

Background: The protein kinase p38α mediates cellular responses to stress and immune signals.
Results: Loss of p38α in epithelial cells results in aberrant activation of multiple protein kinases and disrupts tissue homeostasis.
Conclusion: Epithelial tissue homeostasis requires cross-regulatory interactions between p38α and other protein kinases.
Significance: These findings provide clues about how to prevent the adverse effects of p38 inhibitors.
Keywords: Epithelium, Intestine, Mouse, p38, Skin
Abstract
The epithelium of mucosal and skin surfaces serves as a permeability barrier and affords mechanisms for local immune defense. Crucial to the development and maintenance of a properly functioning epithelium is the balance of cell proliferation, differentiation, and death. Here we show that this balance depends on cross-regulatory interactions among multiple protein kinase-mediated signals and their coordinated transmission. From an investigation of conditional gene knock-out mice, we find that epithelial-specific loss of the protein kinase p38α leads to aberrant activation of TAK1, JNK, EGF receptor, and ERK in distinct microanatomical areas of the intestines and skin. Consequently, the epithelial tissues display excessive proliferation, inadequate differentiation, and sensitivity to apoptosis. These anomalies leave the tissue prone to damage and collapse at the trigger of an environmental insult. The vulnerability of p38α-deficient epithelium predicts adverse effects of long term pharmacological p38α inhibition; yet such limitations could be overcome by concomitant blockade of one or more of the dysregulated protein kinase signaling pathways.
Introduction
Protein kinases are central elements of intracellular signaling pathways involving reversible protein modification. Various physiological cues trigger chain reactions of protein phosphorylation within the cell and alter the activity of the specific proteins at the end points of the biochemical cascades. Such a signaling process commonly entails concurrent activation of multiple protein kinase modules. To ensure an appropriate physiological output, modular signals that are transmitted in parallel must be collectively coordinated as well as individually controlled.
The MAP kinases ERK, JNK, and p38 share structural and enzymatic properties and are activated via similar three-tiered protein kinase cascades (1–3). Identifying the downstream targets has been deemed instrumental in understanding how individual MAP kinases exert their physiological functions, a view positing linearity and independence of their actions. However, in many, if not all, circumstances, the three MAP kinase family members are jointly activated by a single stimulus, and the signals they relay are integrated rather than independently processed. Therefore, it is vital to elucidate how the MAP kinase modules are wired into a regulatory network that enables the flow of signals in tune and balance.
Epithelial tissues in the intestinal mucosa and skin undergo relatively high rates of turnover throughout the life of the organism. To replenish cells lost due to normal shedding and injury, intestinal and skin stem cells cycle to produce transit-amplifying cells, which rapidly proliferate and then differentiate into specialized and functionally competent epithelial cells (4). Cells at distinct stages in this process are located in different areas along the villus-crypt axis and in different layers of the stratified epidermis (5). Perturbed epithelial tissue homeostasis leads to a functional deterioration such as disruption of permeability barrier and impaired immune surveillance. Consequently, the affected tissue incurs risks of inflammatory and neoplastic diseases (6–9).
The ubiquitously expressed protein kinase p38α is one of the four mammalian p38 isoforms, and its activity is sensitive to inhibition by many anti-inflammatory compounds (10, 11). Systemic p38α ablation in mice resulted in embryonic and postnatal developmental defects (12–17), thus precluding precise identification of physiological functions of p38α in adult tissues. Subsequent studies, including our study, reported the generation of mutant mice with cell type-specific deletions of the p38α gene (18–24). Loss of p38α alone was sufficient to suppress inflammatory responses in various disease models, thus accounting for the efficacy of p38 inhibitors against inflammatory pathology and validating p38α as an important target for anti-inflammatory therapies (19–21). Notably, such beneficial effects were afforded even when interference with p38α signaling was targeted to a specific cell type. These promising results notwithstanding, recent clinical studies have revealed a new challenge to therapeutic approaches based on p38 blockade: p38 inhibitors exerted toxic effects and brought about adverse events, which ranged from skin rashes to gastrointestinal reactions to liver damage (25–27). The mechanistic bases for the detrimental effects of p38 blockade remain unclear.
Here, we demonstrate that p38α plays a key role in limiting the activation of multiple protein kinases in diverse cell types. Using conditional gene ablation approaches, we simulate chronic disturbance of p38α function in enterocytes and keratinocytes (epithelial cells of the intestine and skin, respectively), and show that p38α-mediated coordination of protein kinase signaling is essential for tissue homeostasis in the intestinal mucosa and skin. These regulatory mechanisms are likely impaired by long term pharmacological inhibition of p38α, and their restoration may alleviate the adversity of p38 inhibitors.
EXPERIMENTAL PROCEDURES
Animals, Primary Cells, and Cell Lines
All animal studies were performed under the Institutional Animal Care and Use Committee-approved protocols. K-KO (Mapk14fl/fl-K14Cre), M-KO (Mapk14fl/fl-LysMCre), and D-KO (Mapk14fl/fl-CD11cCre) mice were described previously (19, 28). IEC-KO mice were generated by crossing Mapk14fl/fl mice (18) with VilCre mice (29) from The Jackson Laboratory. All animals were on a C57BL/6J background. Primary mouse keratinocytes, macrophages, and dendritic cells were isolated and cultured as described (19, 28). MODE-K mouse intestinal epithelial cells were described previously (30). 293T human kidney epithelial cells and RAW264.7 mouse macrophage cells were obtained from the American Type Culture Collection. Immortalized mouse fibroblasts were derived from a C57BL/6J embryo.
