

Background: Galectins are increased in astrocytes of patients with multiple sclerosis.
Results: TNF up-regulates galectin-9 in primary astrocytes via the TNFR1/JNK/c-Jun pathway and can induce apoptosis of encephalitogenic T-cells.
Conclusion: Astrocytes up-regulate galectin-9 in response to the proinflammatory cytokine TNF.
Significance: Astrocyte-derived galectin-9 may function to restrict encephalitogenic T-cell-mediated inflammation in the CNS.
Keywords: Astrocytes, Galectin, Jun N-terminal Kinase (JNK), Multiple Sclerosis, Neuroinflammation, Tumor Necrosis Factor (TNF), Galectin-9
Abstract
Demyelination and axonal damage in multiple sclerosis (MS) are thought to be a consequence of inflammatory processes that are perpetuated by activated glia and infiltrating leukocytes. Galectin-9 is a β-galactoside binding lectin capable of modulating immune responses and appears to be up-regulated in MS. However, its role in the pathogenesis of MS has yet to be determined. Here, we report that proinflammatory cytokines induce galectin-9 (Gal-9) expression in primary astrocytes and the mechanism by which TNF up-regulates Gal-9. Astrocytes did not express Gal-9 under basal conditions nor did IL-6, IL-10, or IL-13 trigger Gal-9 expression. In contrast, IL-1β, IFN-γ, and particularly TNF up-regulated Gal-9 in astrocytes. TNF-induced Gal-9 expression was dependent on TNF receptor 1 (TNFR1) as TNF failed to induce Gal-9 in TNFR1−/− astrocytes. Blockade of the JNK MAP kinase pathway with the JNK inhibitor SP600125 abrogated TNF-induced Gal-9, whereas p38 and MEK inhibitors had minimal effects. Furthermore, specific knockdown of c-Jun via siRNA in astrocytes before TNF treatment greatly suppressed Gal-9 transcription, suggesting that TNF induces astroglial Gal-9 through the TNF/TNFR1/JNK/cJun signaling pathway. Finally, utilizing astrocytes from Lgals9 mutant (Gal-9−/−) mice as well as a myelin basic protein-specific Tim-3+ encephalitogenic T-cell clone (LCN-8), we found that conditioned medium from TNF-stimulated Gal-9+/+ but not Gal-9−/− astrocytes increased the percentage of apoptotic encephalitogenic T-cells. Together, our results suggest that Gal-9 is induced in astrocytes by TNF via the JNK/c-Jun pathway and that astrocyte-derived Gal-9 may function as an immunoregulatory protein in response to ongoing neuroinflammation.
Introduction
Multiple sclerosis (MS)2 is a central nervous system (CNS) disease that is typified by both demyelination and the progressive decline and eventual loss of neuronal function. Although the initial cause(s) of MS is not yet known, histopathological examination indicates that the symptoms of MS are induced and perpetuated by an aberrant inflammatory reaction that most likely includes the activation of resident glia as well as the infiltration of lymphocytes into the parenchyma from the circulation (1). Although leukocyte infiltration into the CNS as the initiating pathological event in MS lesion formation has been debated (2, 3), the involvement of lymphocytes in the pathogenesis of MS is nearly indisputable, especially when considering the effectiveness of immunomodulatory therapies, such as natalizumab (4–6) and rituximab (7, 8) which limit lymphocyte function and/or trafficking to the CNS.
Astrocytes are the predominant cells in the CNS and are activated in many neurological disorders (9) including MS. As a potent source of immunologically relevant cytokines and chemokines, they play a critical role in modulating CNS immune and inflammatory responses. However, how astrocytes communicate with microglia, the resident macrophages in the CNS, as well as infiltrating lymphocytes including T-cells remains incompletely understood.
Galectin-9 (Gal-9) is a β-galactoside binding lectin (10) capable of influencing both innate and adaptive immunity in part by signaling through receptor Tim-3 present on activated Th1 and Th17 cells, dendritic cells, neutrophils, and microglia (11–13). The involvement of Gal-9 in modulating the neuroinflammatory events in MS are supported by several lines of evidence. First, Gal-9 expression is increased in MS lesions (13, 14). Second, the Gal-9 receptor, Tim-3, is dysregulated on T-cells isolated from the circulation of MS patients, thereby rendering the T-cells less susceptible to the immunoregulatory outcomes after Gal-9 ligation (15). Third, MS patients receiving either IFN-β or glatiramer acetate treatment display partial restoration of Tim-3 function, strongly suggesting that the Tim-3/Gal-9 pathway is a potential target for therapeutic intervention in MS (15). Finally, treatment of mice with a Tim-3-specific antibody before induction of experimental autoimmune encephalomyelitis (EAE), an animal model of MS, results in exacerbated disease as characterized by increased clinical symptoms and neuroinflammation (12). Interestingly, the activation of this pathway in monocytes, dendritic cells, and likely microglial cells, potentiates innate immune responses (13) and thus may potentially modulate CNS inflammation. However, the mechanism underlying Gal-9 up-regulation in the CNS and the consequences of its up-regulation remain elusive.
In this study we identified the molecular mechanism by which Gal-9 is up-regulated in primary astrocytes. We found that Gal-9 was up-regulated in astrocytes after stimulation with the proinflammatory cytokines IL-1β, IFN-γ, and particularly TNF but not with anti-inflammatory cytokines IL-6, IL-10, and IL-13. TNF-induced up-regulation of Gal-9 was mediated through the activation of TNFR1, JNK, and c-Jun. Conditioned media from TNF-stimulated Gal-9+/+ but not Gal-9−/− astrocytes promoted apoptosis of encephalitogenic T-cells in vitro. Together, our data reveal a potentially important role for astrocytes in negatively regulating T-cell responses and concurrently implicate a mechanism by which Gal-9 may contribute to the resolution of adaptive immunity within the CNS.
EXPERIMENTAL PROCEDURES
Mice and Rats
Primary cells (mono-, co-, and mixed glial cultures) were isolated from P1–2 Sprague-Dawley rat pups (Harlan, Houston TX). Mouse cells (mono- and co-cultures) were isolated from wild type (C57BL/6J) or TNFR1−/− mice (The Jackson Laboratory, Bar Harbor, ME). Lgals9 mutant (Gal-9−/−) mice were obtained from the Consortium for Functional Glycomics and had been backcrossed four times to C57BL/6 background. The Gal-9 EGFP mice (Lgals9-EGFP) JF66Gsat/Mmucd strain) were reconstituted from MMRRC. Splenocytes were isolated from SJL mice (Harlan). All animals were housed under constant 12-h light/dark cycles in covered cages and fed with a standard rodent diet ad libitum. The experimental procedures described herein were approved by the Institutional Animal Care and Use Committee and were performed in accordance with guidelines of the National Institutes of Health.
Cell Cultures
Astrocytes, microglia, oligodendrocytes, and mixed glia were prepared by the differential shaking method as described previously (16, 17). In brief, forebrains of 1–2-day-old Sprague-Dawley rats or mice were aseptically dissected. After removal of meninges the brains were minced then incubated at 37 °C for 15 min in Hanks' balanced salt solution containing DNase I (10 μg/ml) and trypsin (0.01%). The cells were washed twice with Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin (D10S) and then filtered through a 70-μm sieve. Cells were plated onto poly-d-lysine-coated T75 flasks or 24-well plates and grown for 7–10 days with media changed every other day. Subsequently, flasks were placed on a rotary shaker, and microglia were isolated after shaking at 200 rpm for 1 h. After the removal of microglia the flasks were shaken again overnight to separate oligodendrocytes from the astrocyte layer. Oligodendrocytes were enriched from residual microglia by pre-plating the cell suspension onto Petri dishes and cultured with serum-free Basal Defined Medium (DMEM containing 0.1% bovine serum albumin, 50 μg/ml human apotransferrin, 5 μg/ml insulin, 30 nm sodium selenite, 10 nm D-biotin, and 10 nm hydrocortisone) supplemented with 10 ng/ml FGF-basic and 10 ng/ml PDGF-AA (PeproTech). Astrocytes were further enriched by subculturing 1–2 times and contained less than 2 and 9% contaminating microglia for rat and mouse cultures, respectively.
