

Background: The mechanisms by which IFNs generate antineoplastic responses remain to be defined.
Results: Type I IFN treatment results in activation of the Mnk/eIF4E pathway in Jak2V617F-transformed cells, and this activation is required for the antineoplastic effect.
Conclusion: Mnk kinases are essential for the antineoplastic effects of IFN.
Significance: This study provides evidence for a key mechanism mediating the effects of IFNs in malignant MPN precursors.
Keywords: Antiviral Agents, Innate Immunity, Interferon, MAP Kinases (MAPKs), Phosphatidylinositol 3-Kinase, RNA, Translation Control, Translation Initiation Factors
Abstract
The mechanisms of generation of the antineoplastic effects of interferons (IFNs) in malignant hematopoietic cells remain to be precisely defined. We examined the activation of type I IFN-dependent signaling pathways in malignant cells transformed by Jak2V617F, a critical pathogenic mutation in myeloproliferative neoplasms (MPNs). Our studies demonstrate that during engagement of the type I IFN receptor (IFNAR), there is activation of Jak-Stat pathways and also engagement of Mnk kinases. Activation of Mnk kinases is regulated by the Mek/Erk pathway and is required for the generation of IFN-induced growth inhibitory responses, but Mnk kinase activation does not modulate IFN-regulated Jak-Stat signals. We demonstrate that for type I IFNs to exert suppressive effects in malignant hematopoietic progenitors from patients with polycythemia vera, induction of Mnk kinase activity is required, as evidenced by studies involving pharmacological inhibition of Mnk or siRNA-mediated Mnk knockdown. Altogether, these findings provide evidence for key and essential roles of the Mnk kinase pathway in the generation of the antineoplastic effects of type I IFNs in Jak2V617F-dependent MPNs.
Introduction
Extensive work during the past four decades has established that IFNs exhibit important biological activities, most prominent of which are their antiviral and antineoplastic effects (1–5). IFNs also exhibit important immunomodulatory properties (1–5). The demonstration that IFNs have antiviral and antitumor properties has led to extensive clinical trials over the years that have established that one type I IFN subtype, IFNα, has major activity for the treatment of hepatitis and certain malignancies (6–10). In addition, IFNβ is used extensively for the treatment of multiple sclerosis (11, 12). It should be noted that beyond the therapeutic properties of IFNs in various malignancies, the IFN system appears to be involved in the pathophysiology of some autoimmune diseases such as Sjögren syndrome and systemic lupus erythematosus (13–15) as well as in the immune pathophysiology of aplastic anemia (16), underscoring the importance and physiological relevance of regulation of the IFN system in humans (13–15).
The type I IFN receptor (IFNAR)4 activates multiple signaling cascades that control the generation of IFN biological effects via coordinated engagement of downstream effectors (5, 17). These include Jak-Stat pathways (reviewed in Refs. 18 and 19), Map kinase pathways (reviewed in Refs. 5, 20, and 21), PKC-dependent pathways (reviewed in Ref. 22), and mTOR signaling cascades (reviewed in Refs. 23 and 24). In recent years, a better understanding of the relevance of the different products of IFN-stimulated genes, regulated by these specific signaling pathways, has defined their specific roles in the generation of IFN responses (25, 26). It appears that the coordinated function of Jak-Stat pathways and cascades that control mRNA translation ultimately result in production of proteins with tumor suppressor or antiviral properties to generate antineoplastic and antiviral responses, respectively. There is accumulating evidence that mTOR-initiated signals constitute key effector pathways that regulate translation of IFN-stimulated genes (23, 24, 27–31). Utilization of mTOR pathways downstream of IFNAR is of substantial interest, as mTOR-controlled signals are also utilized to regulate mRNA translation of genes, the expression of which is controlled by growth factors, oncogenes, and other transformation signals (23). Similarly, there is evidence that other pathways known to be involved in growth factor and pro-oncogenic signaling, such as the Mek/Erk pathway, are also involved in the control of IFN-dependent mRNA translation of IFN-stimulated genes by engaging Mnk kinases and regulating phosphorylation of the eIF4E in a Mnk-dependent manner (24, 32, 33).
Philadelphia chromosome negative (Ph−) myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell disorders and include polycythemia vera (PV), essential thrombocytosis, and myelofibrosis (34–36). PV and other MPNs are characterized by high sensitivity to the antiproliferative effects of type I IFNs, and this has resulted in the introduction of IFNα for the treatment of patients suffering from these diseases (34–36). However, despite the therapeutic effectiveness of IFNα in the treatment of MPNs, their mechanism(s) of action related to their antineoplastic activity in MPNs remains largely unknown. In the present study, we undertook a systematic analysis to define the role of Mnk kinase pathways in Jak2V617F-transformed MPN cells. The Jak2 mutation V617F is pathogenic in MPNs (34). Our data provide the first evidence that IFN-dependent activation of Mnk kinases downstream of the Mek/Erk pathway is required for generation of IFN-induced growth inhibitory responses in MPN cells. Remarkably, transformation of normal hematopoietic precursors with Jak2V617F results in enhanced IFN sensitivity, whereas Mnk kinase activity is essential for the suppressive effects of IFNα on primary malignant progenitors from patients with PV, establishing a critical role for the Mnk pathway in the generation of the antineoplastic effects of type I IFNs.
MATERIALS AND METHODS
Cell Lines, Reagents, and Antibodies
HEL cells were grown in RPMI supplemented with 10% (v/v) fetal bovine serum and antibiotics. Recombinant human IFNα was from Hoffman LaRoche, and recombinant human and mouse IFNβ were from Biogen Idec. The antibodies against p-Mnk1 (Thr197/202), p-eIF4E (Ser209), eIF4E, pSTAT1 (Tyr701), p-STAT3 (Tyr705), STAT3, p-Erk1/2 (Thr202/Tyr204), Erk1/2, MEK1, and JAK2 were obtained from Cell Signaling Technology (Danvers, MA). The antibody against STAT1 was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The antibody for GAPDH was obtained from Millipore (Billerica, MA). The Mnk inhibitor (Mnk-I) was obtained from Calbiochem (Darmstadt, Germany). Ba/F3-EpoR cells and Ba/F3-EpoR cells expressing WT Jak2 or Jak2V617F were kindly provided by Dr. Gary Gililand. For gene silencing by siRNA, cells were transfected with control non-targeting or double-stranded RNA oligonucleotides (Dharmacon) directed to Mnk1 (SMARTpool L-004879-00-0005), Mnk2 (SMARTpool L-004908-0005) MEK1 (L-003571-00-0005), Erk1 (L-003592-00-0005), and Erk2 (L-003555-00-0005) using the Amaxa Biosystems Nucleofection kit per the manufacturer's instructions.
