

Background: The intracellular signaling cascades through which corticosterone rapidly alters neuronal activity are poorly defined.
Results: Corticosterone alters glutamatergic transmission by activating diverse GPCR-dependent signaling pathways.
Conclusion: Corticosterone-induced changes in neuronal excitability are initiated at the plasma membrane.
Significance: The sequential recruitment and integration of diverse signaling cascades by corticosterone adds to the understanding of how steroids rapidly alter neuronal function.
Keywords: G Protein-coupled Receptors (GPCR); Glucocorticoids; Glutamate Receptors Ionotropic (AMPA, NMDA); Neurons; Nonreceptor-tyrosine Kinase; Hippocampus
Abstract
Despite numerous descriptions of rapid effects of corticosterone on neuronal function, the intracellular mechanisms responsible for these changes remain elusive. The present comprehensive analysis reveals that signaling from a membrane-located G protein-coupled receptor activates PKC, Akt/PKB, and PKA, which subsequently trigger the phosphorylation of the tyrosine kinases Pyk2, Src, and Abl. These changes induce rapid cytoskeletal rearrangements (increased PSD-95 co-clustering) within the post-synaptic density; these events are accompanied by increased surface NMDA receptor expression, reflecting corticosterone-induced inhibition of NMDA receptor endocytosis. Notably, none of these signaling mechanisms require de novo protein synthesis. The observed up-regulation of ERK1/2 (downstream of NMDA receptor signaling) together with the fact that c-Abl integrates cytoplasmic and nuclear functions introduces a potential mechanism through which rapid signaling initiated at the plasma membrane may eventually determine the long term integrated response to corticosterone by impacting on the transcriptional machinery that is regulated by classical, nuclear mineralocorticoid, and glucocorticoid receptors.
Introduction
Glucocorticoids (GCs)5 are essential for immediate and long term behavioral and physiological adaptations to stress. Generally, the adaptations triggered by GC secretion are considered to result from activation of glucocorticoid (GR) and/or mineralocorticoid (MR) receptors, ligand-activated transcription factors (or nuclear receptors) that regulate gene expression. On the other hand, GCs can induce rapid changes in neural activity: alterations in neuronal activity (1, 2), cell signaling (3), glutamate release (4), NMDAR-mediated Ca2+ currents (5), behavior (6–9) and neuroendocrine regulation (10) are observable within second to minutes of treating animals or ex vivo brain slice preparations with the major GCs in rodents corticosterone (CORT) (11). In addition, GCs rapidly increase hippocampal neuron spine density (12) and modulate network dynamics (13). Moreover, functional neuroimaging in rats has revealed that CORT rapidly modulates the coordinated activity of the forebrain-hippocampal-hypothalamic circuitry and thus probably facilitates early cognitive and behavioral responding to exogenous stimuli (14).
A common finding in previous studies was that gene transcription and protein translation are not required for the rapid actions of CORT on neuronal function (11, 12, 15). Notwithstanding some exceptions (1, 8), the fast neuronal actions of CORT do not appear to be mediated by nuclear MR and GR. The present study used an approach that was not constrained by a priori assumptions about the nature of the receptor that mediates the rapid effects of CORT. Specifically, we screened for the involvement of non-receptor-tyrosine kinases, a strategy chosen in light of the widespread involvement of these kinases in signaling modules from several types of receptors and their ability to serve as a convergence point for multiple pathways (16–19). Our investigations reveal that CORT recruits a series of diverging and converging signaling pathways that ultimately influence postsynaptic structure and function. Briefly, we show that fast CORT signaling relies on a core of tyrosine kinase (TK) including Pyk2, Abl, and Src that are activated by converging PKC, PKA, and Akt/PKB pathways downstream of a putative G protein-coupled receptor (GPCR) coupled to Gi/o proteins. These results offer new mechanistic insights into fast CORT-induced signaling in hippocampal neurons and provide a framework to better understand how membrane-proximal mediators integrate diverse signals and link with nuclear receptors to elicit molecular and cellular responses to facilitate physiological and behavioral adaptation.
EXPERIMENTAL PROCEDURES
Primary Neuronal Cultures
Trypsin-dissociated primary hippocampal cell cultures were prepared from 4-day-old rats, as described previously (20). Cells were grown at a density of 450–500 cells/mm2. Experiments were started after 10–13 days in vitro (10–13).
Pharmacological Agents
The following drugs were obtained from Sigma and used at the final concentrations indicated in square brackets: AG1478 (selective inhibitor of EGF receptor kinase) [10 μm]; 2-aminoethyl diphenyl borinate (modulator of intracellular inositol trisphosphate-induced calcium release) [100 μm]; BAPTA-AM (selective chelator of intracellular Ca2+) [13 μm]; cycloheximide (CHX; protein synthesis inhibitor) [10 μm]; Gö6983 (Gö69, PKC inhibitor) [5 μm]; K252a (selective and potent inhibitor of Trk) [1 μm]; RU 38486 (mifepristone; nuclear GR antagonist) [100 nm]; water-soluble CORT (2-hydroxy-propyl-β-cyclodextrin corticosterone). The following compounds were purchased from Tocris (Bristol, UK): API-2 (selective Akt/PKB inhibitor) [30 μm]; eplerenone (selective nuclear MR antagonist [100 nm]; MK801 (dizocilpine; non-competitive NMDAR antagonist) [10 μm]; NMDA (prototypic NMDAR agonist) [100 nm]; RU 28318 (RU 283; oxprenoate potassium nuclear MR antagonist) [100 nm]; SCH202676 (general inhibitor of agonist and antagonist binding to GPCR) [1 μm]; spironolactone (competitive nuclear MR antagonist) [100 nm]; U73122 (PLC inhibitor) [5 μm]; W7 (calmodulin antagonist) [50 μm]; wortmannin (selective PI3K inhibitor) [4 μm]. The calmodulin kinase II inhibitor [10 μm], inhibitor of PKA (H-89) [1 μm], Janus tyrosine kinase (JAK) inhibitor [1 μm], the Src family inhibitor and its negative control analog (PP2 and PP3, respectively) [1 μm], the potent Src family inhibitor (SU6656) [1 μm], and the inhibitor of spleen tyrosine kinase (Syk) [1 μm] were obtained from Calbiochem. Cell-impermeable CORT-BSA (corticosterone-3-O-carboxymethyloxime-BSA; used at 100 nm) was purchased from ABiox (Newberg, OR), pertussis toxin (PTX; Gi and Go protein inhibitor) [500 mg/liter] was from List Biologicals (Campbell, CA), PF431396 (potent pyrimidine-based Pyk2 inhibitor) [3 μm] was from Symansis (Timaru, New Zealand), and STI-571 (imatinib, selective inhibitor of c-Abl) [1 μm] was from Cayman (Ann Arbor, MI). The selective GR antagonist J2700 (a gift from Jenapharm, Jena, Germany) was used at 100 nm.
Immunocytochemistry and Image Analysis
Immunolocalization studies were performed on paraformaldehyde-fixed neurons that were incubated (4 °C, 18 h) with antibodies against MAP2a/b (Sigma; 1:500), NR2B subunit (Sigma; 1:200), PSD-95 (Acris, Herford, Germany; 1:400), Pyk2 (Tyr(P)-402, Invitrogen; 1:200), and synapsin 1 (Sigma; 1:750). Surface NR1 expression was performed by quickly cooling cells to 4 °C and incubation with anti-NMDAR1 (BD Biosciences; 1:100 in Neurobasal medium, 15 min) before fixation, blocking, and permeabilization (5% BSA and 0.001% Triton X-100) and incubation with anti-synapsin-I (1:750; 4 °C, 18 h). Immunosignals were visualized after extensive washing and incubation (1 h, room temperature) with appropriate secondary antibodies (mouse, conjugated with Alexa-488, or rabbit conjugated with Alexa-594, both from Invitrogen). Images were acquired with an Olympus FluoView1000 confocal microscope using a plan-apochromat 63×/1.2 water lens at a resolution of 1024 × 1024 in 8-bit format. Phospho-Pyk2, NMDAR1, NR2B, and PSD-95 cluster size were analyzed using ImageJ software after intensity thresholding at the arbitrary values of 100, 80, 150, and 180, respectively. Only clusters juxtaposed to synapsin puncta (manually selected by an investigator who was blind to the treatments) were evaluated; surface areas were measured with ImageJ software. Clusters separated by 1 pixel were considered to represent distinct clusters. Only clusters ≥3 pixels were included in the analysis (noise reduction); although this criterion possibly introduced a bias toward smaller differences in treated versus untreated groups in cases where treatment caused a shrinkage of a substantial proportion of clusters to <3 pixels, it would rather lead to an overestimation of average cluster size in the treated groups without undermining the statistical significance of detected differences. The cluster size analysis was complemented by independent evaluation of the images by a second investigator (also blind to treatments) who ranked images according to cluster size; in all cases there was a 100% match between these latter qualitative evaluations and the quantitative analysis.