Neurons were isolated from embryonic day 16 rat brains as described previously (18). Briefly, meninges were removed from fetal forebrains, and the tissue was digested with papain (20 unit/ml) in Hanks' balanced salt solution containing 1% penicillin-streptomycin, 10 mm HEPES, 0.8 mg/ml l-cysteine, pH 7.4, for 6 min at 37 °C. After washing, cell suspension was passed through a 70-μm sieve and plated at a density of 1.2 × 105 cells per cm2 in NBB27 medium (Neurobasal medium with 2% B27, 1 mm glutamine, and 1% penicillin/streptomycin) containing 25 μm glutamic acid. On day in vitro 4, the medium was changed to NBB27 without glutamic acid. For highly enriched neurons, mitotic inhibitor 5-fluorodeoxyuridine (10 μm) was added at day in vitro 2 to inhibit glial cell proliferation. Two days later the medium was changed back to NBB27. The neuron cultures underwent a total of 3 cycles of 5-fluorodeoxyuridine treatment and were cultured in NBB27 medium for 2–3 weeks. The purity of our primary cultures was characterized previously (16, 19, 20) and assessed again from two independent experiments in this study by immunohistochemistry using antibodies specific for astrocytes (GFAP), microglia (Iba-1), oligodendrocytes (O4), and neurons (MAP2) and DAPI counterstain. Astrocyte cultures were greater than 94% GFAP+, oligodendrocytes were greater than 92% O4+, and microglia cultures were greater than 95% Iba-1+. Because of the 5-fluorodeoxyuridine treatment to kill mitotic glial cells, the neuron cultures contained much DAPI+ debris. None of nuclei debris was positive for Iba-1 or O4. The only contaminating live cells found in the neuronal cultures were a few type II astrocytes, and the neuron cultures were consistently greater than 90% MAP2+.
Encephalitogenic T-cell Clone
LNC-8 cells are a myelin basic protein (MBP)-specific T-cell clone that was derived from the popliteal lymph nodes of an SJL mouse immunized with porcine MBP and capable of inducing EAE upon passive transfer (22). LNC-8 cells were cultured in RPMI 1640 medium containing 10% FBS, 4 mm l-glutamine, and 1 mm sodium pyruvate, 50 μm β-mercaptoethanol, and 1% penicillin and streptomycin. Upon thawing, cells were fed 20 units/ml IL-2 (eBioscience, San Diego, CA). After 5 days of culture, irradiated syngeneic feeder cells were added, and cells were stimulated with bovine MBP (20 μg/ml; Sigma) and IL-2 (20 units/ml; eBioscience). Cells were subsequently given IL-2 once per week and stimulated with antigen and feeders biweekly.
Cytokine Stimulation and Cell Viability Assay
For transcriptional analysis, astrocytes were stimulated with increasing concentrations of species-specific recombinant TNF, IL-1β, IFN-γ, IL-6, IL-10, or IL-13 (R&D Systems, Minneapolis, MN) for 7 h at 37 °C. Where indicated, cell viability after cytokine stimulation was determined by measuring lactate dehydrogenase activity from culture supernatants according to the manufacturer's instructions (Roche Applied Science).
Oxygen Glucose Deprivation
Mixed glial cultures were washed three times with Basal Defined Medium media devoid of glucose, placed in a hypoxia chamber (Billups-Rothenberg, Del Mar, CA), and subjected to hypoxic conditions (95% N2 and 5% CO2) for 2 h at 37 °C. Sister cultures in the normoxia group were washed with normal Basal Defined Medium medium. After 2 h cells from both conditions were either left untreated or were stimulated with LPS (1.0 μg/ml; O111:B4, Sigma) for at least 12 h. Supernatants were sampled and stored at −80 °C until TNF levels could be determined by ELISA. RNA was isolated to examine Gal-9 expression determined by RT-PCR.
RNA Extraction and RT-PCR
RNA extraction and RT-PCR was conducted as described previously (17) with slight modifications. In brief, RNA was extracted using Qiagen RNeasy kits (Qiagen; Valencia, CA); contaminating DNA was removed by digestion with DNase I, and RNA was reverse-transcribed using a Promega AMV kit according to the manufacturer's instructions (Promega, Madison, WI). To ensure no residual DNA was present, samples that were reverse-transcribed in the absence of reverse transcriptase were then tested for amplification. Galectin-9 was amplified from 100 ng of cDNA using the following primers: rat Gal-9, forward (GGCATACCCCACCCCAGCCT) and reverse (CAGGCAGGCTTCGCTCCTCG); mouse Gal-9 forward (GCAGAACGGACAGTGGGGGC) and reverse (GCAGCCGGAGACAGCGATGG); β-actin, forward (AGACTTCGAGCAGGAGATGG) and reverse (CCATCATGAAGTGTGACGTTG). For quantitative PCR, 40 ng of cDNA per reaction was amplified using SYBR Green according to the manufacturer's instructions using a 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA). All samples were run in duplicate or triplicate. Gene expression was normalized to β-actin, and -fold expression was calculated using the formula 2−ΔΔCt.
Western Blot
The activation of p38 MAPK, ERK1/2, and JNK pathways after cell treatments was determined by Western blot. Briefly, cells were lysed on ice in lysis buffer (1% Triton X-100, 20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 10 mm Na2P2O7, 10 mm NaF, 1 mm Na3VO4, 1 mm PMSF, proteinase inhibitor mixture I (Roche Applied Science), and sonicated. Cellular debris was removed by centrifugation at 12,000 × g for 15 min at 4 °C, and protein concentration of the cell lysates was determined by the Bradford assay. Total protein (30 μg) was separated on 10% polyacrylamide gels by SDS-PAGE, transferred to PVDF membranes, blocked with TBST (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% Tween 20) containing 5% nonfat dry milk, and probed with antibodies against P-p38 MAPK (rabbit, dilution 1/1000), p38 MAPK (rabbit, 1/1000), P-ERK1/2 (rabbit, 1/2000), ERK1/2 (rabbit; 1/1000), P-JNK (rabbit, 1/1000), JNK (rabbit, 1/1000), P-c-Jun (rabbit, 1/1000), c-Jun (rabbit, 1/1000; all from Cell Signaling Technology, Danvers, MA) or anti-β-actin (mouse 1/8000; Sigma) overnight at 4 °C. Galectin-9 expression was also determined by Western blot. Tissue samples isolated from Gal-9-EGFP, Gal-9+/+, Gal-9+/−, or Gal-9−/− mice were homogenized on ice in 1 ml of lysis buffer. After sonication, the cellular debris was removed by centrifugation, and total protein was quantitated and separated as above. After transfer, the PVDF membranes were blocked with TBST containing 5% horse serum, and galectin-9 expression was determined by probing with anti-GFP (chicken, 1/1000, Invitrogen) or anti-Gal-9 (goat, 1/1000, R&D Systems) overnight at 4 °C. After washing, membranes were incubated for 1 h with HRP-conjugated secondary antibodies and visualized by chemiluminescence using the West Pico ECL reagent (Thermo Scientific, Rockford, IL). Western blot images were acquired with a Bio-Rad Chemidoc XRS gel documentation system and quantified with Quantity One software.
Small Interfering RNA
Astrocytes were seeded into poly-d-lysine-coated 6-well plates at a density of 2 × 105 per well. After reaching 60% confluency, the culture media (D10S) was changed to one devoid of antibiotics. The following day astrocytes were transfected with a nonspecific (NS) siRNA control (20 nm; Ambion; catalog no. 4390843) or target-specific siRNAs (c-Jun, 20 nm; sense (GGCACAGCUUAAACAGAAAtt) and antisense (UUUCUGUUUAAGCUGUGCCac); p38 MAPK, 20 nm sense (GGACCUCCUUAUAGACGAAtt) and antisense (UUCGUCUAUAAGGAGGUCCct); Ambion, Austin, TX) using Lipofectamine 2000 in Opti-MEM medium according to the manufacturer's instructions (Invitrogen). After incubating for 6 h at 37 °C, the medium was changed to normal growth medium (D10S), and the cells were cultured for an additional 48 h. The astrocytes were then treated with or without TNF (0–50 ng/ml) for 7 h and total RNA extracted to examine Gal-9 expression as described above. Sister cultures were treated in parallel and subjected to Western blot analysis to determine the efficiency of siRNA knockdown of specific targets.
