

Background: Chemotherapeutic sensitivity in ovarian cancer is dependent on effective apoptosis signaling.
Results: Piceatannol enhances cisplatin sensitivity by modulating p53, XIAP (X-linked inhibitor of apoptosis protein), and mitochondrial fission in vitro and in vivo.
Conclusion: Piceatannol is a potent enhancer of cisplatin-induced apoptosis.
Significance: Piceatannol exhibits potential for clinical development for the treatment of ovarian cancer.
Keywords: Chemoresistance, Mitochondria, Ovarian Cancer, p53, XIAp, Piceatannol, Cisplatin
Abstract
Resistance to cisplatin (CDDP) in ovarian cancer (OVCA) arises from the dysregulation of tumor suppressors and survival signals. During genotoxic challenge, these factors can be influenced by secondary agents that facilitate the induction of apoptosis. Piceatannol is a natural metabolite of the stilbene resveratrol found in grapes and is converted from its parent compound by the enzyme CYP1BA1 p450. It has been hypothesized to exert specific effects against various cellular targets; however, its ability to influence CDDP resistance in cancer cells has not been investigated to date. Here, we show that piceatannol is a potent enhancer of CDDP sensitivity in OVCA, and this effect is achieved through the modulation of several major determinants of chemoresistance. Piceatannol enhances p53-mediated expression of the pro-apoptotic protein NOXA, increases XIAP degradation via the ubiquitin-proteasome pathway, and enhances caspase-3 activation. This response is associated with an increase in Drp1-dependent mitochondrial fission, leading to more effective induction of apoptosis. In vivo studies using a mouse model of OVCA reveal that a number of these changes occur in association with a greater overall reduction in tumor weight when mice are treated with both piceatannol and CDDP, in comparison to treatment with either agent alone. Taken together, these findings demonstrate the potential application of piceatannol to enhance CDDP sensitivity in OVCA, and it acts on p53, XIAP, and mitochondrial fission.
Introduction
Despite considerable research investments, ovarian cancer (OVCA)2 remains the most lethal gynecological malignancy. As a result of late diagnosis and chemoresistant recurrent disease, a dismal 5-year survival rate of ∼20% has persisted for years (1). At advanced stages of the disease (III and IV), current therapeutic strategies become increasingly ineffective (2). Derivatives of cisplatin (CDDP: cis-diamminedichloroplatinum) are the first line treatments for OVCA, often administered together with paclitaxel (3). CDDP is genotoxic in vivo and functions by cross-linking DNA, resulting in cell-cycle arrest and ultimately triggering apoptosis. However, its broad mechanism of action causes side effects including nausea, nephrotoxicity, and hemolytic anemia (4, 5).
Chemoresistance is thought to arise from the alteration of genetic and epigenetic mechanisms responsible for detecting genotoxic insults and making appropriate cell fate decisions (6). Evasion from apoptosis can arise from the dysregulation of specific tumor suppressors and survival signals, contributing to a loss in sensitivity to chemotherapeutic agents (7, 8). As a master regulator of cell cycle progression, DNA repair, and apoptosis, the tumor suppressor p53 plays a central role in this process (9–11). After nuclear activation, p53 up-regulates multiple pro-apoptotic factors including NOXA, which localizes to the mitochondria and interacts with anti-apoptotic Bcl-2 family members (12). The subsequent release of pro-apoptotic factors including SMAC (second mitochondria-derived activator of caspases) and cytochrome c plays a major role in caspase-dependent apoptosis (13, 14). The efficiency at which apoptosis is induced can be influenced by mitochondrial fission, part of a dynamic process that involves cleavage of individual mitochondria (15). Fission is a key event that occurs prior to the induction of apoptosis and involves cleavage of the organelle in response to various stimuli, including cell stress. Dynamin-related protein 1 (Drp1) is a cytosolic GTPase activated upon dephosphorylation by calcineurin and oligomerizes to provide the mechanical strength needed for fission to occur (16, 17).
XIAP (X-linked inhibitor of apoptosis protein), another determinant of chemoresistance in OVCA (7), blocks apoptosis signaling in its final stages by inhibiting caspases that would otherwise be activated through the actions of pro-apoptotic mediators like NOXA. The successful induction of mitochondrial-mediated apoptosis therefore relies on a complex but coordinated interplay of signaling events, the dysregulation of which can give rise to chemoresistance. Bioactive natural compounds that exert influences on these pathways and tip the cellular balance in favor of apoptosis may potentially be useful for novel OVCA therapeutic strategies.
Phytochemicals are a major class of functional food compounds, some of which are known to exert highly specific effects on key regulators of apoptosis. The phytoalexin resveratrol is a stilbene found in grapes and mulberry, widely known for its anti-cancer properties in red wine extract (18). Although it has been shown to inhibit ovarian tumor growth in mouse xenograft models and to sensitize cancer cells to CDDP and doxorubicin (19), recent studies have shed doubts on the clinical utility of resveratrol for the prevention and treatment of human cancers (20). Resveratrol is metabolized after ingestion by the CYP1BA1 p450 enzyme into a number of products, one of which is the phenolic compound piceatannol (21). Further research on piceatannol has revealed its superior and potent bioactive properties, including inhibitory effects on platelet-derived growth factor-BB (22), altered gene expression resulting in the delay of adipogenesis, and cell cycle inhibition in colorectal cancer cells (23, 24). However, the effects of piceatannol on CDDP sensitivity in cancer cells has not previously been investigated.
The objective of the present study was to determine the effects of piceatannol on OVCA growth when treated alone and in combination with CDDP. We hypothesize that piceatannol enhances CDDP sensitivity in OVCA cells by exerting specific influences on key regulators of apoptosis. Our findings indicate that piceatannol enhances the apoptotic action of CDDP in OVCA through nuclear activation and stabilization of p53, proteasome-dependent XIAP down-regulation, and the enhancement of Drp1-dependent mitochondrial fission.
EXPERIMENTAL PROCEDURES
Reagents
CDDP, Me2SO, Hoechst 33258, lactacystin, and epoxomicin were purchased from Sigma-Aldrich. Piceatannol was purchased from Tocris Bioscience (Bristol, UK). Mouse monoclonal p53 antibodies (DO-1), MDM2, and PARP were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit monoclonal anti-Ser(P)15-p53, anti-poly(ADP-ribose) polymerase (PARP) antibodies and siRNA constructs were from Cell Signaling Technology (Beverly, CA). Rabbit polyclonal anti-NOXA and anti-XIAP antibodies, as well as mouse monoclonal anti-GAPDH and anti-caspase-3 antibodies were from Abcam (Cambridge, MA). Peroxidase-conjugated goat anti-mouse and goat anti-rabbit immunoglobulin were purchased from Bio-Rad. Alexa Fluor® 488 and 594 secondary antibodies, Lipofectamine 2000 transfection reagent, RNase A, TEMED, RPMI 1640, and DMEM/F12 culture media and fetal bovine serum were from Invitrogen. Complete Mini Protease inhibitor mixture tablets and PhosStop phosphatase inhibitor mixture tablets were obtained from Roche Applied Sciences. CMV-wt-p53 and CMV-GFP adenoviral constructs were synthesized by Vector Biolabs (Philadelphia, PA). The MTS assay kit was purchased from Promega (Thermo Fisher Scientific).
Cell Lines and Culture
CDDP-sensitive (OV2008 (wt-p53) and A2780s (wt-p53)) and CDDP-resistant (C13* (wt-p53), A2780cp (p53 mutant), OVCAR-432 (p53 mutant), and SKOV3 (p53 null)) human OVCA cell lines were gifts from Drs. Rakesh Goel and Barbara Vanderhyden (Ottawa Hospital Cancer Center, Ottawa, Canada) and were cultured as previously reported (25). OV2008 cells and their resistant counterpart C13* are of ovarian endometrioid adenocarcinoma origin with squamous differentiation. A2780s/A2780cp and OCC-1 cells are from undifferentiated ovarian carcinoma tumors. SKOV3 cells are of clear cell carcinoma origin (26).
