eluted proteins were purified using heparin affinity columns (GE Healthcare) and eluted with buffer containing 20 mm Tris (pH 8.0) and 600 mm NaCl. Then the proteins were loaded on Superdex200 (GE Healthcare) with 20 mm Tris (pH 8.0) and 500 mm NaCl. The purified proteins were concentrated to 10 mg/ml in 20 mm Tris (pH 8.0), 300 mm NaCl, 50 mm imidazole, and 1 mm DTT.
Crystallization and Data Collection
All crystals suitable for x-ray diffraction were obtained using the sitting drop vapor diffusion method at 4 °C. N-terminal truncated Ms6564 (1 μl, residues 9–189) was mixed with 1 μl of reservoir solution containing 0.2 m sodium citrate tribasic dehydrate, 0.1 m HEPES sodium (pH 7.5), and 30% (±)-2-methyl-2,4-pentanediol. The mixture was equilibrated against 120 μl of reservoir solution for 7 days. The crystal was soaked in a reservoir supplemented with a stepped concentration (first 10%, then 15%, and finally 20%) of glycerol and flash-cooled in liquid nitrogen. The SeMet-Ms6564 (residues 9–189) crystal was obtained by the same procedure. The cryoprotection of SeMet-Ms6564 was achieved by raising the glycerol concentration stepwise to 20% with a 5% increment in each step.
To crystallize the Ms6564-DNA complex, SeMet-Ms6564 (residues 9–189) was mixed with brominated 31-bp DNA (5′-TCATAAACGAGACGGTACGTCTCGTCTTGTG-3′) at a molar ratio of ∼1.5:1 (Ms6564 dimer:DNA duplex) and incubated at 4 °C for 1 h. Crystals were obtained using the sitting drop vapor diffusion method at 4 °C. The reservoir solution contained 10% (w/v) PEG 3000, 100 mm imidazole/HCl (pH 8.0), and 200 mm lithium sulfate. The crystals were soaked in mother liquor containing 20% ethylene glycol and flash-frozen in liquid nitrogen.
We used the brominated DNA to identify the DNA bases. SeMet-Ms6564 was mixed with 31-bp brominated DNA (5′-TCATAAACGAGACGGTACGTCTCGTCTTGTG-3′). To avoid the effect of bromine atoms on the DNA binding ability of Ms6564, we chose the three underlined bases for bromination according to the electron density of the Ms6564-DNA complex. The crystallization of SeMet-Ms6564/brominated DNA complex was performed using the procedures described above for the native Ms6564-DNA complex.
Structure Determination and Refinement
The x-ray diffraction data were collected using Beamline 3W1A with a mounted MAR-165 CCD detector at Beijing Synchrotron Radiation Facility. All of the data were processed and scaled using the program HKL2000. The structure of SeMet-Ms6564 was determined by single anomalous dispersion. Three selenium sites were located and used to obtain the original experimental phases using Phenix.autosol, which located the selenium atoms and built the initial model; ∼90% of the residues of the whole peptide were traced. The remaining part was manually built using the program COOT. The intact model was refined in Phenix.refine. The crystal structure of Ms6564 (9–189) was determined by molecular replacement, and the structure of SeMet-Ms6564 (11–189) was used as the searching model in Phaser. Iterations of refinement using Phenix.refine, and manual refinement in COOT led to the final native model with excellent geometrical characteristics (see Table 1). The structure of the SeMet-Ms6564-DNA complex also was determined using the single anomalous dispersion method as described above. To determine the precise position of base pairs, DNA was bromine-labeled, and diffraction data at the bromine absorption edge were collected. Two bromine atom sites were identified based on the anomalous Patterson map. Refinement of the complex also was carried out using Phenix.refine. The double-stranded DNA in the final structure of the SeMet-protein-DNA complex did not contain bromine atoms to avoid the bromine effects on protein binding.
TABLE 1.
X-ray data collection and refinement statistics (values in parentheses stand for the parameters of the highest resolution)
| Ms6564 | Ms6564-DNA complexa | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.9793 | 0.9793 |
| Space group | C2221 | P31 |
| Unit cell parameters | a = 58.26, b = 118.29, c = 49.98 Å | a = b = 100.95, c = 99.86 Å |
| Resolution (Å) | 1.80 (1.83–1.80) | 2.50 (2.54–2.50) |
| Number of unique reflections | 16,377 (784) | 39,254 (1887) |
| Completeness (%) | 99.6 (95.7) | 99.5 (99.1) |
| Redundancy | 14.1 (9.70) | 6.30 (5.30) |
| Mean I/σ(I) | 51.1 (2.28) | 34.6 (2.37) |
| Molecules in asymmetric unit | 1 | 6 |
| Rmerge (%) | 4.50 (55.9) | 11.2 (61.0) |
| Structure refinement | ||
| Resolution range (Å) | 29.6–1.80 | 35.5–2.50 |
| Rwork/Rfree (%) | 21.9/26.9 | 22.5/28.1 |
| Number of atoms | ||
| Residues | 179 | 721 |
| Protein | 1361 | 5517 |
| Nucleotide | 1272 | |
| Waters | 114 | 65 |
| Average B factor (Å2) | ||
| Protein | 31.9 | 60.8 |
| Nucleic acid | 66.9 | |
| Waters | 34.5 | 51.2 |
| Ramachandran plot (%) | ||
| Most favored | 95.5 | 96.1 |
| Allowed | 4.5 | 3.9 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.007 | 0.009 |
| Bond angles (°) | 1.037 | 1.332 |
a SeMet-labeled protein.