Immunoblotting
Neurons were lysed by brief sonication in complete radioimmune precipitation assay buffer (with protease phosphatase inhibitor cocktails) before clearing by centrifugation (12,000 g, 10 min). Cleared lysates (40 μg) were fractionated by electrophoresis on 8% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes, blocked (5% nonfat dried milk powder and 0.2% Tween 20 in PBS), and incubated with antibodies against c-Abl (1:1000), NMDAR-Tyr(P)-1472-NR2B (1:1000), pERK1/2 (p44/42, 1:1000), Tyr(P)-100 (phosphotyrosine 100; 1:1000), Pyk2 (1:2000), Src Tyr(P)-416 (1:1000), and Src Tyr(P)-527 (1:1000) from Cell Signaling (Danvers, MA); Pyk2 Tyr(P)-402 (1:1000), Pyk2 Tyr(P)-579 (1:1000), Pyk2 Tyr(P)-580 (1:1000), and Pyk2 Tyr(P)-881 (1:1000; Invitrogen), c-Abl Tyr(P)-412 (1:1000; Novus Littleton, CO), and actin (1:5000; Millipore, Billerica, MA). Anti-PTP-PEST pSer39 (1:1000) was a generous gift from Dr. K. Mashima (Rikkyo University, Japan). Antigens were detected by enhanced chemiluminescence (GE Healthcare) after incubation with the appropriate horseradish peroxidase-IgG conjugates (GE Healthcare). Blots were scanned and quantified (TINA 3.0 Bioimaging software, Raytest, Straubenhardt, Germany) after subtraction of local background. The phosphotyrosine blots were not intended to provide high resolution separation of single bands and were, therefore, analyzed by averaging the density of 10 large bands after subtracting the corresponding local background. Linearity was routinely checked before semiquantification of all blots. Normalized data are expressed as the percentage of controls.
Statistics
Numerical data are depicted as the mean ± S.D. (3–5 independent experiments). Immunofluorescence data derive from evaluation of a minimum of 600 synapses in each of 8–10 neurons. Data were analyzed for statistical significance using analysis of variance and appropriate post hoc tests (Student-Keuls or Kruskal-Wallis multiple comparison procedures, as appropriate) where p < 0.05 was set as the minimum level of significance.
RESULTS
Corticosterone Activates the Src-family Kinase Signaling Pathway
TKs are expressed in neurons and are particularly enriched in synapses (21–24) where they phosphorylate and activate a range of substrates (25). A first set of experiments investigated the overall activation of tyrosine kinases after exposure of hippocampal neurons to CORT. Analysis of dose-response curves (1–1000 nm CORT) showed that tyrosine phosphorylation was most effectively triggered by CORT at a dose of 10 nm (not shown). Immunoblot analysis of the global levels of phosphotyrosine revealed a large number of bands, with molecular mass ranging from 20 to 200 kDa. Levels of tyrosine phosphorylation were significantly increased within 5 min of application of CORT and peaked at 20 min (p ≤ 0.05) (Fig. 1A); accordingly, all further analysis was confined to the first 20 min after treatment to focus on dissection of the early signaling events initiated by CORT. As shown in Fig. 1B, blockade of protein synthesis with cycloheximide did not interfere with the effect of CORT on tyrosine phosphorylation (p ≥ 0.05), excluding the involvement of translational mechanisms.
FIGURE 1.

CORT rapidly induces phosphorylation of protein-tyrosine kinases. Numerical data are expressed as the mean ± S.D. -fold changes relative to control values (dotted line). A, levels of phosphorylated protein-tyrosine kinase (Tyr(P)-100) were significantly increased within 5 min of CORT treatment (10 nm) (CORT, 1.31 ± 0.11, p < 0.05 versus control) with a peak response seen at 20 min after treatment initiation (1.88 ± 0.33, p < 0.05 versus control). B, CORT-induced (10 nm, 20 min) phosphorylation of protein-tyrosine kinase (1.78 ± 0.25, p < 0.001 versus control) was not affected by CHX (10 μm, 30-min pretreatment; CHX alone, 1.12 ± 0.30, p = 0.2 versus control; CHX+CORT, 1.76 ± 0.30, p < 0.001 versus control), indicating that de novo protein synthesis is not essential for the rapid effects of CORT. C, hippocampal neuronal cultures were pretreated with various non-receptor protein kinase inhibitors (inh) for 30 min before application of CORT (10 nm, 20 min; 1.82 ± 0.08, p < 0.001 versus control). PP2 (SFK inhibitor, 1 μm) significantly abrogated the ability of CORT to induce tyrosine phosphorylation (0.53 ± 0.08, p < 0.001 versus CORT). The Abl inhibitor (STI571, 1 μm, 1.33 ± 0.08, p < 0.02 versus CORT) and JAK-STAT inhibitor (1 μm, 1.41 ± 0.14, p < 0.05 versus CORT) caused a smaller but significant decrease in phosphotyrosine levels, whereas the Syk inhibitor (1 μm, 1.55 ± 0.22, p = 0.2 versus CORT) failed to block tyrosine phosphorylation by CORT. Quantified data shown in (A–C) represent the intensities of the 10 most prominent bands between 170 and 60 kDa; the values shown were computed after subtracting local background for each single band, the average change in intensity for each band (across time points or treatments) was calculated, and the mean value of change in the 10 pooled bands was plotted. D, CORT-induced (10 nm, 20 min) tyrosine phosphorylation (1.77 ± 0.14, p < 0.001 versus control) was also resistant to inhibition by two receptor protein kinase inhibitors, AG1478 (EGF receptor inhibitor, 10 μm; AG1478 alone, 1.11 ± 0.16, p = 0.1 versus control; AG1478 + CORT, 1.87 ± 0.36, p = 0.3 versus CORT) and K252a (selective Trk inhibitor 1 μm; K252a alone, 1.14 ± 0.12, p = 0.06 versus control; K252a + CORT, 2.06 ± 0.27 p = 0.1 versus CORT). E, TK phosphorylation by 10 nm CORT (1.87 ± 0.36, p = 0.03) was not blocked by PP3 (an inactive analog of the SFK inhibitor PP2; PP3 alone (1.01 ± 0.11) versus PP3 + CORT (1.93 ± 0.35)) but was significantly reduced (=0.03) in the presence of the Src-family inhibitor SU6656 (SU6656, 0.99 ± 1.23). F, CORT (10 nm) significantly increased phosphorylation Src at tyrosine 416 within 10 min (1.62 ± 0.27, p < 0.001 versus control). There was a gradual dephosphorylation of Src at Tyr-527 with time. Note that CORT did not influence the levels of total Src protein. The blots shown are representative of at least three independent experiments.
Because post-mitotic primary neuronal cultures are not easily amenable to efficient transfection, pharmacological antagonism was used to identify the TK recruited by CORT signaling; specifically, we performed a screening with small molecule TK inhibitors that target the Src-family kinase (SFK), Abl, and inhibitors of the JAK-STAT and of Syk pathways. In addition, the efficacy of CORT signaling was tested in the presence of inhibitors of two abundant receptor-tyrosine kinases (RTK), namely, TrkB and epidermal growth factor (EGF) receptor kinase. The SFK inhibitor PP2 markedly attenuated the up-regulation of Tyr(P) by CORT (p ≤ 0.05), whereas inhibitors of c-Abl (ST1571) and JAK produced weaker, albeit significant effects (Fig. 1C); inhibitors of TrkB (K252a) and EGF receptor kinase (AG1478) did not significantly influence the effects of CORT on Tyr(P) levels (Fig. 1D). Confirming the essential role of SFK, we observed that the Src-family inhibitor SU6656 inhibits CORT-induced TK phosphorylation (p ≤ 0.05) in a fashion that is comparable with that seen with the structurally unrelated compound PP2 (Fig. 1E); on the other hand, PP3, an inactive analog of PP2, did not exert any effect (Fig. 1E).
Corroboration of the above evidence of SFK involvement in CORT-induced TK phosphorylation was obtained by analysis of the phosphorylation state of Src in whole-cell extracts from CORT-treated hippocampal neuronal cultures. To this end, antibodies directed against pSrc (Tyr-416; located in the kinase domain responsible for activation of Src) and the autoinhibitory site pSrc (Tyr-527) were employed. We observed that CORT significantly up-regulated phosphorylation of Src at the Tyr-416 site (p ≤ 0.05) within 10 min of drug application, with peak levels of activation being seen after 30 min (Fig. 1F). Conversely, a significant decrease (p ≤ 0.05) in pSrc Tyr-527 was observed within 30 min of CORT application (Fig. 1E). Together, the above findings identify Src as a major mediator of a non-genomic signaling cascade initiated by CORT.
Pyk2 Phosphorylation Mediates CORT-induced Src Activation
This set of experiments aimed at identifying CORT-triggered signaling events that occur upstream of the SFK members. Numerous stimuli and mechanisms contribute to the activation of SFK (26); among these, the TK scaffold protein Pyk2 has emerged as a prominent and direct activator of Src in hippocampal neurons (21). Here, pretreatment with PF431396, a specific Pyk2 inhibitor, abolished CORT-induced activation of SFK (Fig. 2A). Consistent with this finding, Pyk2 was activated by CORT, evidenced by increased phosphorylation of its Tyr-402 residue; this effect was significant within 10 min of CORT application and progressively increased for at least 60 min thereafter (Fig. 2B). CORT-induced phosphorylation of Pyk2 was further confirmed by immunostaining of hippocampal neurons with anti-pPyk2 Tyr-402 and synapsin I (to identify synaptic sites). At base line, pPyk2 Tyr-402 was detectable at low levels in neuronal cell bodies and nuclei, with very low levels of pPyk2 being detectable along major dendrites; only rarely were pPyk2 puncta found adjacent to synapsin I puncta. However, pPyk2 immunoreactivity (fluorescence intensity) was markedly increased (3-fold) 20 min after CORT application; pPyk2 appeared as puncta along dendrites and was largely co-localized with synapsin I, indicating selective Pyk2 phosphorylation at synaptic sites (Fig. 2C).