Reagents
Cultured cells were treated with mouse recombinant tumor necrosis factor (TNF#; a gift from C. Libert, Ghent University), human recombinant TNF (R&D Systems), LPS (Sigma-Aldrich), Pam3CSK4 (InvivoGen), recombinant mouse EGF (PeproTech), the p38 inhibitors SC409 and SB202190 (EMD Millipore), the peptide JNK inhibitor D-JNKi (EMD Millipore), the ERK inhibitor PD98059 (EMD Millipore), and the proteasome inhibitor MG132 (EMD Millipore). Antibodies against the following proteins were used in immunoblotting, immunoprecipitation, and immunostaining: phosphorylated (p-) p38 (9211), ERK (9102), p-ERK (9101), p-JNK (9251), p-MAPKAPK-2 (p-MK2; 3007), EGF receptor (EGFR; 4267),2 p-EGFR (4407), p-c-Jun (9261), Lys-48-linked polyubiquitin chain (4289; all from Cell Signaling Technology); p38α (sc-535; Santa Cruz Biotechnology); JNK (554285; BD Biosciences); transforming growth factor β-activated kinase 1 (TAK1; a gift from P. Cohen, University of Dundee); p-TAK1 (31); ubiquitin (MMS-257P), K14 (PRB-155P), HA (MMS-101P; all from Covance); Ki67 (M7249; Dako); K1 (ab9286; Abcam); and actin (A4700; Sigma-Aldrich).
Plasmid DNA and siRNA Transfection
Plasmid vectors expressing TAK1 and the constitutively active p38α variant (p38αCA), p38α D176A/F327S, were described previously (32, 33). Control siRNA was an siRNA negative control duplex with medium GC content (Invitrogen). Mouse p38α siRNA was from the Stealth RNAi collection (Invitrogen) and a duplex of the following synthetic oligonucleotides: sense, 5′-ccagcaaccuagcugugaacgaaga-3′; and antisense, 5′-ucuucguucacagcuagguugcugg-3′. Cell transfection with plasmid DNA and siRNA was performed using FuGENE HD (Roche) and Lipofectamine RNAiMAX (Invitrogen) transfection reagents, respectively. Cells were used for subsequent analyses 48 h after transfection.
Protein Analysis
Whole cell lysates were prepared and analyzed by immunoblotting as described (34). HA-TAK1 protein was immunoprecipitated from whole cell lysates with anti-HA antibody and protein G-agarose beads (Cell Signaling Technology). To immunoprecipitate ubiquitinated proteins, whole cell lysates were incubated with anti-ubiquitin antibody in the presence of 0.5% 3-[(cholamidopropyl)dimethylammonio]-1-propanesulfonate.
Chemically Induced Inflammation
To induce intestinal injury and inflammation, dextran sulfate sodium (DSS; 3.5% in drinking water) was orally administered to mice for 7 days. Survival and body weight were monitored daily over a period of 14 days. Colon tissue samples were collected for analysis on day 7 from independent groups of animals. To irritate skin, 12-O-tetradecanoylphorbol-13-acetate (TPA; 10 μg in acetone) was applied to the shaved back skin of mice on two consecutive days. Skin tissue samples were collected for analysis 2 days after the second TPA treatment.
Histology and Immunofluorescence
Mouse ileum, colon, and skin samples were frozen in OCT medium or formalin-fixed and embedded in paraffin. Sections mounted on slides were stained with hematoxylin and eosin or Alcian Blue and incubated with marker-specific antibodies for immunostaining (19) and analyzed by immunofluorescence microscopy. Freshly prepared frozen colon tissue sections were incubated with 2′,7′-dichlorodihydrofluorescein diacetate (5 μm; Invitrogen) at 37 °C for 30 min for detection of reactive oxygen species by fluorescence microscopy. BrdU and TUNEL staining were performed using the BrdU in situ detection kit (BD Biosciences) and the in situ cell death detection kit (Roche Applied Science), respectively. Fluorescence intensity was determined using ImageJ software (National Institutes of Health).
Statistical Analysis
p values were obtained with the unpaired, two-tailed Student's t test.
RESULTS
The suppression of JNK activation by NF-κB signaling represents a cross-regulatory mechanism in the intracellular signaling pathways downstream of the TNF receptor and explains the apoptotic sensitivity (35–37) and deregulated proliferation (38, 39) of TNF-stimulated cells that are devoid of NF-κB activity. Evidence of a similar role for p38α is now accumulating: genetic ablation of p38α has been shown to result in increased JNK activation in mice (16, 19, 21, 22). Besides, some studies have reported that p38α-deficient cells exhibit higher ERK activation as well (19, 21). To establish the effects of p38α ablation on signaling by other MAP kinase family members, we systematically compared ERK and JNK activation in various types of cells with and without p38α expression.
We first examined TNF-induced signaling in mouse MODE-K intestinal epithelial cells. In MODE-K cells transfected with control siRNA, the amount of phosphorylated ERK and JNK, the active forms of the protein kinases, increased 5, 10, and 15 min after TNF treatment and then declined to the levels in untreated cells by 30 min. Knockdown (KD) of endogenous p38α expression by siRNA resulted in prolonged ERK and JNK phosphorylation up to at least 60 min (Fig. 1A). We previously generated keratinocyte-specific p38α knock-out mice, K-KO and observed efficient p38α ablation in their epidermis (19). ERK and JNK hyperactivation also occurred in K-KO mouse-derived keratinocytes treated with TNF (Fig. 1B). We extended this analysis to bone marrow-derived macrophages and lymph node dendritic cells. These cells were prepared from cell type-specific p38α KO mice, M-KO and D-KO, respectively, in which p38α ablation was targeted to the corresponding cell types (19, 28). Again, loss of p38α in these cells led to elevated and prolonged activation of ERK and JNK in response to the Toll-like receptor agonists LPS and Pam3CSK4 (Fig. 1, C and D). Therefore, depending on the cell types tested, p38α ablation brought about increased ERK and JNK signaling in either duration or intensity or both.