DNA Substrate Preparation and EMSA
DNA fragments for the DNA binding activity assays were directly synthesized by Invitrogen or amplified by PCR from the genomic DNA of M. smegmatis mc2155. The DNA substrates were labeled and prepared as described previously (15) and stored at −20 °C until use. Mutant Ms6564 DNA (K47A, Q48A, and K47A/Q48A) were obtained by site-specific mutagenesis using wild type DNA as a template and were cloned into pET28a vectors. Protein expression in E. coli BL21(DE3) was induced with 0.1 mm β-d-1-thiogalactopyranoside at 37 °C for 4 h and purified by Ni2+affinity columns as previously described (15). EMSA experiments using labeled DNA fragments also were performed as previously described (15). Images were acquired using a Typhoon Scanner (GE Healthcare).
RESULTS
Crystal Structure of Ms6564 Alone
We solved the crystal structure of Ms6564 to 1.9 Å and refined it to Rwork (21.9%) and Rfree (26.9%) (Table 1). There is one molecule in the asymmetry unit, and the functional dimer has crystallographic symmetry (Fig. 1A, upper panel). The overall structure of Ms6564 is similar to that of other TFRs (19, 20) and is composed of nine helices: α1 (13–29), α2 (36–43), α3 (47–53), α4 (57–72), α5 (82–98), α6 (100–115), α7 (117–142), α8 (146–167), and α9 (173–187) (Fig. 1A, upper panel). The homodimer can be divided into two domains: the N-terminal DBD and the C-terminal ligand-binding domain (Fig. 1A). The DBD core is composed of helices α1–α3 (residues 13–53), which contain a typical helix-turn-helix motif (α2-α3) and one TFR-featured short recognition helix (α3). Helices α6, α8, and α9 participate in the dimer interface and account for 700 Å2 of the interface surface. Strikingly, Ms6564 was found to contain a 10-residue α4 helix, which is shorter than that of other TFRs, such as QacR (19) (Fig. 1A, lower panel), EthR (23), CmeR (24), AcrR (25), and LfrR (26) but similar to that in the E. coli TetR regulator (17).
FIGURE 1.
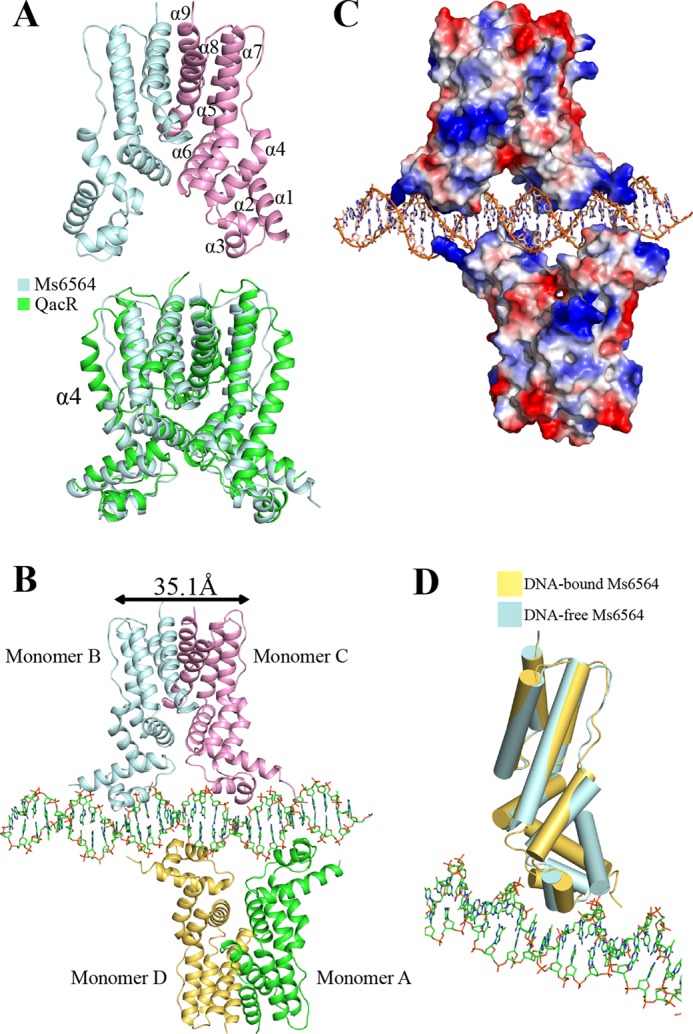
Representation of Ms6564 and the Ms6564-DNA complex. A, structure of the Ms6564 homodimer (upper panel) compared with QacR from S. aureus (lower panel). The secondary structural elements of Ms6564 are labeled. Ms6564 and QacR are displayed in pale cyan and green, respectively. The α4 is noted. B, overall structure of the Ms6564-operator complex. Ribbons represent proteins; sticks represent DNA. The subunits of one dimer are shown as pale cyan and pink, and those of the other as yellow and green. The proximal monomers are pink and yellow, whereas the distal monomers are pale cyan and green. The center to center distance of recognition helices is 35.1 Å, and a 4-bp sequence separates Ms6564 from proximal helices. C, electrostatic surface potentials of Ms6564 upon DNA binding. The blue regions indicate positive electrostatic regions, and red regions indicate negative electrostatic regions. The positively charged N-terminal arms of Ms6564 are noted. The range in electrons between dark red and dark blue is from −77.250 to 77.250. D, structural comparison of Ms6564 with (yellow) and without (pale cyan) DNA binding. Cylinders represent proteins; sticks represent DNA.
