

Background: Actinoporins are pore-forming toxins that damage cellular membranes by α-helices.
Results: An engineered mutant of actinoporin equinatoxin II reveals sequential steps during pore formation.
Conclusion: Pore formation is composed of a succession of ordered steps: fast membrane binding followed by the N-terminal region association with the membrane and oligomerization.
Significance: Equinatoxin II pore formation does not require stable prepore intermediate as is often found in other pore-forming toxins.
Keywords: Erythrocyte, Fluorescence, Kinetics, Membrane, Toxins, Actinoporin, Equinatoxin, Pore Forming
Abstract
Actinoporin equinatoxin II (EqtII) is an archetypal example of α-helical pore-forming toxins that porate cellular membranes by the use of α-helices. Previous studies proposed several steps in the pore formation: binding of monomeric protein onto the membrane, followed by oligomerization and insertion of the N-terminal α-helix into the lipid bilayer. We studied these separate steps with an EqtII triple cysteine mutant. The mutant was engineered to monitor the insertion of the N terminus into the lipid bilayer by labeling Cys-18 with a fluorescence probe and at the same time to control the flexibility of the N-terminal region by the disulfide bond formed between cysteines introduced at positions 8 and 69. The insertion of the N terminus into the membrane proceeded shortly after the toxin binding and was followed by oligomerization. The oxidized, non-lytic, form of the mutant was still able to bind to membranes and oligomerize at the same level as the wild-type or the reduced form. However, the kinetics of the N-terminal helix insertion, the release of calcein from erythrocyte ghosts, and hemolysis of erythrocytes was much slower when membrane-bound oxidized mutant was reduced by the addition of the reductant. Results show that the N-terminal region needs to be inserted in the lipid membrane before the oligomerization into the final pore and imply that there is no need for a stable prepore formation. This is different from β-pore-forming toxins that often form β-barrel pores via a stable prepore complex.
Introduction
A large number of lipid membrane-active proteins and peptides have evolved to affect viability of targeted cells by compromising their membrane integrity. Understanding the kinetics and steps in structural changes upon interaction of these molecules with the lipid membrane is essential for understanding their role in the cell physiological processes. Pore-forming toxins (PFT)2 were found to be a useful model to study many of these processes and provided clues on structural features of membrane proteins, their folding, and protein-protein interactions in the membrane environment. PFT intoxicate cells by a multistep mechanism that involves binding to the cell plasma membrane, oligomerization in the plane of the membrane, and final pore formation (1–3). PFT are a large group of proteins, produced by all major groups of organisms. They are divided primarily by the structural element of the final pore (1). β-Sheet PFT form pores by transmembrane β-barrels (4), whereas α-helical PFT use clusters of amphipathic or hydrophobic helices (2). In general, the details of the pore-forming mechanism of α-PFT are not as well understood as those of the β-PFT. Apart from structural details of the pore formed by cytolysin A from Escherichia coli (5), molecular properties of α-PFT pores are poorly known (i.e. the stoichiometry of the pore, protein regions that participate in the final pore, etc.) (1).
Actinoporins are a unique group of PFT, expressed by sea anemones, and are very important to the understanding of how α-PFT function (6–8). The two most studied examples are equinatoxin II (EqtII) from the sea anemone Actinia equina and sticholysin II from Stichodactyla helianthus. Actinoporins are 20-kDa, mostly basic proteins. Structures of three homologues revealed a conserved fold composed of tightly folded β-sandwich core with two α-helices flanked on each side of the molecule (9–13) (Fig. 1A). Actinoporins form cation-selective pores with a diameter of ∼2 nm, mostly on membranes containing sphingomyelin (SM) (14–16). The initial membrane attachment is achieved by an exposed aromatic cluster situated on the broad loop on the bottom of the molecule and the C-terminal α-helix and by a phosphorylcholine binding site (11, 17–21). The N-terminal region, residues 1–28, was shown to be the only part of the molecule that undergoes a conformational change, and its flexibility is essential for the formation of the final pore (19, 22–24). After membrane binding, the N terminus is detached from the protein core and translocated into the lipid-water interface (22, 25) and finally across the lipid membrane, where it participates in formation of the final pore (25, 26). The molecular details about the final transmembrane pore of actinoporins are still lacking. Based on the kinetic data and cross-linking experiments, three to four monomers were deduced to be part of the final pore (14, 15). The pore was proposed to contain also lipid molecules from the membrane in a toroidal arrangement (27, 28). However, this model of the final pore was recently challenged by the crystal structure of fragaceatoxin (FraC), an actinoporin from the sea anemone Actinia fragacea (12). FraC crystals contain a nonameric arrangement of molecules, and based on the cryo-electron microscopy data, this oligomeric structure was proposed to be the functional unit in lipid membranes. The FraC monomer structure in the nonamer superimposed with EqtII very well, and the N-terminal region remained associated with the β-sandwich. It was proposed that the observed structure is the prepore of actinoporins. Prepores were observed in many β-PFT (4) and were proposed also for some α-PFT (5, 29).
FIGURE 1.

Properties of engineered EqtII mutant. A, three-dimensional model of EqtII. The N-terminal region, which is α-helical in the lipid membrane environment (19, 24), is shown in red. Residues at positions 8, 18, 69, and 179 are shown with spheres and denoted by the residue number. The membrane binding region is located on the bottom of the molecule and is composed of amino acids from the C-terminal helix and large loops at the bottom of the molecule (schematically denoted by a dashed line). B, SDS-PAGE of EqtIIV8C,I18C,K69C-NBD (left) and the same gel under UV light before Coomassie Blue staining (right). 1, wild type EqtII; 2, EqtIIV8C,I18C,K69C-NBD-OX; 3, EqtIIV8C,I18C,K69C-NBD-RED; M, molecular weight marker. EqtIIV8C,I18C,K69C-NBD-OX shows a gel mobility shift due to the disulfide bridge formed between Cys-8 and Cys-69. C, the rate of hemolysis of the wild type (black squares), EqtIIV8C,I18C,K69C (triangles), and EqtIIV8C,I18C,K69C-NBD (circles) was measured turbidimetrically at 630 nm by using a microplate reader. Shown are reduced (solid symbols and green) and oxidized (open symbols and red) variants of EqtIIV8C,I18C,K69C. n = 10–18, average ± S.D. (error bars). Oxidized mutants are unable to transfer the N-terminal helix to the lipid environment, thus preventing their hemolytic activity. D, activity of the EqtIIV8C,I18C,K69C in PLB. PLB were formed of 1,2-diphytanoyl-sn-glycerophosphocholine and 20% (w/w) SM. The buffer was 10 mm Tris-HCl, 100 mm KCl, pH 8.0, on both sides of the membrane. EqtIIV8C,I18C,K69C-RED or EqtIIV8C,I18C,K69C-OX was added at a 2 nm (RED) and 10 nm (OX) final concentration to the cis side, where a constant voltage +40 mV was applied. 1 mm DTT was added into the trans chamber 30 min after the addition of EqtIIV8C,I18C,K69C-OX and to the cis side after another 30-min incubation (denoted by arrows). Representative traces of at least two independent experiments are shown. E, gel filtration chromatography of the wild type (black), EqtIIV8C,I18C,K69C-NBD-OX (red), and EqtIIV8C,I18C,K69C-NBD-RED (green) on a Superdex HR200 column. EqtII mutants eluted as a single peak, showing no sign of aggregation in solution. The buffer used was 20 mm phosphate buffer, 300 mm NaCl, pH 7.2, at 0.5 ml/min. The elution volume of markers is denoted by gray lines. EqtIIV8C,I18C,K69C-NBD-RED was preincubated with DTT, which resulted in the additional peak in the chromatogram. 30 mm DTT injection in the running buffer is shown as a control by the thin gray trace.
In the present work, distinct steps of EqtII pore formation, binding, insertion of the N-terminal α-helix into the membrane, and oligomerization were studied by the use of an engineered EqtII molecule. We show that the insertion of the N-terminal helix in the lipid membrane is dependent on the lipid composition of the membrane and that it immediately follows the initial binding of the toxin on the membranes containing SM. The inactive mutant, which has a membrane-penetrating region attached to the core of the molecule by an introduced disulfide bond, still binds to the lipid membrane and is able to oligomerize on the membrane surface. When such a membrane-bound molecule was treated with reductant dithiothreitol (DTT), the kinetics of the N-terminal insertion and pore forming ability was much slower as compared with the prereduced form. The results imply that the N-terminal region must be inserted into the membrane before the final oligomerization and pore formation occurs. Results imply that stable prepores, observed in almost all cases of β-PFT, are not needed in the pore formation of actinoporins.
EXPERIMENTAL PROCEDURES
Materials
Bovine-brain SM, 1,2-dioleoyl-sn-glycerophosphocholine (DOPC), 1,2-diphytanoyl-sn-glycerophosphocholine and 1-palmitoyl-2-stearoyl-(5-doxyl)-sn-glycero-3-phosphocholine (5-NO-PC) were from Avanti Polar Lipids (Alabaster, AL). All other materials were from Sigma unless specified differently.
Cloning, Expression, and Isolation of the Mutants
The recombinant wild-type EqtII and mutants EqtIII18C, EqtIIA179C, EqtIIV8C,I18C,K69C, and EqtIIV8C,K69C,A179C were prepared as described (30). The expression vectors of the single and triple cysteine mutants were prepared by replacing corresponding EqtII residues with cysteine as described previously (23, 25). All proteins were expressed in an E. coli BL21 (DE3) strain and purified from the bacterial cytoplasm as described elsewhere (30). Wild-type EqtII and mutants were purified to homogeneity as observed on SDS-polyacrylamide gels.