FIGURE 2.

Phosphorylation of Pyk2 occurs rapidly after CORT application to hippocampal neurons. Numerical data are expressed as the mean ± S.D. -fold changes relative to control values (dotted line). A, the Pyk2 inhibitor, PF431396 (3 μm, 30-min pretreatment), abolished CORT-induced (10 nm, 20 min) phosphorylation of Src Tyr-416 (1.91 ± 0.16, p < 0.001 versus control), indicating regulation of Src activation by Pyk2 (PF431396 alone, 0.99 ± 0.03, p = 0.9 versus control; PF431396 + CORT, 1.03 ± 0.15 p < 0.001 versus CORT). B, CORT (10 nm, 20 min) induced the phosphorylation of Pyk2 at the Tyr-402 epitope within 10 min of application (1.61 ± 0.43, p = 0.09) with a peak after 30 min (4.17 ± 0.79, p < 0.01), whereas total levels of Pyk2 remained unchanged (not shown). C, CORT treatment (10 nm, 20 min) caused an increase in the intensity of immunostaining of Pyk2 phosphorylated at Tyr-402. Immunoreactivity was observed in both cell soma and nuclei. The scale bar represents 10 μm. CORT application (10 nm, 20 min) caused a significant increase in the punctuate staining of pPyk2 Tyr-402, suggesting that Pyk2 activation was taking place in synaptic sites. D, the phosphorylation levels of four critical Pyk2 phospho-epitopes were significantly increased after CORT treatment (10 nm, 20 min; Tyr-402, 2.11 ± 0.21; Tyr-579, 2.01 ± 0.12; Tyr-580, 2.01 ± 0.25; Tyr-881, 2.00 ± 0.16; in all cases, p < 0.001 versus control). These events were abolished by pretreatment with the Pyk2 inhibitor PF431396 (PF, 3 μm, 30 min, p < 0.001 versus CORT in all cases). The Src inhibitor PP2 (1 μm, 30 min) did not inhibit phosphorylation of Tyr-402 (p = 0.15 versus CORT alone) but blocked CORT-induced phosphorylation of Tyr-579 (p < 0.01 versus CORT alone) and partially blocked CORT-induced pPyk2 Tyr-580 (p < 0.01 versus CORT alone) and pPyk Tyr-881 (p < 0.01 versus CORT alone) levels. Note that PP3, the inactive analog of PP2, did not exert any effect on any of the potentially phosphorylatable Pyk2 epitopes (not shown). None of the treatments influenced the levels of total Pyk2. Shown blots represent results obtained in at least three independent experiments.
Next, a detailed analysis of Pyk2 activation by CORT exposure was conducted by focusing on four critical phospho-epitopes: the autophosphorylation site Tyr-402 (required for initial kinase activation), the kinase domain sites Tyr-579 and Tyr-580 (required for full kinase activity), and Tyr-881 (located in the C-terminal interaction domain). All epitopes underwent significant phosphorylation when hippocampal neurons were treated with CORT for 20 min (p ≤ 0.05; Fig. 2D). Underlining the crucial role of the Pyk2 autophosphorylation site, we observed that blockade of phosphorylation of Tyr-402 with the Pyk2 inhibitor PF431396 prevented CORT-induced phosphorylation of other Pyk2 phospho-epitopes (Tyr-579, Tyr-580, and Tyr-881). The Src inhibitor PP2 did not interfere with phosphorylation of Tyr-402 but effectively blocked phosphorylation of Pyk2 at Tyr-579, indicating dependence on Src activation. Thus, upon activation by Pyk2, Src “back phosphorylates” Pyk2, amplifying Pyk2 activity. Interestingly, inhibition of Src only partially reduced CORT-induced phosphorylation of Pyk2 at Tyr-580 and Tyr-881, suggesting involvement of other (non-SFK) kinases in the regulation of these sites.
Convergence of PLC-PKC-, PKA-, and Akt/PKB-dependent Pathways Required for CORT-induced Pyk2 Activation
Pyk2 is a signaling hub with inputs from several pathways and second messengers, in particular Ca2+- and PKC-dependent mechanisms (27). It was, therefore, of interest to explore the potential involvement of proximal transduction pathways, specifically PKC, PKA, and Akt/PKB signaling cascades. A pharmacological screen revealed that CORT-induced phosphorylation of the Tyr-402 autophosphorylation site on Pyk2 is blocked in the presence of an inhibitor of PKC (Gö69) but not inhibitors of PKA (H89) or Akt/PKB (AP1–2) (cf. Fig. 3, A and D). Results indicate that the pathway upstream of PKC requires PLC activation and calmodulin function as Pyk2 phosphorylation was prevented when neurons were treated with inhibitors of either PLC (U73122; Fig. 3A) or calmodulin (W7; Fig. 3B). The latter result, pointing to a Ca2+-dependent mechanism, was confirmed by the finding that the CORT effect was prevented when cells were co-treated with either the Ca2+ chelator BAPTA-AM or the inositol trisphosphate receptor antagonist 2-aminoethyl diphenyl borinate, which blocks mobilization of intracellular Ca2+ (Fig. 3C). Notably, verapamil, an L-type voltage-dependent Ca2+ channel blocker, did not affect CORT-induced Pyk2 phosphorylation (not shown), ruling out the requirement of Ca2+ influx through this type of voltage-dependent Ca2+ channel.
FIGURE 3.

CORT-induced events upstream of Pyk2. Numerical data are expressed as the mean ± S.D. -fold changes relative to control values (dotted line). The phosphorylation of Pyk2 at Tyr-402 induced by CORT (10 nm, 20 min, 1.91 ± 0.17, p < 0.001 versus control) was significantly inhibited by pharmacological inhibitors of PLC (U73122, 5 μm; U73122 + CORT, 1.06 ± 0.13, p < 0.001 versus CORT), PKC (Gö69, 5 μm; Gö6969 + CORT, 0.81 ± 0.2, p < 0.001 versus CORT) (A) and calmodulin (W7, 50 μm; W7 + CORT, 0.81 ± 0.15, p < 0.01 versus CORT) (B); all inhibitors were added for 30 min before application of CORT. C, the Ca2+ chelator BAPTA-AM (BAPTA, 13 μm) and inositol trisphosphate receptor antagonist 2-aminoethyl diphenyl borinate (2-APB; 100 μm) prevented CORT-induced phosphorylation of Pyk2 at Tyr-402 (both drugs were applied as a pretreatment for 30 min; BAPTA + CORT, 0.04 ± 0.04 p < 0.001 versus CORT; 2-ABP + CORT, 0.75 ± 0.29, p < 0.001 versus CORT). D, CORT-triggered (10 nm, 20 min) phosphorylation of Pyk2 at Tyr-402 (white bars, 2.24 ± 0.33, p = 0.001 versus control) was significantly inhibited by a PKC inhibitor (Gö69, 5 μm; Gö69 + CORT, 0.70 ± 0.09, p < 0.01 versus CORT) but not by inhibitors of PI3K (wortmannin (WORT), 4 μm; WORT + CORT, 2.04 ± 0.23 p = 0.23 versus CORT); Akt/PKB (API-2, 30 μm; API + CORT, 2.20 ± 0.20, p = 0.43 versus CORT), or PKA (H89, 1 μm; H89 + CORT, 2.13 ± 0.06, p = 0.31 versus CORT). CORT-induced phosphorylation of Pyk2 at Tyr-579 (light gray bars) and Tyr-580 (dark gray bars) were abolished in the presence of Gö69, WORT, API-2, and H89 (p < 0.001 versus CORT in all cases). On the other hand, CORT-induced phosphorylation of Pyk2 at Tyr-881 (black bars) was abolished in the presence of inhibitors of PKC, PI3K, and Akt/PKB (p < 0.001 versus CORT in all cases) but not of PKA (p = 0.2). All drugs were applied for 30 min before CORT application. Blots shown are representative of at least three independent experiments.
As mentioned above, efficient Pyk2 activation and substrate recruitment after its autophosphorylation at Tyr-402 requires phosphorylation of three further tyrosine residues: Tyr-579 and Tyr-580 in the kinase domain and Tyr-881 in the C-terminal interaction domain (27). Consistent with this, the present experiments revealed that although the autophosphorylation of Pyk2 is solely dependent on PKC signaling (Fig. 3D), its phosphorylation at the Tyr-579 and Tyr-580 residues requires PKA and Akt/PKB activity; specifically, phosphorylation at these sites was prevented when cells were treated with inhibitors of PKA (H89), PI3K (wortmannin (WORT)), and Akt/PKB (AP1–2) (Fig. 3D). Interestingly, phosphorylation of the Tyr-881 epitope was sensitive to blockade with wortmannin and API-2 but not H89 (Fig. 3D). Thus, although activation of the PLC-PKC pathway is sufficient to induce autophosphorylation of Pyk2 (Tyr-402), recruitment of additional signaling cascades is required to achieve phosphorylation of Pyk2 at its Tyr-579, Tyr-580, and Tyr-881 residues.