Immunoblotting, Immunoprecipitation, and Ubiquitination Assays
Immunoblotting was performed as previously described (10). Band densities were analyzed and quantified using a Bio-Rad ChemiDoc XRS+ and Image Lab V3.0 (Hercules, CA). For immunoprecipitation, cells were lysed, and supernatant (200 μl) was incubated with protein A Dynabeads (Invitrogen) coated with antibodies targeting HA (2 μg/200 μl; 1 h, room temperature) and immunoprecipitated overnight (4 °C). The beads were pelleted, resuspended in Laemmli sample buffer (2×; 40 μl; Bio-Rad), boiled (10 min), and loaded onto 9% SDS-PAGE. For the XIAP ubiquitination assay, OV2008 cells were transfected with HA-ubiquitin plasmids (1 μg) (Addgene, Cambridge, MA) for 48 h using Lipofectamine 2000.
MTS Assay and Hoechst 33258 Staining
The indicated cell lines were seeded for 15 h in 96-well plates and maintained in culture with piceatannol (10 μm) with or without CDDP (10 μm, 24 h), and tetrazolium compound (Promega, Madison, WI) was added to the cultures for an additional 2–4 h, as per the manufacturer's instructions. Absorbance at 490 nm was determined by a Bio-Rad X-Mark microplate analyzer. Apoptosis was assessed based on stereotypic morphological changes, including chromatin condensation, nuclear fragmentation cytoplasmic shrinkage, and the formation of apoptotic bodies (with nuclear fragments) as assessed in previous studies (11, 27, 28). At least 400 cells/treatment group were counted, and the quantification process was blinded to avoid experiment bias.
Quantitative Real Time PCR, RNA Interference, and Adenoviral Transfection
Quantitative RT-PCR was performed using SYBR Green (Sigma) and a StepOnePlus real time PCR system (Applied Biosystems, Foster City, CA). Relative quantification of gene expression was calculated using the formula 2−ΔΔCt, where ΔΔCt = (Cttarget gene − CtGAPDH)cancerous tissue − (Cttarget gene − CtGAPDH)normal tissue. For RNA interference studies, OV2008 cells were transfected with p53 or control siRNA (100 nm) for 48 h and cultured as previously described (25). A2780cp cells were transfected with adenoviral constructs containing wt-p53 (multiplicity of infection, 10; 6 h) or control GFP, as previously described (29).
Immunofluorescence Microscopy and Quantification of Mitochondrial fission
OV2008 cells were fixed with 4% paraformaldehyde in 8-well chamber slides. The cells were then incubated with appropriate fluorescence-conjugated secondary antibodies and stained with ProLong Gold antifade reagent (Invitrogen) with DAPI (blue nuclear stain). Coverslips were fixed, and the cells were imaged immediately with a Zeiss LSM700 confocal scanning microscope equipped with a Zeiss T-PMT digital camera (Zeiss, Oberkochen, Germany). The quantification of changes in mitochondrial morphology was conducted using the method outlined by Lutz et al. (30). The cells were categorized as one of two types, according to their overall mitochondrial morphology: tubular or fragmented. Cells exhibiting intact tubular mitochondria were classified as tubular. When these tubular structures were visibly disrupted with the appearance of both shortened rod structures and spherical bodies, they were classified as fragmented. The mitochondrial morphology of at least 120 cells/treatment group was determined, with the observer blinded to the identity of treatment groups. Quantifications were derived from three independent experiments. p53 nuclear localization was quantified using the Zeiss confocal microscopy software ZEN lite.
Animals, OVCA Xenografts, and Immunohistochemistry
All animal procedures were conducted in accordance with animal care guidelines provided by Seoul National University. Male athymic nude mice (6–8 weeks old) were purchased from the Institute of Laboratory Animal Resources at Seoul National University. The animals were acclimated for 1 week prior to the study and had free access to food and water. The animals were housed in climate-controlled quarters with a 12-h light/dark cycle. OV2008 (1 × 106) cells in 100 μl of RPMI medium were mixed with 100 μl of Matrigel. Cells implanted subcutaneously in the hind flank of each mouse formed solid and rapidly growing tumors. The mice were treated when tumor volumes reached ∼100 mm3 as measured using calipers, and volumes were estimated using the equation V = π/6 (l × h × w). Piceatannol (20 mg/kg/day, 5 days/week) and CDDP (1.8 mg/kg/week) were administered intraperitoneally, and tumor volumes were measured for 18 days, at which time the experiment was terminated on ethical grounds because of the largest control tumors impeding animal mobility. At sacrifice, tumor tissue was removed, and a portion was fixed in formalin and embedded in paraffin for immunohistochemical analysis. Sections (5 μm thick) from 10% neutral formalin solution-fixed paraffin-embedded tissue were cut and mounted on silane-coated glass slides. After being deparaffinization, the sections were boiled in citric acid buffer (pH 6.0) for antigen retrieval and immunofluorescence analysis. All of the animal procedures were carried out as approved by the Institutional Animal Care and Use Committee of Seoul National University.
CDDP causes toxic effects in humans during normal therapeutic regimens and is an inseparable aspect of its mode of action. As the guidelines from our institutional animal use and care committee stipulate, euthanasia is recommended if excess discomfort is caused by treatment. As standard procedure, we assessed potential toxicity of the treatments during the course of the studies using a number of morbidity criteria including, but not limited to: body weight loss, decreased feeding, mobility impediment, and irregular fecal appearance. We did not observe any of the above phenomena in mice for any treatment group.
Statistical Analysis
Results are expressed as the means ± S.E. of at least three independent experiments. Statistical analysis was carried out by one-way, two-way, or three-way analysis of variance, using SigmaPlot software (versions 12; Systat Software, Chicago, IL). Differences between multiple experimental groups were determined by the Bonferroni post hoc test. Statistical significance was inferred at p < 0.05. To calculate combination index, we used the freely available CalcuSyn software (based on the method described by Chou and Talalay (31)), which calculates combination index from dose-response data.
RESULTS
Piceatannol Enhances the Effects of CDDP in Chemosensitive and Chemoresistant OVCA Cells
We compared the effects of piceatannol (10 μm, 24 h) alone and in combination with CDDP (10 μm, 24 h) on the viability of a number of well established OVCA cell lines in vitro. Piceatannol alone reduced cell viability and markedly enhanced the cytotoxic effects of CDDP in chemosensitive cells (OV2008, A2780s, and OVCAR-432) (Fig. 1, A–C). Piceatannol also induced sensitivity to CDDP in the chemoresistant counterpart of OV2008 containing wild-type p53 (C13*; Fig. 1A). These observations were less apparent in A2780cp (chemoresistant counterpart of A2780s harboring mutant p53) and SKOV3 (p53-null), raising the possibility that p53 status could be a determinant of piceatannol action. However, OVCAR-432 cells (a chemosensitive line harboring mutant p53) also exhibited marked reductions in cell viability. A time course study (Fig. 1B), revealed that piceatannol, when treated in combination with CDDP, accelerated the reduction in cell viability by 24 h.
FIGURE 1.