Protein Labeling
For NBD labeling, EqtIIV8C,I18C,K69C was incubated in 20 mm phosphate buffer, pH 7.2, with the addition of oxidant (0.05 mm 1,10-phenantroline and 0.01 mm CuSO4) at an ambient temperature to obtain oxidation and disulfide bond formation between amino acid residues Cys-8 and Cys-69. After 15 min, protein was separated from oxidant with a PD-10 desalting column (GE Healthcare). This step was omitted for EqtIII18C. Thereafter, N-((2-(iodoacetoxy)ethyl-N-methyl)amino-7-nitrobenz-2-oxi-1,3-diazole (IANBD) (Invitrogen) was added in an 8-fold molar excess in order to label Cys-18. The mixture was incubated on a magnetic stirrer for 1 h at ambient temperature. Labeled protein, EqtIIV8C,I18C,K69C-NBD, was separated from the excess probe by using a 1-ml HiTrap SP HP ion exchange chromatography column (GE Healthcare). Alternatively, EqtIIV8C,I18C,K69C was also incubated in 50 mm Na2HPO4, pH 7.0, overnight at 4 °C to obtain oxidation. IANBD was added in a 20-fold molar excess and incubated on a magnetic stirrer for 4 h at 4 °C. In this case, the labeled protein was separated from the excess probe by using a MonoS ion exchange chromatography column (Amersham Biosciences). The percentage of labeled protein was determined spectrophotometrically from the absorbance at 280 nm (labeled and unlabeled protein) and 478 nm (labeled protein), using molar absorption coefficients of 43,850 m−1 cm−1 for the mutant protein and 25,000 m−1 cm−1 for IANBD (25). A correction factor of 0.084 was used for the absorbance of IANBD at 280 nm.
Similarly, EqtIIA179C and EqtIIV8C,K69C,A179C were labeled at the 179 position with the thiol-reactive dye Alexa Fluor® 488 C5 maleimide (A488; Invitrogen) to obtain EqtIIA179C-A488 and EqtIIV8C,K69C,A179C-A488, respectively. The percentage of labeled protein was determined from the absorbance measured at 280 nm (labeled and unlabeled protein) and 495 nm (labeled protein, A488), using a molar absorption coefficient of 71,000 m−1 cm−1 for A488. A correction factor of 0.125 for the absorbance at 280 nm was used.
Treatment of Mutants with Reductant or Oxidant
The unlabeled or labeled triple cysteine mutants were treated either with 20 mm DTT or 0.5 mm 1,10-phenantroline and 0.1 mm CuSO4 for 30 min at 37 °C to obtain a reduced (e.g. EqtIIV8C,I18C,K69C-NBD-RED) or oxidized (EqtIIV8C,I18C,K69C-NBD-OX) form of the mutant, respectively (23). These concentrations were sufficient to obtain fully oxidized or reduced sample according to SDS-PAGE. Samples were immediately used for measurements. The final concentration of the reductant did not exceed 2 mm and, therefore, had no effect on liposomes or bovine red blood cells (data not shown).
Hemolytic Assay
Hemolytic activity was measured turbidimetrically by the use of a microplate reader (MRX; Dynex Technologies) (18). Thoroughly washed bovine red blood cells (BRBC) were suspended in hemolysis buffer (130 mm NaCl, 20 mm Tris-HCl, pH 7.4) to a dilution of A630 = 0.5. One hundred μl of BRBC suspension were added to 100 μl of 2-fold serially diluted protein. Hemolysis was monitored by measuring the absorbance at 630 nm for 20 min at room temperature.
Planar Lipid Bilayer Experiments
Solvent-free planar lipid bilayers (PLB) were made of 1,2-diphytanoyl-sn-glycerophosphocholine and 20% SM (w/w) (31). EqtIIV8C,I18C,K69C-RED or EqtIIV8C,I18C,K69C-OX was added at 2 and 10 nm concentration, respectively, to stable preformed bilayers on the cis side only (the cis side is where electrical potential was applied, and the trans side was grounded). When using EqtIIV8C,I18C,K69C-OX, 1 mm DTT was added after 30 min of incubation, first into the trans and later into the cis side. All experiments were started in symmetrical conditions, using 10 mm Tris-HCl, 100 mm KCl, pH 8.0, on both sides of the membrane. In addition, when experiments with EqtIIV8C,I18C,K69C-RED were performed, the buffer contained 0.5 mm DTT in order to keep the toxin fully reduced during the experiment. A defined voltage, generally +40 mV, was applied across the membrane. Miniature magnetic stir bars stirred the solutions on both sides of the membrane. The currents across the bilayer were measured, and the conductance (G) was determined as follows (31),
where I is the current through the membrane and U is the applied transmembrane potential.
Macroscopic currents were recorded by a patch clamp amplifier (Axopatch 200, Axon Instruments). A PC equipped with a DigiData 1200 A/D converter (Axon Instruments) was used for data acquisition. The current traces were filtered at 100 Hz and acquired at 500 Hz by the computer using Axoscope 8 software (Axon Instruments). All measurements were performed at room temperature.
For the selectivity measurement, KCl concentration was increased on the trans side only to form a 10-fold gradient (1 m KCl). The potential necessary to zero the transmembrane current (i.e. the reversal potential Urev) was determined. From the reversal potential, the ratio of the cation over anion permeability (P+/P−) was calculated using the Goldman-Hodgkin-Katz equation (32),
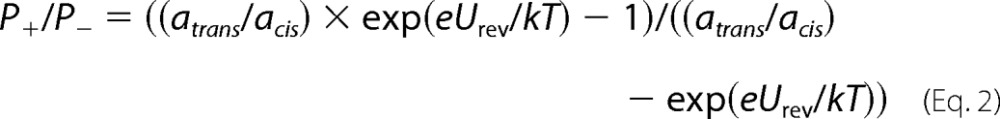 |
where atrans and acis are the activities of KCl in the trans and cis side, respectively. kT/e is ∼25 mV at room temperature.
Lipid Vesicle Preparation
Large unilamellar vesicles (LUV) were prepared from pure DOPC or equimolar mixture of DOPC and brain SM (DOPC/SM). Liposomes used in lipophilic quenching experiments were prepared by replacing 20 mol % DOPC with 5-NO-PC. Lipids were dissolved in chloroform, which was removed from the lipid mixture with a rotary evaporator. The lipid film was suspended in the vesicle buffer (140 mm NaCl, 20 mm Tris-HCl, 1 mm EDTA, pH 8.5) and freeze-thawed six times to create multilamellar vesicles. LUV were extruded through polycarbonate membranes with 100-nm pores by an Avestin lipid extruder (33). Lipid concentration was determined using commercial test LabAssay Phospholipid (Wako Chemicals GmbH) according to the producer's instructions.
Erythrocyte Ghost Preparation
Fresh BRBC were used for preparation of vesicles from red blood cell membranes (ghosts). BRBC were washed 5–7 times in the washing buffer (140 mm NaCl, 5 mm Na2HPO4, pH 8) and centrifuged for 5 min at 2500 rpm and room temperature. Three ml of washed BRBC were then added to 50 ml of an ice-cold lysis buffer (5 mm Na2HPO4, pH 8.0) in order to achieve osmotic lysis. Lysed BRBC were centrifuged at 26,300 × g for 20 min at 4 °C. The lysis cycle was repeated until clean membranes were obtained (7–10 times). The membrane pellet was suspended in ghost buffer (280 mm NaCl, 40 mm Tris-HCl, 2 mm EDTA, pH 8.5) and subjected to 10 freeze-thaw cycles.
Binding to Liposomes
Binding of toxin to liposomes was determined by measuring the residual hemolytic activity of the unbound toxin. 50 nm EqtIIV8C,I18C,K69C-RED was incubated with lipids at the desired molar lipid/toxin (L/T) ratio in a total volume of 100 μl for 10 min at a room temperature. Thereafter, the lipid/toxin mixture was 2-fold serially diluted. One hundred μl of BRBC suspension (A630 = 0.5) was added, and hemolysis was measured as described above.
Fluorescence Spectroscopy
All fluorescence measurements were performed by using a Jasco FP-750 spectrofluorimeter (Jasco Corp.). The sample compartment was equipped with a Peltier thermostated single-cell holder. All experiments were done at 25 °C and at a protein concentration of 250 nm in the vesicle buffer. The excitation and emission slits were set to 5 nm. NBD fluorescence spectra were measured by excitation at 470 nm, and emission was scanned from 500 to 600 nm. Subsequently, the appropriate amount of liposomes, with or without 5-NO-PC, was added at the desired L/T ratio. The spectra were corrected for the dilution factor, and the background was subtracted using the appropriate blank with liposomes. The emission wavelength was set to 538 nm in measurements of the kinetics of NBD fluorescence. The experiment was started with the desired amount of the toxin in the vesicle buffer, and after 1 min, liposomes were added to reach the desired L/T ratio.
Permeabilization of BRBC ghosts loaded with calcein was monitored in a 1.5-ml cuvette. The excitation and emission wavelengths were set to 485 and 520 nm, respectively; both slits were set to 5 nm. Ghosts at the desired concentration were stirred at 25 °C. 50 nm protein was added after 1 min, and the fluorescence was followed for 10–30 min. The maximal release was obtained by the addition of final 2 mm Triton X-100. The percentage of calcein release (i.e. the permeabilizing activity (P (%)) was calculated as follows,
where Fmin and Fmax represent values of fluorescence before toxin addition and after Triton X-100 addition, respectively. FTox is steady-state fluorescence intensity obtained after toxin addition.