Cell Lysis and Immunoblotting
Cells were treated, lysed in phosphorylation lysis buffer containing protease and phosphatase inhibitors, and prepared for immunoblotting as described previously (33, 37). In some experiments, cells were serum-starved overnight and treated with 104 units/ml of IFNα or IFNβ. Immunoblotting using an enhanced chemiluminescence method (Amersham Biosciences) was carried out as described in previous studies (33, 37). Bands were quantified by densitometry using ImageJ software.
Hematopoietic Cell Progenitor Assays
Peripheral blood from patients with PV patients was collected after obtaining consent approved by the Institutional Review Board of Northwestern University, and mononuclear cells were isolated following Histopaque density gradient separation (Sigma). Hematopoietic progenitor colony formation for human erythroid precursors (burst-forming unit erythroid (BFU-E)) was determined in clonogenic assays in methylcellulose as described in our previous studies (32, 37). In some experiments, cells were treated with either MNK inhibitor or DMSO control at a final concentration of 10 μm. Cells were plated in duplicate in complete methylcellulose (Stem Cell Technologies, Vancouver, Canada) and were subsequently cultured for 2 weeks at 37 °C and 5% CO2, and hematopoietic colony formations were scored as described in our previous studies (32, 37, 38).
Evaluation of Erythroid Differentiation and Overexpression of JAK2 Constructs
Differentiation of human CD34+ cells to colony-forming units erythroid (CFU-E) was achieved by culture in a cytokine mixture as described previously (39, 40). Primary normal CD34+ progenitor cells were obtained from Stem Cell Technologies. Expanded CFU-E were nucleofected with JAK2wt-IRES-GFP or JAK2V617F-IRES-GFP vectors (41) following the manufacturer's instructions (Amaxa AG, Cologne, Germany), and the expression of JAK2 was assessed after 48 h.
Cell Viability/Proliferation Assays
Experiments using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide methodology were carried out as described in our previous studies (42).
Mobility Shift Assays
Actively growing cells were treated with 104 international units/ml human IFNβ for 15 min. Equal amounts of nuclear extracts from untreated or IFN-treated cells were analyzed using electrophoretic mobility shift assays with oligonucleotides to detect sis-inducible factor complexes as described in our previous studies (43).
Quantitative Real Time PCR (Taqman)
Cellular mRNA was reverse-transcribed into cDNA using the Omniscript RT kit and oligo(dT) primer (Qiagen) as described previously (33). Quantitative PCR using commercially available FAM-labeled probes and primers (Applied Biosystems) to determine Mnk1 and Mnk2 mRNA expression was used. GAPDH was used for normalization. The mRNA amplification was calculated as described previously (44).
RESULTS
We examined the activation of IFNAR-dependent signaling pathways in cells expressing the Jak2V617F mutation, which is a critical pathogenic mutation in MPNs (35). For these studies, the human erythroleukemia HEL cell line that expresses the Jak2V617F mutation was used. As shown in Fig. 1, type I IFN treatment of HEL cells resulted in phosphorylation/activation of Mnk1 (Fig. 1A) and its downstream effector eIF4E (Fig. 1B). In addition, IFN-activated Jak-Stat pathways were also engaged as reflected by the inducible phosphorylation of Stat1 on tyrosine 701 (Fig. 1C) and Stat3 on tyrosine 705 (Fig. 1D), both of which are classic events in IFN signaling. There was a dose-dependent inhibition of cell viability of HEL cells during IFN treatment (Fig. 1E), indicating that these cells are sensitive to type I IFN-mediated growth inhibition. In further studies, we established that IFN-dependent phosphorylation of eIF4E in HEL cells is Mnk-dependent (Fig. 2A) and that engagement of the Mnk/eIE4E pathway is MEK/Erk dependent, as shown by studies in which MEK1 or Erk1/2 were knocked down using specific siRNAs (Fig. 2, B and C, and supplemental Fig. S1). Consistent with this, engagement of Mnk1 (Fig. 2D) or eIF4E (Fig. 2E) via IFNAR in HEL cells was partially inhibited by pharmacological inhibition of Mek/Erk. Notably, as expected, phosphorylation of Stat1 was Mek/Erk-independent (Fig. 2E). Indeed, Mnk activity did not affect formation of Stat homo- or heterodimers that bind to SIE (Fig. 2, F and G), indicating that the Mnk signaling pathway operates independently of the Jak-Stat pathway in Jak2V617F-expressing cells. Altogether, these studies suggest that IFN activation of IFNAR in Jak2V617F-expressing cells results in activation of the Mnk/eIF4E pathway in a Mek/Erk-dependent manner, with parallel activation of Jak-Stat pathways.
FIGURE 1.

Activation of type I IFN-dependent signaling pathways in human JAK2V617F expressing cells. A and B, HEL cells were treated with human IFNβ for the indicated times. Protein lysates were resolved by SDS-PAGE and immunoblotted with an antibody against phosphorylated Mnk1 (Thr197/202) (A) or an antibody against phosphorylated eIF4E (Ser209) (B). Lysates from the same experiments were analyzed separately and immunoblotted for total Mnk1 (A) or total eIF4E (B). Both of the blots were probed with an antibody against GAPDH. In A, the anti-GAPDH blot for the p-Mnk1 and p-eIF4E is shown, whereas in B, the anti-GAPDH blot for the anti-Mnk1 and anti-eIF4E blots are shown. Bands were quantified by densitometry, and data are expressed as ratios of p-Mnk1/Mnk1 and p-eIF4E/eIF4E, respectively. C and D, HEL cells were treated with human IFNβ for the indicated times. Proteins in lysates were resolved by SDS-PAGE and immunoblotted with antibodies against phosphorylated STAT1 (Tyr701) (C) or phosphorylated STAT3 (Tyr705) (D). The blots were then stripped and reprobed with an antibody against STAT1 or STAT3, respectively. Bands were quantified by densitometry, and data were expressed as ratios of p-STAT1/STAT1 and p-STAT3/STAT3, respectively. E, HEL cells were treated for 7days with the indicated concentrations of IFNβ, and cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays. Data are expressed as percentage control cells and represent means ± S.E. of four independent experiments. IU, international units; 15′, 15 min; 30′, 30 min; 120′, 120 min.
FIGURE 2.