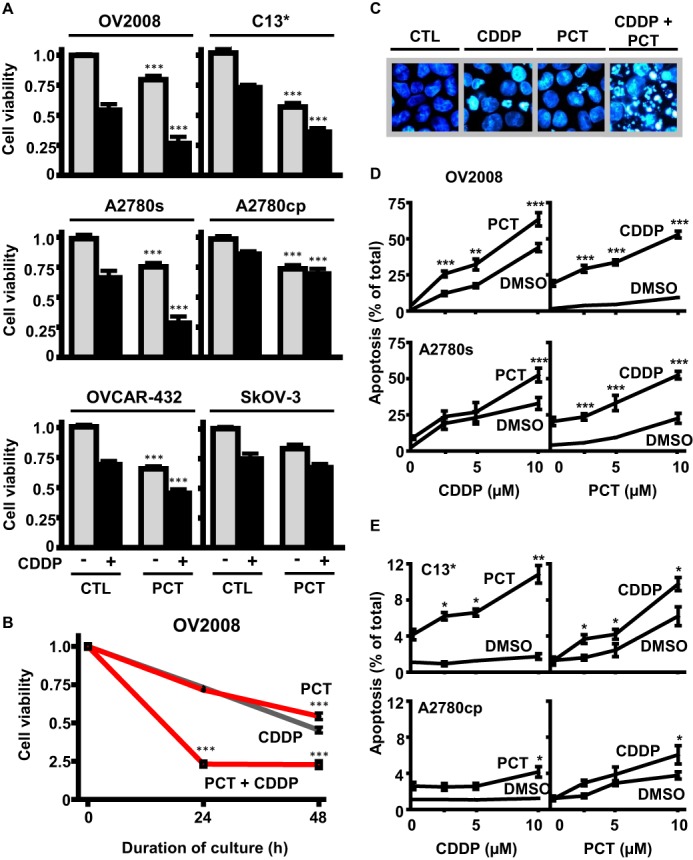
Effects of piceatannol on CDDP treatment in OVCA cell lines. A, reductions in cell viability caused by PCT (10 μm) in combination with CDDP treatment (10 μm, 24 h) in chemosensitive and chemoresistant OVCA. Piceatannol (10 μm) enhances the effects of CDDP (10 μm, 24 h) in OV2008, A2780s, and C13 cells. Although piceatannol did not confer any additional effects on CDDP treatment in p53-null SKOV-3, it enhanced the effects of CDDP (10 μm) in OVCAR-432, a chemosensitive p53-mutant cell line. B, time course study on the effects of PCT (10 μm) and CDDP (10 μm), alone and together, on cell viability of OV2008 cells over 48 h. C, effects of CDDP (5 μm), piceatannol (10 μm), and combined treatment on nuclear morphology in OV2008 cells after 24 h (stained with Hoechst 33258). Dual treatment significantly increases the severity of nuclear condensation and fragmentation in comparison to either agent alone. D, piceatannol treatment (2.5 μm, 24 h) induces a left shift in concentration-response curves for CDDP-induced apoptosis in chemosensitive OV2008 and A2780s. Both cell lines also exhibited a left shift in apoptotic response curves to piceatannol when in the presence of low CDDP concentration (5 μm). E, piceatannol induces a left shift in concentration-response curves for CDDP-induced apoptosis in chemoresistant C13* cells and induces sensitivity in combination with CDDP treatment (10 μm, 24 h) in chemoresistant p53-mutant A2780cp* at 10 μm concentration. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus respective dimethyl sulfoxide (DMSO) control (CTL).
We next investigated whether the cytotoxic activity of piceatannol (Fig. 1A) is associated with the induction of apoptosis. Chemosensitive cells (OV2008 and A2780s) and their chemoresistant counterparts (C13* and A2780cp*, respectively) were cultured in the presence of piceatannol and/or CDDP (0–10 μm, 24 h). Analysis of nuclear morphology using Hoechst 33528 revealed that CDDP (and to a lesser extent piceatannol) caused nuclear condensation and fragmentation typical of apoptosis. This response was markedly enhanced when cells were treated with both CDDP and piceatannol together (0–10 μm, 24 h; Fig. 1C). At a relatively low concentration, piceatannol (2.5 μm) induced a shift in the CDDP concentration-response curve to the left and approximately doubled the total number of OV2008 cells undergoing apoptosis (Fig. 1D). A similar response was observed in A2780s. In C13*, piceatannol (10 μm) promoted CDDP-induced apoptosis by ∼3-fold (Fig. 1E, top panel). A2780cp exhibited more robust chemoresistance, but a concentration of 10 μm piceatannol was observed to induce CDDP sensitivity (10 μm, 24 h; Fig. 1E, bottom panel).
The combination index was calculated (based on the method described by Chou and Talalay (31)), and it was determined that the effect of piceatannol and CDDP on apoptosis is additive. The IC50 values (μm) and the combination index for the main cell line of the study (OV2008) were: CDDP IC50 = 3.6, piceatannol (PCT) IC50 = 29.1, and CDDP + PCT combination index = 1.06.
Piceatannol-induced Cisplatin Sensitivity Is Associated with p53 Activation and NOXA Up-regulation
Recent evidence has highlighted the key interplay between p53 and the mitochondria in CDDP-induced apoptosis (8, 11). We investigated whether NOXA, a mitochondria-related apoptotic factor and a transcriptional target of p53, is involved in this regulation. CDDP-induced apoptosis was associated with NOXA transcriptional up-regulation over time, as evidenced by increased NOXA mRNA abundance and protein content (Fig. 2A). Furthermore, in the presence of piceatannol (10 μm, 24 h), CDDP-dependent up-regulation of NOXA protein content increased in a dose-dependent manner (Fig. 2B). Analysis of p53 expression revealed that submaximal CDDP treatment (5 μm) resulted in p53 activation (increased phospho-p53 (Ser-15)/p53 ratio), a response significantly enhanced by piceatannol (Fig. 2C). MDM2 (mouse double minute 2), which normally functions to maintain low cellular p53 levels, was also down-regulated during co-treatment with CDDP and piceatannol, suggesting that piceatannol may enhance the CDDP response in OVCA by promoting p53 activation and stabilization.
FIGURE 2.
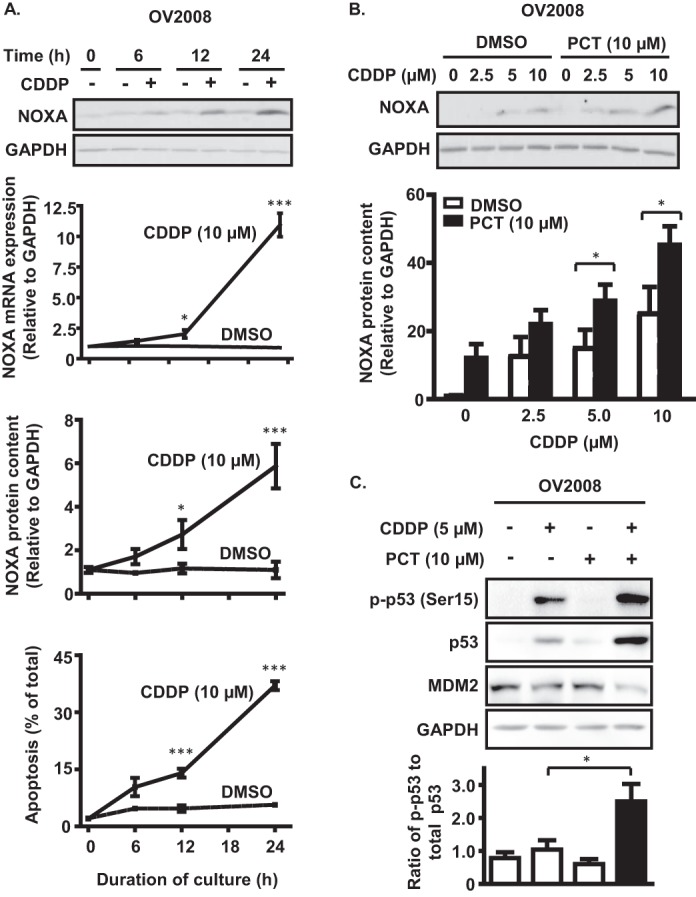
Piceatannol-enhanced CDDP sensitivity is associated with NOXA up-regulation and p53 activation in p53 wild-type OV2008 cells. A, measurements of mRNA abundance and protein content between 0 and 24 h of CDDP treatment (10 μm) reveal up-regulation of NOXA expression. B, piceatannol treatment (10 μm, 24 h) enhances CDDP-dependent up-regulation of NOXA protein content in a concentration-dependent manner. C, immunoblot analysis of the effect of piceatannol on CDDP-dependent p53 expression and phosphorylation at Ser15, as well as down-regulation of MDM2 protein content after 24 h. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus respective dimethyl sulfoxide (DMSO) control.