Stopped-flow Fluorescence Measurements
Experiments were carried out in vesicle buffer at 20 °C. Temperature was controlled by an SC150/A10 bath circulator (Thermo Fisher Scientific). Changes of intrinsic tryptophan fluorescence and NBD or A488 fluorescence upon interaction with DOPC/SM (1:1) LUV were monitored by using a stopped-flow SX-20 apparatus (Applied Photophysics). The protein concentration was 100 nm, and the lipid concentration used was 10 μm, resulting in an L/T molar ratio of 100. The excitation wavelength for tryptophans was 295 nm to avoid tyrosine fluorescence, and a 357/44-nm band pass filter (Semrock) was used for emission. NBD was excited at 470 nm, and emission was monitored with a 495-nm cut-off filter (Schott). NBD fluorescence traces were corrected for NBD-labeled protein in solution alone. For A488 excitation in a self-quenching experiment, 480-nm light was used. A488 emission intensity was monitored by using a cut-off filter at 495 nm (Schott) in combination with a 410–510-nm band pass filter (Schott). The molar ratio of A488/unlabeled protein was 3:7 in order to avoid large signal amplitudes. Self-quenching kinetics was unaffected by the A488 dilution (not shown). Fourteen traces were recorded for intrinsic tryptophan fluorescence, and seven traces were recorded for NBD or A488 experiments. Bad traces were removed by eye, and remaining traces were averaged to obtain a single fluorescent trace. The experimental data were fitted to a single- or double-exponential function,
where F and F0 are fluorescence and fluorescence at time t = 0, A1 and A2 are the amplitudes of the increase, and k1 and k2 are the observed rate constants.
Cross-linking Experiments
Cross-linking experiments were performed with BRBC. Proteins were incubated with 50 μl of BRBC suspension in 30 mm polyethylene glycol 3350 (A630 = 1.0) at a desired concentration and L/T ratio for 10 min at room temperature. Disuccinimidyl suberate (Pierce), prepared freshly as a 2.5 mm stock solution in DMSO, was added to a ∼100 μm final concentration and incubated at room temperature for 5 min. Samples were centrifuged for 10 min at 14,000 × g at room temperature. The supernatant was removed, and pellet was resuspended in 10 μl of SDS-PAGE buffer. Proteins were resolved by a PHAST system (GE Healthcare) on 8–25% gradient gels essentially as described by Anderluh et al. (17). Proteins were resolved by SDS-PAGE, transferred to polyvinylidene fluoride membranes, and blotted with rabbit anti-EqtII serum. Bands were stained with goat anti-rabbit antibodies conjugated with horseradish peroxidase using 4-chloro-1-naphthol/H2O2 as a substrate.
Assaying the Accessibility of the Disulfide Bond
The accessibility of the disulfide bond was assayed by using 2-nitro-5-thiosulfobenzoate (NTSB) as described before (34). Briefly, EqtIIV8C,I18C,K69C-OX or EqtIIV8C,I18C,K69C-OX preincubated for 10 min at room temperature with DOPC/SM (1:1) LUV at an L/T ratio of 100 was assayed in 200 mm Tris, 100 mm Na2SO3, 3 mm EDTA, 0.325 mm NTSB, pH 8.5, at a final 5 μm protein concentration. The absorbance was followed at 412 nm for 600 s at room temperature.
Statistics
Student's two-sample t test was performed for statistical analysis of data, with a confidence interval of 95% and significance level at 0.05 (p < 0.05).
RESULTS
Design of EqtII Triple Cysteine Mutant That Allows Study of Different Steps in Pore Formation
Native EqtII does not possess any cysteine. We have shown previously that the disulfide formation between engineered cysteines at positions 8 and 69 abolished the hemolytic activity. The disulfide prevented the N-terminal region of the protein from transferring to the lipid membrane and participating in the formation of a functional pore (19, 23). Such an EqtII variant might be useful in studying the molecular mechanism of EqtII pore formation. Extrinsic fluorescent probes attached to different parts of the molecule may report the changes in the environment during different steps of the pore forming process, and the presence of the cleavable disulfide bond at the same time enables control over the helix transfer to the lipid membranes. We chose residues 18 and 179 as sites for the probe attachment (Fig. 1A). Labeling of position 18 could report membrane interactions of the N-terminal region. We have previously shown that labeling of the EqtIII18C was sufficiently good and that this residue was transferred to the lipid bilayer during the pore formation (25). The labeling at position 179 has been shown not to affect permeabilizing activity (35), and it is fully exposed to the solvent when the protein is in solution or bound to membranes (17, 35). The C terminus is located on the side opposite that occupied by regions of the molecule that participate in membrane binding (18, 19, 36) or formation of the final pore (23, 25), and any labeling does not interfere with various steps in pore formation. This position would thus be suitable to report oligomerization at the surface of the membrane, when labeled with convenient fluorescent probes.
The resulting triple cysteine mutants EqtIIV8C,I18C,K69C and EqtIIV8C,K69C,A179C were expressed in E. coli and purified in amounts comparable with the wild-type protein (30). EqtIIV8C,I18C,K69C was readily labeled with IANBD at position 18 (to yield EqtIIV8C,I18C,K69C-NBD) (Fig. 1B). The degree of the oxidation was checked by SDS-polyacrylamide gels and measurement of hemolytic activity (Fig. 1, B and C). The oxidized mutant, EqtIIV8C,I18C,K69C-NBD-OX, traveled farther on the SDS-polyacrylamide gels. There were no double bands visible, indicating that the protein prepared in this way was fully oxidized. The pretreatment with DTT fully reduced the disulfide bond formed between residues 8 and 69 (EqtIIV8C,I18C,K69C-NBD-RED; Fig. 1B) and shifted the band to the position of the wild type. The hemolytic activity was evaluated on bovine erythrocytes (Fig. 1C). EqtIIV8C,I18C,K69C-RED and EqtIIV8C,I18C,K69C-NBD-RED were active at a level comparable with that of the wild-type EqtII on BRBC, showing that the mutations and labeling with IANBD did not change hemolytic activity (supplemental Table 1) (c50EqtII = 0.19 ± 0.03 μg/ml, c50EqtIIV8C,I18C,K69C-NBD-RED = 0.18 ± 0.04 μg/ml, p = 0.39) (Fig. 1C). EqtIIV8C,I18C,K69C-OX and EqtIIV8C,I18C,K69C-NBD-OX were not hemolytic up to the highest protein concentration tested of 4 μg/ml. Similar results were obtained also with mutants at position 179, which was labeled with A488 (data not shown).
We also checked the pore forming activity of EqtIIV8C,I18C,K69C by measuring current through pores by the PLB approach. This is considered to be the most suitable approach to directly study permeabilizing activity of proteins (31). EqtIIV8C,I18C,K69C-RED readily formed pores in planar lipid membranes with conductances lower than that of the wild-type pores (GEqtII = 308 ± 85 pS, GEqtIIIV8C,I18C,K69C-RED = 125 ± 65 pS, p = 0.0006) but with the same conductance as the single cysteine mutant EqtIII18C (GEqtIII18C = 145 ± 72 pS, p = 0.705).3 The current-voltage characteristics of the wild-type EqtII and EqtIIV8C,I18C,K69C-RED were similar (data not shown). Cation selectivity, expressed as the ratio of permeabilities of cations over anions, P+/P−, was slightly higher for the cysteine mutant (P+/P−EqtII = 9 ± 1, P+/P−EqtIIV8C,I18C,K69C-RED = 12 ± 1, p = 0.009). The slightly higher cation permeability of the reduced mutant could be ascribed to the removal of the positive Lys-69 at the pore entrance and to the partially charged Cys-8 at this pH, because this cysteine residue is present in the lumen or close to the lumen of the pore and thus affects the selectivity (25). EqtIIV8C,I18C,K69C-OX did not form pores in PLB, even after prolonged, 60-min incubation (data not shown) or when the reductant (final 1 mm DTT) was added into the trans chamber (Fig. 1D). The pores formed only when DTT was added into the cis chamber (Fig. 1D). The conductance of these pores (GEqtIIV8C,I18C,K69C-OX = 138 ± 43 pS, p = 0.158), the cation permeability (P+/P−EqtIIV8C,I18C,K69C-OX = 12 ± 2, p = 0.883, reduced versus oxidized), and the current/voltage characteristics were similar to those of the pores formed by EqtIIV8C,I18C,K69C pretreated with the reductant (see above). On the other hand, the time needed to form pores was significantly longer for EqtIIV8C,I18C,K69C-OX after reductant addition (3–5 min) in comparison with the wild-type EqtII or EqtIIV8C,I18C,K69C-RED addition to planar lipid membranes (5–30 s).
We also checked whether labeling affects aggregation of EqtIIV8C,I18C,K69C in solution. To this end, we performed gel filtration chromatography. EqtII (20 kDa) shows aberrant traveling through gel filtration column because the elution volume was significantly larger as compared with the marker lysozyme (14 kDa). However, both labeled variants, EqtIIV8C,I18C,K69C-NBD-RED and EqtIIV8C,I18C,K69C-NBD-OX, traveled to a similar position as the wild-type protein, and there were no higher molecular weight aggregates visible (Fig. 1E).
According to the results, we concluded that protein prepared in this way has extrinsic fluorescent probe attached at position 18 and a cleavable disulfide bond that can be manipulated by the addition of reductant. Such a protein variant has properties closely resembling those of the previously characterized double cysteine mutant EqtIIV8C,I18C (19, 23) (i.e. there was no activity in the oxidized form, and the reduced form possessed a hemolytic and pore-forming ability closely resembling those of the wild-type EqtII). We then used labeled protein engineered in this way to study the association with the membranes during different steps of pore formation.