FIGURE 1.

Ablation of p38α expression results in ERK and JNK hyperactivation in various cell types. A, MODE-K mouse intestinal epithelial cells were transfected with siRNA specific to p38α mRNA (p38α-KD) and control siRNA (control-KD) and treated with TNF (50 ng/ml). Whole cell lysates were prepared after the indicated durations of stimulation and analyzed by immunoblotting with antibodies against the proteins indicated on the left. B–D, keratinocytes (B), bone marrow-derived macrophages (C), and lymph node dendritic cells (D) were prepared from the indicated mice and treated with TNF (20 ng/ml), LPS (100 ng/ml), and Pam3CSK4 (1 μg/ml). Whole cell lysates were prepared and analyzed as described in A.
To study p38α-dependent regulation of MAP kinase signaling in a physiological context, we investigated the epithelium of mouse intestinal mucosa and skin. To this end, we generated mice lacking p38α in intestinal epithelial cells, designated IEC-KO. Expression of p38α in the ileal and colonic epithelia of IEC-KO mice was almost completely abolished (Fig. 2A). The most salient feature in IEC-KO intestinal epithelia was elongated villi and crypts (Fig. 2B). The numbers of cells circumscribing a villus and a crypt in the ileum of IEC-KO mice were greater than those in WT ileum (Fig. 2C). This hyperplasia was accompanied by a mildly increased number of proliferating epithelial cells (with BrdU incorporation) and drastically reduced abundance of mature, mucus-secreting goblet cells (Fig. 2, D and E). This phenotype was also detected in another independently generated line of intestinal epithelial cell-specific p38α-KO mice (21). A more pronounced imbalance between proliferation and differentiation was detected in the colonic epithelium of IEC-KO mice, which we will describe shortly.
FIGURE 2.

Epithelial-specific p38α ablation leads to a proliferation-differentiation imbalance in the intestinal mucosa. A, whole cell lysates from WT and IEC-KO tissues were analyzed by immunoblotting with antibodies against the proteins indicated on the left. B–E, ileum tissue sections from WT and IEC-KO mice were analyzed by hematoxylin and eosin staining (B), immunostaining with BrdU-specific antibody (D, upper panel), and Alcian Blue staining (D, lower panel). Black and gray lines with double arrows indicate villus and crypt length, respectively (B). Scale bar, 100 μm. The numbers of epithelial cells constituting villus and crypt units (C), and the numbers of BrdU+ cells and Alcian Blue+ goblet cells per crypt (E) were determined. Data represent means ± S.E. *, p < 0.05.
The epidermis of K-KO mice, if left unchallenged, had no discernable anomalies but showed notable parallels with IEC-KO intestinal epithelia when irritated and inflamed by epicutaneous treatment with TPA. TPA-irritated WT and K-KO skin displayed epidermal hyperplasia to similar extents, yet there were a greater number of proliferating cells in K-KO epidermis with multiple layers of Ki67+ keratinocytes whereas cell proliferation in the WT counterpart was confined to a single keratinocyte layer (Fig. 3A). Furthermore, keratinocyte differentiation, as indicated by the expression of the differentiation marker keratin K1, was severely arrested in K-KO epidermis (Fig. 3B). Cultured keratinocytes from K-KO mice exhibited a similar defect in differentiation in vitro as shown by reduced expression of the differentiation marker involucrin (Fig. 3C).
FIGURE 3.
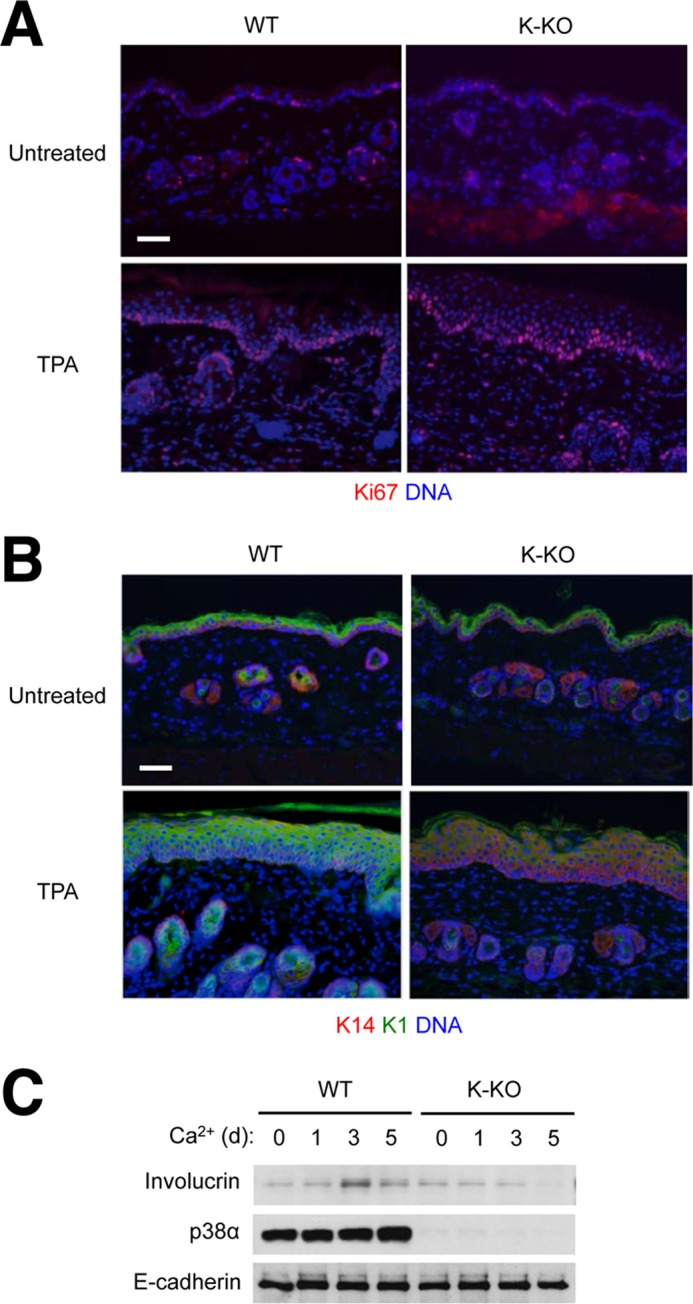
Epithelial-specific p38α ablation leads to a proliferation-differentiation imbalance in the skin. A and B, the shaved back skin of WT and K-KO mice was left untreated or treated with TPA (10 μg/animal) on two consecutive days; 2 days later, skin tissue sections were prepared from untreated and TPA-treated mice and analyzed by immunostaining with Ki67-specific antibody (A) and with Lys-14- and Lys-1-specific antibodies (B). Scale bar, 100 μm. C, keratinocytes from the indicated mice were treated with calcium chloride (2 mm) to induce differentiation. Whole cell lysates were prepared at the indicated time points and analyzed by immunoblotting with antibodies against the proteins indicated on the left. d, day.