The Effects of Piceatannol on CDDP Response Are p53-dependent
We next investigated the functional relevance of the p53 tumor suppressor pathway in the promotion of CDDP sensitivity by piceatannol. CDDP (5 μm) and piceatannol (10 μm) treatment alone increased p53 and NOXA protein content, as well as apoptosis in wild-type p53 chemosensitive OV2008 cells. However, a markedly higher synergistic response in terms of NOXA expression and apoptosis was observed in the presence of both agents, which was significantly attenuated when p53 expression was silenced using siRNA (100 nm; Fig. 3A). In addition, although piceatannol induced apoptosis in p53-mutant chemoresistant OVCA cells (A2780cp), the reconstitution of wild-type p53 in these cells induced CDDP sensitivity and significantly enhanced the apoptotic response to combination treatment (Fig. 3B).
FIGURE 3.

Piceatannol modulates the p53 tumor suppressor pathway. A, p53 silencing by siRNA (100 nm, 24 h) reduces the effect of piceatannol treatment (10 μm, 24 h) on CDDP-induced (5 μm) apoptosis and NOXA expression. B, forced expression of wild-type p53 (multiplicity of infection, 10) in a p53 mutant line (A2780cp*) using an adenoviral delivery system enhances piceatannol-induced NOXA expression and apoptosis. C, quantification of localization data reveals statistically significant effects of piceatannol on CDDP-induced nuclear phospho-p53 (Ser15) content. OV2008 cells were treated with CDDP (5 μm) and piceatannol (10 μm) for 6 h. Red fluorescence indicates Ser(P)15-p53 expression, whereas the nucleus is stained blue (DAPI). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus respective dimethyl sulfoxide control (CTL).
The NOXA mRNA up-regulation observed in response to CDDP in chemosensitive cells (Fig. 2A) raised the possibility that p53 transcriptional activity could be responsible for the enhanced apoptotic response to CDDP and piceatannol treatment. Immunofluorescence analysis revealed that CDDP (5 μm, 6 h) increased phospho-p53 (Ser15) levels within the nucleus (Fig. 3C). We observed a synergistic increase in p53 activation and nuclear translocation in the presence of both CDDP and piceatannol (Fig. 3C). Taken together, these findings indicate a key role for the p53 tumor suppressor pathway in piceatannol-enhanced CDDP sensitivity.
Piceatannol Promotes XIAP Ubiquitination and Proteasome-mediated Degradation
We observed that piceatannol (10 μm, 24 h) enhanced CDDP-induced caspase-3 activation in a concentration-dependent manner in OV2008 cells (Fig. 4A). This hinted at the possibility that the prominent caspase inhibitor XIAP could be involved in piceatannol action. We found that CDDP (5 μm) slightly decreased XIAP content, activated caspase-3, and induced PARP cleavage. However, treatment with piceatannol significantly down-regulated XIAP content to an extent greater than CDDP alone while having minimal effects on caspase-3 activation and PARP cleavage. These responses were greatly enhanced when cells were simultaneously treated with piceatannol and CDDP (Fig. 4B).
FIGURE 4.
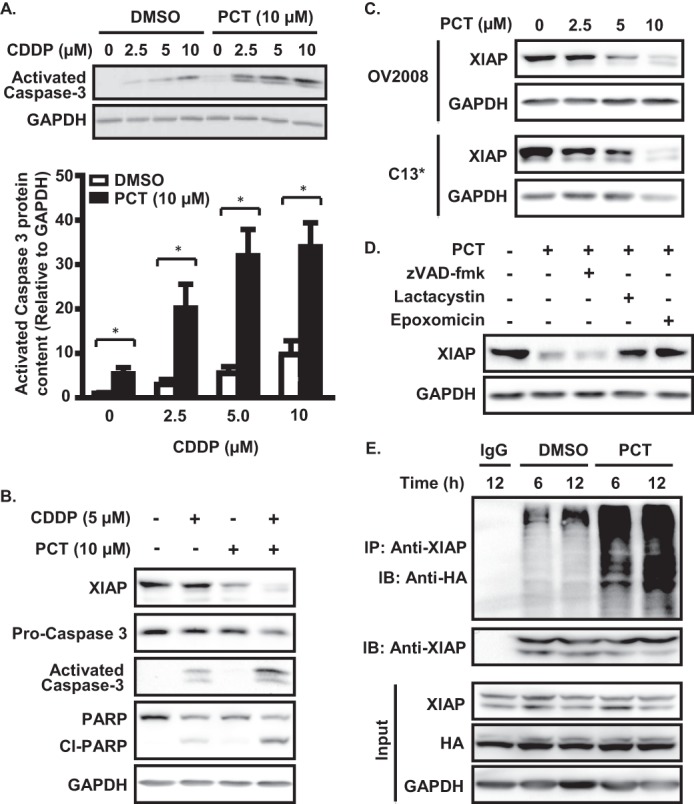
Piceatannol treatment enhances caspase-3 activation in the presence of CDDP and down-regulates XIAP via the ubiquitin-proteasome pathway. A, OV2008 cells were treated with piceatannol (10 μm, 24 h), resulting in enhanced CDDP-induced caspase-3 activation in a concentration-dependent manner. ***, p < 0.001 versus respective dimethyl sulfoxide (DMSO) control. B, effects of 24 h CDDP (5 μm) and piceatannol (10 μm) treatment on activated caspase-3, PARP cleavage and XIAP content. C, piceatannol treatment (24 h) down-regulates XIAP in a concentration-dependent manner in both chemosensitive (OV2008) and chemoresistant (C13*) OVCA cells. D, piceatannol-dependent down-regulation of XIAP expression is rescued by the proteasome inhibitors epoxomicin (15 nm) and lactacystin (10 μm) but not by the pan-caspase inhibitor zVAD-fmk (20 μm). E, immunoprecipitation analysis of piceatannol treatment on XIAP ubiquitination. OV2008 cells were treated for the indicated durations after transfection with ubiquitin-HA constructs (1 μg) in the presence of the proteasome inhibitor epoxomicin (15 nm). IP, immunoprecipitation; IB, immunoblot.
We next sought to investigate whether the 26 S proteasome was involved in the piceatannol-dependent degradation of XIAP. Piceatannol down-regulated XIAP protein content in both chemosensitive (OV2008) and chemoresistant (C13*) OVCA cells in a concentration-dependent manner (0–10 μm, 24 h; Fig. 4C). Co-treatment of chemosensitive OV2008 cells with the specific proteasome inhibitors lactacystin or epoxomicin, but not with the pan-caspase inhibitor z-VAD-fmk, significantly attenuated piceatannol-induced XIAP down-regulation (Fig. 4D). Ubiquitination analysis revealed that piceatannol-induced proteasomal XIAP degradation was associated with polyubiquitination of the protein, indicating that degradation likely occurs via the ubiquitin-proteasome pathway (Fig. 4E).
Piceatannol Enhances CDDP-dependent Apoptosis by Facilitating Drp1-dependent Mitochondrial Fission
Mitochondria are highly dynamic organelles involved in the two opposing processes of fission and fusion (32–34). Although mitochondrial filamentous tubules form numerous small punctate particles when cells undergo cell death (35, 36), the role of fission in the regulation of drug-induced apoptosis is poorly understood (37, 38). Drp1 is a key regulator of mitochondrial fission and its recruitment to the mitochondria is essential for fission, subsequent cytochrome c release, and the activation of apoptosis (39). To investigate whether mitochondrial fission is a determinant of chemosensitivity in OVCA cells and whether the promotion of apoptosis by piceatannol is in part mediated by changes in mitochondrial morphology, we first examined the influence of CDDP and piceatannol (alone and in combination) on mitochondrial morphology in OV2008. As shown in Fig. 5A, cells were assessed overall as having mitochondria that were of tubular or fragmented morphology. Whereas CDDP (5 μm) or piceatannol (10 μm) alone significantly decreased the number of cells exhibiting tubular mitochondria and increased those with fragmented mitochondria, these responses were markedly exaggerated upon co-treatment of the two compounds (Fig. 5B). We observed that piceatannol treatment (10 μm; 0–6 h) resulted in a rapid and time-dependent dephosphorylation of Drp1 (Fig. 5C), an event that precedes the oligomerization of the protein needed for mitochondrial fission to occur (16). To determine whether mitochondrial fission is at least in part involved in CDDP/piceatannol-induced apoptosis, we examined the influence of the specific Drp1 oligomerization inhibitor mDivi-1 (10 μm) on mitochondrial fission and apoptosis induced by the two agents alone and together. The presence of mDivi-1 significantly rescued the number of cells exhibiting fragmented mitochondria in all treated groups (Fig. 5B). We next investigated whether Drp1-dependent mitochondrial fission and its attenuation by mDivi-1 had any influence on the apoptotic response to CDDP and/or piceatannol. Co-treatment with mDivi-1 reduced caspase-3 activation and apoptosis induced by CDDP and piceatannol alone and in combination (Fig. 5D).
