

Background: E2F1-induced apoptosis kills oncogene-stressed cells, and regulators of this process may act as tumor suppressors.
Results: We identified UHRF2 in a functional screen for mediators of E2F1 induced apoptosis.
Conclusion: UHRF2 binds directly to E2F1 and is required for E2F1 induction of apoptosis and expression of several important apoptosis inducers.
Significance: UHRF2 is a suspected tumor suppressor and our work suggests an anti-tumor mechanism.
Keywords: Apoptosis, E2F Transcription Factor, Gene Expression, shRNA, Transcription Target Genes, UHRF2
Abstract
The E2F1 transcription factor is active in many types of solid tumors and can function as either an oncogene or tumor suppressor in vivo. E2F1 activity is connected with a variety of cell fates including proliferation, apoptosis, senescence, differentiation, and autophagy, and these effects are mediated through differential target gene expression. E2F1-induced cell death is an innate anti-cancer mechanism to kill cells with a spontaneous oncogenic mutation that might otherwise form a cancer. Relatively little is known about the molecular circuitry that tips E2F1 balance toward proliferation during normal growth versus apoptosis during oncogenic stress, and which pathways mediate this decision. To further explore these mechanisms, we utilized an unbiased shRNA screen to identify candidate genes that mediate E2F1-induced cell death. We identified the ubiquitin-like with PHD and ring finger domains 2 (UHRF2) gene as an important mediator of E2F1-induced cell death. UHRF2 encodes a nuclear protein involved in cell-cycle regulation. Several of these domains have been shown to be essential for the regulation of cell proliferation, and UHRF2 has been implicated as an oncogene in some settings. Other reports have suggested that UHRF2 causes growth arrest, functions as a tumor suppressor, and is deleted in a variety of tumors. We show that UHRF2 is a transcriptional target of E2F, that it directly interacts with E2F1, and is required for E2F1 induction of apoptosis and transcription of a number of important apoptotic regulators.
Introduction
Mammalian cell proliferation is generally dictated by extracellular signals that activate a cell division gene expression program. During the G1 phase of the cell cycle, this mitogenic stimulation and concomitant gene expression program initiate DNA synthesis (S phase), after which cells are committed to complete the cycle and divide. Passage through START coincides with activation of the E2F transcription factors, which induce expression of genes necessary for DNA replication, metabolism, and synthesis and are key transcriptional regulators of cell cycle progression during normal growth stimulation and cancer. The activity of E2Fs is inhibited by binding of the retinoblastoma (Rb)2 tumor suppressor gene. Rb binding to E2F proteins inhibits transcription by blocking the E2F transactivation region and also by actively recruiting histone deacetylases and other chromatin remodeling factors to repress gene expression (1, 2). Cdk phosphorylation of Rb during mitogenic stimulation, or loss of Rb in pre-neoplastic cells, prevents its inhibiting association with E2Fs and enables E2F-dependent target gene expression and proliferation.
S-phase promotion by deregulated E2F1 also triggers apoptosis, in part through expression of and cooperation with the pro-apoptotic p53 tumor suppressor (3, 4). Apoptosis is a mechanism by which cells respond to different stimuli by triggering a program of cell death for the elimination of redundant, autoreactive, or pre-neoplastic cells. Thus, cells acquiring a pro-proliferatory mutation, such as loss of Rb, can be eliminated via apoptosis unless they carry or acquire another mutation that impairs the apoptotic process. DNA damage also induces E2F1 levels and leads to apoptosis, however, treatment with these agents leads to different post-translational modifications of E2F1 and does not mimic aberrant deregulation during cell cycle entry. E2F1 induces apoptosis in part through the transcription of proapoptotic targets, such as p19ARF, p73, APAF1, SIVA, CASP3, and CASP7 (5–11). Expression of some of these genes appears to be cell type specific and is not absolutely required for E2F1 apoptosis induction. E2F induces the expression of numerous pro-apoptotic Bcl-2 homology 3 (BH3)-only proteins including BIM, BNIP3, BOK, PUMA, and NOXA and represses the expression of the anti-apoptotic MCL1 gene (12–15). The mechanisms suppressing E2F induction of apoptosis during normal cell cycle entry but permitting, or strengthening, cell death under conditions of E2F up-regulation that stem from the Rb pathway loss or other oncogenic mutations are not clearly understood.
To gain further insight into the mechanisms regulating E2F1-mediated cell death, we conducted an unbiased shRNA screen to identify regulators of E2F1 apoptosis induction. We identified the ubiquitin-like plant homeodomain (PHD) and ring finger domains 2 (UHRF2) gene as a mediator of E2F1-induced cell death. UHRF2 (also called NIRF) was previously identified as a ubiquitin ligase that interacts with the cyclin E-CDK2 complex and arrests cells in G1 (16). UHRF2 contains a ubiquitin-like domain, PHD finger domain, a set- and ring-associated (SRA) domain, and a RING finger domain (17). UHRF2 is functionally distinct from the structurally similar UHRF1, as ectopic UHRF2 expression does not rescue the DNA methylation defect in UHRF1 null mouse embryonic stem cells (18, 19). Intriguingly, both UHRF2 and UHRF1 physically associate with the Rb tumor suppressor protein and appear to recognize DNA and histone methylation to control cell cycle progression and gene expression (20, 21).
We demonstrate here that knockdown of UHRF2 impaired E2F1 activation of apoptosis and this effect is reverted by re-addition of a non-degradable form of UHRF2. Furthermore, E2F1 binds to the UHRF2 promoter and transcriptionally induces its expression. We also determined that UHRF2 mRNA and protein levels are induced in shUHRF1 knockdown cells, and conversely that UHRF1 mRNA is induced in shUHRF2 cells, suggesting potential compensatory regulation between these two proteins. We demonstrate that endogenous E2F1 and UHRF2 physically associate by co-immunoprecipitation assays. E2F1-induced gene expression profiles were measured in control and shUHRF2 cells. We determined that the E2F1-induced target genes showing poor induction in shUHRF2 cells were significantly populated with apoptotic target genes like SIVA and BIM. These findings suggest a model whereby E2F1 induces expression of UHRF2, which then binds to E2F1 and positively regulates expression of pro-apoptotic target genes.
EXPERIMENTAL PROCEDURES
shRNA Screen
PLAT-A retrovirus packaging cells were previously obtained from T. Kitamura (22). An Open Biosystems human shRNAmir library was divided into 30 pools with 1000 shRNAs per pool (23). Pooled shRNA plasmids were packaged into retrovirus using PLAT-A cell lines and infected into the U2OS human osteosarcoma cell line (obtained from ATCC). Approximately 2 × 107 U2OS cells were infected by library retroviral shRNAs at a multiplicity of infection of 0.5 and treated with puromycin to stably integrate virus. Cells were brought to quiescence by treatment with 0.25% fetal bovine serum (FBS) in Dulbecco's modified Eagle's medium (DMEM) and then infected with E2F1 expressing adenovirus at a multiplicity of infection (m.o.i.) of 10. This level of infection killed 100% of control (no library shRNAs) infected cells. Rare cells that survived E2F1 treatment after 5 days were pooled and the genomic DNA recovered from them. We PCR amplified recovered genomic DNA, shotgun cloned into TOPO TA vector (Invitrogen) and identified the recovered shRNAs by sequence analysis. We sequenced a total of 2000 clones and listed the recurring clones in Fig. 1B. Around 26% of the entire library was screened in these analyses (8 of 30 pools total).
FIGURE 1.
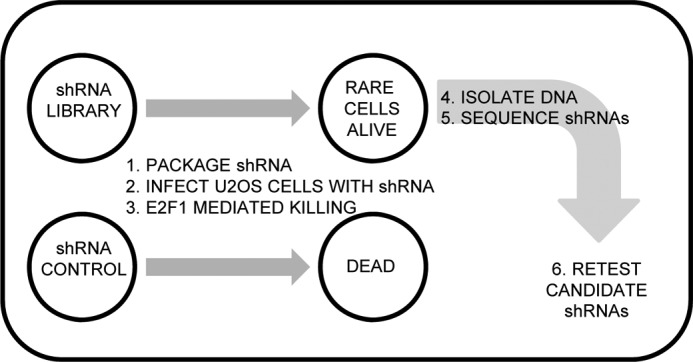
A functional shRNA screen for regulators of E2F1-induced apoptosis. Approach for identifying shRNAs that confer resistance to E2F1-mediated apoptosis in U2OS cells. The Open Biosystems shRNA library was pooled, packaged into retrovirus, and stably selected in U2OS cells. shRNA carrying U2OS cells were infected with E2F1 sufficient to induce apoptosis in 100% of control cells. shRNA carrying cells that resisted E2F1-induced apoptosis were harvested, and genomic DNA was amplified by PCR and cloned to allow sequencing of the region containing the integrated shRNA. Approximately 2000 inserts were sequenced.
