

Background: Angiotensin II is elevated in cachexia and induces skeletal muscle atrophy.
Results: Angiotensin inhibits muscle stem (satellite) cell proliferation via a Notch-dependent mechanism and depletes the satellite cell pool.
Conclusion: Angiotensin prevents skeletal muscle regeneration by suppressing satellite cell function.
Significance: This is the first report to show that satellite cells express angiotensin receptors and that angiotensin inhibits regeneration.
Keywords: Angiotensin II, Myocardial Infarction, Regeneration, Skeletal Muscle, Stem Cells, Cachexia, Satellite Cells
Abstract
Cachexia is a serious complication of many chronic diseases, such as congestive heart failure (CHF) and chronic kidney disease (CKD). Although patients with advanced CHF or CKD often have increased angiotensin II (Ang II) levels and cachexia and Ang II causes skeletal muscle wasting in rodents, the potential effects of Ang II on muscle regeneration are unknown. Muscle regeneration is highly dependent on the ability of a pool of muscle stem cells (satellite cells) to proliferate and to repair damaged myofibers or form new myofibers. Here we show that Ang II reduced skeletal muscle regeneration via inhibition of satellite cell (SC) proliferation. Ang II reduced the number of regenerating myofibers and decreased expression of SC proliferation/differentiation markers (MyoD, myogenin, and active-Notch) after cardiotoxin-induced muscle injury in vivo and in SCs cultured in vitro. Ang II depleted the basal pool of SCs, as detected in Myf5nLacZ/+ mice and by FACS sorting, and this effect was inhibited by Ang II AT1 receptor (AT1R) blockade and in AT1aR-null mice. AT1R was highly expressed in SCs, and Notch activation abrogated the AT1R-mediated antiproliferative effect of Ang II in cultured SCs. In mice that developed CHF postmyocardial infarction, there was skeletal muscle wasting and reduced SC numbers that were inhibited by AT1R blockade. Ang II inhibition of skeletal muscle regeneration via AT1 receptor-dependent suppression of SC Notch and MyoD signaling and proliferation is likely to play an important role in mechanisms leading to cachexia in chronic disease states such as CHF and CKD.
Introduction
Cachexia is typically associated with chronic diseases such as cancer, chronic obstructive pulmonary disease, congestive heart failure (CHF),2 chronic kidney disease, and chronic infections, and it independently worsens outcome (1). Mechanisms are complex and can include anorexia, increased cytokines, insulin resistance, and accelerated muscle protein breakdown (2). There is increasing evidence that angiotensin II (Ang II), the main effector molecule of the renin-angiotensin system, may play an important role in triggering cachexia. Thus, patients with advanced CHF and chronic kidney disease often have elevated circulating Ang II, and treatment with an angiotensin-converting enzyme inhibitor can reduce weight loss (3). There is also interest in the potential role of Ang II in cachexia associated with cancer and chronic obstructive pulmonary disease (4, 5). We have shown that Ang II infusion in rodents decreased body weight through a reduction in appetite and a loss of muscle weight (6–13). In these animals, Ang II disrupted insulin-like growth factor 1 (IGF-1) signaling, resulting in increased muscle protein degradation and apoptosis (6–8). However, it remains unknown whether normal skeletal muscle regenerative responses occur in the setting of Ang II-induced muscle catabolism and wasting. In fact, little is known about the regenerative potential of skeletal muscle in chronic wasting conditions other than that it clearly appears insufficient to sustain normal muscle mass.
Muscle regeneration has been studied using animal models such as the cardiotoxin (CTX)-induced injury model (14). Injury leads to activation and proliferation of mitotically quiescent mononuclear cells, satellite cells, which form myoblasts, differentiate terminally, and fuse to form multinucleated myotubes (15). In adult skeletal muscle, satellite cell number and regenerative capacity remain nearly constant through multiple cycles of regeneration (16, 17). However, there is a depletion of satellite cell number and a decline of muscle tissue regeneration with age (18, 19). In aged satellite cells, the proliferative potential declines because of decreased activation of the Notch pathway and increased TGF-β signaling (20). Increased TGF-β signaling is also implicated in the myopathy and reduced skeletal muscle regeneration that occurs in fibrillin 1 heterozygous mutant mice and in dystrophin-deficient mdx mice (21). Blockade of angiotensin type 1 receptor (AT1R) signaling suppressed TGF-β signaling and improved muscle regeneration and function in these mice (21). Although this report suggests a relation between Ang II and myopathy, the potential direct effects of Ang II on muscle regenerative capacity and mechanisms involved remain unknown. Assessment of the direct effects of Ang II is important because adult skeletal muscle expresses low levels of Ang II receptors (22), and many of the effects of Ang II on adult skeletal muscle are thought to be indirect via intermediate cytokines (8, 22). Here we report that Ang II acts directly via AT1R expressed on satellite cells to prevent their proliferation, which results in attenuated skeletal muscle regeneration.
EXPERIMENTAL PROCEDURES
Animals
Animal protocols were approved by the Institutional Animal Care and Use Committee at Tulane University. 8- to 10-week-old male C57BL/6 mice (Charles River Laboratories, Inc.), AT1aR−/− (B6.129P2-Agtr1atm1Unc/J, The Jackson Laboratory) and Myf5nLacZ/+, were used in this study. Ang II (1.5 μg/kg/min via osmotic minipumps) and candesartan (10 mg/kg/day via drinking water) were administered as described previously (9, 10). CTX was injected as described previously (14).
Flow Cytometry
For the quantification of CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+ satellite cells, mononuclear cells were collected as described previously (23), followed by FACS sorting on the basis of cell surface marker expressions. Briefly, hind limb (gastrocnemius, tibialis anterior, soleus, and extensor digitorum longus) muscles were harvested, minced, and digested with 2 mg/ml collagenase type IV (Worthington) and 1.2 units/ml dispase for 45 min at 37 °C. After filtration and washing, 1 × 105 cells were used for staining. Antibodies used for flow cytometry included anti-mouse CD45 (clone 30-F11, APC conjugate, eBioscience, 1:1600), anti-mouse Sca-1 (clone D7, APC conjugate, eBioscience, 1:800), anti-mouse CD11b (clone M1/70, APC-conjugate, eBioscience, 1:800), anti-mouse CD31 (clone MEC 13.3, APC-conjugate, BD Biosciences, 1:200), anti-mouse CD34 (clone RAM34, PE-conjugate or Alexa Fluor 700 conjugate, BD Biosciences or eBioscience, respectively, 1:50), anti-mouse integrinα7 (clone 3C12, FITC conjugate, MBL, 1:20), and anti-mouse AT1R (rabbit polyclonal, Alomone Labs, 1:100).