The Insertion of the N-Terminal Helix into the Lipid Environment
The fluorescence properties of NBD depend on the environment (25). Therefore, we next checked the fluorescence of EqtIIV8C,I18C,K69C-NBD for both reduced and oxidized states in solution and in the presence of LUV at an L/T molar ratio of 100 (Fig. 2 and Table 1). By measuring the residual hemolytic activity, we estimated that 85% of the protein was associated with liposomes (average of two independent experiments). We also ensured, by using SDS-PAGE, that the reduced and oxidized versions of the labeled triple mutant bound to lipid membranes in comparable degrees (see below). This behavior is in agreement with that of the double cysteine mutant EqtIIV8C,K69C, which in reduced or oxidized form was still able to bind to tethered lipid membrane in comparable nanomolar affinities to the wild-type protein as shown by surface plasmon resonance or by Western blotting using BRBC (19).
FIGURE 2.
NBD steady-state fluorescence emission spectra. NBD fluorescence of ∼250 nm EqtIIV8C,I18C,K69C-NBD in 140 mm NaCl, 20 mm Tris-HCl, 1 mm EDTA, pH 8.5, was measured in solution (solid line) and in the presence of liposomes of different composition at an L/T of 100 (dashed line). The excitation wavelength was set to 470 nm. The spectra were acquired under constant stirring and at 25 °C. The spectra presented are representative of 2–3 experiments. The insets in B and C show normalized spectra of reduced mutant in the absence (solid line) and presence (dashed line) of liposomes and ghosts, respectively. Green, EqtIIV8C,I18C,K69C-NBD-RED; red, EqtIIV8C,I18C,K69C-NBD-OX. A, DOPC LUV; B, DOPC/SM (1:1) LUV and 5-NO-PC/DOPC/SM (2:3:5) (gray); C, BRBC ghosts. a.u., arbitrary units.
TABLE 1.
Properties of NBD steady-state fluorescence spectra presented in Fig. 2
FL and FS represent fluorescence intensity measured at 538 nm in the presence of vesicles and in solution at a lipid/toxin ratio of 100 (for LUV) and 80 (for ghosts). n = 2–4, average ± S.D.
| EqtIIV8C,I18C,K69C-NBD-RED |
EqtIIV8C,I18C,K69C-NBD-OX |
|||
|---|---|---|---|---|
| λmax | FL/FS | λmax | FL/FS | |
| nm | nm | |||
| Solution | 539 ± 1 | 536 ± 1 | 2.8 ± 0.9a | |
| DOPC | 539 ± 2 | 1.0 ± 0.1 | 538 ± 3 | 0.90 ± 0.03 |
| DOPC/SM (1:1) | 530 ± 1 | 3.20 ± 0.04 | 535 ± 1 | 1.1 ± 0.1 |
| Ghosts | 531 ± 1 | 2.3 ± 0.3 | 535 ± 1 | 1.0 ± 0.1 |
| DOPC/5-NO-PC/SM (3:2:5) | 534 ± 1 | 1.5 ± 0.2 | 536 ± 1 | 0.90 ± 0.04 |
a This value reports the ratio between the fluorescence of oxidized and reduced version of the protein in solution.
The EqtIIV8C,I18C,K69C-NBD-RED in solution had a fluorescence emission maximum at 539 nm (Fig. 2 and Table 1), consistent with previous work (25). The fluorescence intensity of EqtIIV8C,I18C,K69C-NBD-OX in solution was increased and blue-shifted to 536 nm in comparison with the reduced form, indicating that the probe was positioned in a slightly more hydrophobic environment. This increase of NBD is not due to aggregation of EqtIIV8C,I18C,K69C-NBD-OX in solution, which could result in probe located in a more hydrophobic environment. The protein was found to be monomeric in solution according to SDS-PAGE and gel chromatography (Fig. 1). Residue 18 is located near the β-sandwich in the solution structure (Fig. 1A), so it is likely that the probe is located in a more hydrophobic environment induced by the disulfide formation in the oxidized state (i.e. between the helix (Cys-8) and the sandwich (Cys-69)). In agreement, the reduction of EqtIIV8C,I18C,K69C-NBD-OX in solution caused a drop in fluorescence and maximum emission shift to values similar to those for the reduced form of the mutant (Table 1), further indicating that the difference in the initial fluorescence intensity between the oxidized and the reduced form of the mutant was due to the orientation of the probe toward the hydrophobic core of the protein.
The fluorescence of EqtIIV8C,I18C,K69C-NBD-RED was increased and blue-shifted when DOPC/SM LUV or red blood cell ghosts were added (Fig. 2). This indicates that the probe was placed in a more hydrophobic lipid membrane environment. Apart from insertion into the lipid bilayer, the changes in fluorescence intensity could also be the result of protein structural rearrangements induced by the lipid membrane that would locate the probe in the more hydrophobic protein environment. The probe could also be located at the interfaces between different toxin monomers in the final pore. A lipophilic collisional quencher (5-NO-PC), which has a nitroxide quenching group located in the region of membrane acyl chains, was used to ascertain that the N-terminal helix indeed inserts into the lipid bilayer. The increase in the fluorescence intensity was considerably lower (∼50%), when 5-NO-PC was incorporated into the LUV (DOPC/5-NO-PC/SM = 3:2:5 molar ratio) (Fig. 2 and Table 1), indicating insertion of the N-terminal helix with the probe into the membrane. EqtII shows poor permeabilizing activity toward DOPC LUV (36), and accordingly, no changes in fluorescence properties of NBD were visible when these vesicles were used as a negative control (Fig. 2 and Table 1). The fluorescence of EqtIIV8C,I18C,K69C-NBD-OX was not changed when any of the tested liposomes or ghosts were added at any L/T ratio. This lack of change in the NBD environment indicates that the N-terminal helix remained fixed on the molecule and was not involved in protein oligomerization at the membrane surface or membrane insertion.
Kinetics of Different Steps in the Pore-forming Mechanism
To obtain better insight into the kinetics of different steps in pore formation induced by actinoporins, we used stopped-flow fluorescence kinetics measurements at similar conditions as in the experiments reported in Fig. 2 (L/T ratio of 100) (Fig. 3A). The EqtII binding can be conveniently monitored by intrinsic tryptophan fluorescence due to the increase in the fluorescence of exposed tryptophans from the aromatic cluster that participate in the membrane association (18, 19, 37). We compared the kinetics of membrane binding with the kinetics of the N-terminal helix transfer to the membrane by monitoring NBD fluorescence. The oligomerization of protein monomers at the surface of the membrane can be monitored by self-quenching of the A488 dye (38, 39) (Fig. 3A).
FIGURE 3.

Comparison of binding, insertion of the N-terminal region, and oligomerization by stopped-flow fluorescence measurements. A, a pore-forming mechanism of EqtII and different steps that were studied. Approximate positions of residues at positions 18 and 179 are presented by blue and red labels, respectively. B, fluorescence traces of different probes as denoted. The proteins that were used in each experiment are noted. The concentration of lipids was 10 μm. DOPC/SM (1:1) LUV in 140 mm NaCl, 20 mm Tris-HCl, 1 mm EDTA, pH 8.5, were used. Protein concentration was 100 nm to achieve an L/T ratio of 100. A minimum of four single injections were averaged to obtain a single fluorescent trace (gray). Residuals of the measured data to an exponential fit are shown below the traces. The red trace shows fit to a mono- or biexponential equation (as reported under “Results”).
The toxin binding was extremely fast. The increase in tryptophan fluorescence could be fitted to a monoexponential curve and was finished in less than 1 s (Fig. 3B). The increase in NBD fluorescence was much slower and could reliably be fitted to a biexponential equation. One of the processes was approximately 10 times slower than the binding, with the observed rate of ∼1 s−1. The other process was much slower (observed rate of ∼0.04 s−1) (Table 2). As expected, EqtIIV8C,I18C,K69C-NBD-RED showed similar kinetics to EqtIII18C-NBD, because a reduced disulfide bond enables an N-terminal conformational change in a fashion similar to that of EqtIII18C-NBD (Table 2). In the oxidized state of the mutant, there was no significant change in the NBD fluorescence, in agreement with the steady state data presented in Fig. 2. Fluorescence traces of A488, which report self-quenching, could be fitted to a monoexponential equation in the case of the wild-type EqtII. The observed rate was similar to the slowest process of the NBD fluorescence (quenching observed rate 0.034 s−1). We also checked self-quenching of the reduced and oxidized triple mutant and found it comparable with that of the wild-type at both conditions. However, in these cases, there were deviations from the monoexponential fit. We also noted that the amplitude of the signal change was larger in the case of the oxidized version in comparison with the wild type and the reduced form. This could indicate that monomers of the oxidized form can come closer to each other at the surface of the lipid membrane.
TABLE 2.