Type I IFN-dependent engagement of Mnk1 and eIF4E in JAK2V617F transformed cells is Mek/Erk-dependent. A–C, HEL cells were transfected with either control (Ctrl) siRNA or Mnk1and Mnk2 siRNA (A) or MEK1 siRNA (B) or Erk1 and Erk2 (C) as indicated, followed by treatment with IFNβ for the indicated times. The cells were lysed, and equal amounts of protein were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. D and E, HEL cells were pretreated with either DMSO or the Mek1/2 inhibitor U0126 (10 μm) for 60 min and followed by treatment with human IFNβ for the indicated times. Equal amounts of protein were resolved by SDS-PAGE and immunoblotted with antibodies against phosphorylated Mnk1 (Thr197/202) (D), phosphorylated Erk1/2 (Thr202/Tyr204) (D), or phosphorylated eIF4E (Ser209) (E) or phosphorylated Stat1 (Tyr701) (E). The blots were stripped and reprobed with antibodies against Mnk1or Erk1/2, respectively (D), and eIF4E or STAT1, respectively (E). F, HEL cells were pretreated with a Mnk-I prior to treatment with IFNβ, as indicated. Nuclear extracts were reacted with 40,000 cpm of 32P-labeled SIE oligonucleotides, and complexes were resolved by native gel electrophoresis and visualized by autoradiography. The migration of the different STAT complexes is indicated by arrows. SIF, sis-inducible factor. G, similar experiment as in F, except that supershifts with an antibody against Stat1 or control RIgG were performed, as indicated.
To examine whether expression of Jak2V617F modulates IFN-signaling pathways, we used Ba/F3 cells expressing WT Jak2 or the Jak2V617F mutant (45). Expression of WT-Jak2 did not modulate type I IFN-dependent phosphorylation of Stat1 on Tyr701 (Fig. 3A), but Jak2V617F expression reduced this phosphorylation (Fig. 3A). Similarly, phosphorylation of Stat3 on Tyr705 was decreased in cells expressing V617F-Jak2 (Fig. 3B), whereas IFN-dependent phosphorylation of Akt was intact (Fig. 3C). When the effects of V617F-Jak2 expression on the Erk/Mnk/eIF4E pathway were examined, we found that there was enhanced phosphorylation/activation in cells expressing Jak2V617F compared with WT-Jak2 (Fig. 3, D and E). Interestingly, Ba/F3-V617FJak2 cells were less sensitive to the growth inhibitory effects of mouse IFNβ (Fig. 4, A and B), but for both WT-Jak2 and Jak2V617F-expressing cells, the Mnk-I pharmacological inhibitor partially reversed IFN-suppressive responses (Figs. 4A and B).
FIGURE 3.
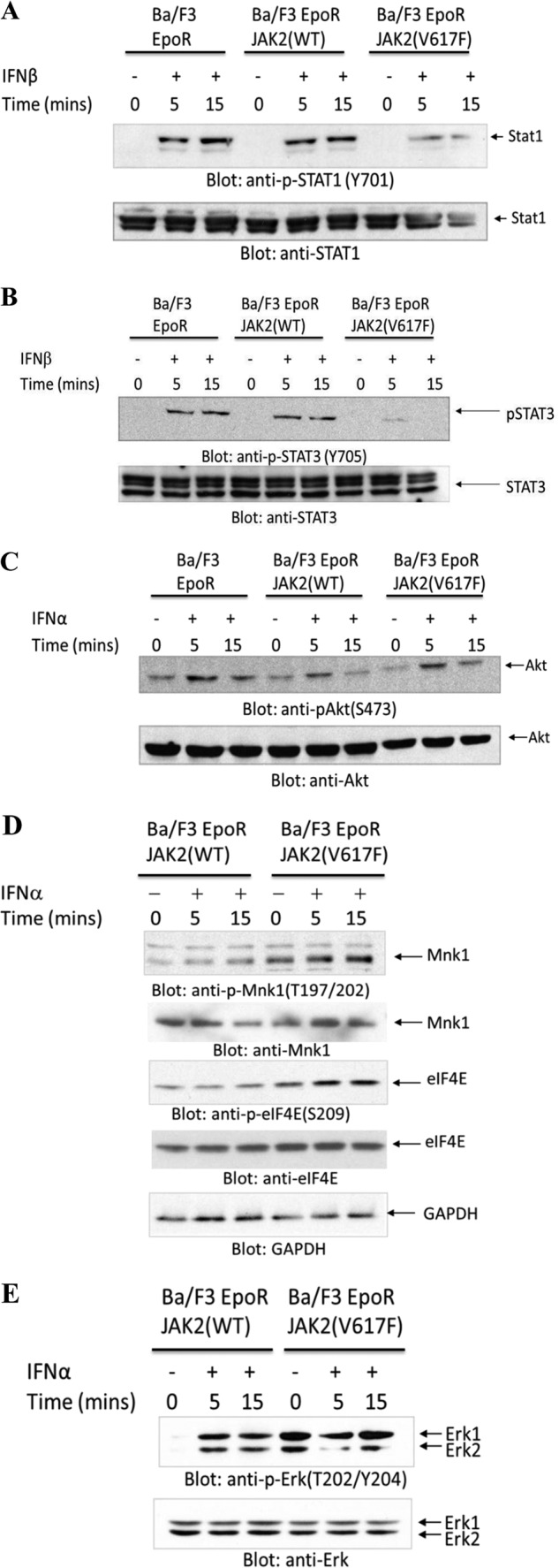
Effects of JAK2V617F expression on IFN-activated signaling pathways. A–C, Ba/F3 cells expressing WT-Jak2 or Jak2V617F and/or EpoR, as indicated, were treated for the indicated times with IFNβ and IFNα, and cell lysates were immunoblotted with antibodies against the phosphorylated form of Stat1 on tyrosine 701 or against Stat1 (A); antibodies against the phosphorylated form of Stat3 on tyrosine 705 or against Stat3 (B); or antibodies against the phosphorylated form of Akt on serine 473 or against Akt (C). D and E, Ba/F3 cells expressing EpoR and WT-Jak2 or Jak2V617F were treated for the indicated times with IFNα, and cell lysates were immunoblotted with antibodies against the phosphorylated form of Mnk1 on threonine 197/202 or against Mnk1 or the phosphorylated form of eIF4E on serine 209 or against eIF4E or against GAPDH (D); or against the phosphorylated form of Erk on threonine 202/tyrosine 204 or against Erk (E) as indicated.
FIGURE 4.