FIGURE 5.

Effects of piceatannol on Drp1-dependent mitochondrial fission and CDDP-induced apoptosis. A, overall mitochondrial morphology (stained with mitochondrial import receptor subunit TOM20, green) was classified as one of two categories: tubular or fragmented. Composite images were obtained using confocal laser microscopy, with at least 16 cross-sectional images spanning the z axis of each cell. The cells were fixed in 4% paraformaldehyde after 4 h treatment, and nuclear regions are indicated in blue (DAPI). B, presence of the specific Drp1 oligomerization inhibitor mDivi-1 (5 μm) reduces the number of fragmented cells induced by CDDP (5 μm) and piceatannol (10 μm) alone or together. Quantification was conducted as described under “Experimental Procedures.” C, effect of piceatannol treatment (10 μm) on levels of phospho-Drp1 (Ser637) over 6 h. D, mDivi-1 significantly but not completely abrogated the influence of picetannol (10 μm) and CDDP (5 μm) on caspase-3 activation and apoptosis after 24 h. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus respective dimethyl sulfoxide (DMSO) control (CTL).
Combination Treatment with Piceatannol and CDDP Increases Mitochondrial Fission and Attenuates Tumor Growth in a Mouse Model of OVCA
To confirm the physiological relevance of the our in vitro observations, we examined the influence of CDDP and piceatannol on OVCA tumor growth in vivo. Over the course of 18 days, we observed that co-treatment with piceatannol (20 mg/kg/day, 5 days/week) and CDDP (1.8 mg/kg/week) significantly reduced tumor growth compared with controls, an effect that was not achieved by either agent alone (the relatively low dose of CDDP for in vivo administration was chosen to avoid excessive cytotoxicity for animals receiving combination treatment; Fig. 6A). Three-way analysis of variance indicated significant effects (p < 0.001) of piceatannol, CDDP, and time and a significant interaction between piceatannol and CDDP (p < 0.05), as evidenced by a greater reduction in tumor volume in the presence of the two compounds. Upon sacrifice, tumors were recovered and immediately weighed (Fig. 6B). Although no significant differences were detected between tumor weights of control mice and those treated with CDDP or piceatannol alone, co-administration of the two compounds caused a statistically significant reduction in tumor weight. Assessment of mitochondrial morphology within the recovered tumors was conducted using immunofluorescence analysis (Fig. 6C). Treatment with CDDP and piceatannol was associated with an increase in mitochondria of fragmented morphology, at the expense of tubular mitochondria. This effect was greatly enhanced in mice receiving the co-treatment, with some tumors appearing to lack tubular mitochondria entirely.
FIGURE 6.
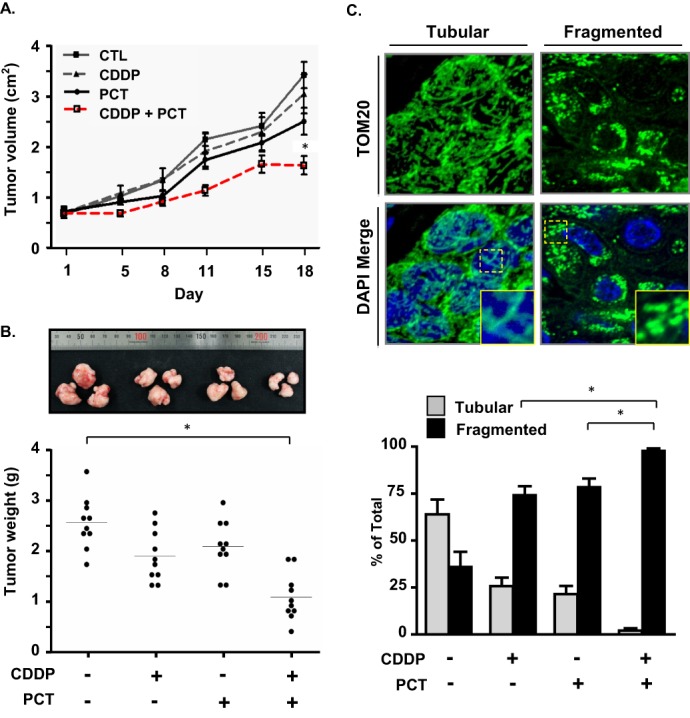
Effects of piceatannol and CDDP treatment on tumor growth in a mouse model of OVCA. A, effect of CDDP (1.8 mg/kg, 1 time/week) and piceatannol (20 mg/kg, 5 times/week) on tumor volume. Tumors were formed by subcutaneous insertion of 1 × 106 OV2008 cells embedded in Matrigel into the hind flanks of athymic nude mice. Tumors were measured over 18 days for the intervals indicated, and volume was calculated using the equation V = π/6(l × h × w). B, measurements of tumor weight on the day of sacrifice. *, p < 0.05. C, effect of CDDP and piceatannol treatment on mitochondrial morphology in recovered tumors. CTL, control.
Piceatannol Enhances CDDP-dependent p53 Activation, XIAP Degradation, and Apoptosis in Vivo
Consistent with our in vitro observations (Fig. 1), tumors recovered from our xenograft studies exhibited higher levels of apoptotic cell death because of combination treatment, as evidenced by TUNEL staining (Fig. 7A). Immunofluorescence analysis of phospho-p53 (Ser-15) (Fig. 7B) revealed expression patterns consistent with our in vitro observations (Fig. 3C). Similarly, piceatannol treatment in combination with CDDP was observed to enhance CDDP-dependent XIAP down-regulation to an extent greater than either agent alone (Fig. 7C). These results further support our hypothesis that piceatannol enhances CDDP-dependent apoptosis in OVCA, at least in part, by regulating key factors related to the p53 tumor suppressor pathway.
FIGURE 7.

Effect of piceatannol and CDDP treatment on p53 activation, XIAP down-regulation, and apoptosis in vivo. A, apoptosis, as examined by immunofluorescent TUNEL staining (yellow), showing enhanced CDDP-dependent apoptosis in the presence of piceatannol. B, effect of piceatannol on nuclear Ser(P)15-p53 localization. Images are representative of overall staining patterns present in three separate tumors. C, piceatannol enhanced CDDP-dependent XIAP (red) down-regulation. Cell nuclei were stained with DAPI (blue). Fluorescence intensity in 200 cells/replicate was quantified using the ImageJ software package. **, p < 0.01; ***, p < 0.001 of combination treated group versus respective CDDP-only treated group. CTL, control.