The finding that PKA and Akt/PKB contribute to the regulation of specific phosphotyrosine residues in Pyk2 by CORT led us to hypothesize the involvement of an intermediate player such as the phosphatase PTP-PEST, a recently identified negative regulator of Pyk2 (28, 29). Previous work showed that PTP-PEST is constitutively active and represses Pyk2 activation under basal conditions and that the phosphatase is inactivated upon phosphorylation at the serine 39 site (30, 31). The present results show induction of phosphorylation of PTP-PEST at Ser-39 within 5 min of exposure of hippocampal neurons to CORT, with peak levels of phosphorylation occurring after 20–30 min (Fig. 4A). The finding that inhibitors of PKA (H89), Akt/PKB (API-2), PI3K (wortmannin), and PKC (Gö69) block the CORT-induced up-regulation of Ser(P)-39 PTP-PEST suggests involvement of all three pathways in the inhibition of PTP-PEST activity and that the ability of PKA and Akt/PKB to regulate the phosphorylation of some Pky2 epitopes depends on PTP inhibition (Fig. 4, B and C).
FIGURE 4.
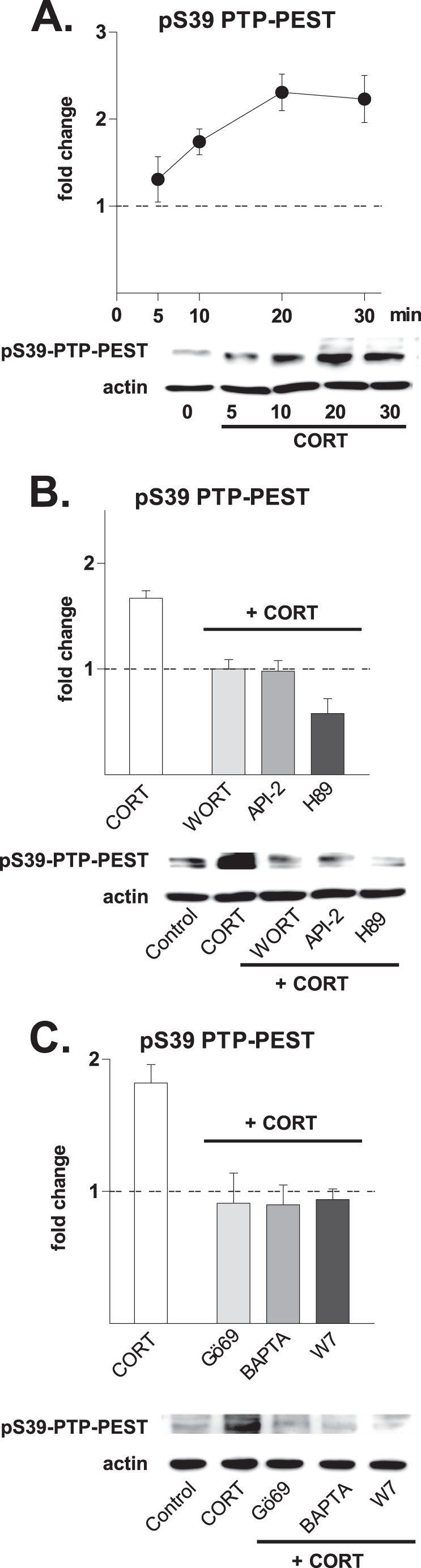
Role of the cytoplasmic protein-tyrosine phosphatase, PTP-PEST, a negative regulator of Pyk2 activity. Numerical data refer to the means ± S.D. Normalized control values are shown as a broken line. A, hippocampal cultures responded to treatment with CORT (10 nm) with increased phosphorylation of PTP-PEST at serine 39 (pS39). The first increase was observed after 5 min (1.31 ± 0.26, p = 0.06), became significant after 10 min (1.74 ± 0.15, p < 0.001), and was maintained at significantly high levels for up to 30 min after the application of CORT (2.23 ± 0.27, p < 0.01). B, the ability of CORT (10 nm, 20 min) to induce phosphorylation of PTP-PEST at Ser-39 (1.67 ± 0.07, p < 0.001) was abolished by inhibitors of PI3K (WORT, 4 μm; WORT + CORT, 1.00 ± 0.09, p < 0.001 versus CORT), Akt/PKB (API, 30 μm; API + CORT, 0.98 ± 0.1, p < 0.001 versus CORT), and PKA (H89, 1 μm; H89 + CORT, 0.58 ± 0.14, p < 0.001 versus CORT). C, CORT-induced phosphorylation of PTP-PEST Ser-39 was also prevented by inhibitors of PKC (Gö69, 5 μm; Gö69 + CORT, 0.91 ± 0.23, p < 0.01 versus CORT), the selective intracellular Ca2+ chelator BAPTA-AM (BAPTA, 13 μm; BAPTA + CORT, 0.90 ± 0.15, p < 0.001 versus CORT), and calmodulin (W7, 50 μm; W7 + CORT, 0.94 ± 0.08, p < 0.001 versus CORT). All drugs were applied as pretreatments (30 min before CORT). The blots shown represent results obtained in at least three independent experiments.
Together, the above data suggest that activation of the PLC-PKC cascade is an absolute requirement in CORT-induced autophosphorylation of Pyk2 at Tyr-402. At the same time, the data show that PKA and Akt/PKB mediate the inhibitory actions of CORT on PTP-PEST, thus allowing complete phosphorylation of Pyk2 at Tyr-579, Tyr-580, and Tyr-881.
CORT Activates the Abl Cascade through Pyk2
Because the Abl inhibitor STI571 reduced CORT-induced Tyr(P) levels (Fig. 1C), and given that c-Abl can be recruited to Pyk2 at Tyr-881 (32, 33), it was of interest to investigate whether c-Abl, alongside Pyk2 and Src, was activated by CORT. The results in Fig. 5A show that CORT leads to robust and sustained autophosphorylation of c-Abl at its Tyr-412 epitope within 20 min of application (p < 0.05). Moreover, inhibition of the upstream kinases SFK and Pyk2 (Fig. 5B) as well as of their upstream transducers (PKA, Akt/PKB, PKC, Fig. 5C) abolished the ability of CORT to activate c-Abl. Blockade of protein synthesis with cycloheximide (Fig. 5D) or NMDAR with MK801 (Fig. 5E) did not interfere with CORT-induced phosphorylation of c-Abl, confirming operation of a non-classical mechanism.
FIGURE 5.

Corticosterone recruits a Pyk2-Abl cascade and leads to actin remodeling. Numerical data refer to means ± S.D. Normalized control values are shown as a broken line. A, exposure of hippocampal cultures to CORT (10 nm) led to an increase in the levels of pAbl Tyr-412 within 20 min (2.10 ± 0.24, p < 0.001 versus control) and a peak response at 60 min (3.19 ± 0.23, p < 0.0001 versus control). B, pretreatment (30 min) with inhibitors of Pyk2 (PF431396, 3 μm) and Src (PP2, 1 μm) abolished the phosphorylation of Abl at Tyr-412 by CORT (CORT, 2.08 ± 0.39, p < 0.01 versus control; PF alone, 0.76 ± 0.19, p < 0.05 versus control; PF + CORT, 0.42 ± 0.14, p < 0.001 versus CORT; PP2 alone, 0.34 ± 0.35, p < 0.05, versus control; PP2 + CORT, 0.61 ± 0.24, p < 0.01 versus CORT). C, CORT-induced Abl phosphorylation at Tyr-412 was abolished by pretreatment (30 min) with inhibitors of pathways lying upstream of Pyk2 (Gö69 + CORT, 1.04 ± 0.12, p = 0.1 versus control; WORT + CORT, 1.10 ± 0.11, p = 0.1 versus control; API-2, 1.12 ± 0.14, p = 0.1 versus control; H89, 94 ± 7%, p = 0.1 versus control). D, the protein synthesis inhibitor CHX (10 μm, 30 min) did not inhibit the phosphorylation of Abl at Tyr-412 by CORT (2.04 ± 0.25, p < 0.001 versus control; CHX alone, 1.06 ± 0.17, p = 0.24 versus control; CHX + CORT, 2.29 ± 0.25, p = 0.1, versus CORT). E, CORT-induced Abl phosphorylation at Tyr-412 was not dependent on NMDAR activation. Pretreatment of hippocampal cells with the broad NMDAR inhibitor MK801 (10 μm, 30 min) did not alter the ability of CORT to phosphorylate Abl (2.13 ± 0.16, p = 0.3 versus control). (The immunoblots shown represent results obtained in at least three independent experiments).
Implication of a Surface-localized GPCR
The engagement of PLC-PKC and PKA pathways by CORT in activating Pyk2 hinted at the potential signaling role of G proteins. Consistent with previous demonstrations that the PLC-PKC pathway can be activated by PTX-insensitive Gαq subunits as well as PTX-sensitive Gβ/γi subunits (34), we observed here that pretreatment with PTX prevents CORT-triggered phosphorylation of Pyk2 and Src (Fig. 6A); these findings implicate the involvement of the β-γ subunits of Gi (34).
FIGURE 6.