Myofiber Explant and Primary Satellite Cell Culture
Single myofibers and satellite cells were collected as described previously (24, 25). Briefly, hind limb muscles (gastrocnemius, soleus, tibialis anterior, and extensor digitorum longus) were incubated with 0.2% (w/v) collagenase type II in DMEM and 10% horse serum for 2 h at 37 °C. Digested muscles were dissociated by triturating with a Pasteur pipette, washed, and further digested with 1 units/ml dispase for 1 h at 37 °C. Cells were filtered, washed, and incubated in proliferation medium (DMEM supplemented with 20% FBS (Mediatech), 10% horse serum, 2% chicken embryo extract (US Biological), 1 mm sodium pyruvate, 2 mm l-glutamine, and penicillin/streptomycin) for 40 min to let the fibroblasts attach onto the plate, and unattached cells were plated onto Matrigel-coated plates in the proliferation medium at the density of 1 × 104 cells/cm2.
Measurement of Cell Proliferation, Viability, and Apoptosis
Satellite cells were collected as described above and plated on Matrigel-coated 96-well plates in the proliferation medium. Cell proliferation, cell viability, and caspase 3/7 activity were measured after 24 h. For the cell proliferation analysis, Ang II (10–1000 nm), candesartan (AstraZeneca, 100 nm), and anti-Notch antibody (Millipore, 1:300) were added in culture together with BrdU (10 μm) and incubated for 24 h. BrdU incorporation was measured using a BrdU cell proliferation ELISA kit (Roche) according to the instructions of the manufacturer. Cell viability and caspase 3/7 were measured using a CellTiter-Glo luminescent cell viability assay (Promega) and Caspase-Glo 3/7 assay (Promega), respectively, in the presence of Ang II (10–1000 nm) according to the instructions of the manufacturer.
Quantitative RT-PCR
Quantitative RT-PCR was performed as described previously (9), and PCR primers used in this study were obtained commercially (SABiosciences, PPM33632F for Pax7, PPM04482A for MyoD, PPM04482A for myogenin, PPM41610F for eMyHC, PPM41621B for Myh1, PPM05162A for AT1aR, PPM31347C for AT1bR, PPM04811A for AT2R, PPM02991B for TGF-β, and PPM03559F for Hprt).
Antibodies
Antibodies used for immunoblotting included anti-MyoD (DAKO, 1:1000), anti-Notch (Abcam, 1:1500), anti-active-Notch (Abcam, 1:500), anti-Numb (Abcam, 1:1000), anti-M-cadherin (BD Biosciences, 1:3000), anti-myogenin (Abcam, 1:500), anti-p27 (Santa Cruz Biotechnology, Inc., 1:200), anti-p57 (Epitomics, 1:1000), anti-p15 (Cell Signaling, 1:1000); anti-p16 (Santa Cruz Biotechnology, Inc., 1:200), anti-cyclin D1 (Santa Cruz Biotechnology, Inc., 1:1000), anti-cyclin E (Santa Cruz Biotechnology, Inc., 1:1000), anti-retinoblastoma protein (BD Biosciences, 1:250), and anti-mouse β-actin (Sigma, 1:10,000). Antibodies used for immunohistochemical staining included anti-Pax7 (Developmental Studies Hybridoma Bank, 1:5), anti-MyoD (DAKO, 1:50), anti-β-galactosidase (Abcam, 1:200), anti-AT1R (Alomone Labs, 1:100), anti-laminin (Abcam, 1:500), anti-mouse IgG-Alexa Fluor 488 (Invitrogen, 1:500), anti-rabbit IgG-Alexa Fluor 594 (Invitrogen, 1:500), and anti-chicken IgG-Alexa Fluor 647 (Invitrogen, 1:500).
Left Anterior Descending (LAD) Artery Ligation
Progressive cardiac dysfunction was induced by ligation of the left coronary artery as described previously (26). Sham-operated animals underwent the same procedure without ligation of the coronary artery. All mice underwent echocardiography and were euthanized after 6 weeks.
RESULTS
Ang II Reduces Skeletal Muscle Regeneration
Although Ang II causes skeletal muscle atrophy (6–13), we have observed very few actively regenerating myofibers with centralized nuclei after Ang II infusion. To determine whether Ang II reduced muscle repair, we analyzed the effect of Ang II on CTX-induced skeletal muscle regeneration (Fig. 1). There was a robust increase in the number of regenerating myofibers in CTX-injected gastrocnemius muscles on d5 and 7, which was markedly attenuated by Ang II (Fig. 1, A–C). Ang II reduced the cross-sectional area of regenerating myofibers on d7 (this could also reflect the atrophic effect of Ang II, Fig. 1D) and decreased muscle weight (Fig. 1E). Consistent with a previous report (14), a number of myogenic transcription factors were up-regulated after CTX injury. Pax7, MyoD, and myogenin mRNA expression reached a maximal level on d3, and Ang II significantly attenuated their increase (Fig. 1, F–H). Embryonic myosin heavy chain (eMyHC) reached a maximal level on d5 and was also suppressed by Ang II (Fig. 1I). These data strongly suggest that Ang II inhibits activation and/or proliferation of satellite cells, resulting in decreased skeletal muscle regeneration. There was a marked decrease of angiotensin type 1a receptor (AT1aR) mRNA expression 3 days after CTX injection, and Ang II further suppressed AT1aR expression throughout the experiment. In contrast, angiotensin type 2 receptor (AT2R) gene expression increased after CTX injection and reached a maximal level on d5, and Ang II significantly suppressed the increase at d3, with a trend to suppression on d5 (p = 0.15).
FIGURE 1.

Ang II infusion suppresses skeletal muscle regeneration. A, representative H&E staining of gastrocnemius muscles after CTX injection. B, experimental model. CTX was injected, and an Ang II infusion (1.5 μg/kg/min) was started on day 0. Gastrocnemius muscles were collected on d3, 5, and 7. For H&E staining, paraffin sections were prepared from the middle of gastrocnemius muscles. For quantitative PCR analysis, RNA was extracted from whole gastrocnemius muscle. C, number of regenerating myofibers (myofibers with centralized nuclei) after CTX injection and Ang II infusion on d0, 3, 5, and 7. The number of regenerating myofibers is shown per section. D, CSA of regenerating myofibers after CTX injection and Ang II infusion on d7. E, gastrocnemius muscle weight 7 days after CTX injection and Ang II infusion. F–I, quantitative RT-PCR analysis of Pax7 (F), MyoD (G), myogenin (H), and eMyHC (I) in the gastrocnemius muscle after CTX injection and Ang II infusion on d0, 3, 5, and 7. n = 5–6, data are mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 between Sham-CTX and Ang II-CTX.