DISCUSSION
Novel strategies that improve chemosensitivity while minimizing undesirable side effects are needed to improve quality of life and therapeutic outcomes for OVCA patients. In the present study, we investigated the effects of the dietary stilbenoid piceatannol on CDDP sensitivity in OVCA cells in vitro and in vivo, with the goal of reducing therapeutic CDDP concentrations needed to induce apoptosis. With this outcome in mind, we selected relatively low doses of both piceatannol and CDDP. Physiologically relevant concentrations of resveratrol are often significantly lower than those used for in vitro studies. Estimates range from as low as 50 nm (40) to 0.1 μm, with 1–100 μm concentrations described as “supraphysiological” (41). Clinical studies on resveratrol bioavailability using human subjects has shown that despite metabolization into glucuronide and sulfate conjugates coupled to renal elimination, oral administration of resveratrol can achieve 0.1–6 μm blood concentrations for several hours, without any apparent side effects (42). We thus sought a balance between concentrations high enough to detect statistical changes caused by treatment across different cell lines, with a dose low enough to maintain physiological relevance. We observed that piceatannol enhances the CDDP response in OVCA by promoting p53 stability and activation, mitochondrial fission, and XIAP degradation (Fig. 8).
FIGURE 8.
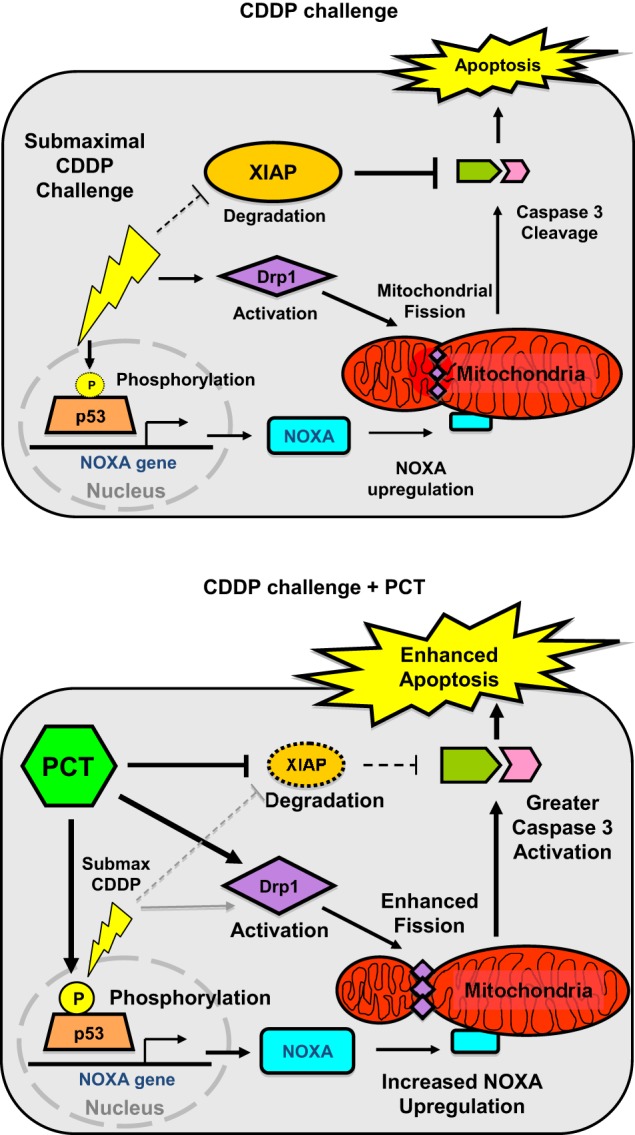
Hypothetical model illustrating multiple actions of piceatannol during CDDP treatment in chemosensitive ovarian cancer cells. Co-treatment with piceatannol leads to Drp1-dependent mitochondrial fission and XIAP down-regulation and enhanced p53-dependent apoptosis.
We have previously identified mechanisms responsible for a number of factors that regulate chemosensitivity in OVCA (8, 11, 34). The tumor suppressor p53 plays a central role in apoptosis, and its activation is critical for sensitivity to genotoxic agents like CDDP (27). Our results show that the increased apoptotic responses to both CDDP and piceatannol appear to be at least partially p53-dependent. The effects of CDDP on cell cycle arrest in cancer cells has been described extensively and involves S phase to early G2 arrest as a result of DNA damage (43). Similarly, we observed that piceatannol induces G2 arrest in chemosensitive OV2008 cells, as well as S phase arrest in chemoresistant C13 cells (data not shown).
CDDP-induced DNA damage activates p53 and the subsequent transcription of gene targets mediated by phosphorylation at Ser15 (44), including NOXA. NOXA targets the mitochondrial outer membrane, where it plays a prominent role in membrane permeabilization (45). Other transcriptional regulators of NOXA also exist, including the transcription factor FoxO1. Although NOXA activation appears to have a minor p53-independent mechanism as seen in the p53-mutant cells (A2780cp), RNA silencing revealed that piceatannol exerts at least some of its effects via p53 (Fig. 3).
XIAP is a potent inhibitor of caspases and apoptotic cell death (46) and is a major determinant of chemoresistance in OVCA (7). As a possible drug target, much interest has been generated in the development of compounds (e.g., SMAC mimetics) that inhibit XIAP activity (47). We chose to focus on piceatannol's effects on XIAP for a number of reasons. XIAP is the most potent of the IAP family and is the only member that inhibits caspases 3, 7 and 9 (48, 49). In addition, the effects of piceatannol on Bcl-2 expression have been characterized in a number of studies, whereas the effects of piceatannol on XIAP have not previously been described (50). Our results demonstrate that piceatannol down-regulates XIAP content in OVCA cells in vitro, as well as in OVCA xenografts in vivo to an extent greater than CDDP alone, despite the fact that CDDP induces higher levels of apoptosis. This may suggest that at least one mechanism of piceatannol action in CDDP sensitivity is to down-regulate apoptotic inhibitors such as XIAP to “clear the way” for increased caspase activation upon genotoxic challenge.
The role of the mitochondrial fission in apoptotic induction has long been established, but only recently have functional food agents been identified that can influence mitochondrial morphology toward this end (51). In the present study, we show for the first time that piceatannol induces Drp1-mediated mitochondrial fission, contributing to higher apoptotic capacity when the cells are treated in combination with CDDP. Our in vivo observations revealed that piceatannol not only decreases ovarian tumor volume and weight in combination with CDDP, but also appears to exert a significant influence on the same factors identified in vitro. We acknowledge that the implantation of human tumor grafts or fresh samples of human ovarian cancer would have added further translational relevance to our findings. However, taking into account practical considerations, we alternatively sought to further test our hypotheses supported by the in vitro observations and thus chose the same cell line for the in vivo experiments.
The broad mechanism of CDDP action in cross-linking DNA causes side effects that range from moderate (including alopecia, nausea, and erythema) to severe (nephrotoxicity, ototoxicity, myelotoxicity, and hemolytic anemia) (52, 53). These factors limit its applicable dose range. We carefully selected our cisplatin dose based on reports in the literature using similar study designs (54, 55) and deliberately selected a slightly lower dose to avoid excess toxicity from combination treatment. In fact, at the dose used in the current study, combination treatment significantly reduced tumor volume and burden, whereas neither CDDP nor PCT alone had any significant effects on these responses.
Although piceatannol influences several major determinants of apoptosis in OVCA, its precise molecular target(s) remain to be determined. Prior studies indicate that piceatannol can bind to and inhibit receptor tyrosine kinases, although the downstream consequences of such inhibition are not fully understood (56). Recent studies have shown that piceatannol can inhibit PI3K, through direct physical binding (22); however, we observed that piceatannol treatment had no effect on Akt activation in OV2008 cells (data not shown). Piceatannol has been reported to act as a receptor antagonist in breast cancer cells, via its estrogen-like activity (57). However, the mode of action of piceatannol is likely to be different in ovarian cancer cell lines because breast cancers (and uterine cancers) are often estrogen-sensitive, whereas ovarian and cervical cancers are relatively estrogen-insensitive (58). The Drp1-dependent increases in mitochondrial fission observed point to the possible involvement of its regulator, the calcium-dependent phosphatase calcineurin. If and how this enzyme is involved in piceatannol action remains to be determined.