Parameters obtained from fitting the stopped-flow data presented in Fig. 3
The traces presented on Fig. 3 were fitted to a monoexponential (for the tryptophan fluorescence and A488 self-quenching traces) or biexponential equation. n = 3–5, average ± S.D. a.u., arbitrary units.
| k1 | A1 | k2 | A2 | |
|---|---|---|---|---|
| s−1 | a.u. | s−1 | a.u. | |
| Tryptophan fluorescence | ||||
| EqtII | 19.3 ± 3.4 | 0.13 ± 0.04 | ||
| NBD fluorescence | ||||
| EqtIII18C-NBD | 0.70 ± 0.05 | 0.14 ± 0.04 | 0.040 ± 0.005 | 0.36 ± 0.07 |
| EqtIIV8C,I18C,K69C-NBD-RED | 0.60 ± 0.06 | 0.26 ± 0.04 | 0.060 ± 0.002 | 0.74 ± 0.08 |
| A488 self-quenching | ||||
| EqtIIA179C-A488 | 0.034 ± 0.012 | −0.45 ± 0.21 | ||
| EqtIIV8C,K69C,A179C-A488-RED | 0.049 ± 0.004 | −0.58 ± 0.13 | ||
| EqtIIV8C,K69C,A179C-A488-OX | 0.074 ± 0.005 | −0.85 ± 0.12 | ||
Membrane-induced Aggregation
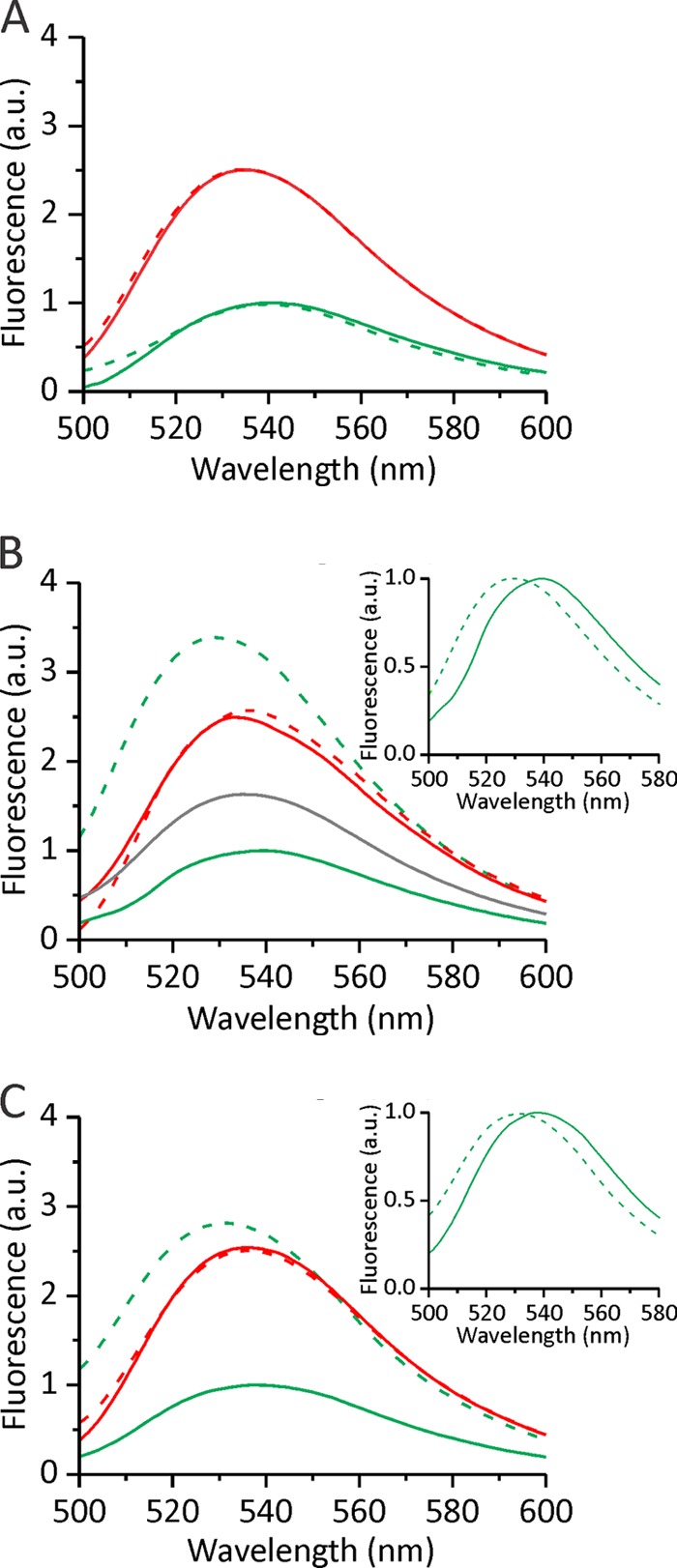
The stopped-flow results are particularly interesting for the oxidized version of the triple mutant because it did not show any changes in NBD fluorescence but obviously has the capacity to oligomerize at the surface of liposomes. To independently assess oligomerization at the plane of the membrane, we employed cross-linking of the protein bound to the membrane. It was previously shown that it is possible to cross-link monomeric EqtII at the surface of LUV (14) to provide qualitative data on its membrane binding and oligomerization. We therefore used disuccinimidyl suberate (DSS) to cross-link EqtII or EqtIIV8C,I18C,K69C via amino groups after binding to bovine erythrocytes. All proteins bound to membranes at comparable levels (Fig. 4). A minor amount of dimers was visible together with predominate monomeric form when proteins were incubated with DSS in the absence of membranes. However, in the presence of the membranes and DSS, the monomeric form of all of the proteins was largely shifted to large molecular weight forms. Trimers and sometimes even tetramers were enriched in comparison with the monomeric form. Some of the protein was found as large high molecular weight aggregates at the site of the application of samples; however, the amounts were very low in comparison with other forms (denoted as Agg in Fig. 4). In these tests, the oxidized version of EqtIIV8C,I18C,K69C behaved identically as the wild-type protein or the reduced version, indicating that membrane-induced aggregation is not prevented if movements of the N-terminal region are restricted by a disulfide formation.
FIGURE 4.
Oligomerization at the surface of the erythrocytes. 0.25 nmol of protein was incubated with BRBC suspension. After a 10-min incubation and the addition of DSS, BRBC with bound cross-linked toxin were pelleted. Electrophoresis buffer was added to the pelleted erythrocytes, and proteins were resolved by SDS-PAGE and blotted with rabbit anti-EqtII serum. Mon, monomer; Dim, dimer; Tri, trimer; Tet, tetramer; Agg, high molecular weight aggregates; wt, wild-type EqtII; Red, EqtIIV8C,I18C,K69C-RED; Ox, EqtIIV8C,I18C,K69C-OX. The left panel shows EqtII cross-linked in solution in the absence of BRBC. The gel was stained with Coomassie Blue.
Helix Must Insert into the Membrane Prior to Oligomerization for Efficient Pore Formation
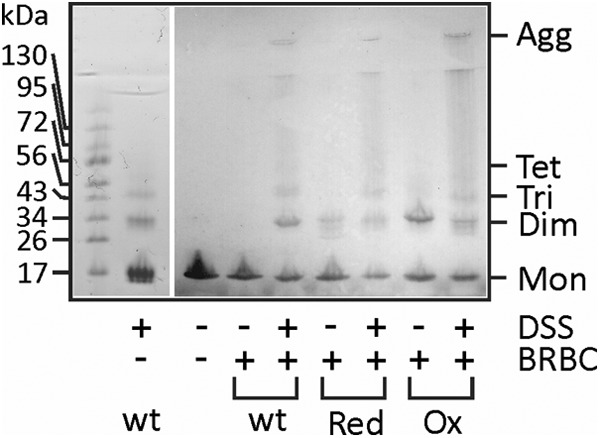
Finally, we tried to activate oxidized triple mutant with the addition of reductant after membrane binding. We therefore added 2 mm DTT to EqtIIV8C,I18C,K69C-NBD-OX after 5 min, when most of the mutant was bound to the membrane (Fig. 5, blue line). The kinetics of the N-terminal insertion was in this case considerably slower and was not finished even after 20 min (Fig. 5A). The differences in fluorescence properties between the reduced and oxidized forms of EqtIIV8C,I18C,K69C-NBD-OX enabled us also to monitor the kinetics of disulfide reduction in the buffer at the same experimental conditions. The mutant is fully reduced in less than 3 min upon the addition of DTT because the NBD fluorescence decreased to the level of EqtIIV8C,I18C,K69C-NBD-RED, much faster than the helix insertion. In order to independently monitor the accessibility of the disulfide between residues 8 and 69, we employed an NTSB assay (34). We found that the rate of the disulfide cleavage was the same when EqtIIV8C,I18C,K69C-NBD-OX was assayed in solution or when bound to LUV (Fig. 5A, inset). These results indicate that the rate of disulfide bond reduction is not the limiting factor in helix insertion of membrane-bound activated EqtIIV8C,I18C,K69C-NBD-OX. Similar results were obtained when using BRBC ghosts (Fig. 5B).
FIGURE 5.
Kinetic measurements of N-terminal helix insertion into the lipid environment. NBD fluorescence of 250 nm EqtIIV8C,I18C,K69C-NBD in reduced and oxidized form was measured at 538 nm. After 60 s, liposomes or ghosts (denoted by a black dot) were added at an L/T ratio of 100. Green, EqtIIV8C,I18C,K69C-NBD-RED in the presence of DOPC/SM LUV or ghosts; red, EqtIIV8C,I18C,K69C-NBD-OX in the presence of DOPC/SM LUV or ghosts; blue, EqtIIV8C,I18C,K69C-NBD-OX in the presence of DOPC/SM LUV or ghosts and reduced with final 2 mm DTT at 300 s (denoted by an arrow); orange, EqtIIV8C,I18C,K69C-NBD-OX in solution and reduced with final 2 mm DTT at 300 s (denoted by an arrow). The N-terminal helix insertion shows slower kinetics when the disulfide bridge is reduced after the protein fully binds to the lipid membrane. A, LUV composed of DOPC/SM; B, BRBC ghosts. The traces are representative of three independent experiments. Inset in A, the rate of the disulfide bond cleavage as monitored by the NTSB assay for EqtIIV8C,I18C,K69C in solution and when bound to LUV. Error bars, S.D. a.u., arbitrary units.