Ang II Depletes the Satellite Cell Pool
The data in Fig. 1 strongly suggest that Ang II prevents skeletal muscle regeneration via suppression of the satellite cell regenerative capacity. It has been shown that the total number of quiescent satellite cells remains constant over repeated cycles of degeneration and regeneration via a process of self-renewal (16, 17) and that the maintenance of satellite cell number is critical for the normal function of skeletal muscle (19). We used X-gal staining to quantify the satellite cell number in sham-infused and Ang II-infused Myf5nLacZ/+ mice, in which the β-gal gene is knocked into the Myf5 locus (27) (Fig. 2A). Approximately 3–4% of total nuclei were positive for β-gal, and there was a 39.4% and 37.1% reduction of the β-gal+ cell number in gastrocnemius and tibialis anterior muscles after 7 days of Ang II infusion, respectively, suggesting that Ang II prevents skeletal muscle regeneration through depletion of satellite cells (Fig. 2, B and C). The β-gal+ cell number was also quantified per myofiber CSA and per myofiber number, and all of these data showed a similar reduction (39.4% reduction by DAPI count, 33.2% reduction by CSA, and 43.4% reduction by myofiber number; p = not significant), indicating that the reduction of the β-gal+ cell number was not caused by infiltration of inflammatory cells or by reduced myofiber CSA. We also analyzed the satellite cell number using flow cytometry to detect cells expressing a combination of markers useful for prospective isolation of satellite cells, namely, CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+ (Fig. 2D) (17). Consistent with the data obtained from Myf5nLacZ/+ mice, there was a decrease in the number of CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+ cells in muscles from Ang II-infused C57BL/6 mice on d7 (Fig. 2E), and this reduction was prevented completely by the AT1R blocker candesartan (E) or in angiotensin type 1a receptor-null (AT1aR−/−) mice (F). These data indicate that Ang II depletes satellite cells through AT1R-mediated signaling.
FIGURE 2.
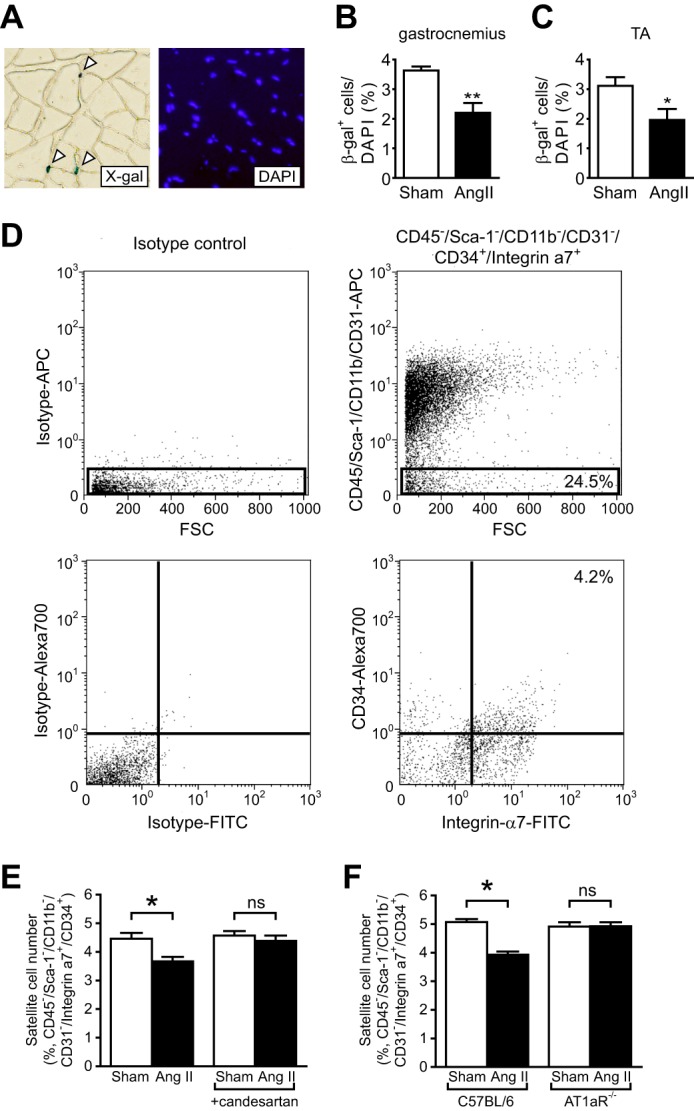
Ang II infusion reduces satellite cell number via an AT1aR-mediated mechanism. A, representative X-gal and DAPI staining of gastrocnemius muscle of Myf5nLacZ/+ mice. White arrowheads, β-gal-positive cells. B and C, number of β-gal-positive cells in non-injured gastrocnemius (B) and tibialis anterior (TA) (C) muscles of Myf5nLacZ/+ mice 7 days after Ang II infusion. The number of β-gal-positive cells was counted and is shown as percentage of DAPI-positive cells. Three areas per section per muscle were analyzed (∼1500 total cells). D, representative flow cytometric analysis of myofiber-associated cells isolated from gastrocnemius muscles. 24.5% of cells were sorted as CD45−/Sca-1−/CD11b−/CD31−, and 17.1% of these cells (4.2% total cells) were sorted as CD34+/Integrinα7+. E, number of CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+cells 7 days after Ang II infusion and candesartan administration in gastrocnemius muscles of C57BL/6 mice. F, number of CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+cells 7 days after Ang II infusion in C57BL/6 mice and AT1aR−/− mice. n = 6, data are mean ± S.E. *, p < 0.05; **, p < 0.01; ns, not significant. APC, allophycocyanin; FSC, forward scatter.