Cell Culture and DNA Plasmids
U2OS (human osteosarcoma) cells from ATCC were cultured in DMEM with 10% fetal calf serum. Apoptotic cells were measured by harvesting floating and adherent cells 40 h post-infection and assayed by measuring anti-active caspase-3 by flow cytometry, or by staining using propidium iodide staining to measure the sub-G1 DNA content (Sigma). For individual shRNA retesting, Open Biosystems shRNA constructs were obtained from the University of Minnesota RNAi core facility, packaged as lentivirus and stably integrated into U2OS cells followed by puromycin selection. The UHRF2 cDNA was kindly provided by Dr. Tsutomu Mori (Fukushima Medical University School of Nursing). To generate the non-degradable UHRF2 allele, we altered the UHRF2 coding cDNA to maintain amino acid coding sequence but prevent degradation by shRNA targeting molecules using the following primers: forward, 5′-CCTGGAGCCCATCCCCTATCTTTCGCTGATGGAAAGTTTTTAAGGCG-3′ and reverse, 5′-CGCCTTAAAAACTTTCCATCAGCGAAAGATAGGGGATGGGCTCCAGG-3′.
The UHRF2 promoter (wild-type and with two putative E2F binding sites mutated) was PCR amplified from human U2OS cells and cloned into pGL3 for luciferase assays, which were performed following the manufacturer's instructions (Promega). A reverse primer was common to both fragments (5′-GACATGTACAAGCTTGAGATCCACGCCGGGCTCCG-3′) and was paired with either wild-type primer (5′-GTCGTAGACGCTAGCGCCTGGAGCTGCCGCCTCCG-3′) or 2x-mutant E2F binding sites (5′-GTCGTAGACGCTAGCGCCTGGAGCTGCCGCCTCCGCCAGGCGACGGGAAACCCTCGGAAGTGGGTGCGGCCGCGAAAGTAGCATTGCGGCCAGGCGGCCGCCGTGTTCGCGAAGCAGGAGGG-3′).
RNA Isolation, Real-time PCR, and Microarray Analysis
We used Qiagen QIAshredder and RNeasy Midi Kits to isolate cellular RNA and the QuantiTect SYBR Green RT-PCR kit from Qiagen for our quantitative real-time PCR. Each experimental condition used 200 ng of RNA for reverse transcription and RT-PCR and was performed in triplicate and normalized against GAPDH expression levels. Analysis was done with a StepOnePlus real-time PCR system (Applied Biosystem). For microarrays, RNA was harvested from infected cells and analyzed on Illumina Human WG-6 version 3 beadchips in duplicate. The GATHER (Gene Annotation Tool to Help Explain Relationships) website was used to perform GO and TRANSFAC analysis. The following primers were used for RT-PCR: UHRF2, forward, 5′-TTGCTGCTGATGAAGACGTT-3′ and reverse, 5′-TTCTGCATCAAACCAGAATCC-3′; MIA2, forward 5′-ATTTGGCGTTCACAGAATCC-3′ and reverse, 5′-TCGGCAGTCAGGTCCTCTAT-3′; WDFY3, forward, 5′-ACGCCTTAGGACTGATGCAC-3′ and reverse, 5′-TTGTTTGATGCTCTCCTTCG-3′; CCNA2, forward, 5′-CTCCAAGAGGACCAGGAGAA-3′ and reverse, 5′-TGAACGCAGGCTGTTTACTG-3′; AZIN1, forward, 5′-GCCTTGGGTATTTCTGCTCA-3′ and reverse, 5′-GCACTTCTCCTAGGCCCTCT-3′; EIF5A2, forward 5′-TTCCCACGGAAAAACTACCA-3′ and reverse, 5′-TTTGCCCGTGAAAATATCAA-3′; KBTBD7, forward, 5′-GAGGGTGAGTCAGCCACTGT-3′ and reverse, 5′-GGACTGCATGGTGGAGATG-3′; GPR45, forward, 5′-ATTTCTGTCCCAGCTCCAAG-3′ and reverse, 5′-GGCCTCTGGTACACGATGAT-3′; MSH6, forward, 5′-AAGGCGAAGAACCTCAACG-3′ and reverse, 5′-TGTTGGGCTGTCATCAAAAA-3′; ADAMTLS3, forward, 5′-CCAGCCCCATTTAACTTCG-3′ and reverse, 5′-AGGAAGCCATGGGGTCTAGT-3′; UHRF1, forward, 5′-GCCTGCAGAGGCTGTTCTAC-3′ and reverse, 5′-GTGGGTGGAGGACTTGTCTG-3′; TRAF3, forward, 5′-CGCTAAAGCTGCACACTGAC-3′ and reverse, 5′-AACGATGCTCTCTTGACACG-3′; LY6H, forward, 5′-CTGCCTGCAGCCATGAAG-3′ and reverse, 5′-GATTCGGACACTGGCACAC-3′; AKT1-V3, forward, 5′-ATGAGCGACGTGGCTATTGT-3′ and reverse, 5′-CGCCACAGAGAAGTTGTTGA-3′; SIVA, forward, 5′-GTCCATTGCCTGTTCCTCAT-3′ and reverse, 5′-CTGGTGCACAGCACTTTCTC-3′; JAG2, forward, 5′-GGAGGTTCTGCGATGAGTGT-3′ and reverse, 5′-CACAGTTCCTGCCCGAGTA-3′; NFKBIB, forward, 5′-ACTCCCGACACCAACCATAC-3′ and reverse, 5′-CGGACCATCTCCACATCTTT-3′; CCNE1, forward, 5′-CCTCGGATTATTGCACCATC-3′ and reverse, 5′-AGAATTGCTCGCATTTTTGG-3′; PDCD5, forward, 5′-AGGGGCTGCGAGAGTGAC-3′ and reverse, 5′-CTGTGCTTTGCTTCCTGTTG-3′; HRK, forward, 5′-CAGGCGGAACTTGTAGGAAC-3′ and reverse, 5′-CCCAGTCCCATTCTGTGTTT-3′; PPP1R13B, forward, 5′-GACTCTCCCCGCGATGAT-3′ and reverse, 5′-TCCCCTCCACACTTCAGCTA-3′; FOXO3, forward, 5′-ACAAACGGCTCACTCTGTCC-3′ and reverse, 5′-TCTTGCCAGTTCCCTCATTC-3′; FOXO1, forward, 5′-AAGAGCGTGCCCTACTTCAA-3′ and reverse, 5′-CTGTTGTTGTCCATGGATGC-3′; CBX7, forward, 5′-CATGGAGCTGTCAGCCATC-3′ and reverse, 5′-CCATCCTTTCCACTTCACCA-3′; BCL211, forward, 5′-CCACCACTTGATTCTTGCAG-3′ and reverse, 5′-GTTGCTTTGCCATTTGGTCT-3′.
Protein Immunoblotting and Co-immunoprecipitation
U2OScells were harvested 40 h post-infection into microcentrifuge tubes and re-suspended in boiling sample buffer. Equivalent amounts of protein were separated by SDS-PAGE, transferred to an Immobilon-P (Millipore) membrane, and blocked in T-TBS containing 5% nonfat dry milk. Blots were then incubated with primary antiserum (1:1000) at room temperature for 4 h, washed three times with T-TBS buffer, and then incubated with the appropriate secondary antiserum (1:2000) for 1 h at room temperature. Blots were processed using the ECL system (Amersham Biosciences). Antiserum against E2F1 (c-20) and anti-Myc epitope (9E10) for immunoblot analysis was purchased from Santa Cruz Biotechnology. Antisera against UHRF2 (U6884) was obtained from Sigma. Anti-HA antisera was purchased from Roche Applied Science (3F10).
For co-immunoprecipitations, cells were lysed into lysis buffer (50 mm Tris, pH 7.4, 300 mm NaCl, 5 mm EDTA, 0.5% Nonidet P-40, 50 mm NaF, 1 mm Na3VO4, 1 × protease inhibitor mixture) and precipitated with primary antibody and Protein A/G beads. Pellets were washed 3 times with ice-cold washing buffer (50 mm HEPES, pH 7.3, 150 mm NaCl, 2.5 mm EGTA, 10% glycerol, 0.1% Tween 20, 1 mm NaF, 1 mm DTT, 0.1 mm Na3VO4) and analyzed by SDS-PAGE.