We sought to determine whether the dysregulation of MAP kinase signaling in IEC-KO intestinal mucosa occurs throughout the p38α-deficient epithelium or in a spatially restricted pattern. To locate the tissue area of ERK activation, we analyzed WT and IEC-KO colon sections by immunostaining for phosphorylated ERK. A strong signal representing the active form of ERK was detected mainly in the crypt base in both WT and IEC-KO mice. Tissue areas showing ERK activation was, however, expanded in IEC-KO compared with WT colons (Fig. 4A). The areas with active ERK signaling overlapped with those containing proliferating epithelial cells (Fig. 4B). As in ileum tissue, IEC-KO colonic tissue contained an increased number of proliferating cells than the WT counterpart (Fig. 4C).
FIGURE 4.

Intestinal epithelial cells lacking p38α exhibit ERK hyperactivation at the crypt base and proliferate at higher rates in response to EGF. A–C, colon tissue sections from WT and IEC-KO mice were analyzed by immunostaining with p-ERK- and BrdU-specific antibodies (A and B, respectively). LS, luminal surface; CB, crypt base. Gray lines with double arrows indicate crypt length (B). Scale bar, 100 μm. The number of BrdU+ cells per crypt was determined (C). Data represent means ± S.E. **, p < 0.01. D–H, MODE-K cells were transfected with siRNA as described in Fig. 1 and treated with EGF (10 ng/ml) and PD98059 (PD; 20 μm) as indicated. After addition to the cells, EGF was present in the culture media throughout the incubation period. Whole cell lysates were prepared after the indicated durations of stimulation and analyzed by immunoblotting with antibodies against the proteins indicated on the left (D and F). Cell proliferation (E) and p-EGFR and EGFR amounts (G and H) were determined at the indicated time points after stimulation. The relative protein amount denotes the percentage of the protein intensity relative to the maximum in each immunoblot. Data represent means ± S.E. **, p < 0.01 (PD98059-treated versus untreated); *, p < 0.05 (p38α-KD versus control KD).
Given the spatial overlap of ERK hyperactivation and epithelial hyperproliferation in the crypt base of IEC-KO mice, we sought to test the causality between the two phenomena. The crypt base harbors intestinal stem cells and displays high EGFR signaling (40). It has been shown that intestinal stem cell proliferation is driven by EGFR and downstream ERK activation in both mammals and fruit flies (40–42). We therefore chose EGF as an inducer of ERK activity and cell proliferation for our in vitro model. MODE-K cells were stimulated with EGF in a serum-deprived condition to prevent high basal ERK phosphorylation in the presence of serum. This condition of EGF treatment caused a transient induction of phosphorylation of p38 and ERK but not JNK. KD of p38α resulted in enhanced ERK activation (Fig. 4D) and increased proliferation (Fig. 4E) of EGF-stimulated MODE-K cells. Blockade of ERK activation by chemical inhibitor treatment prevented the effects of both EGF and p38α KD on cell proliferation (Fig. 4E), confirming ERK activity as crucial for EGF-dependent epithelial cell proliferation and suggesting hyperactivated ERK as a likely driver of the enhanced proliferation of p38α-deficient cells.
Several studies have shown that p38α provides cues for degradation (43, 44) and endocytosis (45, 46) of EGFR by either directly phosphorylating it or acting through other regulatory molecules. Consistent with these findings, the abundance of active EGFR with phosphorylation at tyrosine 1173 and, to a lesser extent, total EGFR was increased in p38α-deficient MODE-K cells (Fig. 4, F-H), which accounts at least in part for the enhanced ERK activation caused by epithelial p38α ablation.