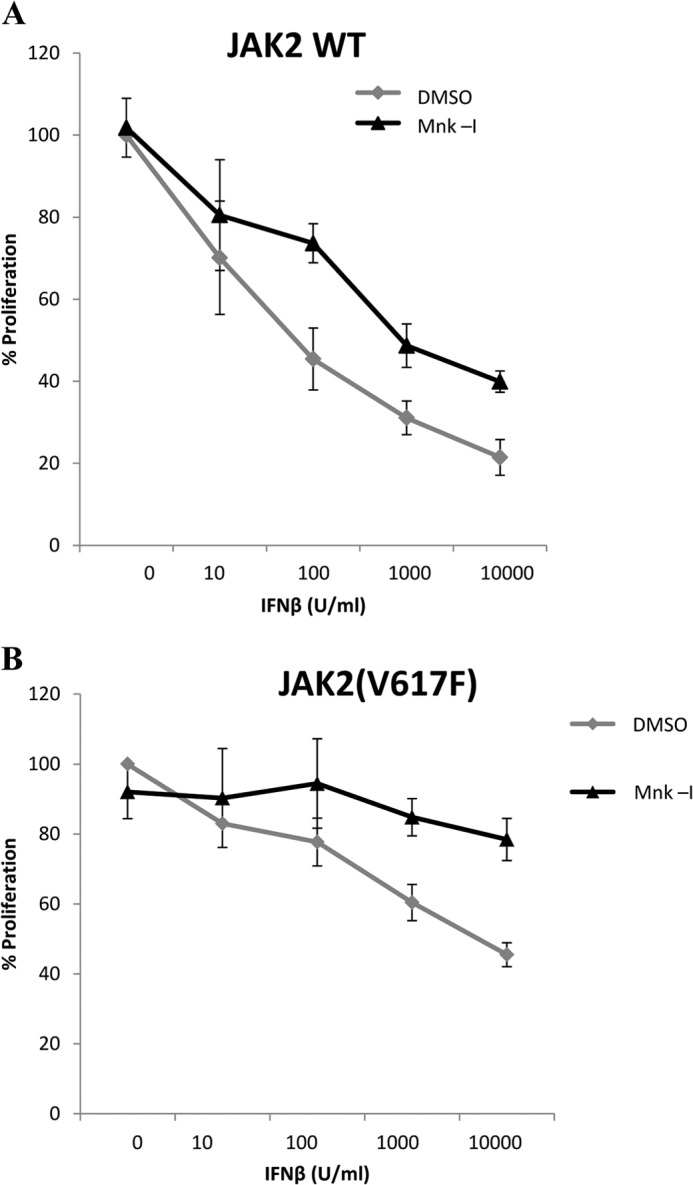
Requirement of Mnk kinase activity for the antiproliferative effects of IFNb. A and B, Ba/F3 cells expressing the EpoR and either WT-JAK2 (A) or Jak2V617F (B) were pretreated with either DMSO or Mnk-I and then treated for 7 days with the indicated concentrations of IFNβ. Cell viability was determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide assays. Data are expressed as the percentage of control cells and represent means ± S.E. of three experiments.
IFNs are potent suppressors of normal hematopoiesis (17, 21), and previous studies have implicated Mnk pathways in the generation of the inhibitory effects of type I IFNs in normal hematopoietic precursors (32, 33). In studies examining the expression of Mnk1 and Mnk2 during differentiation of normal human bone marrow cells, we found that there is a gradual increase of both Mnk1 and Mnk2 mRNA during erythroid differentiation, with the highest expression seen on days 13/14 (Fig. 5, A and B). This suggests an important role for Mnk kinases during erythroid development and led us to further studies to examine their roles in the generation of the effects of type I IFNs on malignant erythropoiesis caused by the Jak2V617F mutation. To determine whether Mnk-generated signals are required for the generation of the antineoplastic effects of type I IFNs in cells transformed by Jak2V617F, we transduced normal human bone marrow-derived CD34+ cells with retroviral vectors expressing either WT Jak2 or mutant V617F Jak2 (Fig. 5C). When the inhibitory effects of IFNα were assessed on transduced CD34+-derived hematopoietic precursors, we found that IFNα exhibited suppressive effects in both myeloid (CFU-GM) and erythroid (BFU-E) precursors (Fig. 5D). Mnk-I treatment reversed the effects of IFNα on both BFU-E and CFU-GM in human hematopoietic cells expressing WT-Jak2 (Fig. 5D). Notably, Mnk-I treatment of cells transduced with Jak2V617F resulted in reversal of the effects of IFNα on transformed BFU-E progenitors but had minimal effects on CFU-GM precursors (Fig. 5D), suggesting relative selectivity in malignant erythroid progenitors and the involvement of additional signaling pathways acting independently of Mnk/eIF4E.
FIGURE 5.

Expression of Mnk1 and Mnk2 during normal human bone marrow erythroid differentiation and their roles as mediators of Type I IFN-dependent suppression of JAK2V617F transformed CD34+-derived human erythroid progenitors. A and B, bone marrow-derived human CD34+ cells were allowed to differentiate toward the erythropoietic lineage, using the methodology described under “Materials and Methods.” mRNA expression of Mnk1 (A) and Mnk2 (B) genes was assessed at the indicated days of hematopoietic cell differentiations by quantitative RT-PCR (Taqman), using GAPDH for normalization. Data are expressed as fold induction over corresponding samples obtained at day 3 and represent means ± S.E. of three independent experiments. C, expanded CFU-E cells were nucleofected with JAK2wt-IRES-GFP or JAK2V617F-IRES-GFP expressing vectors for 48 h. Equal amounts of protein were separated by SDS-PAGE and then immunoblotted with antibodies against JAK2 or GAPDH. D, normal CD34+ bone marrow-derived cells were transfected with JAK2wt-IRES-GFP or JAK2V617F-IRES-GFP vectors, respectively. These cells were then incubated in the presence or absence of IFNα, along with DMSO or Mnk-I, in clonogenic assays in methylcellulose as indicated. Myeloid (CFU-GM) and early erythroid (BFU-E) progenitor colonies were scored after 14 days in culture. Data are expressed as percentage control (CTRL) colony formation from untreated cells and represent means ± S.E. of four independent experiments.
Taken altogether, our studies implicate Mnk pathways in the generation of the antineoplastic effects of type IFNs on malignant erythroid precursors. To define the role of the pathways in a pathophysiologically relevant system, studies were carried out to determine the effects of the Mnk pathways in the induction of IFN responses in primary malignant erythroid precursors from patients with PV. As expected, treatment with IFNα suppressed primary malignant BFU-E hematopoietic precursors from PV patients (Fig. 6A) in clonogenic assays in methylcellulose. This suppression was partially reversed by Mnk-I (Fig. 6A), indicating a requirement for Mnk kinase activity for induction of IFN responses in these cells. In addition, reversal was seen when Mnk1 or Mnk2 was knocked down using specific siRNAs (Fig. 6, B and C), definitively establishing a requirement for Mnk kinases in the generation of IFN responses in malignant erythroid precursors.