ELISA
Levels of secreted IFN-γ, TNF, and IL-1β from supernatants were determined using species-specific ELISA kits according to the manufacturer's instructions (eBioscience).
Astrocyte-conditioned Media and T-cell Co-culture Experiments
Mouse astrocytes were obtained from various genotypes and plated into poly-d-lysine coated 6-well plates at a density of 5.0 × 105 cells per well. After reaching 80% confluency, the astrocytes were treated with or without recombinant mouse TNF (100 ng/ml) for 24 h in a total volume of 700 μl. TNF in empty wells containing only 700 μl of medium served as TNF alone controls. After the treatment the conditioned media were immediately transferred to an empty 24-well plate, and 0.5–1.0 × 106 LCN-8 T-cells were then added to each well containing either TNF alone or the astrocyte-conditioned medium. T-cell apoptosis was determined 72 h later by flow cytometry. In co-culture experiments, astrocytes were plated into poly-d-lysine-coated 96-well plates at a density of 5.0 × 104 cells per well. After 48 h, 1 × 105 T-cells were added and stimulated with recombinant mouse TNF (0–100 ng/ml) for 24 h. The culture was then fixed with 4% paraformaldehyde in PBS at room temperature for 10 min. Cells were then blocked and permeabilized with PBST (PBS containing 0.1%Triton X-100) and 5% goat serum for 1 h at room temperature and immunostained for CD3 (1/100, clone 17A2; eBioscience) overnight at 4 °C. After washing three times with PBS-Tween, the cells were incubated with Alexa Fluor594-conjugated goat anti-rat antibody (1/1000, Invitrogen). Fluorescence images were acquired with an Olympus DP70 digital camera mounted on an Olympus IX71 microscope. The number of CD3+ cells from three representative 20× fields per well (nine fields per condition) was determined.
Flow Cytometry
Tim-3 expression on LNC-8 encephalitogenic T-cells was determined by flow cytometry as described previously (23). Briefly, 1 × 106 cells were blocked on ice for 20 min with anti-CD16/CD32 in staining buffer (clone 93) and were stained for 1 h at 4 °C with anti-CD4 (clone GK1.5; FITC) and anti-Tim-3 (clone RMT3–23; phycoerythrin) or an isotype control (all from eBioscience). For cell viability determination, T-cells were stained with antibodies for 30 min on ice as described above in annexin-V binding buffer and then with annexin V-FITC (eBioscience). 7-Aminoactinomycin D was added immediately before flow cytometric analysis. The percentages of cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences) and FlowJo software (TreeStar, Inc., Ashland, OR). For each condition the percent cell death was determined by subtracting the percentage of cell death observed after stimulation with control-conditioned media (containing no TNF) from those observed after stimulation with conditioned media containing TNF.
Statistics
All data are presented as the means ± S.E. Data were analyzed using two-tailed Student's t tests for comparisons between two groups. When analyzing differences between multiple groups, multiple level analysis of variance followed by Bonferroni's post hoc test was employed using GraphPad Prism 4 (GraphPad Software, San Diego, CA). Differences were considered significant when p < 0.05.
RESULTS
Proinflammatory but Not Anti-inflammatory Cytokines Promote Gal-9 Production in Astrocytes
Human astrocytes have been reported to express Gal-9 after stimulation with IL-1β (24), an inflammatory cytokine that is produced by reactive microglia/macrophages. To determine if other inflammatory cytokines were capable of inducing Gal-9 in astrocyte cultures, we stimulated primary rat astrocytes with recombinant cytokines that are known to be prevalent in MS. We found that both IL-1β and TNF robustly up-regulated Gal-9 expression, whereas IFN-γ, a potent inducer of Gal-9 expression in human endothelial cells, only modestly increased Gal-9 expression in astrocytes (Fig. 1A). In contrast, IL-6, IL-10, and IL-13 failed to induce Gal-9 transcription (Fig. 1A). As with IL-1β and IFN-γ, TNF triggered Gal-9 transcription in a concentration-dependent fashion (Fig. 1, B–D). Of the cytokines examined, TNF appeared to be the most potent Gal-9 inducer in astrocytes and caused Gal-9 up-regulation in a time-dependent manner (Fig. 1E). It should be noted that stimulation of astrocytes with TNF, even at high concentrations, was not toxic and did not result in detectible IL-1β secretion as determined by ELISA (data not shown). Moreover, hypoxia coupled with glucose deprivation also failed to increase Gal-9 expression in mixed glial cultures, although LPS stimulation readily induced Gal-9 expression as well as TNF secretion (Fig. 1F), indicating that Gal-9 up-regulation is tightly controlled and is not just a stress-associated response.
FIGURE 1.

Proinflammatory cytokines TNF, IL-1β, and IFN-γ up-regulate Gal-9 in astrocytes. A–E, primary rat astrocytes were stimulated with recombinant cytokines for 7 h, and Gal-9 up-regulation was determined by quantitative real-time-PCR. A, shown is induction of Gal-9 after stimulation with recombinant TNF, IL-1β, IFN-γ, IL-6, IL-10, and IL-13 (50 ng/ml each). Results are the combined means ± S.E. from 3–5 independent experiments. B–D, stimulation of astrocytes with 0–50 ng/ml recombinant IFN-γ (B), IL-1β (C), or TNF (D) dose-dependently increased Gal-9 transcription. Results are the means ± S.E. of duplicate samples and are representative of 3–5 independent experiments for each cytokine. E, shown is the time-dependent increase in Gal-9 transcription after stimulation of astrocytes with TNF (5 ng/ml). F, mixed glial cultures were subjected to oxygen/glucose deprivation (OGD) for 2 h then stimulated with or without LPS (1.0 μg/ml) for 7 h. The effects of oxygen/glucose deprivation on Gal-9 expression (top) as determined by quantitative real-time-PCR as well as TNF production as determined by ELISA (bottom) are shown. Results are the combined means of triplicate wells and are representative of two independent experiments. *, p < 0.05; ***, p < 0.001.
To determine whether TNF induces Gal-9 in other cells of the CNS, we prepared highly enriched neuron, oligodendrocyte, and microglia cultures and challenged them with TNF. Unstimulated astrocytes, neurons, and oligodendrocytes contained negligible Gal-9 mRNA, and microglia appeared to constitutively transcribe Gal-9 as determined by RT-PCR (Fig. 2). However, in stark contrast to the robust induction of Gal-9 in astrocytes, TNF did not induce Gal-9 transcription in neurons, oligodendrocytes, or microglia despite that these cells are known to possess TNF receptors (Fig. 2).
FIGURE 2.
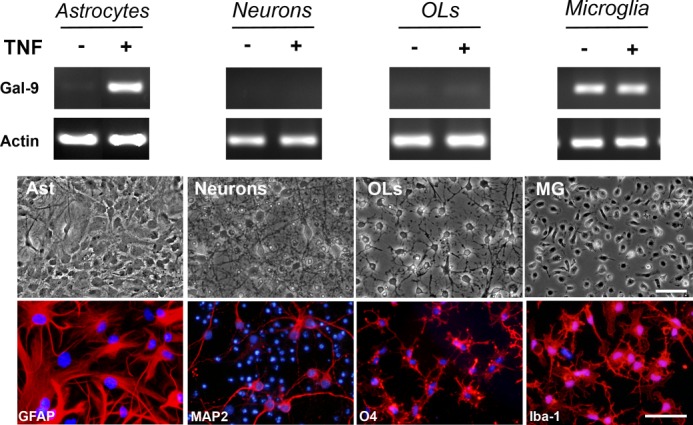
TNF induces Gal-9 transcription in cultured astrocytes. Enriched primary cultures of astrocytes, neurons, oligodendrocytes (OLs), and microglia were stimulated with or without TNF (5 ng/ml) for 7 h, and induction of Gal-9 mRNA was determined by RT-PCR. Phase contrast and immunocytochemistry images of representative cell cultures immunostained for cell-specific marker GFAP (Astrocytes (Ast)), MAP2 (Neurons), O4 (OLs), or Iba-1 (Microglia) are shown. Scale bars, 50 μm. Results are representative of two-three independent experiments.