In contrast to the localization of active ERK to the crypt base, the immunostaining signals of the active forms of JNK and the JNK substrate c-Jun were detected in the colonic luminal surface (Fig. 5, A and B). The signals were substantially stronger in IEC-KO than WT colon sections. JNK signaling promotes various cellular processes, ranging from proliferation to differentiation to death, depending on the cell type and physiological context. Most notably, JNK hyperactivation has been linked to TNF-induced apoptosis (47). We examined whether the chronically elevated JNK activity in IEC-KO colon tissue enhanced epithelial apoptosis during DSS-induced acute colitis, a condition producing a TNF-rich tissue environment. IEC-KO mice developed more severe and longer-lasting inflammatory disease and incurred broader epithelial damage compared with WT animals during and after DSS administration (Fig. 5, C and D). The affected colon tissue in IEC-KO mice exhibited a collapse in epithelial integrity and increased apoptosis (Fig. 5E) and produced higher amounts of reactive oxygen species (Fig. 5F). To verify whether p38α deficiency leads to cell-intrinsic sensitivity to TNF-induced apoptosis in a JNK-dependent manner, we subjected MODE-K cells to p38α KD and TNF stimulation without and with JNK inhibitor treatment. This experiment confirmed the link between prolonged JNK activation following TNF exposure (Fig. 1A) and apoptotic sensitivity (Fig. 5, G and H) in p38α-deficient intestinal epithelial cells.
FIGURE 5.

Intestinal epithelial cells lacking p38α exhibit JNK hyperactivation at the luminal surface and predispose to inflammation-induced apoptosis. A and B, colon tissue sections from WT and IEC-KO mice were analyzed by immunostaining with p-JNK- and p-c-Jun-specific antibodies (A and B, respectively). LS, luminal surface; CB, crypt base. Arrowheads indicate areas where specific immunostaining is detected. Scale bar, 10 μm. C–F, WT and IEC-KO mice were orally administered DSS (3.5% in the drinking water) for 7 days. Changes in body weight were monitored daily (C). Colonic tissue sections were prepared on day 7 and analyzed by hematoxylin and eosin staining and TUNEL staining (E, upper and lower panels, respectively). Colonic tissue sections from untreated and DSS-treated mice were incubated with 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and analyzed by fluorescence microscopy (F). Clinical scores indicate the extent of inflammation (leukocyte infiltration and edema) and epithelial damage (D). Data represent means ± S.E. G and H, MODE-K cells were transfected with siRNA and treated with TNF (10 ng/ml) as described in Fig. 1 in the absence and presence of D-JNKi (1 μm). The rate of apoptosis was analyzed by agarose gel electrophoresis and ethidium bromide staining of genomic DNA obtained from the cells at the indicated time points (G) and TUNEL staining 16 h after TNF treatment (H). The number within each image indicates percentage of TUNEL+ cells and represents mean ± S.D.
JNK hyperactivation resulting from p38α deficiency was a phenomenon shared by several different cell types examined (Fig. 1). We explored signaling events upstream of JNK to trace the cause of this dysregulation and found that p38α ablation dramatically increased the amplitude and duration of activation of the kinase TAK1, which relays signals from cytokine receptors and Toll-like receptors to JNK (48). TAK1 hyperactivation in the absence of p38α expression occurred in TNF-treated epithelial cells and Toll-like receptor agonist-treated macrophages and dendritic cells (Fig. 6, A–D). Furthermore, we observed increased intensity and thicker layers of immunostaining for active TAK1 in the luminal surface of DSS-treated IEC-KO colons and the epidermis of TPA-treated K-KO skin (Fig. 6E).
FIGURE 6.

Ablation of p38α expression results in TAK1 hyperactivation in various cell types. A–D, MODE-K cells (A), keratinocytes (B), bone marrow-derived macrophages (C), and lymph node dendritic cells (D) were prepared, treated, and analyzed by immunoblotting as described in Fig. 1. E, colon tissue sections from DSS-treated WT and IEC-KO mice were prepared as described in Fig. 5 (left). Skin tissue sections from TPA-treated WT and K-KO mice were prepared as described in Fig. 3 (right). Both types of tissue sections were analyzed by immunostaining with p-TAK1-specific antibodies. Arrowheads indicate areas where specific immunostaining is detected. Scale bar, 100 μm. F and G, MODE-K cells (F) and immortalized mouse embryonic fibroblasts (G) were transfected with control and constitutively active p38α (p38αCA)-expressing plasmid vectors, treated with TNF 24 h after transfection and analyzed as described in Fig. 1. Anti-p-p38 antibody detects both transfected (gray arrow) and endogenous p38 (black arrow). H and I, 293T cells were transfected and treated with TNF as described in F. A plasmid vector expressing HA epitope-tagged TAK1 was included in all transfection samples. Whole cell lysates (Extracts) were further subjected to immunoprecipitation with anti-HA antibody. Extracts and immunoprecipitated samples (IP) were analyzed by immunoblotting (H). The ratio of ubiquitinated and total HA-TAK1 amount in the immunoprecipitated proteins was determined by densitometry (I). (Ub)n-TAK1, polyubiquitinated (poly-Ub) TAK1.
We tested whether an enforced increase in p38α kinase activity in cells would exert an effect opposite to that of p38α ablation. A constitutively active p38α variant, p38αCA, when expressed in MODE-K cells and immortalized mouse embryonic fibroblasts, suppressed basal as well as TNF-induced TAK1 activity (Fig. 6, F and G). In 293T cells, active p38α promoted lysine 48-linked ubiquitin chain conjugation to TAK1 (Fig. 6H), a process marking the protein for proteasomal degradation. Curiously, total TAK1 amounts were increased rather than decreased by p38αCA coexpression (Fig. 6H). Therefore, the increase in TAK1 ubiquitination in p38αCA-expressing 293T cells may be at least attributable in part to greater amounts of total TAK1. However, the ratio of ubiquitin-conjugated and total TAK1 amount was still higher with p38αCA coexpression (Fig. 6I). Hence, the mechanism of p38α regulation of TAK1 activation is possibly related to an accelerated turnover of activated TAK1 without proportionately reducing the pool of total TAK1.