Differentiation-dependent Ang II Receptor Expression in Satellite Cells
We have shown previously that skeletal muscle and cultured myocytes do not express significant levels of AT1R (22). Thus, Ang II-induced skeletal muscle atrophy results, in large part, from indirect effects mediated via increased glucocorticoids (8), interleukin 6, and serum amyloid A (22) rather than from direct effects of Ang II on myofibers. However, it is not known whether the satellite cell population expresses Ang II receptors. To determine the potential expression of Ang II receptors in satellite cells and to test the hypothesis that Ang II regulates satellite cell number via direct AT1R-mediated effects, we analyzed AT1R cell surface expression on CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+ satellite cells and found that these cells express AT1R (Fig. 3A). Immunohistochemical staining of intact myofibers cultured for 2 days indicated colocalization of AT1R with the satellite cell marker MyoD (Fig. 3B) and with β-gal (in myofibers from Myf5nLacZ/+ mice, C). We also analyzed Ang II receptor mRNA levels by quantitative RT-PCR in FACS-sorted CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+ satellite cells, and thereafter, these cells were cultured and induced to differentiate for 48 h. Expression of the satellite cell marker Pax7 was significantly higher, and expression of adult MyHC (Myh1) was much lower in these cells than in control gastrocnemius muscle, indicating efficient enrichment of the satellite cell population with no presence of muscle debris (Fig. 3, D and E). The expression of AT1aR and AT1bR was significantly higher in satellite cells compared with whole gastrocnemius muscle. Both AT1aR and AT1bR expression decreased after satellite cells were induced to differentiate, consistent with the low expression in whole gastrocnemius muscle. Although AT2R expression was not detectable in satellite cells, it increased robustly after these cells were induced to differentiate (Fig. 3I). The increased expression of AT2R was associated with abundant expression of eMyHC in differentiating satellite cells (Fig. 3F). Taken together, these data indicate that satellite cells express much higher levels of AT1R compared with mature myofibers and that AT2R is expressed primarily in differentiating satellite cell progenies.
FIGURE 3.

Expression of Ang II receptors in satellite cells. A, AT1R expression in satellite cells. CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrin α7+ cells were sorted from gastrocnemius muscles, and AT1R expression on these cells was analyzed by flow cytometry. FSC, forward scatter. B and C, immunohistochemical staining of AT1R in isolated myofibers. Isolated myofibers were cultured for 2 days in the proliferation medium and costained with laminin and MyoD in C57BL/6 (B) and β-galactosidase in Myf5nLacZ/+ mice (C). D–I, quantitative RT-PCR analysis of Pax7 (D), Myh1 (E), eMyHC (F), AT1aR (G), AT1bR (H), and AT2R (I) in gastrocnemius muscle (GM), FACS-sorted CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrin α7+ cells (SC), and FACS-sorted CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrin α7+ cells differentiated in vitro (SC-d). n = 5, data are mean ± S.E. **, p < 0.01; ***, p < 0.001; ns, not significant.
Ang II Regulates Cell Cycle Protein and Reduces Satellite Cell Proliferation via a Notch-dependent Pathway
To determine the effect of Ang II on satellite cell activation and/or proliferation, we analyzed satellite cell marker expression both in vivo and in vitro and satellite cell proliferative activity in vitro. Satellite cells can be collected with high efficiency by collagenase II and dispase digestion from hind limb muscles (24). 95.3 and 74.1% of satellite cells collected using this method expressed Pax7 and MyoD after 24 h of culture, respectively. Also, the formation of myotubes after induction of differentiation was confirmed with desmin and eMyHC staining. Satellite cells were collected from hind limb muscles after CTX injection, and Ang II infusion on d1–7, proliferation/differentiation marker, and cell cycle regulator expression were analyzed by immunoblotting (Fig. 4A). MyoD, active-Notch (Notch signaling plays an important role in satellite cell proliferation (24, 25)), M-cadherin, and myogenin expression reached a maximal level 3 days after CTX injection, which is consistent with a previous report (14, 25, 28) and with our whole-muscle quantitative RT-PCR data (Fig. 1, F–I), and Ang II infusion significantly suppressed expression of these markers. There was a significant decrease in levels of the chronic kidney disease inhibitors p27, p57, and p16 after CTX injection, which is consistent with a previous report (20). Although Ang II had little effect on chronic kidney disease inhibitors in the presence of CTX, there was an increase of p16 and a decrease of p57 after Ang II infusion in muscles not injected with CTX. Cyclin D1 and cyclin E expression and phosphorylation of retinoblastoma protein were increased after CTX injection, and Ang II suppressed their increase. To analyze potential direct effects of Ang II on satellite cell proliferation and/or viability, we treated cultured satellite cells with Ang II in vitro. Satellite cells were activated spontaneously and started proliferating in our culture conditions. MyoD, active-Notch, and M-cadherin expression increased 24–48 h after initiation of the culture, whereas myogenin expression was not detected until 96 h (Fig. 4B). In the presence of Ang II (10–1000 nm), MyoD, active-Notch, and M-cadherin expression were suppressed significantly at 48 h, consistent with Ang II-mediated inhibition of satellite cell activation/proliferation. Cell proliferation was measured by BrdU incorporation, and results indicated that Ang II dose-dependently suppressed satellite cell proliferation and that candesartan blocked this effect, consistent with an AT1R-mediated effect (Fig. 4C). On the other hand, Ang II had no effect on cell viability or activity of caspase 3/7 (Fig. 4, D and E). Because we found that Ang II reduces active-Notch (Fig. 4, A and B), we measured BrdU incorporation in satellite cells exposed to Ang II in the presence or absence of the activator of Notch signaling (Notch-1-specific antibody (24)) (Fig. 4F). We found that Notch activation completely abrogated the inhibitory effect of Ang II on BrdU incorporation (Fig. 4F), strongly suggesting that Ang II inhibits satellite cell proliferation via prevention of Notch activation.
FIGURE 4.

Ang II-mediated suppression of satellite cell proliferation in vivo and in vitro. A, immunoblotting of satellite cell markers and cell cycle regulators in satellite cells collected from hind limb muscles after CTX injection and Ang II infusion on d0, 1, 2, 3, 5, and 7. Rb, retinoblastoma protein; pRb, phospho-retinoblastoma protein. B, immunoblotting of satellite cell markers and cell cycle regulators in satellite cells cultured in vitro in the presence of Ang II (10–1000 nm). C–F, cell proliferation (C), cell viability (D), and caspase 3/7 activity (E) in satellite cells were measured 24 h after culture. F, cell proliferation of satellite cells in vitro with or without Ang II (100 nm) and anti-Notch antibody. n = 7, data are mean ± S.E. **, p < 0.01.