Overall Structure of the Ms6564-Operator Complex
We further solved the crystal structure of the Ms6564-operator complex to 2.5 Å and refined it to Rwork (22.5%) and Rfree (28.1%) (Table 1). As shown in Fig. 1B, the crystallographic complex is comprised of four Ms6564 monomers and a 31-bp palindromic DNA substrate that is part of the M. smegmatis promoter. Four similar monomers form two dimers: one is composed of distal monomer A and proximal D, and the other is composed of monomer distal B and proximal C (Fig. 1B). The root mean square deviation between the two dimers is 1.18 Å, which indicated they are similar to each other. The operator DNA is recognized by the DBD of Ms6564 and by two dimers on the opposite sides of the DNA (Fig. 1, B and C). When Ms6564 binds DNA, the N-terminal domain is bent toward the DNA, and the positively charged N-terminal arm further inserts into the minor groove (Fig. 1D). Calculation consistently reveals that the root mean square deviation between the DNA-free and DNA-binding structures is 1.8 Å, which indicates the DNA binding induces a significant change in orientation between these terminal domains. Thus, Ms6564 undergoes a conformation change upon binding DNA. Compared with QacR, the symmetry axes of Ms6564 dimers lie in the same plane and antiparallel to each other (Fig. 2, A and B). Ms6564 binds DNA only flexibly, and its recognition helix inserts slightly into the DNA major groove (Fig. 2C, upper panel). This is strikingly different from the case of the QacR-DNA complex, in which recognition helices sink deep into the major groove floor (Fig. 2C, lower panel).
FIGURE 2.
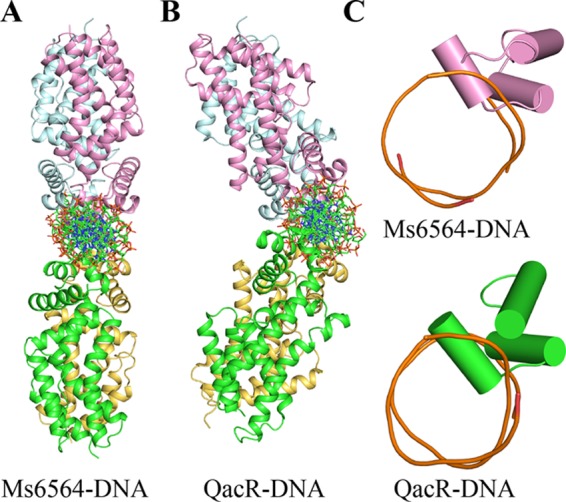
Comparison of two TetR family members bound to cognate DNA. A, structure of the Ms6564-operator complex. This view is that of Fig. 1B rotated by 90°. Ribbons represent proteins; sticks represents DNA. B, structure of the QacR-DNA complex. This view is same as that in A. C, comparison of the relative depths of two recognition helices into the DNA major groove. In the Ms6564-DNA complex (upper panel), the recognition helix is inserted only slightly into the major groove. In contrast, the recognition helix of QacR sinks deeply into the major groove floor (lower panel). Cylinders represent proteins, and tubes represent DNA.