We also assessed the permeabilizing activity of the triple mutant in different oxidation states by assaying hemolytic activity and calcein release from ghosts (Fig. 6). The time needed to reduce absorbance of BRBC suspension by 50% (t50) was less than 1 min for the wild-type (t50 EqtII = 45 ± 5 s, n = 6) and around 2 min for EqtIIV8C,I18C,K69C-NBD-RED (t50 EqtIIV8C,I18C,K69C-NBD-RED = 124 ± 9 s, n = 6). EqtIIV8C,I18C,K69C-NBD-OX was not active in this assay (green trace in Fig. 6A). The activity was restored after the reductant was added to the suspension of BRBC. However, the kinetics of hemolysis was much slower when compared with prereduced mutant (Fig. 6A); t50 was 39 min (t50 EqtIIV8C,I18C,K69C-NBD-OX = 39 ± 8 min, n = 19). In addition, we checked the permeabilizing activity of EqtIIV8C,I18C,K69C-NBD on ghosts loaded with calcein (Fig. 6B). EqtIIV8C,I18C,K69C-NBD-OX did not show permeabilizing activity on ghosts (Fig. 6B). Its activity was restored when 2 mm final DTT was added to the ghost-bound protein (Fig. 6B). The final values of permeabilization were similar in both cases (PEqtIIV8C,I18C,K69C-NBD-RED (%) = 46.6 ± 2.6% (n = 3), PEqtIIV8C,I18C,K69C-NBD-OX + DTT (%) = 37.1 ± 0.3% (n = 3), p = 0.3). However, the kinetics was considerably slower in the case of the DTT addition (Fig. 6B). To summarize, efficient pore formation is linked to movement of the N-terminal region of the protein to the lipid membranes, and this can proceed only if this region of the protein is free. Membrane-bound oxidized oligomerized mutant could be activated with the addition of the reductant; however, the kinetics of the N-terminal insertion and pore formation was considerably slower.
FIGURE 6.
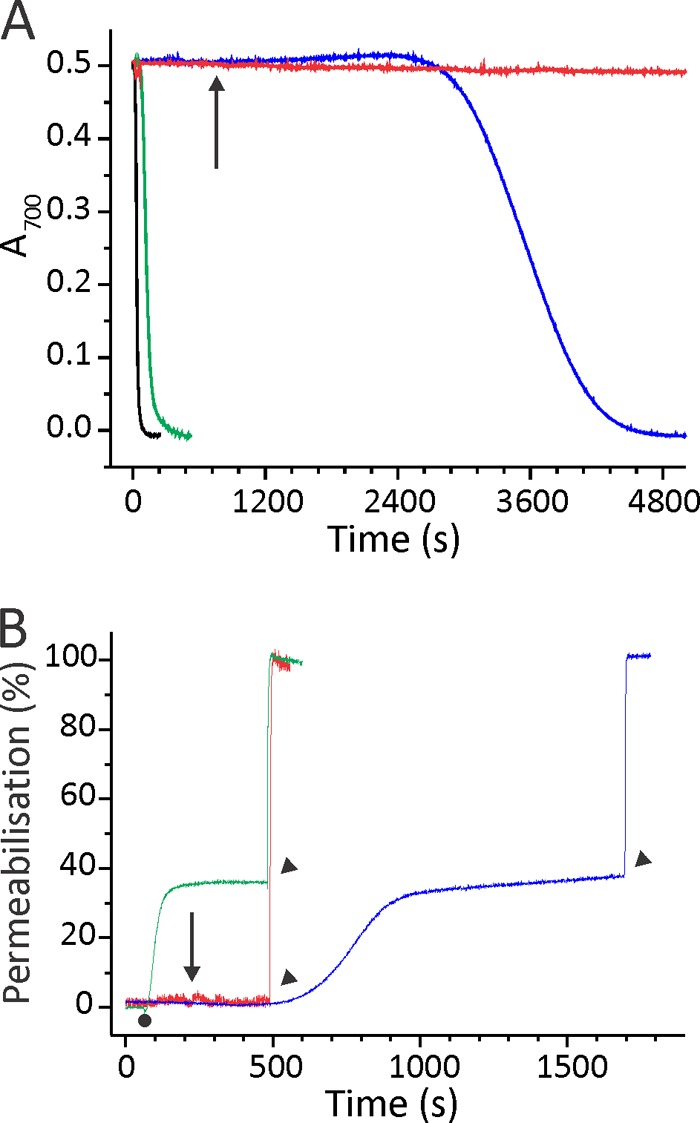
Permeabilizing activity of EqtIIV8C,I18C,K69C-NBD. A, the restoration of hemolytic activity of EqtIIV8C,I18C,K69C-NBD-OX by the DTT addition. BRBC were stirred in hemolysis buffer at 25 °C. Proteins were added to a final concentration of 7.5 nm. The figure is representative of at least six independent experiments. The reductant DTT was added at a final 2 mm concentration to EqtIIV8C,I18C,K69C-NBD-OX already bound to the membrane (denoted by an arrow). B, permeabilizing activity on calcein-loaded ghosts. Ghosts were stirred in vesicle buffer at 25 °C. The excitation and emission wavelengths were set to 485 and 520 nm, respectively. The addition of 50 nm EqtIIV8C,I18C,K69C-NBD-RED or EqtIIV8C,I18C,K69C-NBD-OX is denoted by a small dot. The percentage of permeabilization was determined at the end of the assay by comparing the fluorescence with the maximal one, obtained by the addition of 2 mm final Triton X-100 (denoted by arrowheads). EqtIIV8C,I18C,K69C-NBD-OX was also reduced after binding to ghosts with final 2 mm DTT (denoted by an arrow) to show that permeabilizing activity is slower in comparison with the protein reduced before binding to ghosts. The figures are representative of four independent experiments. The color code is the same for both panels: green, EqtIIV8C,I18C,K69C-NBD-RED; red, EqtIIV8C,I18C,K69C-NBD-OX; blue, EqtIIV8C,I18C,K69C-NBD-OX reduced with final 2 mm DTT after the protein was bound to the membranes; black, the wild type EqtII.
DISCUSSION
In this paper, we studied the separate steps in the pore-forming mechanism of EqtII by employing an engineered mutant protein (Fig. 7). The results allow us to refine a current model of the actinoporin pore-forming mechanism (6–8) by providing two novel important insights. First, we have shown that the N-terminal region insertion in the membrane is relatively fast in comparison with oligomerization, and second, the N-terminal helical region needs to be inserted into the membrane prior to oligomerization in order to allow fast and effective pore formation.
FIGURE 7.
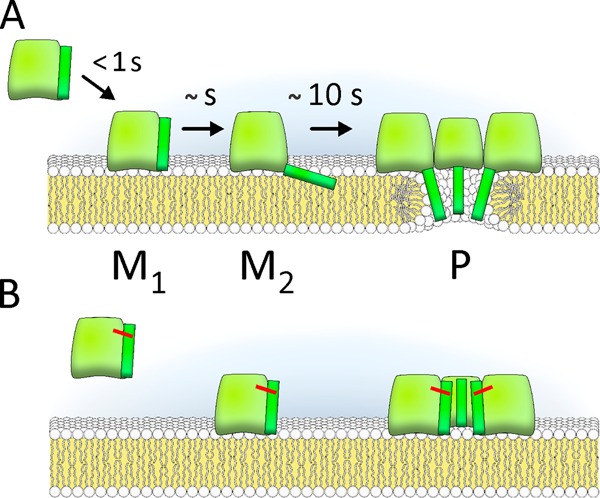
Pore-forming mechanism of EqtII. A, pore formation proceeds through membrane binding (M1 state (25)), N-terminal helix insertion in the membrane (M2), and oligomerization to a final pore (P). B, mutant in which cysteines at positions 8 and 69 locked N-terminal to the body of the molecule could bind to the membrane and oligomerize but could not form the pore.
The engineered fluorescently labeled EqtII mutant allowed us to compare the kinetics of three different steps in the pore-forming mechanism. Results presented in Fig. 3 showed that initial membrane association mediated by exposed aromatic amino acids is rapid (less than 1 s in our system) and followed closely by the N-terminal region insertion in the membrane as monitored by the NBD fluorescence (seconds). However, in this case, the change of the signal was more complex, and apart from the faster component, there was also the slower one (tens of seconds). This second component was as fast as the self-quenching that reports oligomerization, which was monitored by A488 fluorescence. As we and others have previously proposed, there are several membrane-bound forms of EqtII at the membrane, and the increase in the NBD fluorescence reflects the two forms: one where helix lies more or less parallel to the membrane (M2 in Fig. 7) (8, 20, 25) and the other one in the pore form (P), which may further change fluorescent properties of the NBD dye due to additional helix rearrangements. By the use of three different probes, we were therefore able to resolve three different forms of the protein at the surface of the lipid membrane (Fig. 7).
Membrane-bound trapped intermediates are useful in studying mechanisms of pore formation at the membrane surface (40, 41), especially when accessibility of the disulfide bonds is maintained in the bound form. For example, introduction of the disulfide bond between two domains of perfringolysin O blocks pore formation at the prepore state (40). Upon the addition of reductant, the rate of pore formation was much faster compared with the wild-type perfringolysin O because binding and oligomerization steps were already accomplished. In our case, we observed exactly the opposite; upon the addition of reductant, the N-terminal region membrane insertion and pore formation was much slower than normally observed with the wild-type EqtII (Fig. 6).