Chromatin Immunoprecipitation
We followed a protocol described by Bomsztyk for ChIP analysis (24). U2OS cells were treated with serum-deprived media to more closely resemble cellular situations with increased E2F1 levels but lacking growth factor-induced proliferation stimuli. Two 15-cm plates of U2OS cells for each condition were fixed with formaldehyde for 15 min at room temperature and then quenched with glycine (125 mm) for 5 min. Cells were collected, washed, and resuspended with 300 μl of lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris, pH8.0), and then diluted 10-fold with dilution buffer (1% Triton X-100, 150 mm NaCl, 2 mm EDTA, 20 mm Tris, pH 8.1). We resuspended the DNA pellet in IP buffer and sonicated chromatin to an approximate length of 500–600 nucleotides. We performed immunoprecipitations using mock IP, anti-E2F1 (C-20 Santa Cruz), or anti-UHRF2 (U6884, Sigma) antibodies overnight at 4 ºC and then treated with protein A-agarose beads for 60 min. Complexes were centrifuged and washed three times with ice-cold IP buffer (150 mm NaCl, 5 mm EDTA, 0.5% Nonidet P-40, 1% Triton X-100, 50 mm Tris, pH 7.5). DNA precipitation was facilitated using Chelex 100 (Bio-Rad) by adding 100 μl of 10% to DNA samples before ethanol precipitating. Precipitated DNA was quantified by reverse transcription followed by SYBR Green qPCR.
Plasmid ChIP was performed the same as described above, except that U2OS cells were electroporated with 1 μg/106 cells of either wild-type or the 2x-mutant UHRF2 promoter construct plasmid (in pGL3) and harvested 48 h later. The C-20 antibody was used to precipitate E2F1, and precipitated plasmid was detected by RT-PCR using primers that bound pGL3 plasmid (5′-CTAGCAAAATAGGCTGTCCC-3′) and one binding internally to the UHRF2 promoter (5′-GCTCCCGCCACGCGGCC-3′) that functions with both constructs. E2F1 ChIP was compared with pulldown by control IgG antibody.
RESULTS
shRNA Screen Identifies Genes That Mediate E2F1-induced Apoptosis
We utilized an unbiased shRNA screening approach to identify genes that mediate E2F1-induced apoptosis (Fig. 1). Our experimental design hypothesized that integration of shRNAs conferring resistance to E2F1-induced apoptosis should allow those cells to survive a dosage of E2F1 that efficiently kills control cells. We chose to activate cell death by ectopically expressing E2F1 because it more closely mimics the up-regulation of E2F1 seen following RB pathway mutation than that caused by DNA damage treatment, which leads to differential phosphorylation of E2F1. These cells could then be recovered and integrated shRNAs identified. We observed rare surviving cells following infection with E2F1 expressing adenovirus, and these cells were harvested so we could identify recovered shRNAs. We sequenced around 2000 clones and listed the top recovered genes in Table 1.
TABLE 1.
List of gene targets identified by shRNA screening for regulators of E2F1-induced apoptosis. Ten recovered genes are listed with gene names and the number of times identified (Hits)
| Gene | Full name | Hits |
|---|---|---|
| EIF5A2 | Eukaryotic translation initiation factor 5A2 | 9 |
| AZIN1 | Antizyme inhibitor 1 | 4 |
| UHRF2 | Ubiquitin-like with PHD and ring finger domains 2, E3 ubiquitin protein ligase | 3 |
| MSH6 | mutS homolog 6 (Escherichia coli) | 2 |
| ADAMTSL3 | ADAMTS-like 3 | 2 |
| KBTBD7 | Kelch repeat and BTB (POZ) domain containing 7 | 2 |
| CCNA2 | Cyclin A2 | 2 |
| GPR45 | G protein-coupled receptor 45 | 2 |
| WDFY3 | WD repeat and FYVE domain containing 3 | 2 |
| MIA2 | Melanoma inhibitory activity 2 | 2 |
Frequently identified genes were individually knocked down in U2OS cells by shRNA and tested for their ability to inhibit E2F1-induced apoptosis. We analyzed each of these generated cell lines for resistance to E2F1-induced apoptosis. Each cell line was treated with 0.25% FBS containing media (serum deprivation) followed by infection with E2F1 expressing adenovirus at an m.o.i. of 10. Cells were harvested 48 h post-infection and cleaved caspase-3 levels were measured by flow cytometry. Levels of apoptosis in control cells were designated as 100% and apoptotic levels of experimental cell lines were expressed as a % fraction relative to control (Fig. 2A). Paired t tests were performed to determine which of the results were statistically significant. Seven of the knockdown cell lines tested (UHRF2, MIA2, WDFY3, CCNA2, AZIN1, EIF5A2, and KBTBD7) displayed a significant reduction in apoptosis, ranging from 55 to 30%, compared with control cells. The three other lines tested (GPR45, MSH6, and ADAMTSL3) did not show significant apoptotic differences in response to E2F1 induction. Knockdown of UHRF2 had the greatest inhibition of E2F1-induced apoptosis, reducing levels by over 50%. qPCR was used to measure mRNA levels between vector control and shRNA cell lines to determine knockdown efficiency. Knockdown results are displayed in Fig. 2B as “% remaining expression” and range from 3 to 56% of the remaining expression, presented in the order of genes as shown in Fig. 2A.
FIGURE 2.
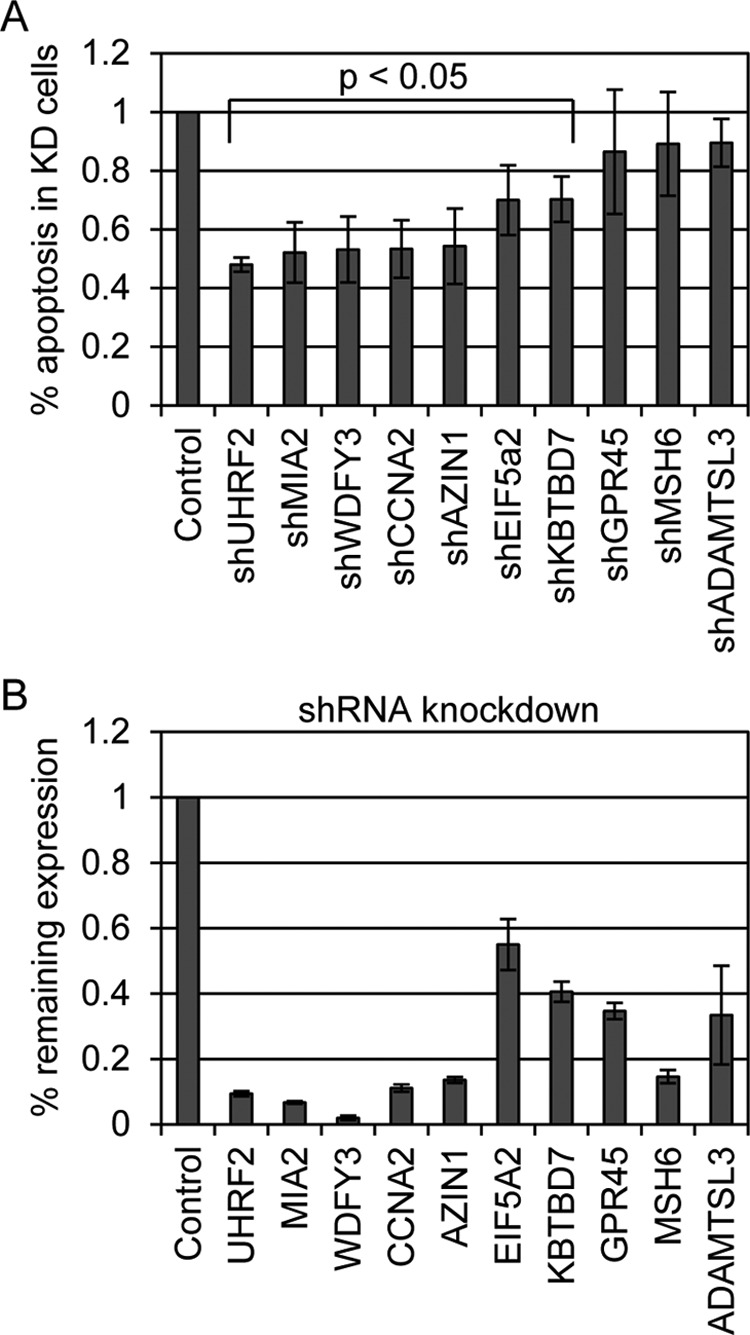
Identification of mediators of E2F1-induced apoptosis. We targeted each of the genes recovered by our screening by shRNA to retest for prevention of E2F1-induced apoptosis. Individual shRNAs listed in Table 1 were stably integrated into U2OS cells. A, serum deprived control and shRNA target cells were infected with E2F1 expressing adenovirus and later measured for apoptosis by quantifying cleaved caspase-3 levels. The level of apoptosis induction by E2F1 compared with control in non-shRNA carrying cells was set to 100%. Three of the 10 shRNA lines (GPR45, MSH6, and ADAMTSL3) did not display reduced E2F1-induced apoptosis. The other seven lines, however, recapitulated the findings of the screen and significantly reduced E2F1 induction of apoptosis from 25–55%. Student's t tests were performed and indicate that the reduced apoptosis seen in these seven cell lines was significant (p < 0.05). B, target gene knockdown was measured by qPCR, comparing levels in vector control to levels in shRNA knockdown lines. Results are reported as % remaining expression in target cells and are listed in the order presented in A.