DNA Deformation Occurs in the Ms6564-DNA Complex
We observed clear evidence of DNA deformation upon Ms6564 binding, although the DNA displays typical B-form DNA with average global roll and twist angles of 2.9 and 34.7°. In the Ms6564-DNA complex, the mean width of the major groove decreased to 10.5 Å compared with 11.4 Å for canonical B-DNA. Interestingly, the recognition helices contact all four regions where the DNA major groove became narrow. In contrast, the average minor groove width is 7.4 Å, which represents a significant increase compared with 5.9 Å for canonical B-form DNA (Fig. 3). Thus, conformation of the DNA changed in the regulator-DNA complex, but the DNA retains the conformation of B-form DNA.
FIGURE 3.

DNA deformation in the Ms6564-DNA complex. A, Ms6564-bound DNA is highlighted by blue, and canonical B-DNA is highlighted by purple. B, the major and minor groove width of the Ms6564-bound DNA. The values for canonical B-DNA are included for comparison (upper panel). The roll and twist angle for each base pair step of the Ms6564-bound DNA is shown in the lower panel.
Eleven Water Molecules Are Involved in Bridging the Protein-DNA Interaction
Previous reports do not describe water molecules that participate in protein-DNA interactions in TFRs (19–21). Unexpectedly, we found that seven water molecules bridge the contacts between Ms6564 and DNA base pairs and four water molecules mediate hydrogen bonds between protein and the DNA backbone (Fig. 4A). The residues Glu-37, Lys-47, Gln-48, Thr-49, and Tyr-51 participate in water-mediated interactions with base pairs. In the Ms6564-DNA complex, two water molecule-mediated hydrogen bonds form between monomer D and the base pairs (Fig. 4D). Only one water molecule contributes to the DNA binding in monomer A or B (Fig. 4, B and E), but three water molecules participate in indirect base pair binding in monomer C (Fig. 4C).
FIGURE 4.

Water-mediated interactions between Ms6564 and DNA. A, 11 water molecules are incorporated into the protein-DNA interface. Red spheres represent water molecules that participate in base pair recognition. The water molecules that bridge the contacts between side chains and the DNA backbone are displayed as blue spheres. The water molecules are numbered. The views of water-mediated base pair interactions between each monomer and DNA are shown in B–E. Each monomer is colored as in Fig. 1B, and the hydrogen bonds mediated by water molecules are represented by dotted lines. Electron density maps (2Fo − Fc) are contoured at the 1.1 σ level (blue mesh).
Only Two DBD Residues Directly Interact with Bases of the Cognate DNA
The DNA operator has a total of 10 bases and 39 phosphates that make direct contact with the two Ms6564 dimers (Fig. 5A). Six residues of short recognition helix (α3, positions 47–53), which is similar to QacR (19), participate in DNA recognition. Within the DBD of Ms6564, two residues, Lys-47 and Gln-48, were observed to directly interact with bases of the cognate DNA (Fig. 5B, left panel). Although Lys-47 interacts only with G, Gln-48 recognizes the base with lower specificity and can interact with cytosine (Fig. 5B, middle panel) or adenine (Fig. 5B, right panel). The nitrogen atom at zeta position of Lys-47 forms hydrogen bonds with the O6 atom and N7 atom of guanine 13. Compared with the two hydrogen bonds between Lys-47 and guanine, Gln-48 forms only one hydrogen bond with its target base, between OE1 of Gln-48 and N4 of cytosine 11 (in monomers A, B, and C, Gln-48 makes contacts with cytosine), or between NE2 of Gln-48 and N7 of adenine 16 (monomer D) (Fig. 5B, middle and right panels). Interestingly, the positively charged Lys-47 and the negatively charged Glu-37 interact with each other upon DNA binding of Ms6564 (Fig. 5B, left panel).
FIGURE 5.

Interactions between Ms6564 and its operator. A, schematic representation of Ms6564-DNA contacts. Rectangles, bases; P, phosphate; sugar ring, deoxyribose; gray circles, water molecules. The sugar ring is numbered. Hydrogen bonds between amino acids and base pairs are represented by red arrows. Hydrogen bonding interactions between amino acids and the DNA backbone are represented by green arrows. The dotted arrows represent hydrogen bonds mediated by water molecules. The base pairs in gray indicate the DNA sequence motif. B, close-up views of direct base pair recognition in the major groove. After DNA binding, the positively charged Lys-47 and negatively charged Glu-37 residues interact with each other (left panel). The hydrogen bonds (dotted lines) between Lys-47 and guanine and between Gln-48 and cytosine, are shown in the middle. Although Lys-47 exhibits specific recognition of guanine, Gln-48 can also make hydrogen bonds with adenine (right panel). The dashed lines represent hydrogen bonds, and only one DNA strand is shown for simplicity. Electron density maps (2Fo − Fc) are contoured at the 1.5 σ level (blue mesh).