Global epidemiological data demonstrates that OVCA risk follows regional patterns, suggesting that cultural and lifestyle factors are at least partially responsible (59). A major lifestyle factor is diet, with multiple lines of evidence pointing to a link between a diet rich in phytochemicals and reduced overall cancer risk (60). Resveratrol has been reported to inhibit the growth of a wide range of cancer cells, including OVCA (18). However, convincing evidence for the therapeutic anti-cancer activity of resveratrol in clinical trials remains elusive and underlines the need for further aspects to be taken into consideration (20). Further studies are needed into the pharmacodynamic properties of piceatannol and how the compound might be modified for higher efficacy.
In conclusion, we have shown for the first time that piceatannol increases CDDP sensitivity in OVCA both in vitro and in vivo by modulating several key determinants of chemoresistance (Fig. 7). Co-treatment with piceatannol in both chemoresistant and chemosensitive cells effectively reduces the concentrations of CDDP needed for the equivalent effect of higher doses. Functional food compounds such as piceatannol represent attractive options for the development of novel treatment strategies for OVCA, because of their long history of human consumption and relative ease of manufacture.
Acknowledgments
We thank Mu-Gyong Han, Wha-Sun Lim, and Jong-Hee Choi for technical assistance and Art Michalak for critical reading of the manuscript.
This work was supported by World Class University Program Grant R31-10056 through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology and Canadian Institutes of Health Research Grant M0P-126144.
- OVCA
- ovarian cancer
- CDDP
- cisplatin
- PARP
- poly ADP-ribose polymerase
- PCT
- piceatannol
- z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- XIAP
- X-linked inhibitor of apoptosis protein.
REFERENCES
- 1. Cancer Facts & Figures (2012) American Cancer Society, Atlanta [Google Scholar]
- 2. Harries M., Gore M. (2002) Part II: Chemotherapy for epithelial ovarian cancer-treatment of recurrent disease. Lancet Oncol. 3, 537–545 [DOI] [PubMed] [Google Scholar]
- 3. Covens A., Carey M., Bryson P., Verma S., Fung Kee Fung M., Johnston M. (2002) Systematic review of first-line chemotherapy for newly diagnosed postoperative patients with stage II, III, or IV epithelial ovarian cancer. Gynecol. Oncol. 85, 71–80 [DOI] [PubMed] [Google Scholar]
- 4. Loehrer P. J., Einhorn L. H. (1984) Drugs five years later. Cisplatin. Ann. Intern. Med. 100, 704–713 [DOI] [PubMed] [Google Scholar]
- 5. Canpolat C., Pearson P., Jaffe N. (1994) Cisplatin-associated hemolytic uremic syndrome. Cancer 74, 3059–3062 [DOI] [PubMed] [Google Scholar]
- 6. Fraser M., Leung B., Jahani-Asl A., Yan X., Thompson W. E., Tsang B. K. (2003) Chemoresistance in human ovarian cancer. The role of apoptotic regulators. Reprod. Biol. Endocrinol. 1, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sasaki H., Sheng Y., Kotsuji F., Tsang B. K. (2000) Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res. 60, 5659–5666 [PubMed] [Google Scholar]
- 8. Yang X., Fraser M., Moll U. M., Basak A., Tsang B. K. (2006) Akt-mediated cisplatin resistance in ovarian cancer. Modulation of p53 action on caspase-dependent mitochondrial death pathway. Cancer Res. 66, 3126–3136 [DOI] [PubMed] [Google Scholar]
- 9. Brown J. M., Wouters B. G. (1999) Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 59, 1391–1399 [PubMed] [Google Scholar]
- 10. Abedini M. R., Qiu Q., Yan X., Tsang B. K. (2004) Possible role of FLICE-like inhibitory protein (FLIP) in chemoresistant ovarian cancer cells in vitro. Oncogene. 23, 6997–7004 [DOI] [PubMed] [Google Scholar]
- 11. Woo M. G., Xue K., Liu J., McBride H., Tsang B. K. (2012) Calpain-mediated processing of p53-associated Parkin-like cytoplasmic protein (PARC) affects chemosensitivity of human ovarian cancer cells by promoting p53 subcellular trafficking. J. Biol. Chem. 287, 3963–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., Tokino T., Taniguchi T., Tanaka N. (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 [DOI] [PubMed] [Google Scholar]
- 13. Chai J., Du C., Wu J.-W., Kyin S., Wang X., Shi Y. (2000) Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406, 855–862 [DOI] [PubMed] [Google Scholar]
- 14. Yang J., Liu X., Bhalla K., Kim C. N., Ibrado A. M., Cai J., Peng T. I., Jones D. P., Wang X. (1997) Prevention of Apoptosis by Bcl-2. Release of cytochrome c from mitochondria blocked. Science 275, 1129–1132 [DOI] [PubMed] [Google Scholar]
- 15. Youle R. J., Karbowski M. (2005) Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 6, 657–663 [DOI] [PubMed] [Google Scholar]
- 16. Cribbs J. T., Strack S. (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cereghetti G. M., Stangherlin A., Martins de Brito O., Chang C. R., Blackstone C., Bernardi P., Scorrano L. (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jang M., Cai L., Udeani G. O., Slowing K. V., Thomas C. F., Beecher C. W., Fong H. H., Farnsworth N. R., Kinghorn A. D., Mehta R. G., Moon R. C., Pezzuto J. M. (1997) Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275, 218–220 [DOI] [PubMed] [Google Scholar]
- 19. Rezk Y. A., Balulad S. S., Keller R. S., Bennett J. A. (2006) Use of resveratrol to improve the effectiveness of cisplatin and doxorubicin. Study in human gynecologic cancer cell lines and in rodent heart. Am. J. Obstet. Gynecol. 194, e23–e26 [DOI] [PubMed] [Google Scholar]
- 20. Aggarwal B. B., Bhardwaj A., Aggarwal R. S., Seeram N. P., Shishodia S., Takada Y. (2004) Role of resveratrol in prevention and therapy of cancer. Preclinical and clinical studies. Anticancer Res. 24, 2783–2840 [PubMed] [Google Scholar]
- 21. Potter G. A., Patterson L. H., Wanogho E., Perry P. J., Butler P. C., Ijaz T., Ruparelia K. C., Lamb J. H., Farmer P. B., Stanley L. A., Burke M. D. (2002) The cancer preventative agent resveratrol is converted to the anticancer agent piceatannol by the cytochrome P450 enzyme CYP1B1. Br. J. Cancer. 86, 774–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choi K. H., Kim J.-E., Song N. R., Son J. E., Hwang M. K., Byun S., Kim J. H., Lee K. W., Lee H. J. (2010) Phosphoinositide 3-kinase is a novel target of piceatannol for inhibiting PDGF-BB-induced proliferation and migration in human aortic smooth muscle cells. Cardiovasc. Res. 85, 836–844 [DOI] [PubMed] [Google Scholar]
- 23. Kwon J. Y., Seo S. G., Heo Y.-S., Yue S., Cheng J.-X., Lee K. W., Kim K. H. (2012) Piceatannol, a natural polyphenolic stilbene, inhibits adipogenesis via modulation of mitotic clonal expansion and insulin receptor-dependent insulin signaling in the early phase of differentiation. J. Biol. Chem. 287, 11566–11578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wolter F., Clausnitzer A., Akoglu B., Stein J. (2002) Piceatannol, a natural analog of resveratrol, inhibits progression through the S phase of the cell cycle in colorectal cancer cell lines. J. Nutr. 132, 298–302 [DOI] [PubMed] [Google Scholar]
- 25. Abedini M. R., Muller E. J., Brun J., Bergeron R., Gray D. A., Tsang B. K. (2008) Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells. Cancer Res. 68, 4511–4517 [DOI] [PubMed] [Google Scholar]
- 26. Shaw T. J., Senterman M. K., Dawson K., Crane C. A., Vanderhyden B. C. (2004) Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 10, 1032–1042 [DOI] [PubMed] [Google Scholar]