FIGURE 6.
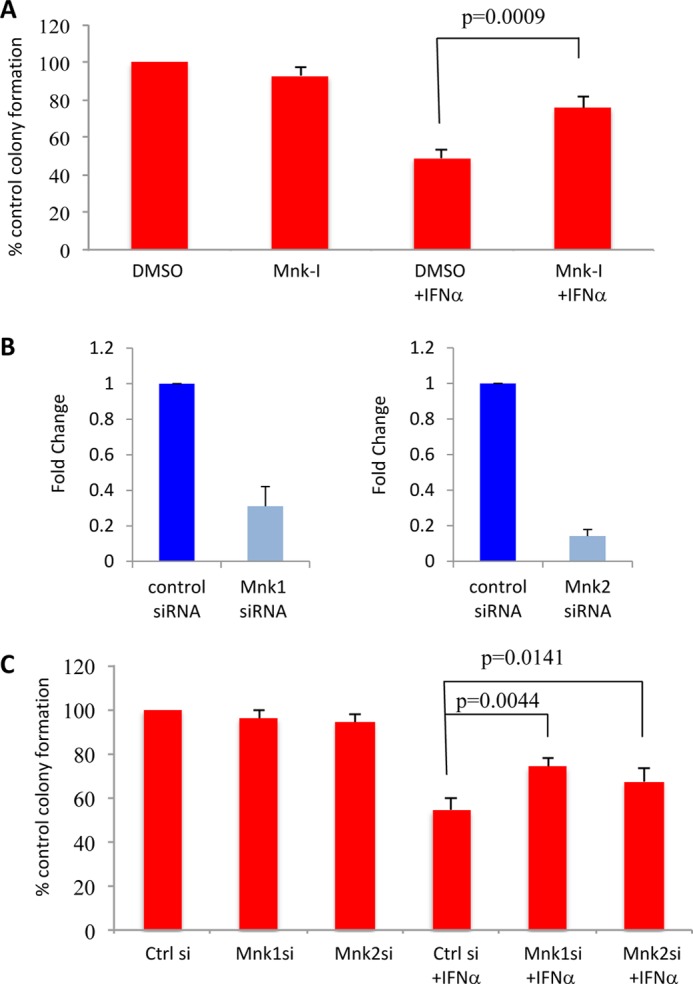
Mnk kinase activity is essential for the inhibitory effects of IFNα on malignant erythroid hematopoietic progenitors from patients with PV. A, mononuclear cells derived from peripheral blood of patients with PV were incubated with DMSO or Mnk-I as indicated and were then plated in a methylcellulose assay system in the absence or presence of human IFNα. Malignant erythroid (BFU-E) progenitor colonies were scored after 14 days in culture. Data are expressed as percentage control colony formation of control, untreated siRNA-transfected cells and represent means ± S.E. of five independent experiments. Paired t test analysis showed p = 0.0009 for the combination of control DMSO and IFNα versus the combination of Mnk-I and IFNα. B, HEL cells were nucleofected with Mnk1 and Mnk2 siRNA (si) for 48 h and Mnk1 and Mnk2 gene expression was tested by quantitative RT-PCR (Taqman), using GAPDH for normalization. Means + S.E. of three experiments are shown. C, mononuclear cells derived from peripheral blood of patients with PV were transfected with the indicated siRNAs and were then plated in a methylcellulose assay system, in the absence or presence of human IFNα. BFU-E progenitor colonies were scored after 14 days in culture. Data are expressed as percentage control (Ctrl) colony formation of the untreated control siRNA-transfected cells and represent means ± S.E. of four independent experiments. Paired t test analysis showed p = 0.0044 for the combination of control siRNA and IFNα versus the combination of Mnk1 siRNA and IFNα; and p = 0.0141 for the combination of control siRNA and IFNα versus the combination of Mnk2 siRNA and IFNα.
DISCUSSION
In recent years, there has been accumulating preclinical and clinical evidence that IFNα exhibits significant therapeutic activity in the treatment of MPNs in humans. Extensive work has established the relevance and utility of IFN treatment in the management of patients with MPNs (35). This includes activity in various hematological malignancies that result from transformation by the mutated Jak2 protein, including PV (46–49), essential thrombocytosis (48–51), and primary myelofibrosis (49–53). Although IFNα is also effective in other myeloproliferative states and diseases such as BCR-ABL-induced chronic myeloid leukemia (54), its activity in Jak2V617F-caused MPNs has particularly important implications, as strategies for the management of MPNs are much more limited than for chronic myeloid leukemia. Although the mechanisms of action of IFN in myeloproliferative disorders remain to be precisely defined, there has been evidence over the years that type I IFN treatment suppresses the growth of malignant hematopoietic progenitors (17), which may account for induction of remission in malignant myeloid hematopoietic disorders. There is also evidence for other mechanisms that may contribute indirectly to the antineoplastic effects of IFNα such as inhibition of cytokine secretion in the bone marrow microenvironment (55), inhibition of angiogenesis (56), and immunoregulatory effects (57).
There has been emerging evidence that activated Mnk kinases downstream of IFN receptors play important roles in mRNA translation of IFN-stimulated genes and the generation of protein products that mediate important biological responses (32, 33). Also, unique roles for these kinases were demonstrated in the regulation of normal hematopoiesis by IFNs (32, 33), suggesting that both Mnk1 and Mnk2 play essential roles in IFN-inducible growth inhibitory responses in normal cells. Beyond involvement of Mnk pathways, there is evidence that the coordinated functions of other IFNAR-regulated signals have important roles in induction of IFN responses in normal hematopoietic progenitors (5, 33). Although much is now known about the signaling pathways that mediate IFN responses in normal hematopoietic precursors, the pathways and cellular networks, the functions of which are required for the generation of the effects of IFNs in malignant hematopoietic progenitors, remain to be defined. Recent work has also implicated engagement of the p38 MAPK pathway in the suppression of Jak2V617F-positive Ph (−) hematopoietic progenitor cells (58). Interestingly, engagement of this pathway was shown previously to be essential for the antileukemic effects of IFNα in Ph (+) leukemic progenitors from patients with chronic myeloid leukemia (38), suggesting a similarity in the mechanism of action in distinct malignant phenotypes. Other recent work has identified Sprouty proteins as novel regulators with inhibitory properties on the generation of the suppressive effects of IFNα in primary malignant erythropoietic progenitors (37). Interestingly, IFN-dependent phosphorylation/activation of the p38 MAPK pathway was shown to be enhanced in Spry 1/2/4−/− cells in that study (37), suggesting that inhibition of p38 MAPK activation may be one mechanism by which these proteins act as negative feedback regulators in the generation of IFN responses.