Structural information suggests that the base pairs that contact with these two residues may be important for regulator-DNA interaction. To test this idea, we designed several new DNA substrates with mutated base pairs that contact Lys-47, Gln-48, or both (Fig. 6A). When all the nucleotides contacting both residues were mutated from guanine or cytosine to adenine, Ms6564 lost specific DNA binding activity. In comparison, Ms6564 could still bind other mutant DNA. Interestingly, Ms6564 could still bind substrate S2 (Fig. 6, A and B, lanes 5–8), in which a base C, previously omitted from the Ms6564 DNA-binding motif (15), was mutated. This result indicates that the C base is not essential, which is consistent with previous results.
FIGURE 6.
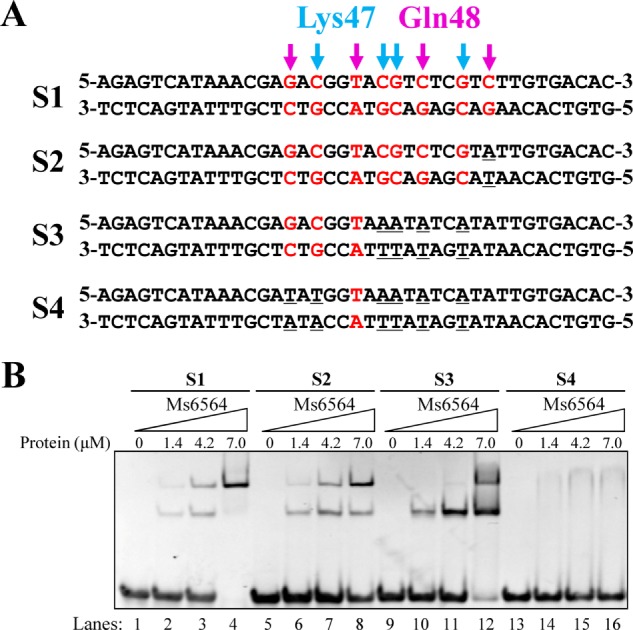
Electrophoretic mobility shift assays for the DNA binding activity of Ms6564 on different DNA substrates. A, DNA substrates designed for the DNA binding activity assays. The bases bound by Ms6564-K47 (indicated by magenta arrows) or Ms6564-Q48 (indicated by blue arrows) in the crystal structure are highlighted. B, EMSA assays for DNA binding activity of Ms6564 on different DNA substrates. 32P-Labeled DNA substrates were co-incubated with increasing amount (1.4–7 μm) of Ms6564 and loaded onto 5% polyacrylamide/bis (37.5:1) gels and run at a constant voltage of 100 V. The concentrations of the proteins are indicated.
Lys-47 Is a Critical Residue for Specific DNA Binding Activity of Ms6564
The present structural data imply that two DBD domain residues, Lys-47 and Gln-48, play important roles in interactions between regulators and DNA. In particular, Lys-47 specifically interacts with guanine through two hydrogen bonds, suggesting that Lys-47 functions as a primary amino acid residue for DNA binding specificity. This hypothesis is confirmed by further mutation and EMSA experiments. Lys-47 and Gln-48 are situated at the positive electrostatic surface of Ms6564 (Fig. 7A). When an increasing amount (1.4–7 μm) of protein is co-incubated with DNA, no obvious shifted bands are observed for the K47A or K47A/Q48A mutant proteins (Fig. 7B, lanes 1–6), which indicated that Lys-47 is essential for the specific DNA binding activity of Ms6564. In contrast, clear shifted bands are still observed for the Q48A mutant protein (Fig. 7B, lanes 3–9). These data indicate that Lys-47, but not Gln-48, is essential for the specific interaction between Ms6564 and its cognate DNA. Interestingly, further alignment analysis indicates that Lys-47 is highly conserved in the Ms6564, QacR, and CgmR proteins (Fig. 7C). Taken together, we have characterized Lys-47 as a critical residue for specific binding of Ms6564 to its cognate operator DNA.
FIGURE 7.

The conserved Lys-47 residue interacts directly with DNA base and is critical for specific DNA binding activity of Ms6564. A, electrostatic surface potential of Ms6564. Electrostatic surface representation of Ms6564 with blue and red regions indicating positive and negative electrostatic regions, respectively. The amino acid residues Glu-37, Lys-47, and Gln-48 are shown as sticks. Note the positively charged Lys-47 and negatively charged Glu-37. B, assays for the DNA binding activity of K47A and Q48A variants of Ms6564. 32P-Labeled DNA substrates were co-incubated with increasing amounts (1.4–7 μm) of wild type or mutant Ms6564 protein and loaded onto 5% polyacrylamide/bis (37.5:1) gels and run at a constant voltage of 100 V. Lane 1, substrate only; lanes 2–4, wild type Ms6564; lanes 5–7, K47A mutant variant of Ms6564; lane 8–10, Q48A mutant variant of Ms6564; lane 11–13, K47Q48A mutant variant of Ms6564. No obviously shifted bands were observed for the K47A variant, whereas clearly shifted bands were observed for the Q48A variant of Ms6564. C, alignment of the amino acid sequence of Ms6564 with those of QacR and CgmR. Conserved residues are boxed and highlighted in red; the conserved lysine located on the N terminus of the recognition helix is marked by a triangle.