To gain further mechanistic insight into the regulation of TAK1 activation by p38α, we examined LPS-induced intracellular signaling in RAW264.7 mouse macrophage cells. Inhibition of p38α kinase activity with two different chemical compounds, SC409 (49) and SB202190 (10), led to efficient suppression of signaling downstream of p38α, as indicated by decreases in MK2 phosphorylation. The p38α inhibitors, particularly SC409, recapitulated the aberrant activation of ERK, JNK, and TAK1 caused by genetic loss of p38α (Fig. 7A). Of note, extra TAK1 forms of higher than predicted molecular weight were detected in RAW264.7 cells. Similar multiple TAK1 forms were also generated when TAK1 was produced from a transfected expression vector (Fig. 7B). Inhibition of p38α kinase activity decreased incorporation of lysine 48-linked ubiquitin into high-molecular weight TAK1 forms without causing changes in total TAK1 amount (Fig. 7, B and C).
FIGURE 7.

Inhibition of p38α kinase activity results in TAK1 hyperactivation via affecting TAK1 ubiquitination and turnover. A, RAW264.7 mouse macrophage cells were preincubated with dimethyl sulfoxide (DMSO; 0.1%), SC409 (10 μm), and SB202190 (10 μm) before treatment with LPS (100 ng/ml). Whole cell lysates were prepared after the indicated durations of stimulation and analyzed by immunoblotting with antibodies against the proteins indicated on the left. Anti-p-TAK1 antibody and anti-TAK1 antibody detect proteins of higher apparent molecular weights (filled circles) in addition to those of the expected size (open circles). Numbers on the right indicate corresponding molecular weight marker positions. B and C, RAW264.7 cells were transfected with an HA-TAK1-expressing plasmid vector and treated with dimethyl sulfoxide, SC409, and LPS as described in A. Whole cell lysates were prepared and subjected to immunoprecipitation with anti-HA antibody and analyzed by immunoblotting (B). The ratio of ubiquitinated and total HA-TAK1 amount in the immunoprecipitated proteins was determined by densitometry (C). D and E, RAW264.7 cells were preincubated with dimethyl sulfoxide and MG132 (20 μm) before treatment with LPS. Whole cell lysates were prepared and analyzed by immunoblotting (D) as described in A. Whole cell lysate from cells preincubated with MG132 and treated with LPS for 60 min (corresponding to the eighth lane in D; In, Input) was subjected to immunoprecipitation with control (−) and anti-ubiquitin (Ub) antibody and analyzed by immunoblotting (E). poly-Ub, polyubiquitin.
Given the disproportionate effects of p38α inhibition on TAK1 ubiquitination and on total TAK1 abundance, we reasoned that active TAK1 comprised only a small proportion of total TAK1 pool in LPS-treated RAW264.7 cells and that this small TAK1 subset was preferentially targeted for proteasomal degradation. In support of this idea, TAK1 and JNK activation was elevated and sustained in RAW264.7 cells treated with LPS in conjunction with the proteasome inhibitor MG132 (Fig. 7D). Importantly, there was an accumulation of phosphorylated TAK1 with molecular mass above 150 kDa in MG132-treated cells. These high-molecular weight phosphorylated forms were also ubiquitin-conjugated (Fig. 7E), suggesting a role for ubiquitination and proteasomal activity in the elimination of activated TAK1. ERK and p38 activation was somewhat prolonged but not increased in intensity by MG132 treatment. The differential effects of inhibition of proteasomal activity on the three MAP kinase cascades illustrate a hitherto unknown complexity in this regulatory circuitry.
DISCUSSION
Our findings reveal that p38α serves important regulatory functions in the protein kinase signaling circuitry underlying tissue homeostasis. Multiple protein kinase modules did not operate with restraint and harmony when p38α was not available to control the magnitude and timing of their activation. The failure of limiting and coordinating the activation of EGFR, ERK, TAK1, and JNK led to complex physiological consequences. Loss of epithelial p38α tipped the balance of tissue homeostasis in favor of cell proliferation at the expense of differentiation. This anomaly, along with the increased apoptotic sensitivity, left the affected epithelium vulnerable to damage following exposure to external injurious stimuli, and to self-destructive inflammatory responses.
p38α has been shown to phosphorylate TAK1-binding protein (TAB1 (transforming growth factor β-activated kinase 1-binding protein)), an event having a negative effect on TAK1 activation (50). We observed p38α-dependent TAB1 phosphorylation in ultraviolet radiation B-exposed keratinocytes but not convincingly in TNF-treated epithelial cells or Toll-like receptor agonist-stimulated macrophages. It is therefore unclear whether TAB1 phosphorylation is mechanistically related to TAK1 ubiquitination and turnover. It remains a possibility that p38α regulates TAK1 activation through multiple mechanisms in a cell type- and signaling pathway-specific manner.
The tissue disturbances and susceptibility to inflammatory damage that arise from genetic ablation of p38α expression lend a likely explanation of the adverse effects resulting from pharmacological inhibition of p38α in clinical studies. It is conceivable that simultaneous inhibition of one or more of the hyperactivated protein kinases can alleviate the toxicity of p38α inhibitors. Alternatively, therapeutic targeting of molecular events downstream of p38α but unrelated to the dysregulated signaling pathways may prove equally efficacious as p38α inhibitors while circumventing their adverse effects.
Acknowledgments
We thank J.-H. Shim for the TAK1 expression vector and technical advice; D. Engelberg for the p38αCA expression vector; P. Cohen for the TAK1-specific antibody; C. Libert for mouse recombinant TNF; and A. H. Lee for advice on the use of MODE-K cells.
This work was supported by National Institutes of Health Grant AI074957 (to J. M. P.).
- EGFR
- EGF receptor
- DSS
- dextran sulfate sodium
- p38αCA
- constitutively active p38α variant
- TAK1
- transforming growth factor β-activated kinase 1
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- KD
- knockdown.