E2F1 is believed to trigger apoptosis through the expression of pro-apoptotic target genes. It is possible that the genes we identified in this screen could be apoptotic target genes transcriptionally induced by E2F1 that when knocked down lower the apoptotic threshold. Alternatively, or in addition to the first possibility, some of the identified genes may regulate the function of E2F1 to promote apoptotic transcriptional output when knocked down, and in this case, E2F1 apoptotic output would be diminished. We used qPCR to determine whether any of the recovered genes are E2F1-induced transcriptional targets (Fig. 3). U2OS cells were infected with control or E2F1-expressing adenovirus at 10 m.o.i. Gene expression levels were compared between control and E2F1-infected cells by qPCR and are displayed as fold-induction changes in Fig. 3. We determined that 4 of the 7 genes we identified that affect E2F1 apoptosis were indeed transcriptionally induced by E2F1. EIF5A2 induced the highest (7-fold), UHRF2 and CCNA2 induced around 6-fold, and AZIN1 around 3-fold (p < 0.05). The genes WDFY3, KBTBD7, and MIA2 did not display mRNA changes following E2F1 expression.
FIGURE 3.

UHRF2 is an E2F1-induced target gene. A, serum deprived U2OS cells were infected with E2F1 expressing adenovirus and mRNA isolated 24 h post-infection for qPCR analysis. Each of the seven genes that significantly reduced E2F1-induced apoptosis in Fig. 2 were tested for transcriptional induction by E2F1. Three of the genes, EIF5A2, UHRF2, and CCNA2, were each induced around 5–8-fold by E2F1. AZIN1 was induced around 3-fold, and the other genes were uninduced. Results are shown as fold-induction compared with induction by control adenovirus in U2OS cells. Student's t tests were performed and indicate that the increased expression of EIF5A2, UHRF2, CCNA2, and AZIN1 was significant (p < 0.05). B, we performed chromatin immunoprecipitations to determine whether endogenous E2F1 binds directly to the native UHRF2 promoter. Chromatin was prepared from serum-deprived wild-type U2OS cells and immunoprecipitated with control IgG or anti-E2F1 antisera. qPCR was used to measure precipitated DNA. Units represent fold-induced increase in binding, comparing binding between anti-E2F1 and control IgG. ChIP analyses indicate that E2F1 binds directly to the endogenous UHRF2 promoter. C, mutations were introduced into two putative E2F sites in the human UHRF2 promoter. The first site, located between −520 and −527 relative to translation start, was mutated from “TTTCCGCG” to “TgcggcCG.” The second site, located between −451 and −458, was altered from “TTTCCCGC” to “gcggCCGC.” The wild-type and 2x mutant E2F UHRF2 promoters were PCR amplified and cloned upstream of luciferase in the pGL-3 vector. D, HEK293 cells were co-transfected with either wild-type or mutant luciferase plasmids and either control or E2F1 expressing plasmid and processed for luciferase activity assays 24 h post-transfection. Our results were that E2F1 induced the wild-type construct 6-fold and the 2x mutant by 3-fold. Thus, mutating the two putative E2F sites reduced responsiveness to E2F1 activity by 50% (p = 0.01). E, U2OS cells were electroporated with either the wild-type or 2x mutant E2F UHRF2-luciferase construct and harvested for ChIP assay 48 h later for immunoprecipitation with IgG or anti-E2F1 (C-20) antisera. We observe a 3.5-fold increased precipitation of the wild-type UHRF2 promoter by anti-E2F1 antisera compared with control IgG. Mutating two putative E2F binding sites significantly reduced pulldown by E2F1 compared with IgG, indicating that E2F1 binding has been perturbed by mutating the candidate E2F binding sites.
Other studies have indicated that UHRF1, like UHRF1, is also transcriptionally induced by E2F1 (25). We tested if E2F1 binds directly to the UHRF2 promoter by ChIP and quantified a 6-fold increase in E2F1 binding compared to control IgG (Fig. 3B). We scanned the UHRF2 promoter for putative E2F recognition motifs. The analysis picked up a number of potential E2F binding sites, and we mutated two putative motifs that gave a high score by TRANSFAC. We sequenced the final construct to verify that the potential E2F binding sites were mutated (Fig. 3C). Our results indicated that E2F1 induced the wild-type construct 6-fold and the 2x mutant by 3-fold (Fig. 3D). Thus, mutating the two putative E2F sites reduced responsiveness to E2F1 activity by 50% (p = 0.01). We think that the most likely conclusion was that the UHRF2 promoter contains more than these two E2F responsive elements.
We performed plasmid ChIP assays to test if mutating these putative E2F binding motifs reduced E2F1 binding to the UHRF2 promoter (Fig. 3E). Wild-type and mutant UHRF2-luciferase constructs were electroporated into U2OS cells and later harvested for ChIP assay. We observed a 3.5-fold increase in wild-type UHRF2 pulldown by anti-E2F1 compared with anti-IgG control antisera. However, mutation of the two putative E2F motifs resulted in a failure of anti-E2F1 to immunoprecipitate the plasmid, compared with IgG. We conclude that E2F1 induces expression of UHRF2, binds to its promoter in vivo and in vitro, and that mutating the putative E2F motifs blocks induction and E2F1 binding in vitro.
Furthermore, UHRF2 and UHRF1 are structurally similar and it is not entirely clear to what extent these proteins share overlapping or distinct functions. We targeted UHRF1 for shRNA degradation in U2OS cells to gain a better understanding of the potential synergistic control of E2F1 induced apoptosis by UHRF1 with UHRF2. Surprisingly, we observed a greater than 2-fold increase in UHRF2 levels in shUHRF1 knockdown cells by qPCR, compared with levels in control cells (Fig. 4A). Furthermore, UHRF1 mRNA levels increased around 2.5-fold in the shUHRF2 cell lines, compared with control. We also generated U2OS cells carrying both shUHRF1 and shUHRF2. Levels of both UHRF1 and UHRF2 were reduced by 40 and 60%, respectively, compared with levels in control cells. We asked whether the potential compensatory changes in UHRF2 mRNA levels also occurred at the protein level. We observed a decrease in UHRF2 protein levels in the shUHRF2 cell lines and also noted a 2–3-fold increase in UHRF2 protein in the shUHRF1 cells (Fig. 4B). We tested if E2F1-induced apoptosis is also affected by UHRF1 knockdown in Fig. 4C. Control, shUHRF1, shUHRF2, and combined shUHRF1/2 U2OS cells were infected with E2F1 expressing adenovirus and harvested later for quantification of levels of apoptotic cells. UHRF1 knockdown blocked E2F1 induction of apoptosis around 40% compared with control cells. As seen previously, E2F1-induced apoptosis was reduced around 50% in shUHRF2 cells. Combined knockdown of UHRF1 and UHRF2 in the same cell line led to a greater decrease in the number of apoptotic cells after E2F1 infection (70% reduction).
FIGURE 4.