DISCUSSION
In recent years, several master regulators that extensively regulate the expression of many genes have been characterized. However, the structural basis for broad transcriptional regulation remains unclear. In this study, we determine the crystal structure of a TFR master regulator, Ms6564, and the Ms6564-operator complex. Although we reveal a general similarity to typical TFR proteins, Ms6564 contacted DNA more loosely, and many disordered water molecules participated in the interface between Ms6564 and DNA. This is the first case in which water molecules have been found to participate in the interaction of a TFR regulator and DNA. In addition, only two amino acid residues in DBD of Ms6564, namely Lys-47 and Gln-48, directly interact with bases of the DNA. These findings enhance our understanding of the mechanisms of protein-DNA interaction and transcriptional regulation.
Overall crystallographic structure of the Ms6564-DNA complex is generally similar to that of typical TFRs. For example, it contains four similar monomers that together comprise two dimers. The operator DNA was recognized by two dimers on the opposite sides of the DNA. However, distinct differences from other TFRs are evident. First, in the Ms6564-DNA complex, the symmetry axes of two dimers lie in the same plane, antiparallel to each other. This is strikingly different from that of QacR in which its two dimers do not lie in the same plane but form a triangular cavity between each dimer, and the angle between the two dimers is nearly 130° (Fig. 2B) (19). The DNA binding mode of Ms6564 might increase its flexibility and selectivity to interact with the target operator DNA, and therefore, the master regulator can more easily play an extensive regulatory function. Second, compared with only 10 bp in QacR, the proximal recognition helices (from monomer C and monomer D) in Ms6564 are separated by only 4 bp. The center to center distance between the recognition helices of each monomer in a Ms6564 dimer is 35.1 Å (measured by the distance between the amide nitrogens of Gln-48). In contrast, this distance is 37 Å in QacR, wider than that of Ms6564, implying that a major DNA deformation was induced by QacR. This hypothesis is consistent with our observation that the DNA has still retained a B-form conformation despite being deformed in the Ms6564-DNA complex. Third, a key distinction between Ms6564 and many TRFs (such as TetR, QacR, and CgmR), or even many DNA-binding proteins, is the location of recognition helix. For example, in the QacR-DNA complex, the major groove is widened significantly throughout the entire binding site (19). Instead of sinking deeply into the DNA major groove (17, 19, 20), the recognition helix of Ms6564 inserts slightly into the DNA major groove. Fourth, hydrophobic amino acid residues Ile-50 and Trp-53 are far from the interaction interface, and no hydrophobic interactions are observed between the recognition helix and DNA major groove in the Ms6564-DNA complex. In contrast, the strong hydrophobic interactions exist between the recognition helix and DNA in TFR- and CgmR-DNA complexes (17, 20), which might push the water molecular out of the protein-DNA interface. Taken together, these findings suggest that Ms6564 makes flexible contact with DNA and may thus slide on the DNA more easily compared with other TFRs.
The water molecule has been reported to play important roles for interaction between protein and DNA (1, 27, 28). One example is that in the trp repressor-operator complex, where three highly ordered water molecules mediate interactions between the base pairs of half-operators and half-repressors (28). However, water molecules are not reported to participate in regulating interactions between TFR proteins and DNA (17, 19, 20, 29). In the current study, a significant number of water molecules unexpectedly participated in the Ms6564-DNA interface. Moreover, compared with the well ordered water molecules in the trp repressor-DNA interface, these 11 water molecules existed in a disorderly manner within the crystal structure. These water molecules obviously contribute to the DNA binding affinity of Ms6564. This finding is consistent with the observation that Ms6564 inserts only slightly into DNA and that its recognition helix makes flexible contacts with the DNA major groove (Fig. 2C, upper panel). This structure leaves a suitable space for water molecules to be incorporated into the Ms6564-DNA interface. In contrast, with other typical TetR regulator-DNA complexes, such as the QacR-DNA complex, the regulator is tightly bound to the DNA substrate, and the recognition helices of the protein sink deeply into the major groove floor (Fig. 2C, lower panel), leaving no space for water molecules to enter the interface of the protein-DNA complex. Therefore, our study reveals a novel structure model in which disordered water molecules can participate in the interaction between TFRs and DNA. This finding enhances our understanding of the mechanisms of regulator-DNA interaction for the TetR family of transcriptional factors.