Mnk kinases are effector kinases of MAPK pathways and play important regulatory roles in cells by controlling phosphorylation of the eIF4E (reviewed in Ref. 60). Because of their involvement in promalignant/promitogenic cellular pathways and the fact that their target eIF4E is deregulated in several malignancies, there has been substantial interest in targeting these kinases for the treatment of various tumors (61–64). In fact, preclinical studies investigating cercosporamide, a novel Mnk inhibitor in different types of malignancies, are encouraging and suggest that Mnk targeting may provide an effective antitumor approach in the future (63, 64). Surprisingly, however, there is also emerging evidence that in the case of IFNs, the Mnk-eIF4E pathway plays a positive role and is required for mRNA translation of genes regulated by IFN activation of the type I IFN receptor.
Type I IFNs are cytokines that suppress cell proliferation and several of the genes that they induce exhibit proapoptotic properties (4). Accordingly, we sought to determine whether the Mnk-eIF4E pathway is implicated in the generation of the inhibitory effects of IFNs in MPN cells that express the Jak2V617F mutation. Our studies establish that the Mnk-eIF4E pathway is activated in Jak2V617F-transformed cells in a Mek-Erk-dependent manner and that its function is essential for the generation of the inhibitory effects of IFNα in malignant hematopoietic progenitors from PV patients. These findings establish that the Mnk/eIF4E pathway plays key and essential regulatory roles in the generation of the antineoplastic effects of type I IFNs, whereas it remains to be seen whether beyond eIF4E, other putative downstream effectors of Mnk kinases, such PSF (65) and hnRNPA1 (59), are also involved and have roles in the generation of IFN responses. Independent of the precise downstream elements involved in the generation of signals for IFN-inducible, Mnk-mediated responses, the results of this work have direct translational implications as they suggest that Mnk pathways are positive effectors for IFN antineoplastic responses in MPNs. As there is substantial interest in targeting Mnk as a novel approach in the treatment of different tumors, our findings suggest that combining Mnk inhibitors with IFNα for the treatment of MPNs should probably be avoided, as these combinations may impair induction of IFN responses.
Supplementary Material
This work was supported by National Institutes of Health Grants CA155566, CA77816, and CA161796 (to L. C. P.); by a Merit Review grant from the Department of Veterans Affairs (to L. C. P.). D. P. B. is an employee of Biogen Idec and an owner of Biogen Idec stock.

This article contains supplemental Fig. S1.
- IFNAR
- type I IFN receptor
- MPN
- myeloproliferative neoplasm
- PV
- polycythemia vera
- Mnk-I
- Mnk inhibitor
- BFU-E
- burst-forming unit erythroid
- DMSO
- dimethyl sulfoxide
- CFU-E
- colony-forming unit erythroid.
REFERENCES
- 1. Stark G. R., Kerr I. M., Williams B. R., Silverman R. H., Schreiber R. D. (1998) How cells respond to interferons. Annu. Rev. Biochem. 67, 227–264 [DOI] [PubMed] [Google Scholar]
- 2. Pestka S., Langer J. A., Zoon K. C., Samuel C. E. (1987) Interferons and their actions. Annu. Rev. Biochem. 56, 727–777 [DOI] [PubMed] [Google Scholar]
- 3. Pestka S. (2007) The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 282, 20047–20051 [DOI] [PubMed] [Google Scholar]
- 4. Borden E. C., Sen G. C., Uze G., Silverman R. H., Ransohoff R. M., Foster G. R., Stark G. R. (2007) Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6, 975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Platanias L. C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 [DOI] [PubMed] [Google Scholar]
- 6. Parmar S., Platanias L. C. (2003) Interferons: mechanisms of action and clinical applications. Curr. Opin. Oncol. 15, 431–439 [DOI] [PubMed] [Google Scholar]
- 7. Lamers M. H., Kirgiz Ö. Ö., Heidrich B., Wedemeyer H., Drenth J. P. (2012) Interferon-α for patients with chronic hepatitis delta: a systematic review of randomized clinical trials. Antivir. Ther. 17, 1029–1037 [DOI] [PubMed] [Google Scholar]
- 8. Tai A. W., Chung R. T. (2009) Treatment failure in hepatitis C: mechanisms of non-response. J. Hepatol. 50, 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scagnolari C., Antonelli G. (2013) Antiviral activity of the interferon α family: biological and pharmacological aspects of the treatment of chronic hepatitis C. Expert Opin. Biol. Ther. 5, 693–711 [DOI] [PubMed] [Google Scholar]
- 10. Tarhini A. A., Gogas H., Kirkwood J. M. (2012) IFN-α in the treatment of melanoma. J. Immunol. 189, 3789–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karussis D. (2013) Immunotherapy of multiple sclerosis: the state of the art. BioDrugs 27, 113–148 [DOI] [PubMed] [Google Scholar]
- 12. Inoue M., Shinohara M. L. (2013). The role of IFNβ in the treatment of MS and EAE - in the perspective of inflammasomes. Immunology 139, 11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yao Y., Liu Z., Jallal B., Shen N., Rönnblom L. (2013) Type I interferons in Sjögren's syndrome. Autoimmun. Rev. 12, 558–566 [DOI] [PubMed] [Google Scholar]
- 14. Elkon K. B., Wiedeman A. (2012) Type I IFN system in the development and manifestations of SLE. Curr. Opin. Rheumatol. 24, 499–505 [DOI] [PubMed] [Google Scholar]
- 15. Rönnblom L., Eloranta M. L. (2013) The interferon signature in autoimmune diseases. Curr. Opin. Rheumatol. 25, 248–253 [DOI] [PubMed] [Google Scholar]
- 16. Young N. S., Calado R. T., Scheinberg P. (2006) Current concepts in the pathophysiology and treatment of aplastic anemia. Blood 108, 2509–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Platanias L. C., Fish E. N. (1999) Signaling pathways activated by interferons. Exp. Hematol. 27, 1583–1592 [DOI] [PubMed] [Google Scholar]