TNFR1 Activation of the JNK Pathway Is Required for TNF-induced Gal-9 Expression
To investigate the mechanism by which TNF induces Gal-9 in astrocytes, we first determined whether TNFR1 was required. Astrocytes were isolated from C567BL/6 (WT) and TNFR1−/− mice and were stimulated with TNF or IFN-γ (positive control). In WT astrocytes both TNF and IFN-γ increased Gal-9 expression (Fig. 3A). However, only IFN-γ induced Gal-9 expression in TNFR1−/− astrocytes, indicating that TNFR1 is essential for Gal-9 induction after TNF stimulation (Fig. 3A).
FIGURE 3.

TNFR1/JNK pathway activation mediates Gal-9 up-regulation in astrocytes. A, TNF failed to induce Gal-9 in TNFR1-deficient astrocytes. Astrocytes isolated from WT or TNFR1−/− mice were stimulated with recombinant mouse TNF (100 ng/ml) or IFN-γ (100 ng/ml) for 7 h, and Gal-9 up-regulation was determined by RT-PCR. Results are representative of three independent experiments. Ctl, control. B–F, TNF transiently activated the MAPK pathways in astrocytes. Rat astrocytes were stimulated with TNF (50 ng/ml), and activation of the JNK, p38, and MEK pathways were determined by Western blot. TNF stimulation caused a transient phosphorylation of both JNK (B and D) and c-Jun (C) as well as p38 MAPK (E) and ERK1/2 (F). D–I, the JNK-specific inhibitor, but not p38- or MEK-specific inhibitors, abolished TNF-induced Gal-9 in astrocytes. Astrocytes were pretreated with JNK inhibitor SP600125 (20 μm), p38 MAPK inhibitor SB203580 (20 μm), or MEK1/2 inhibitor PD98059 (20 μm) for 1 h followed by TNF stimulation. Pretreatment with SP600125 (D) or PD98059 (F) significantly inhibited TNF-induced phosphorylation of JNK or ERK1/2, respectively, at all time points. Pretreatment with SB203580 had no effect on p38 MAPK phosphorylation. Nearly identical effects were observed with 5 ng/ml TNF. Results are representative of three independent experiments. Densitometry results in D–F are the combined means ± S.E. of three independent experiments. G–I, astrocytes were pretreated with JNK (SP600125; 20 μm) (G)-, p38 MAPK (SB203580; 20 μm) (H)-, or MEK1/2 (PD98059; 20 μm) (I)-specific inhibitors for 1 h and subsequently stimulated with TNF (5 ng/ml) for 7 h. The effect of each inhibitor on Gal-9 expression was determined by quantitative real-time-PCR. Results are the combined means ± S.E. from 4–7 independent experiments.
Next, we examined the intracellular signaling pathways required for TNF/TNFR1-dependent Gal-9 up-regulation. As TNF is capable of activating all three MAPK kinases pathways, we first examined the kinetics of TNF-induced activation of JNK, p38, and ERK1/2. Stimulation of astrocytes with TNF caused transient phosphorylation of JNK and its downstream substrate c-Jun (Fig. 3, B and C) and activation of ERK1/2 and p38 (Fig. 3, E and F). We then utilized a pharmacological approach to delineate the signaling pathway required for TNF-induced Gal-9 up-regulation. Pretreatment of astrocytes with the JNK-specific inhibitor SP600125 before TNF stimulation markedly prevented JNK phosphorylation (Fig. 3D) and subsequent induction of Gal-9 (Fig. 3G). In contrast, the p38 MAPK inhibitor SB203580 and the MEK inhibitor PD98059 had minimal effect on TNF-induced Gal-9 expression despite that PD98059 completely blocked ERK1/2 activation (Fig. 3, E–I), suggesting that the MEK and p38 MARK pathways are dispensable for TNF-induced Gal-9 expression. Because the p38 inhibitor SB203580 inhibits p38 catalytic activity (25) instead of the phosphorylation status of p38 MAPK (Fig. 3E), to confirm the above result with this inhibitor, we specifically knocked down p38 and found that deletion of p38 had no effect on TNF-induced Gal-9 transcription (Fig. 4F). Together, these results demonstrate that the activation of the JNK pathway is required for TNF-induced Gal-9 expression.
FIGURE 4.

c-Jun activation is required for TNF-induced Gal-9 transcription in astrocytes. A–C, astrocytes were treated with JNK (SP600125; 20 μm)-, p38 MAPK (SB203580; 20 μm)-, or MEK1/2 (PD98059; 20 μm)-specific inhibitors for 1 h before TNF stimulation, and the effects were determined by Western blot. Treatment with the JNK inhibitor SP600126 before TNF (50 ng/ml) stimulation inhibited phosphorylation of c-Jun (A), whereas SB203580 (B) or PD98059 (C) had no effect on c-Jun phosphorylation. Western blots are representative of three independent experiments. Densitometry results represent the means ± S.E. of 3 independent experiments. D and E, c-Jun is needed for TNF-induced Gal-9 transcription. Astrocytes were transfected with either control or c-Jun-specific siRNA and were stimulated with TNF (5 ng/ml) 48 h later as described. D, Western blot analysis demonstrates the specificity and efficiency of c-Jun knockdown. Results are the means ± S.E. of three independent experiments. E, the effect of siRNA mediated c-Jun knockdown on TNF-induced Gal-9 transcription is shown. Results are the means ± S.E. of triplicate wells and are representative of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. F, TNF-induced activation of p38 is not required for Gal-9 up-regulation. Astrocytes were transfected with NS or target specific siRNAs as indicated for 48 h, stimulated with or without TNF (5 ng/ml) for 7 h, and then subjected to Western blot or quantitative real-time-PCR (qPCR) analysis. Results are representative of three-four independent experiments in triplicate. **, p < 0.01.
c-Jun Activation Mediates TNF-induced Gal-9 Expression in Astrocytes
Consistent with the time-dependent activation of JNK signaling pathway in TNF-stimulated astrocytes, c-Jun was activated/phosphorylated in a similar time-dependent fashion (Fig. 4, A–C). TNF-induced phosphorylation of c-Jun was markedly blocked by the JNK inhibitor SP600125, which also prevented Gal-9 induction (Fig. 3G), but not by p38 inhibitor SB203580 nor by MEK inhibitor PD98059 (Fig. 4, A–C), confirming the specificity of the inhibitors used. To definitively determine whether c-Jun is indeed the downstream target of the JNK activation that induces Gal-9 transcription, we specifically knocked down c-Jun in astrocytes. Transfection with c-Jun-specific siRNA resulted in ∼70% down-regulation of total c-Jun protein levels compared with NS siRNA controls and significantly abolished TNF-induced Gal-9 up-regulation (Fig. 4, D and E). In contrast, neither nonspecific siRNA nor p38-specific siRNA prevented TNF-induced Gal-9 transcription (Fig. 4F). These data demonstrate that the activation of c-Jun is needed for Gal-9 transcription in astrocytes after TNF stimulation.
To determine whether Gal-9 is also up-regulated at the protein level, we first validated a commercially available Gal-9 antibody using Gal-9-EGFP reporter mice that express EGFP under the control of the endogenous Gal-9 promoter. Consistent with a previous report on Gal-9 mRNA expression in multiple tissues but not in the brain (10), similar Gal-9 expression patterns were found in the EGFP reporter mice (Fig. 5A). Western blot analysis confirmed that GFP expression was low in the nervous system but prominent in the intestine, lung, liver, and thymus (Fig. 5B). Probing the same membrane with the Gal-9 antibody demonstrated a good correlation between activation of Gal-9 transcription as judged by GFP co-expression and Gal-9 protein production in most tissues except the intestine, which expresses a different isoform of Gal-9 (long isoform) (10).