Another interesting observation we made is that only two residues in the DBD of Ms6564 are involved in direct interaction with DNA. In contrast, both QacR and CgmR have four amino acid residues that engage in direct DNA binding (19, 20), and three amino acid residues are responsible for DNA binding in the E. coli TetR-DNA complex (17). In addition, a transcriptional repressor, MogR, has seven amino acid residues involved in direct interaction with DNA bases (30). We report that Lys-47 and Gln-48 residues in Ms6564 directly interact with bases of the cognate DNA. Lys-47 is specifically associated with guanine through bifurcated hydrogen bonds in each monomer. However, Gln-48 recognizes bases with lower specificity only through a hydrogen bond. Previous studies indicate that a single hydrogen bond usually does not contribute to base specificity (1, 31). Consistent with our interpretation, our mutation experiments indicated that Lys-47, but not Gln-48, is essential for DNA binding specificity of Ms6564. M. smegmatis is a fast growing and nonpathogenic mycobacterium whose genome has a high GC percentage of nearly 65%. The GC-rich genome thus provides large numbers of potential bases that can be recognized by Lys-47. This could be a possible mechanism by which Ms6564 regulates expression of many target genes and functions as a master regulator in M. smegmatis.
In summary, we report the crystal structure of the Ms6564-DNA complex and that the conformations of both the regulator and the DNA change upon their interaction. Compared with other TFR proteins, the recognition helix of Ms6564 inserts only slightly into the DNA major groove, and numerous disordered water molecules unexpectedly bridge the interface of TFR-DNA. Furthermore, the symmetry axes of two Ms6564 dimers lie on the same plane, and the DNA still retains a B-form conformation in the complex. These data imply that Ms6564 can bind DNA with strong affinity but makes flexible contacts with DNA. This function may permit the regulator to slide more easily along the genomic DNA and extensively regulate the expression of diverse genes in M. smegmatis.
This work was supported by National Natural Science Foundation of China Grants 31121004, 31025002, and 30930003; Fundamental Research Funds for the Central Universities Grant 2011PY140; the Creative Research Groups of Hubei; and the Hubei Chutian Scholar Program (to H. Z.-G.).
The atomic coordinates and structure factors (codes 4JKZ and 4JL3) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TFR
- TetR family regulator
- DBD
- DNA-binding domain
- SeMet
- selenomethionine-labeled.
REFERENCES
- 1. Rohs R., Jin X., West S. M., Joshi R., Honig B., Mann R. S. (2010) Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 79, 233–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garvie C. W., Wolberger C. (2001) Recognition of specific DNA sequences. Mol. Cell 8, 937–946 [DOI] [PubMed] [Google Scholar]
- 3. Rohs R., West S. M., Sosinsky A., Liu P., Mann R. S., Honig B. (2009) The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Müller C. W. (2001) Transcription factors. Global and detailed views. Curr. Opin. Struct. Biol. 11, 26–32 [DOI] [PubMed] [Google Scholar]
- 5. Bolla J. R., Do S. V., Long F., Dai L., Su C. C., Lei H. T., Chen X., Gerkey J. E., Murphy D. C., Rajashankar K. R., Zhang Q., Yu E. W. (2012) Structural and functional analysis of the transcriptional regulator Rv3066 of Mycobacterium tuberculosis. Nucleic Acids Res. 40, 9340–9355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miller D. J., Zhang Y. M., Subramanian C., Rock C. O., White S. W. (2010) Structural basis for the transcriptional regulation of membrane lipid homeostasis. Nat. Struct. Mol. Biol. 17, 971–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sawai H., Yamanaka M., Sugimoto H., Shiro Y., Aono S. (2012) Structural basis for the transcriptional regulation of heme homeostasis in Lactococcus lactis. J. Biol. Chem. 287, 30755–30768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nair S. K., Burley S. K. (2003) X-ray structure of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 112, 193–205 [DOI] [PubMed] [Google Scholar]
- 9. Maris A. E., Sawaya M. R., Kaczor-Grzeskowiak M., Jarvis M. R., Bearson S. M., Kopka M. L., Schröder I., Gunsalus R. P., Dickerson R. E. (2002) Dimerization allows DNA target site recognition by the NarL response regulator. Nat. Struct. Biol. 9, 771–778 [DOI] [PubMed] [Google Scholar]
- 10. Baikalov I., Schröder I., Kaczor-Grzeskowiak M., Cascio D., Gunsalus R. P., Dickerson R. E. (1998) NarL dimerization? Suggestive evidence from a new crystal form. Biochemistry 37, 3665–3676 [DOI] [PubMed] [Google Scholar]
- 11. Baikalov I., Schröder I., Kaczor-Grzeskowiak M., Grzeskowiak K., Gunsalus R. P., Dickerson R. E. (1996) Structure of the Escherichia coli response regulator NarL. Biochemistry 35, 11053–11061 [DOI] [PubMed] [Google Scholar]