Potential involvement of a G protein-mediated mechanism. Numerical data are expressed as mean ± S.D. -fold changes relative to control values (dotted line). A, CORT-induced (10 nm, 20 min) pSrc Tyr-416 and pPyk2 Tyr-402 levels was abolished in the presence of the Gi/o protein inhibitor PTX (500 ng/ml, added 120 min before the application of CORT) (pSrc Tyr-416: CORT, 2.08 ± 0.23, p < 0.001; PTX, 0.85 ± 0.17 p = 0.1 versus control; PTX + CORT, 0.85 ± 0.19, p < 0.001 versus CORT) and (Pyk2 Tyr-402: CORT, 1.91 ± 0.17, p < 0.001; PTX, 1.09 ± 0.19, p = 0.2 versus control; PTX + CORT, 1.32 ± 0.03p < 0.01 versus CORT). B, CORT (10 nm, open bars) as well as membrane-impermeable CORT-BSA conjugate (10 nm, dashed bars) induced phosphorylation of Pyk2 at Tyr-402 within 20 min (CORT, 2.33 ± 0.12, p < 0.01 versus control; CORT-BSA, 2.00 ± 0.26, p < 0.01 versus control). These effects were not prevented by pretreatment (30 min) with antagonists of either nuclear GR (RU486 and J2700, 100 nm each, gray bars) or nuclear MR (RU28318, eplerenone (EPLE) and spironolactone (SPIRO), 100 nm each, black bars). RU486 + CORT-BSA, 2.17 ± 0.23 p < 0.001 versus control; J2700 + CORT-BSA, 2.23 ± 0.39, p < 0.01 versus control; RU28318 +CORT-BSA, 2.31 ± 0.25, p < 0.001 versus control; eplerenone + CORT-BSA, 2.27 ± 0.41, p < 0.01; SPIRO + CORT-BSA, 2.23 ± 0.12 p < 0.001 versus control). C, pretreatment of neurons with SCH20267, a general inhibitor of agonist and antagonist binding to GPCR (1 μm), blocked the ability of CORT (10 nm, 20 min) to phosphorylate Pyk2 at Tyr-402 (CORT, 1.94 ± 0.19, p < 0.001 versus control; SCH20267 alone, 1.28 ± 0.1, p = 0.06 versus control; SCH20267 + CORT, 0.91 ± 0.05, p < 0.001 versus CORT). D, like CORT, a cell-impermeable CORT-BSA conjugate (10 nm, 20 min) rapidly induced the phosphorylation of Pyk2 at Tyr-402 (CORT, 2.33 ± 0.12; CORT-BSA, 2.17 ± 0.15, p > 0.05 versus CORT). These effects were prevented by pretreatment with the Gi/o protein inhibitor PTX (500 ng/ml, 120 min) (PTX + CORT-BSA, 1.30 ± 0.24, p < 0.01 versus CORT-BSA) and the inhibitor of PKC Gö69 6983 (Gö6969; 5 μm, 30 min; Gö69+CORT-BSA, 1.28 ± 0.13, p < 0.001 versus CORT-BSA). Blots shown are representative of at least three independent experiments.
To test the possibility that fast CORT signaling originates at the plasma membrane, a cell-impermeable CORT-BSA conjugate was used to exclude binding to intracellular (nuclear) MR and GR. Exposure of hippocampal neurons to this conjugate produced the same degree of Pyk2 phosphorylation (at Tyr-402) as CORT (Fig. 6B, first and second lanes). Moreover, the actions of both CORT and CORT-BSA were insensitive to other, structurally unrelated antagonists of nuclear GR (RU38486, J2700) and nuclear MR (RU28318, eplerenone, and spironolactone) (Fig. 6B). Importantly, pretreatment of hippocampal cultures with a general GPCR inhibitor (SCH20267) abolished CORT activation of Pyk-2 (Fig. 6C). Investigation of further upstream events revealed that the actions of both CORT-BSA and unconjugated CORT were sensitive to PTX and Gö69, the PKC inhibitor (Fig. 6D). These observations indicate that CORT-BSA and CORT initialize an identical signaling cascade, and their pharmacological profile suggests that the fast actions of both compounds are mediated by a membrane-localized entity that uses a putative GPCR-mediated transduction pathway.
CORT Induces Phosphorylation of NMDAR through Pyk2 and Src
We next explored the synaptic consequences of fast CORT signaling through TK, focusing on the most abundant synaptic phosphotyrosine proteins (25), namely the NR2A and NR2B subunits of NMDAR. Given the above-described effects of CORT on TK signaling and the known role of TK in preventing NMDAR endocytosis through phosphorylation of the NR2B subunit at Tyr-1472 (26, 35, 36), we examined the influence of CORT on the dynamic processes that regulate NMDAR expression at the neuronal surface. Expression of surface NR1 under non-permeabilizing conditions was monitored using an antibody directed against an extracellular epitope. As shown in Fig. 7A, CORT treatment up-regulated NR1 immunoreactivity at the cell surface, an effect that was sensitive to inhibition of Pyk2 and SKF; interestingly, inhibition of Abl did not interfere with the actions of CORT on NR1 expression. These data were confirmed by monitoring phosphorylation of the Tyr-1472 on the NR2B cytoplasmic tail (35, 36) by immunoblotting of whole cell extracts (Fig. 7C). Together, these observations indicate that Pyk2 and Src contribute to the regulation of the surface expression of NMDAR by CORT.
FIGURE 7.

CORT induces phosphorylation of NMDAR through Pyk2 and Src. Numerical data refer to means + S.D. Normalized control values are shown as a broken line. A, surface synaptic levels of NR1 subunits were significantly increased after CORT treatment (10 nm, 20 min). This event was sensitive to inhibition by pretreatment with inhibitors of Pyk2 (PF431396 (PF), 3 μm, 30 min) and Src (PP2; 1 μm) but not to the Abl inhibitor, STI-571 (STI, 1 μm). B, CORT (10 nm, 20 min) increased the size of PSD-95 clusters in dendritic spines of hippocampal neurons; this effect was markedly attenuated by the Pyk2 inhibitor PF431396 (3 μm), the Src inhibitor (PP2, 1 μm), and the Abl inhibitor, STI-571 (STI, 1 μm). C, treatment of hippocampal neuronal cultures with CORT (10 nm, 20 min) led to an increase in the levels of pNR2B Tyr-1472, an effect that was abolished by pretreatment (30 min) with the Pyk2 inhibitor PF431396 (3 μm) and Src inhibitor PP2 (1 μm), but not STI-571, the inhibitor of Abl (STI, 1 μm) (CORT, 1.78 ± 0.08, p < 0.0001 versus control; PF431396 + CORT, 0.89 ± 0.14, p < 0.001 versus CORT; PP2 + CORT, 0.51 ± 0.26, p = 0.001 versus CORT; STI + CORT, 1.99 ± 0.47, p = 0.2 versus CORT). D, CORT (10 nm) induced pERK1/2 Thr-202/Tyr-204 within 5 min of application (1.31 + 0.15, p < 0.01 versus control), and maximum effects were observed after 20 min (2.28 ± 0.34, p < 0.001 versus control). E, the activation of ERK 1/2 was completely abrogated when neurons were pretreated (30 min) with the NMDAR antagonist, MK801 (10 μm; CORT, 2.38 ± 0.36, p < 0.01 versus control; MK801 alone, 0.28 ± 0.25, p < 0.001 versus CORT; MK801 + CORT, 0.44 ± 0.18, p < 0.001 versus CORT). F, the activation of ERK1/2 by CORT was dose-dependent. CORT (20 min) induced ERK1/2 phosphorylation at 1 nm (1.27 ± 0.15, p < 0.05 versus control) and 10 nm (2.74 ± 0.11, p < 0.001 versus control), but higher concentrations (100 nm, 1 μm, 10 μm) were progressively less effective (2.15 ± 0.24, 1.69 ± 0.12, 1.16 ± 0.14, respectively). All inhibitors were applied 30 min before the addition of CORT to culture medium. Immunoblots shown represent results obtained in at least three independent experiments.
Scaffold proteins play an important modulatory role in NMDAR surface localization and signaling (37, 38). Accordingly, we monitored the pattern of clustering of PSD-95, a scaffold protein found in the neuronal post-synaptic density (20) after treating hippocampal neurons with CORT (10 nm) for 30 min. Immunostaining for PSD-95 and synapsin (to mark synapses) revealed that whereas PSD-95 in dendritic spines is largely punctate and occurs in close apposition to synapsin-positive puncta under baseline conditions, PSD-95 cluster size increases within 20 min of CORT application. Importantly, the latter effect is blocked in the presence of inhibitors of Pyk2 and Src as well as Abl (Fig. 7B). Thus, although NR2B phosphorylation does not require Abl (cf. Fig. 7A), the signaling pathway that regulates PSD-95 clustering is critically dependent on Abl.
A functional correlate of the observed CORT-induced increase in surface-localized NMDAR was obtained by examining phosphorylation of the MAP kinase ERK1/2. Consistent with previous work showing that NMDAR activation results in the phosphorylation of ERK1/2 (p44/p42) (39), we observed here a significant increase in pERK1/2 levels within 20 min of CORT application to hippocampal neurons (Fig. 7D); abrogation of this effect by MK801, a non-competitive NMDAR antagonist (Fig. 7E), demonstrated dependence on glutamatergic transmission. As depicted in Fig. 7F, an inverted U-shaped dose-response curve was obtained when levels of pERK1/2 were measured in neurons that were exposed to CORT; a similar dose-response curve was observed when neurons were treated with membrane-impermeable CORT-BSA conjugate (not shown). Together these sets of data show that tyrosine phosphorylation of NR2B leads to increased surface expression of NMDAR and PSD-95 clustering; in turn, these events activate the ERK1/2 pathway.