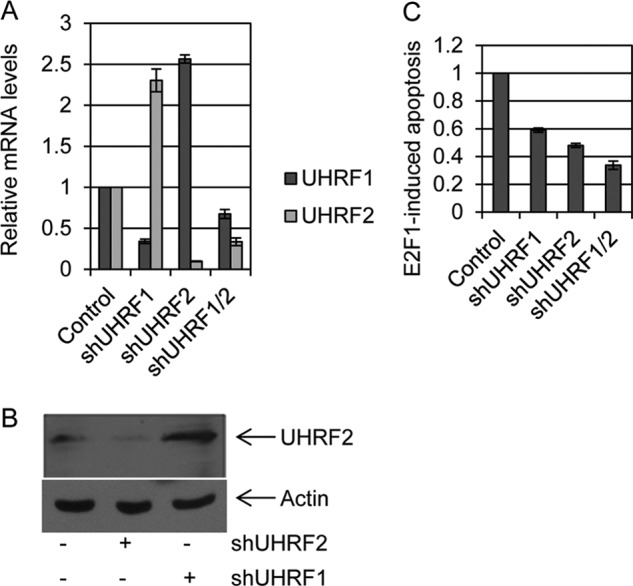
Compensatory expression of UHRF2 following loss of UHRF1. A, UHRF1 and UHRF2 were knocked down individually, or both together in U2OS cells. qPCR was used to measure expression levels of both UHRF1 and UHRF2 in these cell lines. We observe that knockdown of UHRF1 caused a significant reduction in UHRF1 levels, but interestingly caused over a 2-fold induction in UHRF2. Similarly, UHRF2 knockdown effectively reduced UHRF2 mRNA levels, whereas simultaneously inducing UHRF1 around 2-fold. Simultaneous knockdown of both UHRF1 and UHRF2 led to reductions in mRNA of both genes. B, endogenous UHRF2 protein levels were assessed by immunoblotting in control, shUHRF2 and shUHRF1 cell lines. UHRF2 protein levels are elevated in shUHRF1 cell lines. Actin immunoblotting serves as a control for equal protein loading. C, E2F1-induced apoptosis was measured in each of the knockdown cell lines. We found that knockdown of either UHRF1 or UHRF2 led to decreased E2F1-mediated apoptosis induction, and knockdown of both genes further reduced apoptosis induction by E2F1.
To confirm that the effect of shUHRF2 on E2F1-induced apoptosis was not due to off-target shRNA effects, we constructed a non-degradable (nd) allele of UHRF2 that maintained proper amino acid coding sequence but altered mRNA to prevent its degradation by the shRNA. This allele of UHRF2 was fused to an amino-terminal HA tag. The HA-ndUHRF2 or control plasmids were transfected into shUHRF2 U2OS cells and harvested 24 h post-transfection for anti-HA immunoblot analysis (Fig. 5a). We observed an accumulation of stabilized HA-UHRF2 following transfection of this construct. For apoptosis assays, control and shUHRF2 U2OS cells were transfected with control or HA-ndUHRF2 plasmids (46) and then infected 24 h later with control or E2F1-expressing adenovirus (Fig. 5B). Cells were harvested 48 h post-infection for measurement of apoptosis levels. E2F1 infection induced apoptosis in 65% of control cells, and shUHRF2 knockdown reduced the percent of apoptotic cells to 28%. Transfection of the HA-ndUHRF2 plasmid led to an E2F1-dependent restoration of apoptotic levels to 53%. These results indicate that E2F1-induced apoptosis is affected by UHRF2 degradation and not by off-target shRNA effects.
FIGURE 5.
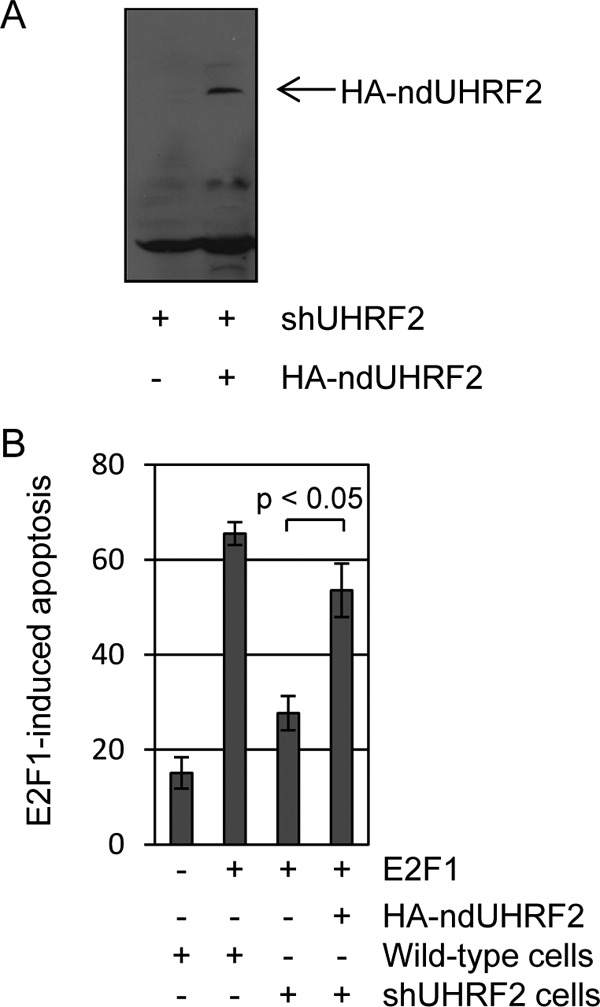
Non-degradable UHRF2 restores E2F1-induced apoptosis. A, an HA-tagged UHRF2 cDNA was mutated to prevent degradation by shRNA but not alter protein coding sequence. This or control cDNA was transfected into shUHRF2 U2OS stable cell lines and harvested 48 h later for HA-immunoblot analysis. HA-UHRF2 protein levels are substantially restored by the ND-UHRF2 allele. B, wild-type or shUHRF2 U2OS cells were deprived of serum and transfected with control or ND-UHRF2. Transfected cells were then infected with either control or E2F1 expressing adenovirus and harvested 48 h later for apoptosis analysis. Addition of the non-degradable form of UHRF2 restored levels of E2F1-induced apoptosis to 83% of that seen in control transfected U2OS cells. Student's t tests were performed and indicate that the increased apoptosis observed upon ND-UHRF2 introduction was significant (p < 0.05). These findings indicate that the ability of shUHRF2 on reducing E2F1 apoptosis induction is not due to off-target shRNA effects.
We showed that UHRF2 is an E2F1 transcriptional target and is bound by E2F1 at its promoter region. It is possible that UHRF2 facilitates apoptosis induction through E2F1 either by directly interacting with E2F1, or by performing other activities downstream of its induction by E2F1. We used a variety of co-immunoprecipitation assays to determine whether E2F1 and UHRF2 directly physically interact (Fig. 6). First, plasmids encoding HA-tagged E2F1 or Myc-tagged UHRF2 were co-transfected into HEK293 cells (Fig. 6A). Extracts were co-immunoprecipitated with control or anti-Myc monoclonal antisera (9E10) and immunoblotted with anti-HA antisera to detect precipitated E2F1. We observed a significant HA-E2F1 immunoprecipitation by Myc-UHRF2, compared with IgG control. In Fig. 6B, the reciprocal immunoprecipitation was performed. Extracts, prepared as in Fig. 6A, were immunoprecipitated with control IgG, anti-Myc, or anti-HA antisera. Immunoblotting with anti-Myc demonstrated a strong UHRF2 IP-Western blot (lane 3). Furthermore, HA-E2F1 immunoprecipitation also co-precipitated Myc-UHRF2 (lane 4). In Fig. 6, C and D, we asked if endogenous E2F1 interacts with the Myc-tagged UHRF2 protein. HEK293 cells were transfected with Myc-UHRF2 and endogenous E2F1 was co-precipitated (Fig. 6C) and then immunoblotted with anti-Myc to detect precipitated UHRF2. In Fig. 6D, extracts were immunoprecipitated with anti-Myc antisera and blotted for endogenous E2F1. Fig. 6, C and D, demonstrate that endogenous E2F1 associates with Myc-UHRF2. Finally, we tested the direct interaction of endogenous E2F1 and UHRF2 in Fig. 6E. HEK293 cell extracts were immunoprecipitated with control IgG or anti-E2F1 antisera and immunoblotted with anti-UHRF2 antisera. UHRF2 was co-precipitated with anti-E2F1 but not the control antisera.
FIGURE 6.