- 18. Stark G. R., Darnell J. E., Jr. (2012) The JAK-STAT pathway at twenty. Immunity 36, 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Shea J. J., Plenge R. (2012) JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36, 542–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Katsoulidis E., Li Y., Mears H., Platanias L. C. (2005) The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J. Interferon Cytokine Res. 25, 749–756 [DOI] [PubMed] [Google Scholar]
- 21. Platanias L. C. (2003) Map kinase signaling pathways and hematologic malignancies. Blood 101, 4667–4679 [DOI] [PubMed] [Google Scholar]
- 22. Redig A. J., Platanias L. C. (2007) The protein kinase C (PKC) family of proteins in cytokine signaling in hematopoiesis. J. Interferon Cytokine Res. 27, 623–636 [DOI] [PubMed] [Google Scholar]
- 23. Beauchamp E. M., Platanias L. C. (2012) The evolution of the TOR pathway and its role in cancer. Oncogene, DOI 10.1038/onc.2012.567 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24. Joshi S., Kaur S., Kroczynska B., Platanias L. C. (2010) Mechanisms of mRNA translation of interferon stimulated genes. Cytokine 52, 123–127 [DOI] [PubMed] [Google Scholar]
- 25. Diamond M. S., Farzan M. (2013) The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13, 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang D., Zhang D. E. (2011) Interferon-stimulated gene 15 and the protein ISGylation system. J. Interferon Cytokine Res. 31, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaur S., Sassano A., Dolniak B., Joshi S., Majchrzak-Kita B., Baker D. P., Hay N., Fish E. N., Platanias L. C. (2008) Role of the AKT pathway in mRNA translation of interferon (IFN) stimulated genes. Proc. Natl. Acad. Sci. U.S.A. 105, 4808–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panaretakis T., Hjortsberg L., Tamm K. P., Björklund A. C., Joseph B., Grandér D. (2008) Interferon α induces nucleus-independent apoptosis by activating extracellular signal-regulated kinase 1/2 and c-Jun NH2-terminal kinase downstream of phosphatidylinositol 3-kinase and mammalian target of rapamycin. Mol. Biol. Cell 19, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kroczynska B., Kaur S., Katsoulidis E., Majchrzak-Kita B., Sassano A., Kozma S. C., Fish E. N., Platanias L. C. (2009) Interferon-dependent engagement of eukaryotic initiation factor 4B via S6 kinase (S6K)- and ribosomal protein S6K-mediated signals. Mol. Cell Biol. 29, 2865–2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kroczynska B., Sharma B., Eklund E. A., Fish E. N., Platanias L. C. (2012) Regulatory effects of programmed cell death 4 (PDCD4) protein in interferon (IFN)-stimulated gene expression and generation of type I IFN responses. Mol. Cell Biol. 32, 2809–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaur S., Sassano A., Majchrzak-Kita B., Baker D. P., Su B., Fish E. N., Platanias L. C. (2012) Regulatory effects of mTORC2 complexes in type I interferon IFN signaling and generation of IFN responses. Proc. Natl. Acad. Sci. U.S.A. 109, 7723–7728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Joshi S., Kaur S., Redig A. J., Goldsborough K., David K., Ueda T., Watanabe-Fukunaga R., Baker D. P., Fish E. N., Fukunaga R., Platanias L. C. (2009) Type I interferon (IFN)-dependent activation of Mnk1 and its role in the generation of growth inhibitory responses. Proc. Natl. Acad. Sci. U.S.A. 106, 12097–12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joshi S., Sharma B., Kaur S., Majchrzak B., Ueda T., Fukunaga R., Verma A. K., Fish E. N., Platanias L. C. (2011) Essential role for Mnk kinases in type II interferon (IFNγ) signaling and its suppressive effects on normal hematopoiesis. J. Biol. Chem. 286, 6017–6026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tefferi A., Vainchenker W. (2011) Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J. Clin. Oncol. 29, 573–582 [DOI] [PubMed] [Google Scholar]
- 35. Kiladjian J. J., Mesa R. A., Hoffman R. (2011) The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 117, 4706–4715 [DOI] [PubMed] [Google Scholar]
- 36. Stein B. L., Tiu R. V. (2013) Biologic rationale and clinical use of interferon in the classical BCR-ABL negative myeloproliferative neoplasms. J. Interferon Cytokine Res. 33, 145–153 [DOI] [PubMed] [Google Scholar]
- 37. Sharma B., Joshi S., Sassano A., Majchrzak B., Kaur S., Aggarwal P., Nabet B., Bulic M., Stein B. L., McMahon B., Baker D. P., Fukunaga R., Altman J. K., Licht J. D., Fish E. N., Platanias L. C. (2012) Sprouty proteins are negative regulators of interferon (IFN)-signaling and IFN-inducible biological responses. J. Biol. Chem. 287, 42352–42360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mayer I. A., Verma A., Grumbach I. M., Uddin S., Lekmine F., Ravandi F., Majchrzak B., Fujita S., Fish E. N., Platanias L. C. (2001) The p38 MAPK pathway mediates the growth inhibitory effects of interferon-α in BCR-ABL-expressing cells. J. Biol. Chem. 276, 28570–28577 [DOI] [PubMed] [Google Scholar]
- 39. Redig A. J., Sassano A., Majchrzak-Kita B., Katsoulidis E., Liu H., Altman J. K., Fish E. N., Wickrema A., Platanias L. C. (2009) Activation of protein kinase Cη by type I interferons. J. Biol. Chem. 284, 10301–10314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kang J. A., Zhou Y., Weis T. L., Liu H., Ulaszek J., Satgurunathan N., Zhou L., van Besien K., Crispino J., Verma A., Low P. S., Wickrema A. (2008) Osteopontin regulates actin cytoskeleton and contributes to cell proliferation in primary erythroblasts. J. Biol. Chem. 283, 6997–7006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berkofsky-Fessler W., Buzzai M., Kim M. K., Fruchtman S., Najfeld V., Min D. J., Costa F. F., Bischof J. M., Soares M. B., McConnell M. J., Zhang W., Levine R., Gilliland D. G., Calogero R., Licht J. D. (2010) Transcriptional profiling of polycythemia vera identifies gene expression patterns both dependent and independent from the action of JAK2V617F. Clin. Cancer Res. 16, 4339–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Katsoulidis E., Carayol N., Woodard J., Konieczna I., Majchrzak-Kita B., Jordan A., Sassano A., Eklund E. A., Fish E. N., Platanias L. C. (2009) Role of Schlafen 2 (SLFN2) in the generation of interferon α-induced growth inhibitory responses. J. Biol. Chem. 284, 25051–25064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uddin S., Majchrzak B., Woodson J., Arunkumar P., Alsayed Y., Pine R., Young P. R., Fish E. N., Platanias L. C. (1999) Activation of the p38 mitogen-activated protein kinase by type I interferons. J. Biol. Chem. 274, 30127–30131 [DOI] [PubMed] [Google Scholar]