Reduced Satellite Cell Number in an Animal Model of CHF
Our findings that Ang II suppresses satellite cell proliferation and skeletal muscle regeneration suggest that, in conditions of high Ang II levels, such as CHF, a reduction in the satellite cell pool could blunt normal muscle regenerative responses and provide an additional mechanism together with the known proteolytic effect (7, 8), whereby Ang II induces muscle wasting. To investigate this hypothesis and to corroborate our findings from the Ang II infusion model, we analyzed muscle wasting and satellite cell number in mice 6 weeks following LAD coronary artery ligation, an animal model of CHF in which plasma Ang II levels are increased (29, 30). LAD artery ligation caused an increase in left ventricular end-systolic and end-diastolic diameters and a decrease in fractional shortening (FS) (Fig. 5, A–D). Consistent with a previous report (31), LAD-ligated mice showed significantly lower gastrocnemius muscle weight after 6 weeks (Fig. 5E). In these mice, the number of satellite cells (CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+) in gastrocnemius muscle was decreased significantly after LAD artery ligation (Fig. 5F). Candesartan administration blocked the reduction of gastrocnemius weight (Fig. 5E) and the decrease in satellite cell number (F), whereas there is no protective effect on cardiac function (A–D). These data suggest that Ang II-mediated depletion of the satellite cell pool and inhibition of skeletal muscle regeneration contribute to mechanisms of muscle wasting in conditions of Ang II excess, such as CHF.
FIGURE 5.

Muscle wasting and reduced satellite cell number in LAD artery-ligated mice. A–C, left ventricular end-systolic diameter (LVESd, A), left ventricular end-diastolic diameter (LVEDd, B), and fractional shortening (FS, C) of LAD artery-ligated and sham-operated mice were measured by echocardiography after 6 weeks. D, systolic blood pressure at 6 weeks of LAD artery ligation. E, gastrocnemius muscle weight at 6 weeks of LAD artery ligation. F, the number of satellite cells (CD45−/Sca-1−/CD11b−/CD31−/CD34+/Integrinα7+) in gastrocnemius muscle after 6 weeks of LAD ligation (% total cells). n = 7–9, data are mean ± S.E. *, p < 0.05; **, p < 0.01; ns, not significant.
DISCUSSION
Here we show that systemic Ang II infusion significantly reduces satellite cell number (Fig. 2) and prevents CTX injury-induced skeletal muscle regeneration (Fig. 1, A–E). Ang II reduces satellite cell expression of MyoD, myogenin, M-cadherin, and active-Notch (Figs. 1, F–I, and 4A) and inhibits satellite cell proliferation via an AT1R- and Notch-dependent mechanism (Fig. 4, B–F). AT1R is highly expressed on satellite cells, but AT2R is expressed only after satellite cells are induced to differentiate in culture (Fig. 3). There is a marked reduction of AT1aR expression in hind limb muscles after CTX injection, followed by a robust increase of AT2R expression, consistent with a switch from AT1R- to AT2R-mediated signaling as satellite cells differentiate. Satellite cell number is also reduced in mice following LAD ligation. Importantly, the reduction in satellite cell number in Ang II-infused animals and LAD-ligated animals were both rescued by candesartan administration (Figs. 2E and 5F). Although it is difficult to separate out the effect of Ang II on the satellite cell pool and its effect to reduce satellite cell proliferation and the relative contribution of each in vivo to the inhibition of regeneration, these data suggest that, in pathophysiological conditions of Ang II excess, normal muscle regenerative responses are blunted via an AT1R-mediated mechanism. It has been reported that AT1R blockers in LAD artery-ligated rodents have either no effect or modest effects on cardiac function (32–35). Our data indicate that there was no significant improvement in cardiac function in response to candesartan in our experimental setting (Fig. 5, A–D), although the reduction of satellite cell number was prevented. This suggests that the ability of candesartan to inhibit the reduction of satellite cell number was likely not mediated via its effects on cardiac function.
Previous reports indicate that angiotensin-converting enzyme inhibitors impaired skeletal muscle growth following CTX-induced injury (36) and attenuated overload-induced skeletal muscle hypertrophy (37, 38), suggesting that Ang II may be a positive regulator of muscle growth and regeneration. However, our data clearly show that Ang II prevents muscle regeneration as early as 3 days after injury, at which time there is reduced expression of MyoD and myogenin and attenuation of Notch activation (Figs. 1, F and G, and 4A). Johnston et al. (36) reported that Ang II increased Myf5, MyoD, Pax7, cyclin D1, and AT1R mRNA levels in C2C12 cells but that these cells express very low levels of Ang II receptors (22), and the dose of Ang II used in this study (10 μm) was supraphysiological (39). In our experimental setting, 100 nm Ang II was sufficient to reduce MyoD and myogenin expression, Notch activation, and satellite cell proliferation in vitro (Fig. 4B). Discrepancies in results between Johnston et al. (36) and our study might also be explained by differences in methods to prepare primary myoblasts and/or length of cells in culture. We used a two-step enzymatic digestion method (17, 24) yielding a high enrichment of satellite cells (> 90%) and used these cells without further passage. Satellite cells cultured in vitro spontaneously start expressing myogenic transcription factors (Fig. 4B), suggesting that the time point of analysis is critical to assess cell activation, proliferation, and differentiation. We have shown that myofibers and cultured myoblasts (C2C12 and L6 cells) express very low levels of Ang II receptors (22) and that Ang II wasting effects on skeletal muscle are mediated in significant part via intermediates such as glucocorticoids (8), serum amyloid A, and interleukin-6 (IL-6) (22). Although IL-6 is thought to promote satellite cell proliferation (40), our data clearly show that the net effect of systemic Ang II increase in vivo is to reduce muscle regeneration. Importantly, our findings that SCs express high levels of AT1R and that Ang II reduced satellite cell proliferation in vitro strongly suggest that an increase in circulating Ang II suppresses muscle regeneration via direct effects on SCs, not via other intermediate factors as in the case of Ang II-induced muscle atrophy (22).