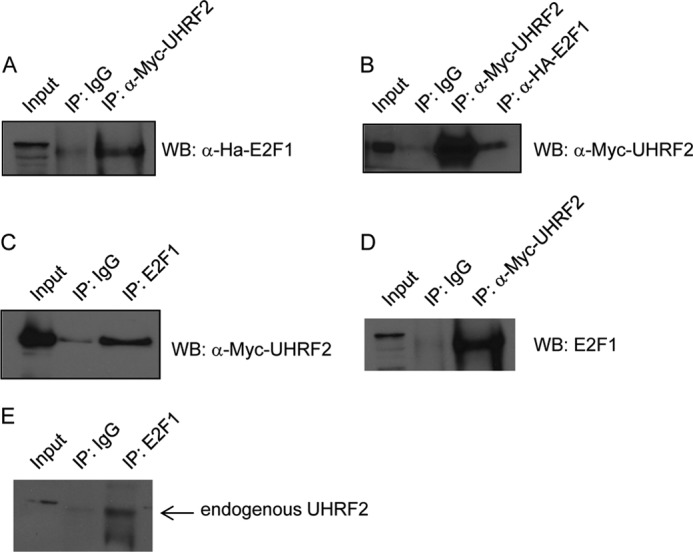
UHRF2 interacts with E2F1 by co-immunoprecipitation. We probed for an interaction between E2F1 and UHRF2 using several complementary approaches. First, HA-tagged E2F1 and Myc-tagged UHRF2 were co-transfected into 293T cells. Protein extracts were prepared and used for co-immunoprecipitation assays. A, extracts were immunoprecipitated with anti-Myc antibodies (9E10) and immunoblotted with anti-HA to detect input and precipitated HA-E2F1. Alternatively, in B, extracts were immunoprecipitated with anti-Myc or anti-HA and then immunoblotted with anti-Myc to detect input and pelleted Myc-UHRF2. C, 293T cells were transfected with Myc-tagged UHRF2 and then endogenous E2F1 was immunoprecipitated followed by anti-Myc immunoblotting to detect input and precipitated UHRF2. D, extracts from 293T cells transfected with Myc-UHRF2 were immunoprecipitated using anti-Myc monoclonal followed by immunoblotting with anti-E2F1 antisera to detect co-precipitated endogenous E2F1. E, extracts from unaltered 293T cells were subjected to immunoprecipitation with control or anti-E2F1 antisera to test for an interaction between endogenous E2F1 and UHRF2. Precipitated pellets were immunoblotted with anti-UHRF2 antisera, and this indicated stronger pulldown of UHRF2 with anti-E2F1 than control IgG antisera. WB, Western blot.
The data thus far point to a model where E2F1 induces expression UHRF2, which then binds to E2F1 to induce apoptosis. Because E2F1 promotes apoptosis primarily through transcription of apoptotic target genes, we asked if disrupting the E2F1-UHRF2 complex perturbed target gene induction. To assess this possibility, we performed microarray analysis comparing gene expression caused by E2F1 expression in control and shUHRF2 U2OS cells. Control and shUHRF2 knockdown U2OS cells were deprived of serum for 48 h and infected with 10 m.o.i. control (CMV) or E2F1 expressing adenovirus. RNA was harvested from cells 24 h post-infection and processed for analysis on Illumina Human WG-6 v3 beadchips. Results were normalized using GeneSpring and analyzed for expression differences again using either GeneSpring software or Microsoft Excel.
Genes were initially sorted in Excel based on the strategy outlined in Fig. 7. To do this we compared gene expression in control cells infected with control or E2F1-expressing adenovirus. We focused on genes induced at least 2-fold by E2F1 in control U2OS cells, which totaled 2445 genes out of a total of 48,806, or roughly 5%. These E2F1-induced genes were then further sorted based on the effect that shUHRF2 had on their expression. We did this by comparing gene expression levels between E2F1-infected control versus shUHRF2 cells. Genes identified as being induced by E2F1 and that partially require UHRF2 for full expression are listed in Table 2. “F.I.” refers to fold-induction by E2F1, and “shUHRF2” is the ratio between the expression level of the gene in E2F1-infected shUHRF2 cells divided by the level in E2F1-infected control cells. The majority of E2F1-induced genes were unaffected by UHRF2 depletion. Around 6% of the E2F1-induced targets were reduced 50% or more following loss of UHRF2.
FIGURE 7.
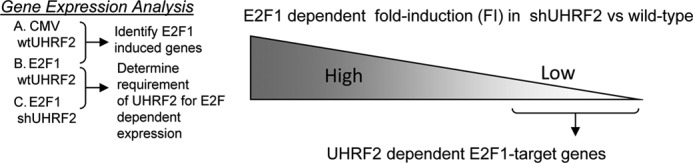
Strategy to identify E2F1 target genes requiring UHRF2 for expression. We utilized a microarray strategy to determine which E2F1-induced targets require UHRF2 to mediate expression. Control and shUHRF2 U2OS cells were infected with equal m.o.i. of control or E2F1-expressing adenovirus. RNA was harvested from infected cells and analyzed on Illumina Human WG-6 v3 beadchips. We identified E2F1-induced targets in U2OS cells by comparing gene induction levels between control or E2F1-infected cells. Genes that had been induced at least 2-fold by E2F1 were then further sorted based on the effect that UHRF2 knockdown had on E2F1-dependent expression.
TABLE 2.
Identification of E2F1 target genes requiring UHRF2 for expression
The E2F-induced genes that display the greatest failure in induction upon UHRF2 depletion are shown. F.I. refers to “fold-induction” by E2F1 compared with CMV infection in control U2OS cells. shUHRF2 is the percent remaining expression of the gene in shUHRF2 U2OS cells compared with E2F1-induction of the gene in control cells.
| Gene | F.I. | shUHRF2 | Definition |
|---|---|---|---|
| SLC32A1 | 9.6 | 30% | Solute carrier family 32 (GABA vesicular transporter), member 1 (SLC32A1), mRNA. |
| FHDC1 | 3.3 | 31% | FH2 domain containing1 (FHDC1), mRNA. |
| CKB | 3.8 | 35% | Creatine kinase, brain (CKB), mRNA. |
| LY6H | 3.2 | 36% | Lymphocyte antigen 6 complex, locusH (LY6H), mRNA. |
| AKT1 | 4.0 | 36% | v-akt murine thymoma viral oncogene homolog1 (AKT1), transcript variant 3, mRNA. |
| ZNF672 | 3.3 | 39% | Zinc finger protein 672 (ZNF672), mRNA. |
| LHX3 | 8.3 | 39% | LIM homeobox3 (LHX3), transcript variant 1, mRNA. |
| CRIP2 | 5.8 | 41% | Cysteine-rich protein2 (CRIP2), mRNA. |
| ADIPOR2 | 2.5 | 41% | Adiponectin receptor2 (ADIPOR2), mRNA. |
| PHF21B | 4.1 | 45% | PHD finger protein 21B (PHF21B), mRNA. |
| NFKBIB | 8.3 | 45% | Nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor, β |
| TRAF3 | 2.8 | 46% | TNF receptor-associated factor3 (TRAF3), transcript variant 3, mRNA. |
| JAG2 | 10.1 | 48% | Jagged2 (JAG2), transcript variant 1, mRNA. |
| PPP1R13B | 4.2 | 49% | Protein phosphatase 1, regulatory (inhibitor) subunit 13B (PPP1R13B), mRNA. |
| SIVA | 3.6 | 52% | CD27-binding (Siva) protein (SIVA), transcript variant 1, mRNA. |
| CBX7 | 4.2 | 53% | Chromobox homolog7 (CBX7), mRNA. |
| CCNE1 | 7.1 | 56% | Cyclin E1 (CCNE1), transcript variant 2, mRNA. |
| CSE1L | 3.6 | 56% | CSE1 chromosome segregation 1-like (yeast) (CSE1L), transcript variant 2, mRNA. |
| MOAP1 | 2.5 | 57% | Modulator of apoptosis1 (MOAP1), mRNA. |
| PDCD5 | 3.5 | 58% | programmed cell death5 (PDCD5), mRNA. |
| BCL2L11 | 32.0 | 65% | BCL2-like 11 (apoptosis facilitator) (BCL2L11), transcript variant 6, mRNA. |
| HRK | 15.1 | 70% | Harakiri, BCL2 interacting protein (contains only BH3 domain) (HRK), mRNA. |
| FOXO3 | 4.4 | 74% | Forkhead box O3 (FOXO3), transcript variant 2, mRNA. |
| FOXO1 | 9.8 | 77% | Forkhead box O1 (FOXO1), mRNA. |
We used gene ontology (GO) annotations to better understand if the UHRF2-regulated E2F1 target genes could be grouped into functional categories. We selected the top 150 genes whose induction by E2F1 was most impaired by shUHRF2 and analyzed them by GO annotations using GATHER (Gene Annotation Tool to Help Explain Relationships). These genes showed a reduction in induction by E2F1 between 40 and 70% in shUHRF2 cells compared with control. GO analysis of these genes indicated that the functional category most highly represented in this group was apoptosis (GO:0006915) followed by programmed cell death (GO:0012501) with p values of 0.01 for each category (Table 3). TRANSFAC analysis using the GATHER website indicated that E2F1 was the most highly represented transcription factor binding site in the 150 promoters analyzed (p value <0.0001) (Table 3). These results suggest that E2F1-UHRF2 complexes may regulate expression of a higher frequency of apoptotic target genes. This is in agreement with our finding that UHRF2 knockdown impairs apoptosis induction.