REFERENCES
- 1. Chang L., Karin M. (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40 [DOI] [PubMed] [Google Scholar]
- 2. Johnson G. L., Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912 [DOI] [PubMed] [Google Scholar]
- 3. Dong C., Davis R. J., Flavell R. A. (2002) MAP kinases in the immune response. Annu. Rev. Immunol. 20, 55–72 [DOI] [PubMed] [Google Scholar]
- 4. Simons B. D., Clevers H. (2011) Strategies for homeostatic stem cell self-renewal in adult tissues. Cell 145, 851–862 [DOI] [PubMed] [Google Scholar]
- 5. Hsu Y. C., Fuchs E. (2012) A family business: stem cell progeny join the niche to regulate homeostasis. Nat. Rev. Mol. Cell Biol. 13, 103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vereecke L., Beyaert R., van Loo G. (2011) Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol. Med. 17, 584–593 [DOI] [PubMed] [Google Scholar]
- 7. Kubo A., Nagao K., Amagai M. (2012) Epidermal barrier dysfunction and cutaneous sensitization in atopic diseases. J. Clin. Invest. 122, 440–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Medema J. P., Vermeulen L. (2011) Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 474, 318–326 [DOI] [PubMed] [Google Scholar]
- 9. Dotto G. P. (2008) Notch tumor suppressor function. Oncogene 27, 5115–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee J. C., Laydon J. T., McDonnell P. C., Gallagher T. F., Kumar S., Green D., McNulty D., Blumenthal M. J., Heys J. R., Landvatter S. W. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 [DOI] [PubMed] [Google Scholar]
- 11. Han J., Lee J. D., Bibbs L., Ulevitch R. J. (1994) A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265, 808–811 [DOI] [PubMed] [Google Scholar]
- 12. Tamura K., Sudo T., Senftleben U., Dadak A. M., Johnson R., Karin M. (2000) Requirement for p38 in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell 102, 221–231 [DOI] [PubMed] [Google Scholar]
- 13. Adams R. H., Porras A., Alonso G., Jones M., Vintersten K., Panelli S., Valladares A., Perez L., Klein R., Nebreda A. R. (2000) Essential role of p38 MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell 6, 109–116 [PubMed] [Google Scholar]
- 14. Allen M., Svensson L., Roach M., Hambor J., McNeish J., Gabel C. A. (2000) Deficiency of the stress kinase p38 results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J. Exp. Med. 191, 859–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mudgett J. S., Ding J., Guh-Siesel L., Chartrain N. A., Yang L., Gopal S., Shen M. M. (2000) Essential role for p38 mitogen-activated protein kinase in placental angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 97, 10454–10459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hui L., Bakiri L., Mairhorfer A., Schweifer N., Haslinger C., Kenner L., Komnenovic V., Scheuch H., Beug H., Wagner E. F. (2007) p38 suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet. 39, 741–749 [DOI] [PubMed] [Google Scholar]
- 17. Ventura J. J., Tenbaum S., Perdiguero E., Huth M., Guerra C., Barbacid M., Pasparakis M., Nebreda A. R. (2007) p38 MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet. 39, 750–758 [DOI] [PubMed] [Google Scholar]
- 18. Nishida K., Yamaguchi O., Hirotani S., Hikoso S., Higuchi Y., Watanabe T., Takeda T., Osuka S., Morita T., Kondoh G., Uno Y., Kashiwase K., Taniike M., Nakai A., Matsumura Y., Miyazaki J., Sudo T., Hongo K., Kusakari Y., Kurihara S., Chien K. R., Takeda J., Hori M., Otsu K. (2004) p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 24, 10611–10620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim C., Sano Y., Todorova K., Carlson B. A., Arpa L., Celada A., Lawrence T., Otsu K., Brissette J. L., Arthur J. S., Park J. M. (2008) The kinase p38α serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat. Immunol. 9, 1019–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang G., Wang Y., Vogel P., Kanneganti T. D., Otsu K., Chi H. (2012) Signaling via the kinase p38α programs dendritic cells to drive TH17 differentiation and autoimmune inflammation. Nat. Immunol. 13, 152–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Otsuka M., Kang Y. J., Ren J., Jiang H., Wang Y., Omata M., Han J. (2010) Distinct effects of p38α deletion in myeloid lineage and gut epithelia in mouse models of inflammatory bowel disease. Gastroenterology 138, 1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heinrichsdorff J., Luedde T., Perdiguero E., Nebreda A. R., Pasparakis M. (2008) p38α MAPK inhibits JNK activation and collaborates with IκB kinase 2 to prevent endotoxin-induced liver failure. EMBO Rep. 9, 1048–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guma M., Hammaker D., Topolewski K., Corr M., Boyle D. L., Karin M., Firestein G. S. (2012) Antiinflammatory functions of p38 in mouse models of rheumatoid arthritis: advantages of targeting upstream kinases MKK-3 or MKK-6. Arthritis Rheum. 64, 2887–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kang Y. J., Chen J., Otsuka M., Mols J., Ren S., Wang Y., Han J. (2008) Macrophage deletion of p38α partially impairs lipopolysaccharide-induced cellular activation. J. Immunol. 180, 5075–5082 [DOI] [PubMed] [Google Scholar]
- 25. Cohen S. B., Cheng T. T., Chindalore V., Damjanov N., Burgos-Vargas R., Delora P., Zimany K., Travers H., Caulfield J. P. (2009) Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 60, 335–344 [DOI] [PubMed] [Google Scholar]
- 26. Damjanov N., Kauffman R. S., Spencer-Green G. T. (2009) Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 60, 1232–1241 [DOI] [PubMed] [Google Scholar]