According to the increase in NBD fluorescence intensity, the N-terminal membrane insertion of bound EqtIIV8C,I18C,K69C-NBD-OX occurred only when the reductant was added (Fig. 5). However, in this case, the kinetics of the N-terminal membrane insertion was significantly slower. This agrees with hemolytic assays and calcein release from red blood cell ghosts (Fig. 6), where activity of initially bound and subsequently reduced EqtIIV8C,I18C,K69C-NBD-OX was compared with the activity of EqtIIV8C,I18C,K69C-NBD-RED. Although the kinetics of the pore formation is much slower once the toxin is attached to the membrane, the final pores have the same characteristics, as shown by planar lipid bilayer experiments (Fig. 1D), and proceed to the same final release of calcein in the BRBC ghost assay (Fig. 6B). The kinetics of EqtIIV8C,I18C,K69C-NBD-OX disulfide bond reduction in solution is not the limiting factor for the observed slow kinetics because the protein is fully reduced in less than 3 min (Fig. 5A). The DTT may act on a toxin not associated with the lipid membrane. However, as shown in Fig. 4, and consistent with (19, 23), comparable amounts of reduced and oxidized forms are bound to the membrane. The accessibility of the disulfide bond should be the same for the protein in solution or bound to the membrane unless (i) EqtII tilts upon membrane binding, posing the helix more parallel to the membrane, or (ii) EqtII rapidly oligomerizes on the membrane, thus preventing the N-terminal region from dissociating from the body of the molecule due to steric reasons. The former possibility would imply a form of the membrane-bound EqtII not yet anticipated (i.e. none of the mutants from that side of the β-sandwich, that were used in a cysteine-scanning mutagenesis, were shown to interact with the lipid membrane) (17). Furthermore, in the current model of actinoporin pore formation, the membrane binding site is exactly opposite the site of the disulfide bond between residues 8 and 69 (Fig. 1A, dashed line) (6–8, 20, 21). This implies that the disulfide remains accessible on the membrane-bound form, which we confirmed by using an NTSB assay. In the case of tilting, EqtIIV8C,I18C,K69C-NBD-OX should also move position 18 closer to the membrane, causing an increase in NBD signal, but this is not the case (Figs. 2 and 3). Finally, the reason for the slow kinetics may be the aggregation of the oxidized version of the toxin on the membrane plane similar to the wild type or the reduced form, possibly to form a porelike state by a weak association of monomers. In this case, the helices could not be transferred to the lipid-water interface after the disulfide reduction due to steric reasons. Disassembly of the molecules in the pore would allow insertion and consequent formation of functional pores but slowed down the insertion of the N-terminal region to the lipid membrane and consequently hemolysis or calcein release. Consistent with this proposition, cross-linking experiments (Fig. 4) confirmed the ability of the oxidized form of EqtIIV8C,I18C,K69C-NBD to oligomerize in the presence of erythrocytes. One conclusion from the cross-linking study is also that the region covered by the N-terminal part of the toxin does not participate in protein-protein interactions because the aggregation pattern of the oxidized form was no different from the wild-type EqtII or the reduced form of the mutant.
In summary, the results in this study hint at a fundamental difference in the pore-forming process of actinoporins from that of PFT, which form pores by a prepore intermediate. Clearly, the major conformational change in the molecule occurs immediately after the binding in the case of actinoporins and not after oligomerization, as is the case in some other examples of PFT. The N-terminal region of EqtII must be transferred to the membrane surface before the oligomerization to the final pore occurs because there is not enough space to coordinately transfer four helices from the membrane surface across the membrane to the other side through the lumen of a pore of 2 nm in diameter (11, 16). The proposed mechanism for actinoporin pore formation (Fig. 7) is different from the one proposed by Mechally et al. (12), where the N-terminal insertion occurs only after oligomerization in a prepore composed of nine monomers. Our observation is also in contrast to β-PFT that often form non-lytic prepore complexes. Prepores are stable associations of toxin subunits on the lipid membrane, which allow the formation of hydrogen bonds between the adjacent β-strands and a coordinate conformational change that brings them across the membrane as a β-barrel to form the final pores (4, 42–44). The resulting β-barrel structure does not leave any free non-hydrogen-bonded edge exposed to the hydrophobic milieu of the lipid membrane. In contrast, the actinoporin α-helix that will cross the lipid bilayer can exist at the lipid-water interface and in the lipid membrane core as single units because the stable hydrogen bond network is formed within the helix itself only, and therefore, there is no requirement for a stable prepore (45).
Supplementary Material
Acknowledgments
G. V. and M. D. S. thank the Laboratory of Biomolecular Sequence and Structure Analysis for Health for technical support.
This work was supported by grants from the Slovenian Research Agency.

This article contains supplemental Table 1.
G. Viero, unpublished observations.
- PFT
- pore-forming toxin(s)
- 5-NO-PC
- 1-palmitoyl-2-stearoyl-(5-doxyl)-sn-glycero-3-phosphocholine
- BRBC
- bovine red blood cell(s)
- DOPC
- 1,2-dioleoyl-sn-glycerophosphocholine
- DSS
- disuccinimidyl suberate
- EqtII
- equinatoxin II
- IANBD
- N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole
- NBD
- 7-nitrobenz-2-oxa-1,3-diazole
- L/T
- lipid to toxin molar ratio
- LUV
- large unilamellar vesicle(s)
- NTSB
- 2-nitro-5-thiosulfobenzoate
- PLB
- planar lipid bilayer(s)
- SM
- sphingomyelin
- A488
- Alexa Fluor® 488 C5 maleimide
- pS
- picosiemens.
REFERENCES
- 1. Parker M. W., Feil S. C. (2005) Pore-forming protein toxins. From structure to function. Prog. Biophys. Mol. Biol. 88, 91–142 [DOI] [PubMed] [Google Scholar]
- 2. Anderluh G., Lakey J. H. (2008) Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 33, 482–490 [DOI] [PubMed] [Google Scholar]
- 3. Dalla Serra M., Tejuca Martinez M. (2011) Pore-forming toxins. eLS, DOI: 10.1002/9780470015902.a0002655.pub2 [DOI] [Google Scholar]
- 4. Heuck A. P., Tweten R. K., Johnson A. E. (2001) β-barrel pore-forming toxins. Intriguing dimorphic proteins. Biochemistry 40, 9065–9073 [DOI] [PubMed] [Google Scholar]
- 5. Mueller M., Grauschopf U., Maier T., Glockshuber R., Ban N. (2009) The structure of a cytolytic α-helical toxin pore reveals its assembly mechanism. Nature 459, 726–730 [DOI] [PubMed] [Google Scholar]
- 6. Anderluh G., Maček P. (2002) Cytolytic peptide and protein toxins from sea anemones (Anthozoa: Actiniaria). Toxicon 40, 111–124 [DOI] [PubMed] [Google Scholar]
- 7. Kristan K. C., Viero G., Dalla Serra M., Maček P., Anderluh G. (2009) Molecular mechanism of pore formation by actinoporins. Toxicon 54, 1125–1134 [DOI] [PubMed] [Google Scholar]
- 8. García-Ortega L., Alegre-Cebollada J., García-Linares S., Bruix M., Martínez-Del-Pozo A., Gavilanes J. G. (2011) The behavior of sea anemone actinoporins at the water-membrane interface. Biochim. Biophys. Acta 1808, 2275–2288 [DOI] [PubMed] [Google Scholar]
- 9. Athanasiadis A., Anderluh G., Maček P., Turk D. (2001) Crystal structure of the soluble form of equinatoxin II, a pore-forming toxin from the sea anemone Actinia equina. Structure 9, 341–346 [DOI] [PubMed] [Google Scholar]
- 10. Hinds M. G., Zhang W., Anderluh G., Hansen P. E., Norton R. S. (2002) Solution structure of the eukaryotic pore-forming cytolysin equinatoxin II. Implications for pore formation. J. Mol. Biol. 315, 1219–1229 [DOI] [PubMed] [Google Scholar]
- 11. Mancheño J. M., Martín-Benito J., Martínez-Ripoll M., Gavilanes J. G., Hermoso J. A. (2003) Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure 11, 1319–1328 [DOI] [PubMed] [Google Scholar]
- 12. Mechaly A. E., Bellomio A., Gil-Cartón D., Morante K., Valle M., González-Mañas J. M., Guérin D. M. (2011) Structural insights into the oligomerization and architecture of eukaryotic membrane pore-forming toxins. Structure 19, 181–191 [DOI] [PubMed] [Google Scholar]
- 13. Pardo-Cea M. A., Castrillo I., Alegre-Cebollada J., Martínez-del-Pozo Á., Gavilanes J. G., Bruix M. (2011) Intrinsic local disorder and a network of charge-charge interactions are key to actinoporin membrane disruption and cytotoxicity. FEBS J. 278, 2080–2089 [DOI] [PubMed] [Google Scholar]