TABLE 3.
GO and TRANSFAC analysis of genes co-regulated by E2F1 and UHRF2
The top 150 E2F1-induced genes displaying the greatest reduction in expression in shUHRF2 cells were analyzed for over-represented GO categories or putative transcription factor-binding sites using the GATHER website. Apoptosis and programmed cell death were the top represented functional categories, and E2F1 binding sites were the most prevalent represented TRANSFAC site. E2F target genes were least induced in shUHRF2 cells.
| No. | p value | BF | |
|---|---|---|---|
| GO annotation | |||
| 1 | GO:0006915 [6]: apoptosis | 0.01 | 1 |
| 2 | GO:0012501 [5]: programmed cell death | 0.01 | 1 |
| 3 | GO:0009896 [6]: positive regulation of catabolism | 0.01 | 1 |
| 4 | GO:0016572 [9]: histone phosphorylation | 0.01 | 1 |
| 5 | GO:0040023 [6]: nuclear positioning | 0.01 | 1 |
| 6 | GO:0045732 [7]: positive regulation of protein catabolism | 0.01 | 1 |
| 7 | GO:0007097 [7]: nuclear migration | 0.01 | 1 |
| 8 | GO:0015849 [5]: organic acid transport | 0.01 | 1 |
| 9 | GO:0046942 [6]: carboxylic acid transport | 0.01 | 1 |
| 10 | GO:0008219 [4]: cell death | 0.01 | 1 |
| 11 | GO:0016265 [3]: death | 0.01 | 1 |
| TRANSFAC | |||
| 1 | V$E2F1_Q3_01 | <0.0001 | 16 |
| 2 | V$KROX_Q6 | <0.0001 | 15 |
| 3 | V$MYCMAX_B: c-Myc:Max binding sites | 0.0001 | 5 |
| 4 | V$CEBPDELTA_Q6 | 0.0001 | 5 |
| 5 | V$E2F1DP2_01: E2F-1:DP-2 heterodimer | 0.0002 | 5 |
| 6 | V$CDX2_Q5 | 0.0003 | 4 |
| 7 | V$E2F1DP1_01: E2F-1:DP-1 heterodimer | 0.0005 | 4 |
| 8 | V$E2F_Q3_01 | 0.0005 | 4 |
We manually tested the effect of UHRF2 loss on E2F1 induction of 14 of these target genes by qPCR. Control and shUHRF2 U2OS cell lines were infected with empty or E2F1-expressing adenovirus and qPCR was used to analyze some of the genes identified by the microarray approach. We determined by what percentage these E2F1-induced targets were reduced following loss of UHRF2, by comparing their E2F1-dependent expression in shUHRF2 with expression levels in control cells. The results are displayed with the genes showing the greatest dependence for their induction by E2F1 on the left of Fig. 8A. For example, TRAF3, which is induced by E2F1 around 16-fold, displays a 70% drop in E2F1-dependent expression in shUHRF2 cells. Other proapoptotic E2F1 targets include SIVA, PDCD5, and HRK, which also showed a significantly reduced induction by E2F1 following UHRF2 knockdown. In total, 9 of 14 genes we tested showed a significant drop in E2F1-dependent expression in shUHRF2 cells compared with induction in control cells. E2F1 mediated fold-induction results presented in Fig. 8B are in the order based on their positions in Fig. 8A. Target genes were induced ranging from 4- (PDCD5) to 40-fold (JAG2).
FIGURE 8.
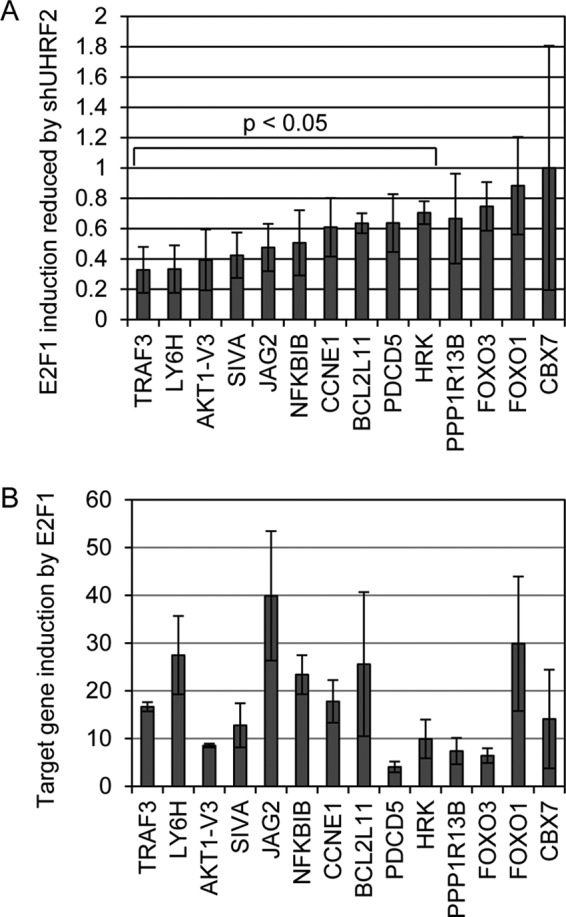
Validation of E2F1/UHRF2 dependent target gene expression. We used qPCR to determine the validity of the microarray results. A, control and shUHRF2 U2OS cells were infected with E2F1 expressing adenovirus, mRNA harvested, and 14 genes were analyzed by qPCR. We compared E2F1-dependent expression levels in shUHRF2 cells compared with levels of induction by E2F1 in wild-type U2OS and presented this as a ratio. Ten of the 14 genes tested showed a significant (p < 0.5) reduction, as determined by Student's t test, ranging from 30 to 70% reduction in E2F1 induced expression in shUHRF2 cells. B, E2F1 induction of these genes was tested by comparing expression levels in U2OS cells infected with either control or E2F1-expressing adenovirus. Target gene expression by E2F1 varies from 4- to 40-fold and is presented in the order of genes in A.
We used chromatin immunoprecipitation (ChIP) to test if UHRF2 and E2F1 both bound to the promoters of co-regulated target genes identified by microarray and confirmed by qPCR. U2OS cells were treated with serum-deprived media to more closely resemble cellular situations with increased E2F1 levels but lacking growth factor-induced proliferation stimuli. Chromatin from wild-type U2OS cells was prepared and immunoprecipitated with control IgG, anti-E2F1, or anti-UHRF2 antisera. Our ChIP analysis indicates that E2F1 binds to the BIM, cyclin E1 (CCNE1), SIVA, TRAF3, and dihydrofolate reductase promoters, but not to the β-actin control (Fig. 9A). Units represent fold-enrichment of PCR product generated from chromatin precipitated with anti-E2F1 and control IgG. In Fig. 9B, anti-UHRF2 ChIP analysis reveals that BIM, CCNE1, SIVA, and TRAF3 are bound by UHRF2, however, neither dihydrofolate reductase nor β-actin is bound by UHRF2. These results indicate that four genes identified by microarray and qPCR as reliant on E2F1 and UHRF2 for expression are bound at their promoters by both E2F1 and UHRF2. Dihydrofolate reductase was not identified in the microarray analysis as reliant on UHRF2 for expression, and it is not bound by UHRF2 by ChIP.
FIGURE 9.