The ability of skeletal muscle to regenerate diminishes and ultimately fails with advancing age, likely as a result of an aging-associated decline of satellite cell number (18, 19, 24, 41). Aged satellite cells show less proliferative potential in response to injury or mitogenic stimulation (24). On the basis of our finding that Ang II reduces the satellite cell pool and blunts muscle regeneration, one can speculate that Ang II effects may play a role in aging-associated reduction in muscle regenerative capacity. In aged skeletal muscle, there is constitutive activation of TGF-β and attenuation of Notch activation after injury. Inhibition of TGF-β signaling or activation of Notch rescues the aging-associated decline of muscle regeneration via suppression of chronic kidney disease inhibitors (20), demonstrating that TGF-β and Notch are key age-related determinants of muscle regenerative potential. In vitro studies (42) have confirmed that TGF-β represses satellite cell proliferation and differentiation. However, the role played by the rapid increase of TGF-β expression after muscle injury (43, 44) in muscle regeneration is unclear. In our model, TGF-β mRNA levels were up-regulated on d3, consistent with previous reports (43, 44). It seems unlikely that TGF-β is a critical determinant of MyoD and myogenin expression at this time point because TGF-β is known to inhibit their expression (45). Nevertheless, it is interesting that Ang II suppressed CTX-induced TGF-β up-regulation. Future studies will be required to dissect the interaction of Ang II, TGF-β, and Notch signaling in muscle regeneration. It has been shown that Numb, a repressor of Notch signaling, asymmetrically segregates to two daughter cells after satellite cell division and has an important role to balance self-renewal and differentiation (46). Our data show that Numb expression is suppressed rapidly after CTX injection (as early as d1, Fig. 4A), consistent with activation and commitment of satellite cells to proliferation. Ang II prevents this reduction of Numb and activation of Notch, suggesting that Ang II down-regulation of Notch signaling prevents satellite cell division and the production of differentiating myoblasts. Our finding that Notch activation reversed the Ang II-induced decrease in satellite cell proliferation is consistent with this hypothesis (Fig. 4F). Furthermore, up-regulation of cyclin D1 and cyclin E and phosphorylation of retinoblastoma protein after CTX injection (Fig. 4A) is consistent with the activation of cell division, and Ang II-mediated suppression of cyclins and phospho-retinoblastoma protein provides further evidence that Ang II suppresses satellite cell division in regenerating skeletal muscle. CTX injection caused a reduction of AT1aR expression on d3, coinciding with an increase of MyoD and myogenin expression and activation of Notch, suggesting that AT1R signaling may inhibit these myogenic pathways (Fig. 1, G and H). On the other hand, the marked up-regulation in AT2R expression after CTX occurred after the reduction in AT1aR and coincided with eMyHC up-regulation (Fig. 1I). Up-regulation of AT2R following injury was consistent with high expression of AT2R in differentiating satellite cells cultured in vitro (Fig. 3I). It has been shown that AT2R signaling inhibits AT1R-mediated functions (47). Thus, the expression patterns of AT1R and AT2R following CTX-induced injury and in satellite cells in culture suggest opposing roles of these receptors in muscle regeneration.
In summary, we show that Ang II acts via the AT1aR on satellite cells to prevent satellite cell proliferation by suppressing MyoD and Notch signaling, thereby inhibiting normal skeletal muscle regeneration. Our findings may have major implications for understanding mechanisms of cachexia and sarcopenia that occur in chronic disease states and with aging. Some studies (but not all (48)) have suggested that Ang II may play a role in reducing muscle mass and function in the elderly (49–51), and angiotensin-converting enzyme inhibitors are reported to reduce the risk of weight loss in patients with CHF. Although the mechanisms of sarcopenia and cachexia are undoubtedly complex (52), the potential contribution of reduced muscle regenerative responses to functional impairment in patients with cachexia is very poorly understood. In patients with CHF and chronic kidney disease, there is a 2- to 5-fold increase in plasma Ang II levels, in many cases even in the presence of angiotensin-converting enzyme inhibitor therapy (53–56). Plasma Ang II levels increase about 3-fold in rats (29) and dogs (30) after LAD artery ligation, and infusion of 1 μg/kg/min Ang II yields a 3.2-fold increase in plasma Ang II (57). Therefore, our use of a dose of 1.5 μg/kg/min is consistent with the high Ang II levels seen in patients with CHF and chronic kidney disease, a population that has a high incidence of muscle wasting. Ang II-mediated inhibition of skeletal muscle regeneration may play a significant role in mechanisms of muscle wasting secondary to chronic diseases and aging.
Acknowledgments
We thank Mr. Alan Tucker (Tulane Flow Cytometry and Cell Sorting Core) for assistance with flow cytometry.
This research was supported, in whole or in part, by NHLBI National Institutes of Health Grants R01-HL070241 and R01-HL080682; National Institutes of Health National Center for Research Resources Grant P20-RR018766; NIGMS National Institutes of Health Grants P20-GM103514, P30-GM103337, and U54-GM104940; NIA National Institutes of Health Grants R01AG021566 and R01AG035377; and NIAMS National Institutes of Health Grant R21AR057794. This work was also supported by Muscular Dystrophy Association Grant 135908.
- CHF
- congestive heart failure
- Ang II
- angiotensin II
- AT1R
- angiotensin type 1 receptor
- LAD
- left anterior descending
- d5
- day 5
- eMyHC
- embryonic myosin heavy chain
- AT1aR
- angiotensin type 1a receptor
- X-gal
- 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside
- SC
- satellite cell
- CSA
- cross-sectional area.
REFERENCES
- 1. Tan B. H., Fearon K. C. (2008) Cachexia. Prevalence and impact in medicine. Curr. Opin. Clin. Nutr. Metab. Care 11, 400–407 [DOI] [PubMed] [Google Scholar]
- 2. Evans W. J., Morley J. E., Argilés J., Bales C., Baracos V., Guttridge D., Jatoi A., Kalantar-Zadeh K., Lochs H., Mantovani G., Marks D., Mitch W. E., Muscaritoli M., Najand A., Ponikowski P., Rossi Fanelli F., Schambelan M., Schols A., Schuster M., Thomas D., Wolfe R., Anker S. D. (2008) Cachexia. A new definition. Clin. Nutr. 27, 793–799 [DOI] [PubMed] [Google Scholar]