- 44. Kaur S., Lal L., Sassano A., Majchrzak-Kita B., Srikanth M., Baker D. P., Petroulakis E., Hay N., Sonenberg N., Fish E. N., Platanias L. C. (2007) Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J. Biol. Chem. 282, 1757–1768 [DOI] [PubMed] [Google Scholar]
- 45. Levine R. L., Wadleigh M., Cools J., Ebert B. L., Wernig G., Huntly B. J., Boggon T. J., Wlodarska I., Clark J. J., Moore S., Adelsperger J., Koo S., Lee J. C., Gabriel S., Mercher T., D'Andrea A., Fröhling S., Döhner K., Marynen P., Vandenberghe P., Mesa R. A., Tefferi A., Griffin J. D., Eck M. J., Sellers W. R., Meyerson M., Golub T. R., Lee S. J., Gilliland D. G. (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387–397 [DOI] [PubMed] [Google Scholar]
- 46. Kiladjian J. J., Cassinat B., Chevret S., Turlure P., Cambier N., Roussel M., Bellucci S., Grandchamp B., Chomienne C., Fenaux P. (2008) Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 112, 3065–3072 [DOI] [PubMed] [Google Scholar]
- 47. Samuelsson J., Hasselbalch H., Bruserud O., Temerinac S., Brandberg Y., Merup M., Linder O., Bjorkholm M., Pahl H. L., Birgegard G., and Nordic Study Group for Myeloproliferative Disorders (2006) A phase II trial of pegylated interferon α-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 106, 2397–2405 [DOI] [PubMed] [Google Scholar]
- 48. Silver R. T. (1988) Recombinant interferon-α for treatment of polycythaemia vera. Lancet. 2, 403. [DOI] [PubMed] [Google Scholar]
- 49. Gowin K., Thapaliya P., Samuelson J., Harrison C., Radia D., Andreasson B., Mascarenhas J., Rambaldi A., Barbui T., Rea C. J., Camoriano J., Gentry A., Kiladjian J. J., O'Connell C., Mesa R. (2012) Experience with pegylated interferon α-2a in advanced myeloproliferative neoplasms in an international cohort of 118 patients. Haematologica 97, 1570–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Langer C., Lengfelder E., Thiele J., Kvasnicka H. M., Pahl H. L., Beneke H., Schauer S., Gisslinger H., Griesshammer M. (2005) Pegylated interferon for the treatment of high risk essential thrombocythemia: results of a phase II study. Haematologica 90, 1333–1338 [PubMed] [Google Scholar]
- 51. Alvarado Y., Cortes J., Verstovsek S., Thomas D., Faderl S., Estrov Z., Kantarjian H., Giles F. J. (2003) Pilot study of pegylated interferon-α 2b in patients with essential thrombocythemia. Cancer Chemother. Pharmacol. 51, 81–86 [DOI] [PubMed] [Google Scholar]
- 52. Ianotto J. C., Kiladjian J. J., Demory J. L., Roy L., Boyer F., Rey J., Dupriez B., Berthou C., Abgrall J. F. (2009) PEG-IFN-α-2a therapy in patients with myelofibrosis: a study of the French Grouped'Etudes des Myelofibroses (GEM) and France Intergroupe des syndromes Myeloproliferatifs (FIM). Br. J. Haematol. 146, 223–225 [DOI] [PubMed] [Google Scholar]
- 53. Silver R. T., Vandris K., Goldman J. J. (2011) Recombinant interferon-α may retard progression of early primary myelofibrosis: a preliminary report. Blood 117, 6669–6672 [DOI] [PubMed] [Google Scholar]
- 54. Verma A., Platanias L. C. (2002) Signaling via the interferon-α receptor in chronic myelogenous leukemia cells. Leuk. Lymphoma 43, 703–709 [DOI] [PubMed] [Google Scholar]
- 55. Peschel C., Aulitzky W. E., Huber C. (1996) Influence of interferon-α on cytokine expression by the bone marrow microenvironment-impact on treatment of myeloproliferative disorders. Leuk. Lymphoma 22, 129–134 [DOI] [PubMed] [Google Scholar]
- 56. Indraccolo S. (2010) Interferon-α as angiogenesis inhibitor: learning from tumor models. Autoimmunity 43, 244–247 [DOI] [PubMed] [Google Scholar]
- 57. Xiong Z., Yan Y., Liu E., Silver R. T., Verstovsek S., Yang F., Wang H., Prchal J., Yang X. (2007) Novel tumor antigens elicit anti-tumor humoral immune reactions in a subset of patients with polycythemia vera. Clin. Immunol. 122, 279–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu M., Zhang W., Li Y., Berenzon D., Wang X., Wang J., Mascarenhas J., Xu M., Hoffman R. (2010) Interferon-α targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp. Hematol. 38, 472–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Buxadé M., Parra J. L., Rousseau S., Shpiro N., Marquez R., Morrice N., Bain J., Espel E., Proud C. G. (2005) The Mnks are novel components in the control of TNF α biosynthesis and phosphorylate and regulate hnRNPA1. Immunity 23, 177–189 [DOI] [PubMed] [Google Scholar]
- 60. Buxade M., Parra-Palau J. L., Proud C. G. (2008) The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases). Front Biosci. 13, 5359–5373 [DOI] [PubMed] [Google Scholar]
- 61. Joshi S., Platanias L. C. (2012) Mnk kinases in cytokine signaling and regulation of cytokine responses. Biomol. Concepts 3, 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hou J., Lam F., Proud C., Wang S. (2012) Targeting Mnks for cancer therapy. Oncotarget 3, 118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Konicek B. W., Stephens J. R., McNulty A. M., Robichaud N., Peery R. B., Dumstorf C. A., Dowless M. S., Iversen P. W., Parsons S., Ellis K. E., McCann D. J., Pelletier J., Furic L., Yingling J. M., Stancato L. F., Sonenberg N., Graff J. R. (2011) Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 71, 1849–1857 [DOI] [PubMed] [Google Scholar]
- 64. Altman J. K., Szilard A., Konicek B. W., Iversen P. W., Kroczynska B., Glaser H., Sassano A., Vakana E., Graff J. R., Platanias L. C. (2013) Inhibition of Mnk kinase activity by cercosporamide and suppressive effects on acute myeloid leukemia precursors. Blood 121, 3675–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Buxadé M., Morrice N., Krebs D. L., Proud C. G. (2008) The PSF.p54nrb complex is a novel Mnk substrate that binds the mRNA for tumor necrosis factor α. J. Biol. Chem. 283, 57–65 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.