DISCUSSION
Glucocorticoids induce rapid changes in the function of both neuronal and non-neuronal cells through signaling mechanisms that are believed to originate at the plasma membrane, which, however, are poorly understood. In this study we undertook a comprehensive analysis of both membrane-proximal and -distal signaling pathways that are activated within 20 min of CORT application to cultured hippocampal neurons.
Our results reveal that fast CORT signaling in hippocampal neurons occurs in three sequentially interconnected steps: events at the plasma membrane that activate serine-threonine kinases, activation of core non-receptor TKs, and finally, alterations in synaptic activity. None of these effects can be blocked in the presence of antagonists of nuclear GR and MR. On the other hand, results showing that both unconjugated CORT and membrane-impermeable CORT-BSA (also see Refs. 2 and 3) elicit identical signaling events, strongly suggest that a membrane-bound receptor for CORT is responsible for mediating the rapid neuronal actions of this steroid. Although the latter was found to be sensitive to Gi/o inhibition (PTX) and to a general inhibitor of GPCR function (SCH20267), the nature of the putative membrane-localized CORT receptor remains unknown. Although some earlier studies observed immunoreactivity corresponding to the classical, nuclear GR in the plasma membrane (12, 15, 40, 42), more recent ones have proposed that the fast actions of CORT in hippocampal neurons are mediated by translocated nuclear MR (7, 43). It is pertinent to note that recent work has implicated an orphan GPCR in fast signaling by two other steroid hormones, estradiol and aldosterone (44, 45); the latter is enzymatically (18-hydroxylase and 18-hydroxysteroid dehydrogenase) derived from CORT.
Downstream of the Gi/o-mediated events, our data show that CORT activates diverging signaling cascades. Expanding the scope of previous reports on CORT activation of PKA and PKC, with subsequent alterations of NMDA receptor trafficking and integration into the postsynaptic density (3, 5), our study shows that recruitment of downstream signaling molecules depends on the simultaneous activation of PKC, Akt/PKB, and PKA. Given that the synaptic actions of many neurotransmitters and neuromodulators occur through activation of PKA, Akt/PKB, and PKC (for example, see Ref. 46), activation of these divergent pathways during the initial phases of the rapid response to CORT provides the cell with a high degree of plasticity, including cross-talk and cross-modulation. Involvement of other signaling cascades is conceivable if the membrane actions of CORT are indeed mediated by a GPCR that results in the assembly of a signalosome. Here, CORT was found to up-regulate pERK in an NMDAR-dependent manner, but CORT-induced changes in JNK and p38 activation were not observed. This suggests that activation of the PKA, Akt/PKB, and PKC pathways may be the most prominent proximal events in rapid CORT signaling initiated at the plasma membrane.
Our experiments show that the PKA, Akt/PKB, and PKC pathways eventually converge on Pyk2, a scaffold tyrosine kinase known to be a key player in GPCR-mediated signaling in neurons (46), in agreement with previous studies (Refs. 21, 47, and 48 and as shown in Fig. 8). Two mechanisms regulate the activation of Pyk2; the first, direct one, that leads to its initial autophosphorylation, is Ca2+-dependent, and the second involves dephosphorylation by various phosphatases (49), the tyrosine phosphatase PTP-PEST being a prime example. Regulation of Pyk2 by PTP-PEST has been described in a number of non-neuronal cellular contexts (29, 50), and PTP-PEST has been shown to control dendritic arborization (52) by dephosphorylating Pyk2 and other proteins of the focal adhesion complex. PTP-PEST affects the phosphorylation status of a large set of proteins (53), and therefore, it is conceivable that PTP-PEST contributes an additional and currently unknown layer of complexity to fast CORT-triggered signaling pathways.
FIGURE 8.

Schematic representation of CORT-initiated signaling cascades at the plasma membrane of hippocampal neurons. The non-classical signaling cascade set in motion by corticosterone originates at a still unidentified membrane-located GPCR. The putative receptor is linked via a PTX-sensitive Gi protein to three diverging proximal cascades, namely, PLC-PKC, PI3K-Akt/PKB, and PKA. In this scheme, PKC and calmodulin induce Pyk2 autophosphorylation, and PKC, Akt/PKB, and PKA signaling converge to phosphorylate and inhibit the Pyk2 phosphatase PTP-PEST; full activation of Pyk2 requires PKC, PKA, and Akt/PKB activities. Pyk2 is responsible for the activation of Src family kinases and c-Abl, which in turn influence the phosphorylation and trafficking of NMDAR as well as the clustering of synaptic PSD-95 and remodeling of actin structures. The full signal transduction cascade uses Pyk2 as major hub at which proximal signaling cascades converge and are integrated and from which tyrosine kinase signaling diverges to modulate final target proteins. CaMKII, calmodulin kinase II.
The activation of Pyk2, a core non-receptor TK represents a major point for integrating up-stream signals and, at the same time, a hub from which divergent downstream cascades can emerge. Although the extent of Pyk2 activation is controlled at several checkpoints (PKC-dependent autophosphorylation and Akt/PKB- and PKA-dependent relief of the inhibitory effect of PTP-PEST), Pyk2 autophosphorylation and the subsequent activation of Src or Fyn, which back-phosphorylates Pyk2, form a positive feedback loop that ensures signal amplification. However, because direct competitive binding of Pyk2 to the SH2 domain of Src (or Fyn) dispenses with the need for full dephosphorylation of its Tyr-527 inhibitory site (54), it is interesting that phosphorylation of Src Tyr-527 is detectable even when the Tyr-416 site is strongly phosphorylated (Fig. 1E). Our results show CORT to be an efficient activator of Pyk2; for example, we observed a 3-fold increase in local pPyk2 levels (Fig. 2B).
Pyk2 as well as Src and Fyn reside in close contact with post-synaptic density (PSD) proteins (23), and Pyk2 is involved in several types of synaptic plasticity (21, 55). The ability of CORT to rapidly activate Pyk2 suggests that this hormone has the potential to modulate a large number of synapses within a very short timeframe. Moreover, because Pyk2 is also implicated in the extra-synaptic regulation of the dendritic cytoskeleton (56), it is likely that CORT can also rapidly influence dendritic architecture. The C terminus of Pyk2 is known to serve as scaffold module on whose phosphotyrosines multiple adaptors (e.g. vav and Grb2; Ref. 57), signaling molecules (e.g. RasGAP and RhoGAP; Refs. 58 and 59), and various other kinases, can be recruited and/or phosphorylated to provide further signaling divergence (Fig. 8).
The present work identifies Abl as a target of fast CORT signaling initiated at the plasma membrane of hippocampal neurons (Fig. 5). Our results indicate that CORT-induced activation of Abl occurs downstream of Pyk2 and independently of NMDAR and that its activation lags behind that of Pyk2 and Src. Moreover, by using a highly specific Abl kinase inhibitor (STI571), we show that Abl signaling increases clustering of PSD-95, consistent with the results of another recent study (24). Because PSD-95 clustering leads to increased autophosphorylation of Pyk2 (23), CORT-induced activation of Abl and PSD-95 clustering provides additional amplification of Pyk2 signaling (beyond and downstream of the Pyk2/Src interaction). Interestingly, by phosphorylating WASP and WAVE complexes, Abl serves as a powerful regulator of the actin cytoskeleton (33); given the crucial role of actin in spine physiology (60), we suggest that the Pyk2-Src-Abl module may explain the previously reported fast actions of CORT on spine morphology (12).
Analysis of morphological correlates of synaptic plasticity revealed that CORT rapidly alters NMDAR localization and PSD-95 clustering. Because plasticity-inducing stimuli are known to regulate cell surface availability of NMDAR through Pyk2 and its SFK-dependent amplification loop (21, 61, 62), we here monitored synaptic NMDAR surface localization in CORT-treated hippocampal neurons. Our results show concomitant increases in the phosphorylation of the Tyr-1472 residue in the cytoplasmic tail of the NR2B subunit of the NMDAR and surface retention of NMDAR after CORT application, providing a molecular explanation for the induction of fast CORT-induced changes in NMDAR-induced Ca2+ currents (5). Previous studies showed that the cytoplasmic tail of the NR2 subunit is a major target of TK, with the Tyr-1472 residue appearing to be a major regulator of NMDAR endocytosis as it provides the binding site for the endocytic adaptor AP-2 (35, 36).
The fast increase in surface NMDAR expression after CORT application was associated with an up-regulation of other, NMDAR-dependent, downstream signaling pathways such as ERK1/2 (and calmodulin kinase II and pCREB; not shown) that influence the transcriptional machinery. In this context it should also be noted that besides their roles in modulation of synaptic structure and activity, Pyk2 and Abl can modulate gene transcription after translocation to the nucleus, at least in non-neuronal cells (19, 51, 63, 64). Thus, the potential of the NMDAR- and Pyk2-Abl-initiated cascades to converge with or “prime” the transcriptional response to activated nuclear GR adds an additional layer of complexity to the mechanisms through which CORT can alter neuronal activity. Interestingly, CORT rapidly modulated or triggered signaling cascades from unrelated receptors. The possibility of such “receptor transactivation” (see Ref. 19) was investigated, but neither EGF receptor nor TrkB was found involved in CORT-initiated cascades (see Fig. 1); notably, a major contribution of CORT-stimulated signaling cascades comes from the recruitment and activation of NMDAR, which leads to a significant induction of ERK phosphorylation. In fact, CORT-induced activation of the ERK pathway appears to be largely, if not only, due to NMDAR activity. Thus, overall rapid CORT signaling may involve extensive overlap and cross-modulation of signaling from other neurotransmitter receptors, with consequences that have yet to be elucidated.