- 3. Anker S. D., Negassa A., Coats A. J., Afzal R., Poole-Wilson P. A., Cohn J. N., Yusuf S. (2003) Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors. An observational study. Lancet 361, 1077–1083 [DOI] [PubMed] [Google Scholar]
- 4. Sanders P. M., Russell S. T., Tisdale M. J. (2005) Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. Br. J. Cancer 93, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shrikrishna D., Astin R., Kemp P. R., Hopkinson N. S. (2012) Renin-angiotensin system blockade. A novel therapeutic approach in chronic obstructive pulmonary disease. Clin. Sci. 123, 487–498 [DOI] [PubMed] [Google Scholar]
- 6. Brink M., Wellen J., Delafontaine P. (1996) Angiotensin II causes weight loss and decreases circulating insulin-like growth factor I in rats through a pressor-independent mechanism. J. Clin. Invest. 97, 2509–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brink M., Price S. R., Chrast J., Bailey J. L., Anwar A., Mitch W. E., Delafontaine P. (2001) Angiotensin II induces skeletal muscle wasting through enhanced protein degradation and down-regulates autocrine insulin-like growth factor I. Endocrinology 142, 1489–1496 [DOI] [PubMed] [Google Scholar]
- 8. Song Y.-H., Li Y., Du J., Mitch W. E., Rosenthal N., Delafontaine P. (2005) Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J. Clin. Invest. 115, 451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshida T., Semprun-Prieto L., Sukhanov S., Delafontaine P. (2010) IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am. J. Physiol. Heart Circ. Physiol. 298, H1565–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshida T., Semprun-Prieto L., Wainford R. D., Sukhanov S., Kapusta D. R., Delafontaine P. (2012) Angiotensin II reduces food intake by altering orexigenic neuropeptide expression in the mouse hypothalamus. Endocrinology 153, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Semprun-Prieto L. C., Sukhanov S., Yoshida T., Rezk B. M., Gonzalez-Villalobos R. A., Vaughn C., Michael Tabony A., Delafontaine P. (2011) Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem. Biophys. Res. Comm. 409, 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tabony A. M., Yoshida T., Galvez S., Higashi Y., Sukhanov S., Chandrasekar B., Mitch W. E., Delafontaine P. (2011) Angiotensin II upregulates protein phosphatase 2Cα and inhibits AMP-activated protein kinase signaling and energy balance leading to skeletal muscle wasting. Hypertension 58, 643–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rezk B. M., Yoshida T., Semprun-Prieto L., Higashi Y., Sukhanov S., Delafontaine P. (2012) Angiotensin II infusion induces marked diaphragmatic skeletal muscle atrophy. PLoS ONE 7, e30276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan Z., Choi S., Liu X., Zhang M., Schageman J. J., Lee S. Y., Hart R., Lin L., Thurmond F. A., Williams R. S. (2003) Highly coordinated gene regulation in mouse skeletal muscle regeneration. J. Biol. Chem. 278, 8826–8836 [DOI] [PubMed] [Google Scholar]
- 15. Yablonka-Reuveni Z., Day K. (2011) Regenerating the Heart: Stem Cells and the Cardiovascular System, pp. 173–200, Springer, New York [Google Scholar]
- 16. McCroskery S., Thomas M., Maxwell L., Sharma M., Kambadur R. (2003) Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell Biol. 162, 1135–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sacco A., Doyonnas R., Kraft P., Vitorovic S., Blau H. M. (2008) Self-renewal and expansion of single transplanted muscle stem cells. Nature 456, 502–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shefer G., Van de Mark D. P., Richardson J. B., Yablonka-Reuveni Z. (2006) Satellite-cell pool size does matter. Defining the myogenic potency of aging skeletal muscle. Dev. Biol. 294, 50–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chakkalakal J. V., Jones K. M., Basson M. A., Brack A. S. (2012) The aged niche disrupts muscle stem cell quiescence. Nature 490, 355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carlson M. E., Hsu M., Conboy I. M. (2008) Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 454, 528–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cohn R. D., van Erp C., Habashi J. P., Soleimani A. A., Klein E. C., Lisi M. T., Gamradt M., ap Rhys C. M., Holm T. M., Loeys B. L., Ramirez F., Judge D. P., Ward C. W., Dietz H. C. (2007) Angiotensin II type 1 receptor blockade attenuates TGF-β-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 13, 204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang L., Du J., Hu Z., Han G., Delafontaine P., Garcia G., Mitch W. E. (2009) IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J. Am. Soc. Nephrol. 20, 604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ieronimakis N., Balasundaram G., Reyes M. (2008) Direct isolation, culture and transplant of mouse skeletal muscle derived endothelial cells with angiogenic potential. PLoS ONE 3, e0001753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Conboy I. M., Conboy M. J., Smythe G. M., Rando T. A. (2003) Notch-mediated restoration of regenerative potential to aged muscle. Science 302, 1575–1577 [DOI] [PubMed] [Google Scholar]
- 25. Conboy I. M., Rando T. A. (2002) The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell 3, 397–409 [DOI] [PubMed] [Google Scholar]
- 26. Michael L. H., Entman M. L., Hartley C. J., Youker K. A., Zhu J., Hall S. R., Hawkins H. K., Berens K., Ballantyne C. M. (1995) Myocardial ischemia and reperfusion. A murine model. Am. J. Physiol. 269, H2147–54 [DOI] [PubMed] [Google Scholar]
- 27. Day K., Shefer G., Shearer A., Yablonka-Reuveni Z. (2010) The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev. Biol. 340, 330–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Irintchev A., Zeschnigk M., Starzinski-Powitz A., Wernig A. (1994) Expression pattern of M-cadherin in normal, denervated, and regenerating mouse muscles. Dev. Dyn. 199, 326–337 [DOI] [PubMed] [Google Scholar]
- 29. Lütken S. C., Kim S. W., Jonassen T., Marples D., Knepper M. A., Kwon T.-H., Frøkiaer J., Nielsen S. (2009) Changes of renal AQP2, ENaC, and NHE3 in experimentally induced heart failure. Response to angiotensin II AT1 receptor blockade. Am. J. Physiol. Renal Physiol. 297, F1678–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jin D., Takai S., Sakaguchi M., Okamoto Y., Muramatsu M., Miyazaki M. (2004) An antiarrhythmic effect of a chymase inhibitor after myocardial infarction. J. Pharmacol. Exp. Ther. 309, 490–497 [DOI] [PubMed] [Google Scholar]
- 31. Schulze P. C., Fang J., Kassik K. A., Gannon J., Cupesi M., MacGillivray C., Lee R. T., Rosenthal N. (2005) Transgenic overexpression of locally acting insulin-like growth factor-1 inhibits ubiquitin-mediated muscle atrophy in chronic left-ventricular dysfunction. Circ. Res. 97, 418–426 [DOI] [PubMed] [Google Scholar]