- 27. Ali A. Y., Abedini M. R., Tsang B. K. (2012) The oncogenic phosphatase PPM1D confers cisplatin resistance in ovarian carcinoma cells by attenuating checkpoint kinase 1 and p53 activation. Oncogene 31, 2175–2186 [DOI] [PubMed] [Google Scholar]
- 28. Asselin E., Wang Y., Tsang B. K. (2001) X-linked inhibitor of apoptosis protein activates the phosphatidylinositol 3-kinase/Akt pathway in rat granulosa cells during follicular development. Endocrinology 142, 2451–2457 [DOI] [PubMed] [Google Scholar]
- 29. Abedini M. R., Muller E. J., Bergeron R., Gray D. A., Tsang B. K. (2010) Akt promotes chemoresistance in human ovarian cancer cells by modulating cisplatin-induced, p53-dependent ubiquitination of FLICE-like inhibitory protein. Oncogene 29, 11–25 [DOI] [PubMed] [Google Scholar]
- 30. Lutz A. K., Exner N., Fett M. E., Schlehe J. S., Kloos K., Lämmermann K., Brunner B., Kurz-Drexler A., Vogel F., Reichert A. S., Bouman L., Vogt-Weisenhorn D., Wurst W., Tatzelt J., Haass C., Winklhofer K. F. (2009) Loss of Parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chou T.-C., Talalay P. (1984) Quantitative analysis of dose-effect relationships. The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22, 27–55 [DOI] [PubMed] [Google Scholar]
- 32. Cerveny K. L., Tamura Y., Zhang Z., Jensen R. E., Sesaki H. (2007) Regulation of mitochondrial fusion and division. Trends Cell Biol. 17, 563–569 [DOI] [PubMed] [Google Scholar]
- 33. Chan D. C. (2006) Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22, 79–99 [DOI] [PubMed] [Google Scholar]
- 34. Detmer S. A., Chan D. C. (2007) Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879 [DOI] [PubMed] [Google Scholar]
- 35. Lee Y. J., Jeong S.-Y., Karbowski M., Smith C. L., Youle R. J. (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 15, 5001–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheridan C., Delivani P., Cullen S. P., Martin S. J. (2008) Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome c release. Mol. Cell. 31, 570–585 [DOI] [PubMed] [Google Scholar]
- 37. Alirol E., James D., Huber D., Marchetto A., Vergani L., Martinou J.-C., Martinou J. C., Scorrano L. (2006) The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol. Biol. Cell 17, 4593–4605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu T., Fox R. J., Burwell L. S., Yoon Y. (2005) Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J. Cell Sci. 118, 4141–4151 [DOI] [PubMed] [Google Scholar]
- 39. Bras M., Yuste V. J., Roué G., Barbier S., Sancho P., Virely C., Rubio M., Baudet S., Esquerda J. E., Merle-Béral H., Sarfati M., Susin S. A. (2007) Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Mol. Cell. Biol. 27, 7073–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takahashi S., Nakashima Y. (2012) Repeated and long-term treatment with physiological concentrations of resveratrol promotes NO production in vascular endothelial cells. Br. J. Nutr. 107, 774–780 [DOI] [PubMed] [Google Scholar]
- 41. Nicholson S. K., Tucker G. A., Brameld J. M. (2008) Effects of dietary polyphenols on gene expression in human vascular endothelial cells. Proc. Nutr. Soc. 67, 42–47 [DOI] [PubMed] [Google Scholar]
- 42. Amiot M. J., Romier B., Dao T.-M., Fanciullino R., Ciccolini J., Burcelin R., Pechere L., Emond C., Savouret J. F., Seree E. (2013) Optimization of trans-resveratrol bioavailability for human therapy. Biochimie 95, 1233–1238 [DOI] [PubMed] [Google Scholar]
- 43. Siddik Z. H. (2003) Cisplatin. Mode of cytotoxic action and molecular basis of resistance. Oncogene 22, 7265–7279 [DOI] [PubMed] [Google Scholar]
- 44. Fraser M., Bai T., Tsang B. K. (2008) Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int. J. Cancer. 122, 534–546 [DOI] [PubMed] [Google Scholar]
- 45. Shibue T., Takeda K., Oda E., Tanaka H., Murasawa H., Takaoka A., Morishita Y., Akira S., Taniguchi T., Tanaka N. (2003) Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17, 2233–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deveraux Q. L., Roy N., Stennicke H. R., Van Arsdale T., Zhou Q., Srinivasula S. M., Alnemri E. S., Salvesen G. S., Reed J. C. (1998) IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 17, 2215–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Probst B. L., Liu L., Ramesh V., Li L., Sun H., Minna J. D., Wang L. (2010) Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-α-dependent manner. Cell Death Differ. 17, 1645–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suzuki Y., Nakabayashi Y., Nakata K., Reed J. C., Takahashi R. (2001) X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes. J. Biol. Chem. 276, 27058–27063 [DOI] [PubMed] [Google Scholar]
- 49. Shiozaki E. N., Chai J., Rigotti D. J., Riedl S. J., Li P., Srinivasula S. M., Alnemri E. S., Fairman R., Shi Y. (2003) Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell. 11, 519–527 [DOI] [PubMed] [Google Scholar]
- 50. Alas S., Bonavida B. (2001) Rituximab inactivates signal transducer and activation of transcription 3 (STAT3) activity in B-non-Hodgkin's lymphoma through inhibition of the interleukin 10 autocrine/paracrine loop and results in down-regulation of Bcl-2 and sensitization to cytotoxic drugs. Cancer Res. 61, 5137–5144 [PubMed] [Google Scholar]
- 51. Raj L., Ide T., Gurkar A. U., Foley M., Schenone M., Li X., Tolliday N. J., Golub T. R., Carr S. A., Shamji A. F., Stern A. M., Mandinova A., Schreiber S. L., Lee S. W. (2011) Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475, 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52. Hesketh P. J., Batchelor D., Golant M., Lyman G. H., Rhodes N., Yardley D. (2004) Chemotherapy-induced alopeci. Psychosocial impact and therapeutic approaches. Support. Care Cancer 12, 543–549 [DOI] [PubMed] [Google Scholar]
- 53. Barabas K., Milner R., Lurie D., Adin C. (2008) Cisplatin. A review of toxicities and therapeutic applications. Vet. Comp. Oncol. 6, 1–18 [DOI] [PubMed] [Google Scholar]
- 54. Ghamande S., Hylander B. L., Oflazoglu E., Lele S., Fanslow W., Repasky E. A. (2001) Recombinant CD40 ligand therapy has significant antitumor effects on CD40-positive ovarian tumor xenografts grown in SCID mice and demonstrates an augmented effect with cisplatin. Cancer Res. 61, 7556–7562 [PubMed] [Google Scholar]
- 55. Plumb J. A., Strathdee G., Sludden J., Kaye S. B., Brown R. (2000) Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 60, 6039–6044 [PubMed] [Google Scholar]
- 56. Geahlen R. L., McLaughlin J. L. (1989) Piceatannol (3,4,3′,5′-tetrahydroxy-trans-stilbene) is a naturally occurring protein-tyrosine kinase inhibitor. Biochem. Biophys. Res. Commun. 165, 241–245 [DOI] [PubMed] [Google Scholar]
- 57. Maggiolini M., Recchia A. G., Bonofiglio D., Catalano S., Vivacqua A., Carpino A., Rago V., Rossi R., Andò S. (2005) The red wine phenolics piceatannol and myricetin act as agonists for estrogen receptor α in human breast cancer cells. J. Mol. Endocrinol. 35, 269–281 [DOI] [PubMed] [Google Scholar]
- 58. Biggar R. J. (2012) Molecular pathways. Digoxin use and estrogen-sensitive cancers. Risks and possible therapeutic implications. Clin. Cancer Res. 18, 2133–2137 [DOI] [PubMed] [Google Scholar]
- 59. Rose D. P., Boyar A. P., Wynder E. L. (1986) International comparisons of mortality rates for cancer of the breast, ovary, prostate, and colon, and per capita food consumption. Cancer. 58, 2363–2371 [DOI] [PubMed] [Google Scholar]
- 60. Surh Y.-J. (2003) Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 3, 768–780 [DOI] [PubMed] [Google Scholar]