- 14. Belmonte G., Pederzolli C., Maček P., Menestrina G. (1993) Pore formation by the sea anemone cytolysin equinatoxin II in red blood cells and model lipid membranes. J. Membr. Biol. 131, 11–22 [DOI] [PubMed] [Google Scholar]
- 15. Tejuca M., Dalla Serra M., Ferreras M., Lanio M. E., Menestrina G. (1996) Mechanism of membrane permeabilization by sticholysin I, a cytolysin isolated from the venom of the sea anemone Stichodactyla helianthus. Biochemistry 35, 14947–14957 [DOI] [PubMed] [Google Scholar]
- 16. Tejuca M., Dalla Serra M., Potrich C., Alvarez C., Menestrina G. (2001) Sizing the radius of the pore formed in erythrocytes and lipid vesicles by the toxin sticholysin I from the sea anemone Stichodactyla helianthus. J. Membr. Biol. 183, 125–135 [DOI] [PubMed] [Google Scholar]
- 17. Anderluh G., Barlič A., Podlesek Z., Maček P., Pungerčar J., Gubenšek F., Zecchini M. L., Dalla Serra M., Menestrina G. (1999) Cysteine-scanning mutagenesis of an eukaryotic pore-forming toxin from sea anemone. Topology in lipid membranes. Eur. J. Biochem. 263, 128–136 [DOI] [PubMed] [Google Scholar]
- 18. Malovrh P., Barlič A., Podlesek Z., Maček P., Menestrina G., Anderluh G. (2000) Structure-function studies of tryptophan mutants of equinatoxin II, a sea anemone pore-forming protein. Biochem. J. 346, 223–232 [PMC free article] [PubMed] [Google Scholar]
- 19. Hong Q., Gutiérrez-Aguirre I., Barlič A., Malovrh P., Kristan K., Podlesek Z., Maček P., Turk D., González-Mañas J. M., Lakey J. H., Anderluh G. (2002) Two-step membrane binding by Equinatoxin II, a pore-forming toxin from the sea anemone, involves an exposed aromatic cluster and a flexible helix. J. Biol. Chem. 277, 41916–41924 [DOI] [PubMed] [Google Scholar]
- 20. Alegre-Cebollada J., Cunietti M., Herrero-Galán E., Gavilanes J. G., Martínez-del-Pozo A. (2008) Calorimetric scrutiny of lipid binding by sticholysin II toxin mutants. J. Mol. Biol. 382, 920–930 [DOI] [PubMed] [Google Scholar]
- 21. Castrillo I., Araujo N. A., Alegre-Cebollada J., Gavilanes J. G., Martínez-del-Pozo A., Bruix M. (2010) Specific interactions of sticholysin I with model membranes. An NMR study. Proteins 78, 1959–1970 [DOI] [PubMed] [Google Scholar]
- 22. Gutiérrez-Aguirre I., Barlič A., Podlesek Z., Maček P., Anderluh G., Gonzǎlez-Mañas J. M. (2004) Membrane insertion of the N-terminal alpha-helix of equinatoxin II, a sea anemone cytolytic toxin. Biochem. J. 384, 421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kristan K., Podlesek Z., Hojnik V., Gutiérrez-Aguirre I., Gunčar G., Turk D., González-Mañas J. M., Lakey J. H., Maček P., Anderluh G. (2004) Pore formation by equinatoxin, a eukaryotic pore-forming toxin, requires a flexible N-terminal region and a stable β-sandwich. J. Biol. Chem. 279, 46509–46517 [DOI] [PubMed] [Google Scholar]
- 24. Drechsler A., Potrich C., Sabo J. K., Frisanco M., Guella G., Dalla Serra M., Anderluh G., Separovic F., Norton R. S. (2006) Structure and activity of the N-terminal region of the eukaryotic cytolysin equinatoxin II. Biochemistry 45, 1818–1828 [DOI] [PubMed] [Google Scholar]
- 25. Malovrh P., Viero G., Dalla Serra M., Podlesek Z., Lakey J. H., Maček P., Menestrina G., Anderluh G. (2003) A novel mechanism of pore formation. Membrane penetration by the N-terminal amphipathic region of equinatoxin. J. Biol. Chem. 278, 22678–22685 [DOI] [PubMed] [Google Scholar]
- 26. Kristan K., Viero G., Maček P., Dalla Serra M., Anderluh G. (2007) The equinatoxin N-terminus is transferred across planar lipid membranes and helps to stabilize the transmembrane pore. FEBS J. 274, 539–550 [DOI] [PubMed] [Google Scholar]
- 27. Valcarcel C. A., Dalla Serra M., Potrich C., Bernhart I., Tejuca M., Martinez D., Pazos F., Lanio M. E., Menestrina G. (2001) Effects of lipid composition on membrane permeabilization by sticholysin I and II, two cytolysins of the sea anemone Stichodactyla helianthus. Biophys. J. 80, 2761–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anderluh G., Dalla Serra M., Viero G., Guella G., Maček P., Menestrina G. (2003) Pore formation by equinatoxin II, a eukaryotic protein toxin, occurs by induction of nonlamellar lipid structures. J. Biol. Chem. 278, 45216–45223 [DOI] [PubMed] [Google Scholar]
- 29. Jiménez-Juárez N., Muñoz-Garay C., Gómez I., Gill S. S., Soberón M., Bravo A. (2008) The pre-pore from Bacillus thuringiensis Cry1Ab toxin is necessary to induce insect death in Manduca sexta. Peptides 29, 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anderluh G., Pungerčar J., Štrukelj B., Maček P., Gubenšek F. (1996) Cloning, sequencing, and expression of equinatoxin II. Biochem. Biophys. Res. Commun. 220, 437–442 [DOI] [PubMed] [Google Scholar]
- 31. Dalla Serra M., Menestrina G. (2000) Characterization of molecular properties of pore-forming toxins with planar lipid bilayers. Methods Mol. Biol. 145, 171–188 [DOI] [PubMed] [Google Scholar]
- 32. Comai M., Dalla Serra M., Coraiola M., Werner S., Colin D. A., Monteil H., Prévost G., Menestrina G. (2002) Protein engineering modulates the transport properties and ion selectivity of the pores formed by staphylococcal γ-haemolysins in lipid membranes. Mol. Microbiol. 44, 1251–1267 [DOI] [PubMed] [Google Scholar]
- 33. MacDonald R. C., MacDonald R. I., Menco B. P., Takeshita K., Subbarao N. K., Hu L. R. (1991) Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta 1061, 297–303 [DOI] [PubMed] [Google Scholar]
- 34. Thannhauser T. W., Konishi Y., Scheraga H. A. (1984) Sensitive quantitative analysis of disulfide bonds in polypeptides and proteins. Anal. Biochem. 138, 181–188 [DOI] [PubMed] [Google Scholar]
- 35. Anderluh G., Barlič A., Križaj I., Menestrina G., Gubenšek F., Maček P. (1998) Avidin-FITC topological studies with three cysteine mutants of equinatoxin II, a sea anemone pore-forming protein. Biochem. Biophys. Res. Commun. 242, 187–190 [DOI] [PubMed] [Google Scholar]
- 36. Bakrač B., Gutiérrez-Aguirre I., Podlesek Z., Sonnen A. F., Gilbert R. J., Maček P., Lakey J. H., Anderluh G. (2008) Molecular determinants of sphingomyelin specificity of a eukaryotic pore-forming toxin. J. Biol. Chem. 283, 18665–18677 [DOI] [PubMed] [Google Scholar]
- 37. Maček P., Zecchini M., Pederzolli C., Dalla Serra M., Menestrina G. (1995) Intrinsic tryptophan fluorescence of equinatoxin II, a pore-forming polypeptide from the sea anemone Actinia equina L, monitors its interaction with lipid membranes. Eur. J. Biochem. 234, 329–335 [DOI] [PubMed] [Google Scholar]
- 38. Ramachandran R., Surka M., Chappie J. S., Fowler D. M., Foss T. R., Song B. D., Schmid S. L. (2007) The dynamin middle domain is critical for tetramerization and higher-order self-assembly. EMBO J. 26, 559–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Whitson K. B., Beechem J. M., Beth A. H., Staros J. V. (2004) Preparation and characterization of Alexa Fluor 594-labeled epidermal growth factor for fluorescence resonance energy transfer studies. Application to the epidermal growth factor receptor. Anal. Biochem. 324, 227–236 [DOI] [PubMed] [Google Scholar]
- 40. Heuck A. P., Hotze E. M., Tweten R. K., Johnson A. E. (2000) Mechanism of membrane insertion of a multimeric β-barrel protein. Perfringolysin O creates a pore using ordered and coupled conformational changes. Mol. Cell 6, 1233–1242 [DOI] [PubMed] [Google Scholar]
- 41. Viero G., Gropuzzo A., Joubert O., Keller D., Prévost G., Dalla Serra M. (2008) A molecular pin to study the dynamics of β-barrel formation in pore-forming toxins on erythrocytes. A sliding model. Cell. Mol. Life Sci. 65, 312–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fang Y., Cheley S., Bayley H., Yang J. (1997) The heptameric prepore of a staphylococcal α-hemolysin mutant in lipid bilayers imaged by atomic force microscopy. Biochemistry 36, 9518–9522 [DOI] [PubMed] [Google Scholar]
- 43. Miller C. J., Elliott J. L., Collier R. J. (1999) Anthrax protective antigen. Prepore-to-pore conversion. Biochemistry 38, 10432–10441 [DOI] [PubMed] [Google Scholar]
- 44. Heuck A. P., Tweten R. K., Johnson A. E. (2003) Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J. Biol. Chem. 278, 31218–31225 [DOI] [PubMed] [Google Scholar]
- 45. Anderluh G., Lakey J. H. (2005) Lipid interactions of α-helical protein toxins. in Protein-Lipid Interactions: From Membrane Domains to Cellular Networks (Tamm L. K., ed) pp. 141–162, Wiley-VCH, Weinheim, Germany [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.