- 32. Segersvärd H., Lakkisto P., Forsten H., Immonen K., Kosonen R., Palojoki E., Kankuri E., Harjula A., Laine M., Tikkanen I. (2013) Effects of angiotensin II blockade on cardiomyocyte regeneration after myocardial infarction in rats. J. Renin Angiotensin Aldosterone Syst., in press [DOI] [PubMed] [Google Scholar]
- 33. Sun C.-K., Chang L.-T., Sheu J.-J., Wang C.-Y., Youssef A. A., Wu C.-J., Chua S., Yip H.-K. (2007) Losartan preserves integrity of cardiac gap junctions and PGC-1 α gene expression and prevents cellular apoptosis in remote area of left ventricular myocardium following acute myocardial infarction. Int. Heart J. 48, 533–546 [DOI] [PubMed] [Google Scholar]
- 34. Ararat E., Brozovich F. V. (2009) Losartan decreases p42/44 MAPK signaling and preserves LZ+ MYPT1 expression. PLoS ONE 4, e5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pourdjabbar A., Parker T. G., Nguyen Q. T., Desjardins J.-F., Lapointe N., Tsoporis J. N., Rouleau J.-L. (2005) Effects of pre-, peri-, and postmyocardial infarction treatment with losartan in rats. Effect of dose on survival, ventricular arrhythmias, function, and remodeling. Am. J. Physiol. Heart Circ. Physiol. 288, H1997–2005 [DOI] [PubMed] [Google Scholar]
- 36. Johnston A. P., Baker J., Bellamy L. M., McKay B. R., De Lisio M., Parise G. (2010) Regulation of muscle satellite cell activation and chemotaxis by angiotensin II. PLoS ONE 5, e15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gordon S. E., Davis B. S., Carlson C. J., Booth F. W. (2001) ANG II is required for optimal overload-induced skeletal muscle hypertrophy. Am. J. Physiol. Endocrinol Metab 280, E150–9 [DOI] [PubMed] [Google Scholar]
- 38. Westerkamp C. M., Gordon S. E. (2005) Angiotensin-converting enzyme inhibition attenuates myonuclear addition in overloaded slow-twitch skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R1223–31 [DOI] [PubMed] [Google Scholar]
- 39. Skurk T., van Harmelen V., Hauner H. (2004) Angiotensin II stimulates the release of interleukin-6 and interleukin-8 from cultured human adipocytes by activation of NF-κB. Arterioscler. Thromb. Vasc. Biol. 24, 1199–1203 [DOI] [PubMed] [Google Scholar]
- 40. Serrano A. L., Baeza-Raja B., Perdiguero E., Jardí M., Muñoz-Cánoves P. (2008) Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 7, 33–44 [DOI] [PubMed] [Google Scholar]
- 41. Shefer G., Rauner G., Yablonka-Reuveni Z., Benayahu D. (2010) Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS ONE 5, e13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ten Broek R. W., Grefte S., Von den Hoff J. W. (2010) Regulatory factors and cell populations involved in skeletal muscle regeneration. J. Cell. Physiol. 224, 7–16 [DOI] [PubMed] [Google Scholar]
- 43. Goetsch S. C., Hawke T. J., Gallardo T. D., Richardson J. A., Garry D. J. (2003) Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol. Genomics 14, 261–271 [DOI] [PubMed] [Google Scholar]
- 44. Smith C. A., Stauber F., Waters C., Alway S. E., Stauber W. T. (2007) Transforming growth factor-β following skeletal muscle strain injury in rats. J. Appl. Physiol. 102, 755–761 [DOI] [PubMed] [Google Scholar]
- 45. Vaidya T. B., Rhodes S. J., Taparowsky E. J., Konieczny S. F. (1989) Fibroblast growth factor and transforming growth factor β repress transcription of the myogenic regulatory gene MyoD1. Mol. Cell Biol. 9, 3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kuang S., Gillespie M. A., Rudnicki M. A. (2008) Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell 2, 22–31 [DOI] [PubMed] [Google Scholar]
- 47. Fyhrquist F., Saijonmaa O. (2008) Renin-angiotensin system revisited. J. Intern. Med. 264, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gray S. L., LaCroix A. Z., Aragaki A. K., McDermott M., Cochrane B. B., Kooperberg C. L., Murray A. M., Rodriguez B., Black H., Woods N. F., and Women's Health Initiative Observational Study (2009) Angiotensin-converting enzyme inhibitor use and incident frailty in women aged 65 and older. Prospective findings from the Women's Health Initiative Observational Study. J. Am. Geriatr. Soc. 57, 297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Onder G., Penninx B. W., Balkrishnan R., Fried L. P., Chaves P. H., Williamson J., Carter C., Di Bari M., Guralnik J. M., Pahor M. (2002) Relation between use of angiotensin-converting enzyme inhibitors and muscle strength and physical function in older women. An observational study. Lancet 359, 926–930 [DOI] [PubMed] [Google Scholar]
- 50. Di Bari M., van de Poll-Franse L. V., Onder G., Kritchevsky S. B., Newman A., Harris T. B., Williamson J. D., Marchionni N., Pahor M., and Health, Aging and Body Composition Study (2004) Antihypertensive medications and differences in muscle mass in older persons. The Health, Aging and Body Composition Study. J. Am. Geriatr Soc. 52, 961–966 [DOI] [PubMed] [Google Scholar]
- 51. Sumukadas D., Witham M. D., Struthers A. D., McMurdo M. E. (2007) Effect of perindopril on physical function in elderly people with functional impairment. A randomized controlled trial. CMAJ 177, 867–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Glass D., Roubenoff R. (2010) Recent advances in the biology and therapy of muscle wasting. Ann. N.Y. Acad. Sci. 1211, 25–36 [DOI] [PubMed] [Google Scholar]
- 53. Masson S., Latini R., Bevilacqua M., Vago T., Sessa F., Torri M., Anesini A., Salio M., Pasotti E., Agnello D., Santoro L., Catania A., Ghezzi P., Moccetti T., Maggioni A. P. (1998) Within-patient variability of hormone and cytokine concentrations in heart failure. Pharmacol. Res. 37, 213–217 [DOI] [PubMed] [Google Scholar]
- 54. Roig E., Perez-Villa F., Morales M., Jiménez W., Orús J., Heras M., Sanz G. (2000) Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur. Heart J. 21, 53–57 [DOI] [PubMed] [Google Scholar]
- 55. Graziani G., Badalamenti S., Del Bo A., Marabini M., Gazzano G., Como G., Viganò E., Ambroso G., Morganti A. (1993) Abnormal hemodynamics and elevated angiotensin II plasma levels in polydipsic patients on regular hemodialysis treatment. Kidney Int. 44, 107–114 [DOI] [PubMed] [Google Scholar]
- 56. Simões e Silva A. C., Diniz J. S., Pereira R. M., Pinheiro S. V., Santos R. A. (2006) Circulating renin Angiotensin system in childhood chronic renal failure. Marked increase of angiotensin-(1–7) in end-stage renal disease. Pediatr. Res. 60, 734–739 [DOI] [PubMed] [Google Scholar]
- 57. Gonzalez-Villalobos R. A., Seth D. M., Satou R., Horton H., Ohashi N., Miyata K., Katsurada A., Tran D. V., Kobori H., Navar L. G. (2008) Intrarenal angiotensin II and angiotensinogen augmentation in chronic angiotensin II-infused mice. Am. J. Physiol. Renal Physiol. 295, F772–9 [DOI] [PMC free article] [PubMed] [Google Scholar]