FIGURE 5.

Blockade of the JNK pathway prevents increased production of Gal-9 protein in TNF-stimulated astrocytes. A–C, shown is validation of the Gal-9-specific antibody. A, shown is a gross examination of individual organs after dissection of Gal-9-EGFP mouse under UV light. B, dissected tissues (Br, brain; SC, spinal cord; Int, intestine; Kid, kidney; Thyr, thyroid; Spl, spleen; Liv, liver; Thym, thymus; Hrt, heart) were homogenized and subjected to Western blot analysis with GFP (top)-, Gal-9 (middle)-, or β-actin (bottom)-specific antibodies. Results were obtained from two adult mice. C, Western blot analyses of brain and thymus tissue homogenates from Gal-9-EGFP (GFP), Gal-9+/+ (WT), Gal-9+/− (Het), or Gal-9−/− (KO) mice with GFP-, Gal-9-, or β-actin-specific antibodies. Het, heterozygous. D, TNF-induced production of Gal-9 protein in astrocytes was abolished by JNK inhibitor. Mouse astrocyte monocultures were treated with the JNK inhibitor SP600125 (20 μm), TNF (100 ng/ml), TNF and SP600125, IL-1β (100 ng/ml), or IFNγ (100 ng/ml) for 24 h and subjected to Western blot analysis. Results are the means ± S.E. of two independent experiments.
The specificity of the Gal-9 antibody was further validated with brain and thymus tissue homogenates from Gal-9 EGFP and Gal-9 mutant mice (Fig. 5C). As expected, GFP expression was low in the brain and abundant in the thymus of Gal-9 EGFP mice. Although a few nonspecific bands were observed, the Gal-9 antibody specifically detected the expected 34-kDa Gal-9 band that was completely absent in Gal-9 knock-out samples (Fig. 5C, middle).
Using enriched mouse astrocytes and the validated antibody, we then determined if Gal-9 protein level was increased after stimulation with proinflammatory cytokines. Consistent with the above quantitative PCR analyses (Fig. 1), TNF and IL-1β prominently increased Gal-9 protein levels, whereas IFN-γ had limited effect (Fig. 5D). Similar to its inhibitory effect on TNF-induced Gal-9 transcription, SP600125 also abolished TNF-induced up-regulation of Gal-9 protein, indicating that the JNK pathway is required for up-regulation of Gal-9 at both transcription and translational levels.
TNF-stimulated Gal-9+/+ but Not Gal-9−/− Astrocytes Decrease Viability of LNC-8 T-cells
Tim-3 was first cloned in an effort to obtain a T-helper 1 (Th1) cell surface marker and has since been assigned an immunoregulatory role. Binding of Gal-9 to Tim-3-expressing Th1 cells results in T-cell apoptosis and suppression of Th1 immunity, whereas abolition of Tim-3 has been shown to enhance the pathology of T-cell-mediated autoimmunity (12). LNC-8 T-cells are an MBP-specific clone that when passively transferred to naïve mice induces EAE by a mechanism that is dependent on the production of the Th1 cytokines Ltα and TNF (22). We first confirmed a dose-dependent production of IFN-γ (Fig. 6A) and TNF (Fig. 6B) by these encephalitogenic T-cells after antigenic stimulation with MBP in the presence of syngeneic irradiated splenocytes as antigen-presenting cells (22). The production of Ltα, TNF, and IFN-γ after MBP stimulation demonstrates that these cells are Th1 cells. Moreover, flow cytometry analysis confirmed surface expression of Tim-3 on ∼90% of these LNC-8 T-cells (Fig. 6C).
FIGURE 6.

TNF-stimulated Gal-9+/+ but not Gal-9−/− astrocytes decreases the viability of LNC-8 T-cells. A and B, LNC-8 T-cell responses to antigenic stimulation are shown. T-cells were stimulated with increasing concentrations of MBP (0–100 μg/ml) for 24 h. The amount of IFN-γ (A) and TNF (B) in supernatants was determined by ELISA. Results are the means ± S.E. of triplicate samples and are representative of two independent experiments. C, LNC-8 T-cells express Tim-3. LNC-8 cells were stained with fluorophore-conjugated anti-CD4 and anti-Tim-3 antibodies and were analyzed by flow cytometry. Top panel, gating of CD4+ cells. SSC, side scatter channel. Bottom panel, frequency and Tim-3 staining intensity chart of unstained cells (filled gray), isotype control (thin black line), and Tim-3 antibody-stained cells (dark black line). Results are representative of four independent experiments. D and E, the experimental scheme examining the effect of astrocyte (Ast)-conditioned media (CM) on LNC-8 cell viability. LNC-8 T-cells were treated with conditioned media from astrocytes for 72 h, stained for Tim-3-phycoerythrin, annexin-V-FITC, and 7-aminoactinomycin D (7-AAD), and analyzed by flow cytometry. Shown are percentage increases in apoptotic Tim-3+ LNC-8 cells after treatment with conditioned media from TNF-stimulated versus vehicle-treated astrocytes. Data represent the means ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01. F–H, LNC-8 T-cells were cultured with Gal-9+/+ (WT) or Gal-9−/− astrocytes in the absence (top) or presence (bottom) of TNF (0–100 ng/ml) for 24 h and then stained for CD3 (red) and nuclei (DAPI, blue). Representative photomicrographs (F) and quantification of percent change in CD3+ cells (G) and the number of mitotic CD3+ cells (H) are shown. Arrows indicate mitotic cells; the inset shows higher magnification of a CD3+ cell in metaphase. Scale bar, 50 μm. Results are the means ± S.E. of triplicate and are representative of two independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine the functional consequence of astrocyte-derived Gal-9, we collected conditioned media from vehicle- and TNF-treated Gal-9+/+, Gal-9+/−, or Gal-9−/− astrocytes, incubated LNC-8 T-cells with the conditioned media, and examined Tim-3+ T-cell viability by flow cytometry (Fig. 6D). Although TNF alone had little effect on Tim-3+ T-cell viability, conditioned media from TNF-stimulated Gal-9+/+ or Gal-9+/− astrocytes caused a significant increase in the percentage of apoptotic T-cells (Fig. 6E). In contrast, conditioned media from TNF-activated Gal-9−/− astrocytes induced significantly less T-cell apoptosis (Fig. 6E). Furthermore, TNF stimulation of Gal-9+/+ astrocyte/T-cell co-cultures caused a moderate decrease in T-cell number, whereas at the same time the number of T-cells in Gal-9−/− astrocyte co-cultures were increased (Fig. 6, F and G). Consistently, TNF appeared to increase the number of T-cells undergoing mitosis in Gal-9−/− astrocyte co-cultures (Fig. 6, F and H). Collectively, these results indicate that TNF-activated astrocytes release factors such as Gal-9 that promote apoptosis of Tim-3+ Th1 cells and that suppress T-cell proliferation.
DISSUSSION
In this study we demonstrate that the proinflammatory cytokines TNF, IL-1β, and IFN-γ, but not the anti-inflammatory cytokines IL-6, IL-10, and IL-13, are capable of inducing galectin-9 expression in primary astrocyte cultures. To our knowledge this represents the first report to demonstrate that TNF promotes the up-regulation of Gal-9. TNF-mediated Gal-9 production in astrocytes is dependent on TNFR1 and activation of JNK and c-Jun, a finding that is further supported by the presence of c-Jun recognition sequences within the Gal-9 promoter (26). Consistent with the known function of Gal-9 in regulating type 1 immunity, we found that conditioned medium from TNF-activated Gal-9+/+ and Gal-9+/− but not Gal-9−/− astrocytes is capable of decreasing cell viability and inducing apoptosis of LNC-8 encephalitogenic T-cells. Together these data indicate that astrocytes are capable of sensing inflammatory events and can respond in an immunoregulatory fashion in part through the production of Gal-9.