- 27. Genovese M. C., Cohen S. B., Wofsy D., Weinblatt M. E., Firestein G. S., Brahn E., Strand V., Baker D. G., Tong S. E. (2011) A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral SCIO-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J. Rheumatol. 38, 846–854 [DOI] [PubMed] [Google Scholar]
- 28. Ritprajak P., Hayakawa M., Sano Y., Otsu K., Park J. M. (2012) Cell type-specific targeting dissociates the therapeutic from the adverse effects of protein kinase inhibition in allergic skin disease. Proc. Natl. Acad. Sci. U.S.A. 109, 9089–9094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Madison B. B., Dunbar L., Qiao X. T., Braunstein K., Braunstein E., Gumucio D. L. (2002) Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283 [DOI] [PubMed] [Google Scholar]
- 30. Vidal K., Grosjean I., evillard J. P., Gespach C., Kaiserlian D. (1993) Immortalization of mouse intestinal epithelial cells by the SV40-large T gene. Phenotypic and immune characterization of the MODE-K cell line. J. Immunol. Methods 166, 63–73 [DOI] [PubMed] [Google Scholar]
- 31. Singhirunnusorn P., Suzuki S., Kawasaki N., Saiki I., Sakurai H. (2005) Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-β-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J. Biol. Chem. 280, 7359–7368 [DOI] [PubMed] [Google Scholar]
- 32. Greenblatt M. B., Shim J. H., Zou W., Sitara D., Schweitzer M., Hu D., Lotinun S., Sano Y., Baron R., Park J. M., Arthur S., Xie M., Schneider M. D., Zhai B., Gygi S., Davis R., Glimcher L. H. (2010) The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Invest. 120, 2457–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Askari N., Diskin R., Avitzour M., Capone R., Livnah O., Engelberg D. (2007) Hyperactive variants of p38α induce, whereas hyperactive variants of p38γ suppress, activating protein 1-mediated transcription. J. Biol. Chem. 282, 91–99 [DOI] [PubMed] [Google Scholar]
- 34. Park J. M., Ng V. H., Maeda S., Rest R. F., Karin M. (2004) Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med. 200, 1647–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Javelaud D., Besançon F. (2001) NF-κB activation results in rapid inactivation of JNK in TNF α-treated Ewing sarcoma cells: a mechanism for the anti-apoptotic effect of NF-κB. Oncogene 20, 4365–4372 [DOI] [PubMed] [Google Scholar]
- 36. De Smaele E., Zazzeroni F., Papa S., Nguyen D. U., Jin R., Jones J., Cong R., Franzoso G. (2001) Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature 414, 308–313 [DOI] [PubMed] [Google Scholar]
- 37. Tang G., Minemoto Y., Dibling B., Purcell N. H., Li Z., Karin M., Lin A. (2001) Inhibition of JNK activation through NF-κB target genes. Nature 414, 313–317 [DOI] [PubMed] [Google Scholar]
- 38. Zhang J. Y., Green C. L., Tao S., Khavari P. A. (2004) NF-κB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev. 18, 17–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maeda S., Kamata H., Luo J. L., Leffert H., Karin M. (2005) IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121, 977–990 [DOI] [PubMed] [Google Scholar]
- 40. Wong V. W., Stange D. E., Page M. E., Buczacki S., Wabik A., Itami S., van de Wetering M., Poulsom R., Wright N. A., Trotter M. W., Watt F. M., Winton D. J., Clevers H., Jensen K. B. (2012) Lrig1 controls intestinal stem-cell homeostasis by negative regulation of ErbB signalling. Nat. Cell Biol. 14, 401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Biteau B., Jasper H. (2011) EGF signaling regulates the proliferation of intestinal stem cells in Drosophila. Development 138, 1045–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang H., Grenley M. O., Bravo M. J., Blumhagen R. Z., Edgar B. A. (2011) EGFR/Ras/MAPK signaling mediates adult midgut epithelial homeostasis and regeneration in Drosophila. Cell Stem Cell 8, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frey M. R., Dise R. S., Edelblum K. L., Polk D. B. (2006) p38 kinase regulates epidermal growth factor receptor downregulation and cellular migration. EMBO J. 25, 5683–5692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swat A., Dolado I., Rojas J. M., Nebreda A. R. (2009) Cell density-dependent inhibition of epidermal growth factor receptor signaling by p38α mitogen-activated protein kinase via Sprouty2 downregulation. Mol. Cell. Biol. 29, 3332–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zwang Y., Yarden Y. (2006) p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. EMBO J. 25, 4195–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Singhirunnusorn P., Ueno Y., Matsuo M., Suzuki S., Saiki I., Sakurai H. (2007) Transient suppression of ligand-mediated activation of epidermal growth factor receptor by tumor necrosis factor-α through the TAK1-p38 signaling pathway. J. Biol. Chem. 282, 12698–12706 [DOI] [PubMed] [Google Scholar]
- 47. Deng Y., Ren X., Yang L., Lin Y., Wu X. (2003) A JNK-dependent pathway is required for TNFα-induced apoptosis. Cell 115, 61–70 [DOI] [PubMed] [Google Scholar]
- 48. Sakurai H. (2012) Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 33, 522–530 [DOI] [PubMed] [Google Scholar]
- 49. Mbalaviele G., Anderson G., Jones A., De Ciechi P., Settle S., Mnich S., Thiede M., Abu-Amer Y., Portanova J., Monahan J. (2006) Inhibition of p38 mitogen-activated protein kinase prevents inflammatory bone destruction. J. Pharmacol. Exp. Ther. 317, 1044–1053 [DOI] [PubMed] [Google Scholar]
- 50. Cheung P. C., Campbell D. G., Nebreda A. R., Cohen P. (2003) Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J. 22, 5793–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]