- 12. Deng D., Yan C., Pan X., Mahfouz M., Wang J., Zhu J. K., Shi Y., Yan N. (2012) Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335, 720–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin P., Deng D., Yan C., Pan X., Xi J. J., Yan N., Shi Y. (2012) Specific DNA-RNA hybrid recognition by TAL effectors. Cell Rep. 2, 707–713 [DOI] [PubMed] [Google Scholar]
- 14. Deng D., Yin P., Yan C., Pan X., Gong X., Qi S., Xie T., Mahfouz M., Zhu J. K., Yan N., Shi Y. (2012) Recognition of methylated DNA by TAL effectors. Cell Res. 22, 1502–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang M., Gao C., Cui T., An J., He Z. G. (2012) A TetR-like regulator broadly affects the expressions of diverse genes in Mycobacterium smegmatis. Nucleic Acids Res. 40, 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le T. B., Schumacher M. A., Lawson D. M., Brennan R. G., Buttner M. J. (2011) The crystal structure of the TetR family transcriptional repressor SimR bound to DNA and the role of a flexible N-terminal extension in minor groove binding. Nucleic Acids Res. 39, 9433–9447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orth P., Schnappinger D., Hillen W., Saenger W., Hinrichs W. (2000) Structural basis of gene regulation by the tetracycline inducible Tet repressor–operator system. Nat. Struct. Biol. 7, 215–219 [DOI] [PubMed] [Google Scholar]
- 18. Schumacher M. A., Miller M. C., Grkovic S., Brown M. H., Skurray R. A., Brennan R. G. (2001) Structural mechanisms of QacR induction and multidrug recognition. Science 294, 2158–2163 [DOI] [PubMed] [Google Scholar]
- 19. Schumacher M. A., Miller M. C., Grkovic S., Brown M. H., Skurray R. A., Brennan R. G. (2002) Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J. 21, 1210–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Itou H., Watanabe N., Yao M., Shirakihara Y., Tanaka I. (2010) Crystal structures of the multidrug binding repressor Corynebacterium glutamicum CgmR in complex with inducers and with an operator. J. Mol. Biol. 403, 174–184 [DOI] [PubMed] [Google Scholar]
- 21. Kim Y., Kim B. S., Park Y. J., Choi W. C., Hwang J., Kang B. S., Oh T. K., Choi S. H., Kim M. H. (2010) Crystal structure of SmcR, a quorum-sensing master regulator of Vibrio vulnificus, provides insight into its regulation of transcription. J. Biol. Chem. 285, 14020–14030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kendall S. L., Withers M., Soffair C. N., Moreland N. J., Gurcha S., Sidders B., Frita R., Ten Bokum A., Besra G. S., Lott J. S., Stoker N. G. (2007) A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol. Microbiol. 65, 684–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carette X., Blondiaux N., Willery E., Hoos S., Lecat-Guillet N., Lens Z., Wohlkönig A., Wintjens R., Soror S. H., Frénois F., Dirié B., Villeret V., England P., Lippens G., Deprez B., Locht C., Willand N., Baulard A. R. (2012) Structural activation of the transcriptional repressor EthR from Mycobacterium tuberculosis by single amino acid change mimicking natural and synthetic ligands. Nucleic Acids Res. 40, 3018–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gu R., Su C. C., Shi F., Li M., McDermott G., Zhang Q., Yu E. W. (2007) Crystal structure of the transcriptional regulator CmeR from Campylobacter jejuni. J. Mol. Biol. 372, 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li M., Gu R., Su C. C., Routh M. D., Harris K. C., Jewell E. S., McDermott G., Yu E. W. (2007) Crystal structure of the transcriptional regulator AcrR from Escherichia coli. J. Mol. Biol. 374, 591–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bellinzoni M., Buroni S., Schaeffer F., Riccardi G., De Rossi E., Alzari P. M. (2009) Structural plasticity and distinct drug-binding modes of LfrR, a mycobacterial efflux pump regulator. J. Bacteriol. 191, 7531–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Otwinowski Z., Schevitz R. W., Zhang R. G., Lawson C. L., Joachimiak A., Marmorstein R. Q., Luisi B. F., Sigler P. B. (1988) Crystal structure of trp repressor/operator complex at atomic resolution. Nature 335, 321–329 [DOI] [PubMed] [Google Scholar]
- 28. Kalodimos C. G., Biris N., Bonvin A. M., Levandoski M. M., Guennuegues M. (2004) Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 305, 386–389 [DOI] [PubMed] [Google Scholar]
- 29. Ramos J. L., Martínez-Bueno M., Molina-Henares A. J., Terán W., Watanabe K., Zhang X., Gallegos M. T., Brennan R., Tobes R. (2005) The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69, 326–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen A., Higgins D. E., Panne D. (2009) Recognition of AT-rich DNA binding sites by the MogR repressor. Structure 17, 769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coulocheri S. A., Pigis D. G., Papavassiliou K. A., Papavassiliou A. G. (2007) Hydrogen bonds in protein-DNA complexes. Where geometry meets plasticity. Biochimie 89, 1291–1303 [DOI] [PubMed] [Google Scholar]