To date, little is known about the transcriptional regulation of Gal-9. Although a few studies have focused on the mechanisms governing Gal-9 up-regulation in endothelial cells (27–31), essentially none has investigated the signaling mechanisms underlying Gal-9 up-regulation in other cell types. In IFN-γ-stimulated human umbilical vein endothelial cells, expression of Gal-9 is regulated by histone deacetylase 3 and PI3K-mediated IRF3 phosphorylation (27, 31). Similar mechanisms may control Gal-9 up-regulation after stimulation with the RNA viral mimic poly(I:C) or infection with Dengue virus in these cells (28–30). In addition, the PI3K pathway has also been implicated in Gal-9 up-regulation in human nasal polyp fibroblast (32). The presence of a partial IRF3 consensus sequence within the mouse Gal-9 promoter adds further support for a role of IRF3 in facilitating Gal-9 transcription (26). However, unlike previous studies our data demonstrate that after TNF stimulation of astrocytes, but not oligodendrocytes, neurons, or microglia, Gal-9 is up-regulated by a mechanism that is distinct from that of the IFN-γ/PI3K/IRF3 pathway and requires the activation of the JNK/c-Jun pathway.
The notion that astrocytes have the capacity to restrict T-cell access into the CNS has been clearly demonstrated by studies that utilized transgenic mice to conditionally ablate astrogliosis after injury (33) or during inflammatory demyelination (34). Transgenic mice in which proliferating, reactive astrocytes were selectively ablated exhibited exacerbated EAE with seemingly unrestricted infiltration of peripheral cells (monocytes/macrophages, T-cells, and neutrophils) into the parenchyma (34). Although the mechanism(s) of the restriction has not been entirely elucidated, astrocyte-derived factors likely contribute to the process and modulate the CNS immune responses. CD95L expressed by astrocytes has been previously shown to induce apoptosis of Th1 and Th2 cells in vitro (35). Although TNF stimulation increases both CD95 (Fas) and CD95L (FasL) expression, the in vivo role of this pathway in contributing to the resolution of CNS inflammation is dampened by the fact that FasL−/− mice develop EAE with similar kinetics compared with controls (36). Aside from FasL, astrocytes, through a yet to be identified secreted factor, can promote CTLA-4 expression on T-cells, thereby limiting T-cell proliferation and cytokine production (37). In addition, conditioned medium from IFN-γ stimulated astrocytes was shown to suppress T-cell proliferation and activation (38). More recently, it has been shown that IFN-γ induces the expression of a protein termed astrocyte-derived immune-suppressor factor (AdiF), which can also promote apoptosis of T-cells (39). Finally, the idea that proinflammatory cytokine production can mitigate ongoing CNS inflammation is also more generally supported by the fact that mice deficient in IFN-γ (40, 41) or IFN-γ receptor (42) develop atypical EAE characterized by a more aggressive phenotype. Our finding that astrocyte-derived Gal-9 increases Th1 cell apoptosis suggests that Gal-9 produced by astrocytes in response to proinflammatory cytokines could potentially act as one of the negative feedback signals to dampen the Th1 immunity in the CNS.
Although we cannot definitively conclude that the T-cells used in this experiment underwent apoptosis in a manner dependent on secreted Gal-9 and Tim-3 ligation, the loss of T-cell viability was nevertheless Gal-9-dependent, as demonstrated by the conditioned medium and astrocyte/T-cell co-culture experiments. The results from both sets of experiments suggest that TNF stimulation of astrocytes induces Gal-9-dependent toxicity to LNC-8 T-cells. Interestingly, our data also indicate that TNF-induced astroglial Gal-9 may inhibit T-cell proliferation as demonstrated by increased numbers of CD3+ cells undergoing mitosis in co-cultures containing Gal-9−/− astrocytes, although it is difficult to ascertain whether this effect is also attributable to cell death. Although Gal-9 does not contain a conventional secretion sequence, it can be released into the extracellular space via exosomes (43), and Gal-9-containing exosomes are in fact capable of inducing apoptosis of Epstein-Barr virus-specific T-cells (44). Furthermore, astrocytes are capable of exosome secretion (45, 46), making exosomal transport of astrocyte-derived Gal-9 a plausible means of export. Nevertheless, it remains possible that secreted Gal-9 is not the only factor responsible for T-cell apoptosis, and intracellular Gal-9-dependent secretion of other immunoregulatory molecules may act on the T-cells.
Given the results of the current study, it is interesting to note that both TNF−/− and TNFR1−/− mice exhibit reduced inflammation and 50% fewer apoptotic T-cells during myelin oligodendrocyte glycoprotein-induced EAE (36, 47). Although the effects of Gal-9 on infiltrating T-cells during acute EAE remain to be established, studies thus far support for such an immunoregulatory mechanism to be in place and potentially activated within the inflamed CNS. In this scheme TNF ligation onto TNFR1 would promote the production of pro-apoptotic Gal-9 from astrocytes, which then functions to alleviate ongoing T-cell responses in the CNS. It is thus tempting to speculate that this pathway may contribute to the limitation of T-cell infiltration into the CNS in MS, a hypothesis that is indirectly supported by two strong pieces of evidence. First, anti-TNF therapy has paradoxically resulted in the exacerbation of MS symptoms (48, 49) and has been associated with the onset of demyelination (50, 51); second, treatment of MS patients with either IFN-β or glatiramer acetate restores the susceptibility of T-cells derived from the peripheral blood to Tim-3-mediated responses (15).
It should be emphasized that the current study was concerned with the mechanism of Gal-9 up-regulation and the apoptotic functions of astrocyte-derived Gal-9. Recent evidence also points to a role for Gal-9 in regulating T-cell responses directly by inducing a state of exhaustion (52–55) or indirectly by promoting the polarization of regulatory T-cells (21, 56–58). To date the immunoregulatory capacity of glial-derived Gal-9 has not been elucidated and is the subject of ongoing investigation. Nevertheless, given the relative immune privilege status of the CNS, it is most likely that glial-derived Gal-9 acts directly on infiltrating T-cells, whereas peripherally produced Gal-9 in the secondary lymphoid tissue influences T-cell polarization.
Comprehending the function of glial involvement in either the promotion or inhibition of innate and adaptive immunity within the central nervous system is of great importance and is at the center of understanding the pathogenesis of CNS infection as well as autoimmunity. Determining the negative feedback loops that are in place to maintain CNS homeostasis or promote repair are also pertinent to understanding many other neurodegenerative diseases that are associated with aberrant inflammation. In this study we have demonstrated that astrocytes express Gal-9 after TNF stimulation via the TNFR1/JNK/c-Jun signaling pathway. Uncovering the molecular mechanism that governs differential induction of Gal-9 in resident CNS cells as well as establishing the functional outcomes of such up-regulation, both in response to infection and autoimmunity, represents the next crucial step needed to understand the role of Gal-9 in neuroinflammation.
Acknowledgments
We thank the Scripps Research Institute and the Consortium for Functional Glycomics for providing Lgals9 mutant mouse breeders, Dr. Nancy Ruddle, Yale University, for the T-cell clones, Dr. Aline Rodrigues for help taking pictures of the Gal-9-EGFP mouse, and Drs. Hisami Koito and Sunja Kim and Chris Gillis for technical support.
This work was supported, in whole or in part, by National Institutes of Health Grant R01NS060017 (to J. L.). This work was also supported by National Multiple Sclerosis Society Research Grants RG3975 and RG4586 (to J. L.) and Postdoctoral Fellowship FG1937 (to A. J. S.) and a College of Veterinary Medicine Texas A&M University postdoctoral research training grant (to A. J. S.).
- MS
- multiple sclerosis
- Gal-9
- Galectin-9
- TNFR1
- TNF-α receptor 1
- EAE
- encephalomyelitis
- MBP
- myelin basic protein
- NS
- nonspecific
- Th1
- T-helper 1
- EGFP
- enhanced GFP
- GFAP
- glial fibrillary acidic protein.