The present results (see Fig. 8) provide a comprehensive analysis of the non-genomic signaling cascades triggered by CORT at the plasma membrane of hippocampal neurons. They demonstrate that divergent pathways downstream of a putative GPCR converge to feed into a major signaling hub (TK) and subsequently influence functional targets such as NMDAR and PSD-95. These mechanisms are likely to explain the rapid effects of CORT on synaptic function (11), behavioral, and endocrine responses (6, 10) and remodeling of neuronal networks (14). Previously, convergence of CORT-triggered signaling pathways was only known in the context of transcriptional regulation (41); the present report showing the convergence of multiple upstream cascades on Pyk2 represents a new level of integration of CORT signaling with other pathways, including the nuclear GR pathway.
Acknowledgments
Zsolt Némethy performed early proof-of-concept experiments, Dieter Fischer and Rainer Stoffel gave valuable technical assistance, Dr. Peter Hutzler provided indispensable help with the confocal imaging, and Carola Hetzel provided administrative and editorial assistance. We thank Therese Riedemann for discussions and helpful suggestions during the execution of these experiments and for critical remarks on a draft version of the manuscript and Professors Florian Holsboer (Munich) and Paolo Livrea (Bari) for encouragement. We also thank Professor K. Mashima and staff (Rikkyo University) for kindly providing the PTP-PEST antibody.
Footnotes
- GC
- glucocorticoid
- GR
- glucocorticoid receptor
- GPCR
- G protein-coupled receptor
- Abl
- Abelson
- CORT
- corticosterone
- CORT-BSA
- corticosterone-BSA conjugate
- CHX
- cycloheximide
- CREB
- cAMP response element-binding protein
- MR
- mineralocorticoid receptor
- NMDAR
- NMDA receptor
- PSD-95
- post-synaptic density protein 95
- PTP
- protein-tyrosine phosphatase
- PTX
- pertussis toxin
- Pyk2
- non-receptor proline-rich tyrosine kinase 2
- RTK
- receptor-tyrosine kinase
- SFK
- Src family kinase
- Syk
- spleen tyrosine kinase
- TK
- tyrosine kinase
- WORT
- wortmannin
- PLC
- phospholipase C.
REFERENCES
- 1. Karst H., Berger S., Erdmann G., Schütz G., Joëls M. (2010) Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc. Natl. Acad. Sci. U.S.A. 107, 14449–14454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Olijslagers J. E., de Kloet E. R., Elgersma Y., van Woerden G. M., Joëls M., Karst H. (2008) Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur. J. Neurosci. 27, 2542–2550 [DOI] [PubMed] [Google Scholar]
- 3. Qiu J., Wang P., Jing Q., Zhang W., Li X., Zhong Y., Sun G., Pei G., Chen Y. (2001) Rapid activation of ERK1/2 mitogen-activated protein kinase by corticosterone in PC12 cells. Biochem. Biophys. Res. Commun. 287, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 4. Venero C., Borrell J. (1999) Rapid glucocorticoid effects on excitatory amino acid levels in the hippocampus. A microdialysis study in freely moving rats. Eur. J. Neurosci. 11, 2465–2473 [DOI] [PubMed] [Google Scholar]
- 5. Takahashi T., Kimoto T., Tanabe N., Hattori T. A., Yasumatsu N., Kawato S. (2002) Corticosterone acutely prolonged N-methyl-d-aspartate receptor-mediated Ca2+ elevation in cultured rat hippocampal neurons. J. Neurochem. 83, 1441–1451 [DOI] [PubMed] [Google Scholar]
- 6. Sato S., Osanai H., Monma T., Harada T., Hirano A., Saito M., Kawato S. (2004) Acute effect of corticosterone on N-methyl-d-aspartate receptor-mediated Ca2+ elevation in mouse hippocampal slices. Biochem. Biophys. Res. Commun. 321, 510–513 [DOI] [PubMed] [Google Scholar]
- 7. Dorey R., Piérard C., Shinkaruk S., Tronche C., Chauveau F., Baudonnat M., Béracochéa D. (2011) Membrane mineralocorticoid but not glucocorticoid receptors of the dorsal hippocampus mediate the rapid effects of corticosterone on memory retrieval. Neuropsychopharmacology 36, 2639–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mikics E., Barsy B., Haller J. (2007) The effect glucocorticoids on aggressiveness in established colonies of rats. Psychoneuroendocrinology 32, 160–170 [DOI] [PubMed] [Google Scholar]
- 9. Radulovic J., Rühmann A., Liepold T., Spiess J. (1999) Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress. Differential roles of CRF receptors 1 and 2. J. Neurosci. 19, 5016–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Evanson N. K., Tasker J. G., Hill M. N., Hillard C. J., Herman J. P. (2010) Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology 151, 4811–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Riedemann T., Patchev A. V., Cho K., Almeida O. F. (2010) Corticosteroids. Way upstream. Mol. Brain 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Komatsuzaki Y., Murakami G., Tsurugizawa T., Mukai H., Tanabe N., Mitsuhashi K., Kawata M., Kimoto T., Ooishi Y., Kawato S. (2005) Rapid spinogenesis of pyramidal neurons induced by activation of glucocorticoid receptors in adult male rat hippocampus. Biochem. Biophys. Res. Commun. 335, 1002–1007 [DOI] [PubMed] [Google Scholar]
- 13. Hu W., Zhang M., Czéh B., Flügge G., Zhang W. (2010) Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology 35, 1693–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferris C. F., Stolberg T. (2010) Imaging the immediate non-genomic effects of stress hormone on brain activity. Psychoneuroendocrinology 35, 5–14 [DOI] [PubMed] [Google Scholar]
- 15. Prager E. M., Brielmaier J., Bergstrom H. C., McGuire J., Johnson L. R. (2010) Localization of mineralocorticoid receptors at mammalian synapses. PLoS ONE 5, e14344s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dushek O., Goyette J., van der Merwe P. A. (2012) Non-catalytic tyrosine-phosphorylated receptors. Immunol. Rev. 250, 258–276 [DOI] [PubMed] [Google Scholar]
- 17. Pawson T. (2004) Specificity in signal transduction. From phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203 [DOI] [PubMed] [Google Scholar]
- 18. McGarrigle D., Huang X. Y. (2007) GPCRs signaling directly through Src-family kinases. Sci. STKE 2007, pe35. [DOI] [PubMed] [Google Scholar]
- 19. Almendro V., García-Recio S., Gascón P. (2010) Tyrosine kinase receptor transactivation associated to G protein-coupled receptors. Curr. Drug Targets 11, 1169–1180 [DOI] [PubMed] [Google Scholar]
- 20. Roselli F., Tirard M., Lu J., Hutzler P., Lamberti P., Livrea P., Morabito M., Almeida O. F. (2005) Soluble β-amyloid1–40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. 25, 11061–11070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang Y., Lu W., Ali D. W., Pelkey K. A., Pitcher G. M., Lu Y. M., Aoto H., Roder J. C., Sasaki T., Salter M. W., MacDonald J. F. (2001) CAKβ/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 29, 485–496 [DOI] [PubMed] [Google Scholar]
- 22. Seabold G. K., Burette A., Lim I. A., Weinberg R. J., Hell J. W. (2003) Interaction of the tyrosine kinase Pyk2 with the N-methyl-d-aspartate receptor complex via the Src homology 3 domains of PSD-95 and SAP102. J. Biol. Chem. 278, 15040–15048 [DOI] [PubMed] [Google Scholar]
- 23. Bartos J. A., Ulrich J. D., Li H., Beazely M. A., Chen Y., Macdonald J. F., Hell J. W. (2010) Postsynaptic clustering and activation of Pyk2 by PSD-95. J. Neurosci. 30, 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perez de Arce K., de Arce K. P., Varela-Nallar L., Farias O., Cifuentes A., Bull P., Couch B. A., Koleske A. J., Inestrosa N. C., Alvarez A. R. (2010) Synaptic clustering of PSD-95 is regulated by c-Abl through tyrosine phosphorylation. J. Neurosci. 30, 3728–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trinidad J. C., Thalhammer A., Specht C. G., Lynn A. J., Baker P. R., Schoepfer R., Burlingame A. L. (2008) Quantitative analysis of synaptic phosphorylation and protein expression. Mol. Cell. Proteomics 7, 684–696 [DOI] [PubMed] [Google Scholar]
- 26. Kalia L. V., Gingrich J. R., Salter M. W. (2004) Src in synaptic transmission and plasticity. Oncogene 23, 8007–8016 [DOI] [PubMed] [Google Scholar]
- 27. Avraham H., Park S. Y., Schinkmann K., Avraham S. (2000) RAFTK/Pyk2-mediated cellular signaling. Cell. Signal. 12, 123–133 [DOI] [PubMed] [Google Scholar]
- 28. Lyons P. D., Dunty J. M., Schaefer E. M., Schaller M. D. (2001) Inhibition of the catalytic activity of cell adhesion kinase β by protein-tyrosine phosphatase-PEST-mediated dephosphorylation. J. Biol. Chem. 276, 24422–24431 [DOI] [PubMed] [Google Scholar]