REFERENCES
- 1. Popescu B. F., Lucchinetti C. F. (2012) Pathology of demyelinating diseases. Annu. Rev. Pathol. 7, 185–217 [DOI] [PubMed] [Google Scholar]
- 2. Barnett M. H., Prineas J. W. (2004) Relapsing and remitting multiple sclerosis. Pathology of the newly forming lesion. Ann. Neurol. 55, 458–468 [DOI] [PubMed] [Google Scholar]
- 3. Henderson A. P., Barnett M. H., Parratt J. D., Prineas J. W. (2009) Multiple sclerosis. Distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 66, 739–753 [DOI] [PubMed] [Google Scholar]
- 4. Kivisäkk P., Healy B. C., Viglietta V., Quintana F. J., Hootstein M. A., Weiner H. L., Khoury S. J. (2009) Natalizumab treatment is associated with peripheral sequestration of proinflammatory T-cells. Neurology 72, 1922–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller D. H., Khan O. A., Sheremata W. A., Blumhardt L. D., Rice G. P., Libonati M. A., Willmer-Hulme A. J., Dalton C. M., Miszkiel K. A., O'Connor P. W. (2003) A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 348, 15–23 [DOI] [PubMed] [Google Scholar]
- 6. Polman C. H., O'Connor P. W., Havrdova E., Hutchinson M., Kappos L., Miller D. H., Phillips J. T., Lublin F. D., Giovannoni G., Wajgt A., Toal M., Lynn F., Panzara M. A., Sandrock A. W. (2006) A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 354, 899–910 [DOI] [PubMed] [Google Scholar]
- 7. Cross A. H., Stark J. L., Lauber J., Ramsbottom M. J., Lyons J. A. (2006) Rituximab reduces B cells and T-cells in cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 180, 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hauser S. L., Waubant E., Arnold D. L., Vollmer T., Antel J., Fox R. J., Bar-Or A., Panzara M., Sarkar N., Agarwal S., Langer-Gould A., Smith C. H. (2008) B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 358, 676–688 [DOI] [PubMed] [Google Scholar]
- 9. Sofroniew M. V., Vinters H. V. (2010) Astrocytes. Biology and pathology. Acta Neuropathol. 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wada J., Kanwar Y. S. (1997) Identification and characterization of galectin-9, a novel β-galactoside-binding mammalian lectin. J. Biol. Chem. 272, 6078–6086 [DOI] [PubMed] [Google Scholar]
- 11. Zhu C., Anderson A. C., Schubart A., Xiong H., Imitola J., Khoury S. J., Zheng X. X., Strom T. B., Kuchroo V. K. (2005) The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6, 1245–1252 [DOI] [PubMed] [Google Scholar]
- 12. Monney L., Sabatos C. A., Gaglia J. L., Ryu A., Waldner H., Chernova T., Manning S., Greenfield E. A., Coyle A. J., Sobel R. A., Freeman G. J., Kuchroo V. K. (2002) Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 [DOI] [PubMed] [Google Scholar]
- 13. Anderson A. C., Anderson D. E., Bregoli L., Hastings W. D., Kassam N., Lei C., Chandwaskar R., Karman J., Su E. W., Hirashima M., Bruce J. N., Kane L. P., Kuchroo V. K., Hafler D. A. (2007) Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318, 1141–1143 [DOI] [PubMed] [Google Scholar]
- 14. Stancic M., van Horssen J., Thijssen V. L., Gabius H. J., van der Valk P., Hoekstra D., Baron W. (2011) Increased expression of distinct galectins in multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 37, 654–671 [DOI] [PubMed] [Google Scholar]
- 15. Yang L., Anderson D. E., Kuchroo J., Hafler D. A. (2008) Lack of TIM-3 immunoregulation in multiple sclerosis. J. Immunol. 180, 4409–4414 [DOI] [PubMed] [Google Scholar]
- 16. Li J., Ramenaden E. R., Peng J., Koito H., Volpe J. J., Rosenberg P. A. (2008) Tumor necrosis factor α mediates lipopolysaccharide-induced microglial toxicity to developing oligodendrocytes when astrocytes are present. J. Neurosci. 28, 5321–5330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steelman A. J., Li J. (2011) Poly(I:C) promotes TNFα/TNFR1-dependent oligodendrocyte death in mixed glial cultures. J. Neuroinflammation 8, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koito H., Li J. (2009) Preparation of rat brain aggregate cultures for neuron and glia development studies. J. Vis. Exp. 31, . 10.3791/1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim S., Steelman A. J., Koito H., Li J. (2011) Astrocytes promote TNF-mediated toxicity to oligodendrocyte precursors. J. Neurochem. 116, 53–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim S., Steelman A. J., Zhang Y., Kinney H. C., Li J. (2012) Aberrant upregulation of astroglial ceramide potentiates oligodendrocyte injury. Brain Pathol. 22, 41–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sehrawat S., Suryawanshi A., Hirashima M., Rouse B. T. (2009) Role of Tim-3/galectin-9 inhibitory interaction in viral-induced immunopathology. Shifting the balance toward regulators. J. Immunol. 182, 3191–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruddle N. H., Bergman C. M., McGrath K. M., Lingenheld E. G., Grunnet M. L., Padula S. J., Clark R. B. (1990) An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J. Exp. Med. 172, 1193–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Steelman A. J., Dean D. D., Young C. R., Smith R., 3rd, Prentice T. W., Meagher M. W., Welsh C. J. (2009) Restraint stress modulates virus specific adaptive immunity during acute Theiler's virus infection. Brain Behav. Immun. 23, 830–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoshida H., Imaizumi T., Kumagai M., Kimura K., Satoh C., Hanada N., Fujimoto K., Nishi N., Tanji K., Matsumiya T., Mori F., Cui X. F., Tamo W., Shibata T., Takanashi S., Okumura K., Nakamura T., Wakabayashi K., Hirashima M., Sato Y., Satoh K. (2001) Interleukin-1β stimulates galectin-9 expression in human astrocytes. Neuroreport 12, 3755–3758 [DOI] [PubMed] [Google Scholar]
- 25. Kumar S., Jiang M. S., Adams J. L., Lee J. C. (1999) Pyridinylimidazole compound SB 203580 inhibits the activity but not the activation of p38 mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 263, 825–831 [DOI] [PubMed] [Google Scholar]
- 26. Lensch M., Lohr M., Russwurm R., Vidal M., Kaltner H., André S., Gabius H. J. (2006) Unique sequence and expression profiles of rat galectins-5 and -9 as a result of species-specific gene divergence. Int. J. Biochem. Cell Biol. 38, 1741–1758 [DOI] [PubMed] [Google Scholar]
- 27. Alam S., Li H., Margariti A., Martin D., Zampetaki A., Habi O., Cockerill G., Hu Y., Xu Q., Zeng L. (2011) Galectin-9 protein expression in endothelial cells is positively regulated by histone deacetylase 3. J. Biol. Chem. 286, 44211–44217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Warke R. V., Xhaja K., Martin K. J., Fournier M. F., Shaw S. K., Brizuela N., de Bosch N., Lapointe D., Ennis F. A., Rothman A. L., Bosch I. (2003) Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells. J. Virol. 77, 11822–11832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ishikawa A., Imaizumi T., Yoshida H., Nishi N., Nakamura T., Hirashima M., Satoh K. (2004) Double-stranded RNA enhances the expression of galectin-9 in vascular endothelial cells. Immunol. Cell Biol. 82, 410–414 [DOI] [PubMed] [Google Scholar]
- 30. Imaizumi T., Yoshida H., Nishi N., Sashinami H., Nakamura T., Hirashima M., Ohyama C., Itoh K., Satoh K. (2007) Double-stranded RNA induces galectin-9 in vascular endothelial cells. Involvement of TLR3, PI3K, and IRF3 pathway. Glycobiology 17, 12C–15C [DOI] [PubMed] [Google Scholar]
- 31. Imaizumi T., Kumagai M., Sasaki N., Kurotaki H., Mori F., Seki M., Nishi N., Fujimoto K., Tanji K., Shibata T., Tamo W., Matsumiya T., Yoshida H., Cui X. F., Takanashi S., Hanada K., Okumura K., Yagihashi S., Wakabayashi K., Nakamura T., Hirashima M., Satoh K. (2002) Interferon-γ stimulates the expression of galectin-9 in cultured human endothelial cells. J. Leukoc. Biol. 72, 486–491 [PubMed] [Google Scholar]
- 32. Park W. S., Jung W. K., Park S. K., Heo K. W., Kang M. S., Choi Y. H., Kim G. Y., Park S. G., Seo S. K., Yea S. S., Liu K. H., Shim E. B., Kim D. J., Her M., Choi I. W. (2011) Expression of galectin-9 by IFN-γ stimulated human nasal polyp fibroblasts through MAPK, PI3K, and JAK/STAT signaling pathways. Biochem. Biophys. Res. Commun. 411, 259–264 [DOI] [PubMed] [Google Scholar]
- 33. Bush T. G., Puvanachandra N., Horner C. H., Polito A., Ostenfeld T., Svendsen C. N., Mucke L., Johnson M. H., Sofroniew M. V. (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23, 297–308 [DOI] [PubMed] [Google Scholar]
- 34. Voskuhl R. R., Peterson R. S., Song B., Ao Y., Morales L. B., Tiwari-Woodruff S., Sofroniew M. V. (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci. 29, 11511–11522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bechmann I., Steiner B., Gimsa U., Mor G., Wolf S., Beyer M., Nitsch R., Zipp F. (2002) Astrocyte-induced T-cell elimination is CD95 ligand-dependent. J. Neuroimmunol 132, 60–65 [DOI] [PubMed] [Google Scholar]
- 36. Bachmann R., Eugster H. P., Frei K., Fontana A., Lassmann H. (1999) Impairment of TNF-receptor-1 signaling but not fas signaling diminishes T-cell apoptosis in myelin oligodendrocyte glycoprotein peptide-induced chronic demyelinating autoimmune encephalomyelitis in mice. Am. J. Pathol. 154, 1417–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gimsa U., ØRen A., Pandiyan P., Teichmann D., Bechmann I., Nitsch R., Brunner-Weinzierl M. C. (2004) Astrocytes protect the CNS. Antigen-specific T helper cell responses are inhibited by astrocyte-induced upregulation of CTLA-4 (CD152). J. Mol. Med. 82, 364–372 [DOI] [PubMed] [Google Scholar]
- 38. Xiao B. G., Diab A., Zhu J., van der Meide P., Link H. (1998) Astrocytes induce hyporesponses of myelin basic protein-reactive T and B cell function. J. Neuroimmunol. 89, 113–121 [DOI] [PubMed] [Google Scholar]
- 39. Hara H., Nanri Y., Tabata E., Mitsutake S., Tabira T. (2011) Identification of astrocyte-derived immune suppressor factor that induces apoptosis of autoreactive T-cells. J. Neuroimmunol 233, 135–146 [DOI] [PubMed] [Google Scholar]
- 40. Krakowski M., Owens T. (1996) Interferon-γ confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol. 26, 1641–1646 [DOI] [PubMed] [Google Scholar]
- 41. Ferber I. A., Brocke S., Taylor-Edwards C., Ridgway W., Dinisco C., Steinman L., Dalton D., Fathman C. G. (1996) Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 156, 5–7 [PubMed] [Google Scholar]
- 42. Willenborg D. O., Fordham S. A., Staykova M. A., Ramshaw I. A., Cowden W. B. (1999) IFN-γ is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue. A possible role for nitric oxide. J. Immunol. 163, 5278–5286 [PubMed] [Google Scholar]
- 43. Keryer-Bibens C., Pioche-Durieu C., Villemant C., Souquère S., Nishi N., Hirashima M., Middeldorp J., Busson P. (2006) Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer 6, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klibi J., Niki T., Riedel A., Pioche-Durieu C., Souquere S., Rubinstein E., Le Moulec S., Moulec S. L., Guigay J., Hirashima M., Guemira F., Adhikary D., Mautner J., Busson P. (2009) Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood 113, 1957–1966 [DOI] [PubMed] [Google Scholar]
- 45. Wang G., Dinkins M., He Q., Zhu G., Poirier C., Campbell A., Mayer-Proschel M., Bieberich E. (2012) Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4). Potential mechanism of apoptosis induction in Alzheimer disease (AD). J. Biol. Chem. 287, 21384–21395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Basso M., Pozzi S., Tortarolo M., Fiordaliso F., Bisighini C., Pasetto L., Spaltro G., Lidonnici D., Gensano F., Battaglia E., Bendotti C., Bonetto V. (2013) Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes. Implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 288, 15699–15711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eugster H. P., Frei K., Bachmann R., Bluethmann H., Lassmann H., Fontana A. (1999) Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur. J. Immunol. 29, 626–632 [DOI] [PubMed] [Google Scholar]
- 48. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group (1999) TNF neutralization in MS. Results of a randomized, placebo-controlled multicenter study. Neurology 53, 457–465 [PubMed] [Google Scholar]
- 49. van Oosten B. W., Barkhof F., Truyen L., Boringa J. B., Bertelsmann F. W., von Blomberg B. M., Woody J. N., Hartung H. P., Polman C. H. (1996) Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 47, 1531–1534 [DOI] [PubMed] [Google Scholar]
- 50. Mohan N., Edwards E. T., Cupps T. R., Oliverio P. J., Sandberg G., Crayton H., Richert J. R., Siegel J. N. (2001) Demyelination occurring during anti-tumor necrosis factor α therapy for inflammatory arthritides. Arthritis Rheum. 44, 2862–2869 [DOI] [PubMed] [Google Scholar]
- 51. Sicotte N. L., Voskuhl R. R. (2001) Onset of multiple sclerosis associated with anti-TNF therapy. Neurology 57, 1885–1888 [DOI] [PubMed] [Google Scholar]
- 52. Abdelbary N. H., Abdullah H. M., Matsuzaki T., Hayashi D., Tanaka Y., Takashima H., Izumo S., Kubota R. (2011) Reduced Tim-3 expression on human T-lymphotropic virus type I (HTLV-I) Tax-specific cytotoxic T lymphocytes in HTLV-I infection. J. Infect Dis. 203, 948–959 [DOI] [PubMed] [Google Scholar]
- 53. Allen S. J., Mott K. R., Zandian M., Ghiasi H. (2010) Immunization with different viral antigens alters the pattern of T-cell exhaustion and latency in herpes simplex virus type 1-infected mice. J. Virol. 84, 12315–12324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang Z. Z., Grote D. M., Ziesmer S. C., Niki T., Hirashima M., Novak A. J., Witzig T. E., Ansell S. M. (2012) IL-12 upregulates TIM-3 expression and induces T-cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J. Clin. Invest. 122, 1271–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou Q., Munger M. E., Veenstra R. G., Weigel B. J., Hirashima M., Munn D. H., Murphy W. J., Azuma M., Anderson A. C., Kuchroo V. K., Blazar B. R. (2011) Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 117, 4501–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lv K., Xu W., Wang C., Niki T., Hirashima M., Xiong S. (2011) Galectin-9 administration ameliorates CVB3-induced myocarditis by promoting the proliferation of regulatory T-cells and alternatively activated Th2 cells. Clin. Immunol. 140, 92–101 [DOI] [PubMed] [Google Scholar]
- 57. Oomizu S., Arikawa T., Niki T., Kadowaki T., Ueno M., Nishi N., Yamauchi A., Hirashima M. (2012) Galectin-9 suppresses Th17 cell development in an IL-2-dependent but Tim-3-independent manner. Clin. Immunol. 143, 51–58 [DOI] [PubMed] [Google Scholar]
- 58. Seki M., Oomizu S., Sakata K. M., Sakata A., Arikawa T., Watanabe K., Ito K., Takeshita K., Niki T., Saita N., Nishi N., Yamauchi A., Katoh S., Matsukawa A., Kuchroo V., Hirashima M. (2008) Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T-cells, and regulates experimental autoimmune arthritis. Clin. Immunol. 127, 78–88 [DOI] [PubMed] [Google Scholar]