- 29. Davidson D., Shi X., Zhong M. C., Rhee I., Veillette A. (2010) The phosphatase PTP-PEST promotes secondary T cell responses by dephosphorylating the protein tyrosine kinase Pyk2. Immunity 33, 167–180 [DOI] [PubMed] [Google Scholar]
- 30. Garton A. J., Tonks N. K. (1994) PTP-PEST. A protein-tyrosine phosphatase regulated by serine phosphorylation. EMBO J. 13, 3763–3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakamura K., Palmer H. E., Ozawa T., Mashima K. (2010) Protein phosphatase 1α associates with protein-tyrosine phosphatase-PEST inducing dephosphorylation of phospho-serine 39. J. Biochem. 147, 493–500 [DOI] [PubMed] [Google Scholar]
- 32. Park D., Jhon D. Y., Lee C. W., Lee K. H., Rhee S. G. (1993) Activation of phospholipase C isozymes by G protein βγ subunits. J. Biol. Chem. 268, 4573–4576 [PubMed] [Google Scholar]
- 33. Zrihan-Licht S., Avraham S., Jiang S., Fu Y., Avraham H. K. (2004) Coupling of RAFTK/Pyk2 kinase with c-Abl and their role in the migration of breast cancer cells. Int. J. Oncol. 24, 153–159 [PubMed] [Google Scholar]
- 34. Colicelli J. (2010) ABL tyrosine kinases. Evolution of function, regulation, and specificity. Sci. Signal. 3, re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roche K. W., Standley S., McCallum J., Dune Ly C., Ehlers M. D., Wenthold R. J. (2001) Molecular determinants of NMDA receptor internalization. Nat. Neurosci. 4, 794–802 [DOI] [PubMed] [Google Scholar]
- 36. Prybylowski K., Chang K., Sans N., Kan L., Vicini S., Wenthold R. J. (2005) The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 47, 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sattler R., Xiong Z., Lu W. Y., Hafner M., MacDonald J. F., Tymianski M. (1999) Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284, 1845–1848 [DOI] [PubMed] [Google Scholar]
- 38. Lin Y., Skeberdis V. A., Francesconi A., Bennett M. V., Zukin R. S. (2004) Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J. Neurosci. 24, 10138–10148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim M. J., Dunah A. W., Wang Y. T., Sheng M. (2005) Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46, 745–760 [DOI] [PubMed] [Google Scholar]
- 40. Liposits Z., Bohn M. C. (1993) Association of glucocorticoid receptor immunoreactivity with cell membrane and transport vesicles in hippocampal and hypothalamic neurons of the rat. J. Neurosci. Res. 35, 14–19 [DOI] [PubMed] [Google Scholar]
- 41. Schmidt P., Holsboer F., Spengler D. (2001) β2-adrenergic receptors potentiate glucocorticoid receptor transactivation via G protein βγ-subunits and the phosphoinositide 3-kinase pathway. Mol. Endocrinol. 15, 553–564 [DOI] [PubMed] [Google Scholar]
- 42. Samarasinghe R. A., Di Maio R., Volonte D., Galbiati F., Lewis M., Romero G., DeFranco D. B. (2011) Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 108, 16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karst H., Berger S., Turiault M., Tronche F., Schütz G., Joëls M. (2005) Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. U.S.A. 102, 19204–19207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu S. B, Zhang N., Guo Y. Y., Zhao R., Shi T. Y., Feng S. F., Wang S. Q., Yang Q., Li X. Q., Wu Y. M., Ma L., Hou Y., Xiong L. Z., Zhang W., Zhao M. G. (2012) G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors. J. Neurosci. 32, 4887–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gros R., Ding Q., Liu B., Chorazyczewski J., Feldman R. D. (2013) Aldosterone mediates its rapid effects in vascular endothelial cells through gper/gpr30 activation. Am. J. Physiol. Cell Physiol 304, C532–C540 [DOI] [PubMed] [Google Scholar]
- 46. Macdonald D. S., Weerapura M., Beazely M. A., Martin L., Czerwinski W., Roder J. C., Orser B. A., MacDonald J. F. (2005) Modulation of NMDA receptors by pituitary adenylate cyclase activating peptide in CA1 neurons requires Gαq, protein kinase C, and activation of Src. J. Neurosci. 25, 11374–11384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Della Rocca G. J., van Biesen T., Daaka Y., Luttrell D. K., Luttrell L. M., Lefkowitz R. J. (1997) Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. Convergence of Gi- and Gq-mediated pathways on calcium/calmodulin, Pyk2, and Src kinase. J. Biol. Chem. 272, 19125–19132 [DOI] [PubMed] [Google Scholar]
- 48. Nicodemo A. A., Pampillo M., Ferreira L. T., Dale L. B., Cregan T., Ribeiro F. M., Ferguson S. S. (2010) Pyk2 uncouples metabotropic glutamate receptor G protein signaling but facilitates ERK1/2 activation. Mol. Brain 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu J., Kurup P., Bartos J. A., Patriarchi T., Hell J. W., Lombroso P. J. (2012) Striatal-enriched protein-tyrosine phosphatase (STEP) regulates Pyk2 kinase activity. J. Biol. Chem. 287, 20942–20956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Souza C. M., Davidson D., Rhee I., Gratton J. P., Davis E. C., Veillette A. (2012) The phosphatase PTP-PEST/PTPN12 regulates endothelial cell migration and adhesion, but not permeability, and controls vascular development and embryonic viability. J. Biol. Chem. 287, 43180–43190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mantovani F., Piazza S., Gostissa M., Strano S., Zacchi P., Mantovani R., Blandino G., Del Sal G. (2004) Pin1 links the activities of c-Abl and p300 in regulating p73 function. Mol. Cell 14, 625–636 [DOI] [PubMed] [Google Scholar]
- 52. Grace E. A., Busciglio J. (2003) Aberrant activation of focal adhesion proteins mediates fibrillar amyloid β-induced neuronal dystrophy. J. Neurosci. 23, 493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davidson D., Veillette A. (2001) PTP-PEST, a scaffold protein-tyrosine phosphatase, negatively regulates lymphocyte activation by targeting a unique set of substrates. EMBO J. 20, 3414–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dikic I., Tokiwa G., Lev S., Courtneidge S. A., Schlessinger J. (1996) A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 383, 547–550 [DOI] [PubMed] [Google Scholar]
- 55. Cheung H. H., Gurd J. W. (2001) Tyrosine phosphorylation of the N-methyl-d-aspartate receptor by exogenous and postsynaptic density-associated Src-family kinases. J. Neurochem. 78, 524–534 [DOI] [PubMed] [Google Scholar]
- 56. Suo L., Lu H., Ying G., Capecchi M. R., Wu Q. (2012) Protocadherin clusters and cell adhesion kinase regulate dendrite complexity through Rho GTPase. J. Mol. Cell Biol. 4, 362–376 [DOI] [PubMed] [Google Scholar]
- 57. Gao C., Blystone S. D. (2009) A Pyk2-Vav1 complex is recruited to β3-adhesion sites to initiate Rho activation. Biochem. J. 420, 49–56 [DOI] [PubMed] [Google Scholar]
- 58. Zrihan-Licht S., Fu Y., Settleman J., Schinkmann K., Shaw L., Keydar I., Avraham S., Avraham H. (2000) RAFTK/Pyk2 tyrosine kinase mediates the association of p190 RhoGAPwith RasGAP and is involved in breast cancer cell invasion. Oncogene 19, 1318–1328 [DOI] [PubMed] [Google Scholar]
- 59. Ying Z., Giachini F. R., Tostes R. C., Webb R. C. (2009) PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler. Thromb. Vasc. Biol. 29, 1657–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bosch M., Hayashi Y. (2012) Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 22, 383–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hsin H., Kim M. J., Wang C. F., Sheng M. (2010) Proline-rich tyrosine kinase 2 regulates hippocampal long-term depression. J. Neurosci. 30, 11983–11993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Heidinger V., Manzerra P., Wang X. Q., Strasser U., Yu S. P., Choi D. W., Behrens M. M. (2002) Metabotropic glutamate receptor 1-induced up-regulation of NMDA receptor current. Mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J. Neurosci. 22, 5452–5461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aoto H., Sasaki H., Ishino M., Sasaki T. (2002) Nuclear translocation of cell adhesion kinase β/proline-rich tyrosine kinase 2. Cell Struct. Funct. 27, 47–61 [DOI] [PubMed] [Google Scholar]
- 64. Sanchez-Arévalo L. V. J., Doni M., Verrecchia A., Sanulli S., Fagà G., Piontini A., Bianchi M., Conacci-Sorrell M., Mazzarol G., Peg V., Losa J. H., Ronchi P., Ponzoni M., Eisenman R. N., Doglioni C., Amati B. (2013). Dual regulation of Myc by Abl. Oncogene 10.1038/onc.2012.621 [DOI] [PMC free article] [PubMed] [Google Scholar]