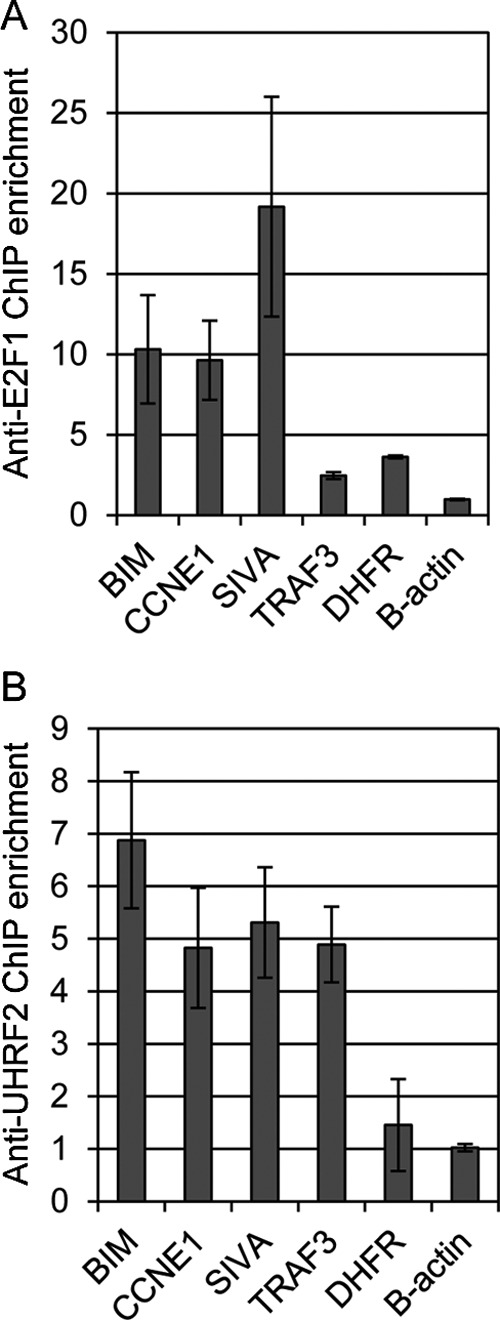
E2F1 and UHRF2 bind to co-regulated targets by ChIP analysis. We performed chromatin immunoprecipitations to determine whether E2F1 and UHRF2 bind to the promoters of co-regulated target genes. Chromatin was prepared from wild-type U2OS cells grown in serum-deprived media and immunoprecipitated with control IgG or anti-E2F1 (A) or anti-UHRF2 (B) antisera and quantified by qPCR. Units represent fold-enrichment of the PCR product corresponding to the promoters under analysis, comparing binding between anti-E2F1 or UHRF2 and control IgG. ChIP analyses indicate that the BIM, CCNE1, SIVA, and TRAF3 promoters are bound by both E2F1 and UHRF2. The dihydrofolate reductase gene is bound by E2F1 but not UHRF2, and the control β-actin is not bound by either E2F1 or UHRF2.
DISCUSSION
The Rb/E2F pathway is deregulated in a wide variety of human solid tumors. Rampant E2F activity caused by loss of Rb promotes unrestrained activation of proliferation target gene expression, critical to tumor cell growth. However, E2F activity also promotes distinct cell fate outcomes such as cell death, senescence, differentiation, and others, but the key regulatory mechanisms underlying these decisions are not clearly understood. We therefore utilized an unbiased shRNA screening procedure to identify regulators of E2F1-induced apoptotic cell death. Overall, the genes we identified as putative mediators of E2F1-induced apoptosis can be classified into three broad functional categories. These categories include cell signaling (MIA2, WDFY3, CCNA2, and AZIN1), protein translation (EIF5A2), and DNA transcription/chromatin state modification (UHRF2 and KBTBD7).
MIA2 (melanoma inhibitory activity 2) is an SH3 containing protein that is down-regulated in hepatocellular carcinomas and its overexpression causes growth arrest in hepatocytes (26). WDFY3 (WD repeat and FYVE domain containing 3) can bind to phosphatidylinositol 3-phosphate (PI3K) and regulate aggregated protein clearance by autophagy (27). CCNA2 (cyclin A2) is a member of the conserved cyclin family of proteins that binds to and controls cyclin-dependent kinases and function as regulators of G1/S and G2/M cell cycle transitions. Interestingly, CCNA2-CDK2 complexes bind to and phosphorylate the amino terminus of E2F1, inhibiting its DNA binding affinity (28, 29). AZIN1 (antizyme inhibitor 1) indirectly regulate polyamine biosynthesis through the stabilization of ornithine decarboxylase, a mediator of the ornithine to putrescine conversion (30). eIF5A2 (eukaryotic translation initiation factor 5A2) is amplified and appears oncogenic in a variety of human tumors (31). eIF5A2, and the family member eIF5A1, are the only eukaryotic proteins that utilize the unique, polyamine-derived amino acid hypusine (32). KBTBD7 (kelch repeat and BTB (POZ) domain containing 7) is a member of a large family of kelch motif containing proteins that are generally involved in protein/protein interactions. KBTB7, when overexpressed, was shown to activate the transcriptional activities of activator protein-1 and serum response element (SRE) factors (33). UHRF2 (ubiquitin-like with PHD and ring finger domains 2, E3 ubiquitin protein ligase) encodes a complex, multidomain nuclear protein with DNA and histone binding capacity and ubiquitin-ligase activity and is involved in cell cycle regulation and potentially tumorigenesis (17, 34).
UHRF2 was originally called NIRF and was cloned through a two-hybrid interaction with PEST-containing nuclear protein and described as highly expressed in proliferating cells but not in the G0/G1 cell cycle phase (34). Subsequent work demonstrated that ectopic UHRF2 expression caused G1 phase arrest concomitant with Cdk2-cyclin E binding and degradation (16). UHRF2 is reported to interact centrally with many cell cycle regulators including CDKs, cyclins, proliferating cell nuclear antigen, p53, and pRB (21). Some of these interactions, such as those with cyclin D1 and E1, destabilize the target proteins. Our work here demonstrated that E2F1 induces expression of UHRF2, that UHRF2 binds directly to the E2F1 protein, and that this complex controls apoptosis induction and expression of some apoptotic target genes, like BIM and SIVA.
UHRF2 is closely related to the structurally similar UHRF1 gene, which also encodes a DNA- and histone-binding protein. Both proteins contain similar domains, like an ubiquitin-like domain, tandem tudor domain, a PHD, a SRA domain, and a RING finger domain (17). In UHRF1, the tandem tudor domain binds heterochromatin tri-methylated histone H3 lysine 9 (H3K9me3), the PHD domain binds methylated histones and other proteins like pRb and DNMT1 (17, 35). Both UHRF1 and UHRF2 contain LXCXE motifs in their PHD domain and physically associate with the pRB protein (20, 21). The SRA domain of UHRF1 preferentially recognizes and binds to hemimethylated DNA, although the UHRF2 SRA domain shows no apparent distinction in binding between hemi- over unmethylated DNA (18).
Common documented functions of UHRF2 and UHRF1 include regulation and inheritance of the epigenetic code during cell division, targeted protein ubiquitination and degradation, cell cycle control, transcriptional regulation, and tumorigenesis (17, 36). UHRF1 plays a role in maintaining DNA methylation in mammalian cells during DNA replication (37, 38). UHRF1 binds to the DNA methyltransferase DNMT1 and through the SRA domain interacts with hemimethylated DNA and executes base-flipping of 5-methylcytosines out of the DNA helix during replication (39–41). UHRF1 null mouse embryos display a severe DNA methylation defect, similar to DNMT1 nulls (42, 43). Intriguingly, ectopic expression of UHRF2 does not rescue this UHRF1 null defect, nor does UHRF2 display preferential binding to hemi-methylated DNA over unmethylated DNA as UHRF1, suggesting distinct functions between these proteins (18, 19).
Both UHRF1 and UHRF2 are implicated in tumorigenesis. UHRF1 expression, like we demonstrate for UHRF2, is induced by E2F1 and is critical for proliferation of various tumor cell lines (25). UHRF1 is highly expressed in a variety of human cancers (44, 45). In contrast, UHRF2 is located at the 9p24 locus, a site of frequent deletion in human tumors. Indeed, analysis in Oncomine indicates a reduced copy number of UHRF2 in non-small cell lung carcinomas, which correlates with the reduced UHRF2 mRNA in subtypes of this cancer (21). The mechanisms by which UHRF2 suppresses various tumor types, whether this involves suppression of Rb/E2F1 induction of apoptosis and/or G1 arrest, and how UHRF2 controls E2F1 induction of target gene induction are critical questions for future studies.
Acknowledgement
We thank Dr. Tsutomu Mori (Fukushima Medical University School of Nursing, Fukushima, Japan) for graciously providing tagged UHRF2 plasmids.
This work was supported in part by Children's Cancer Research Fund, Minneapolis, MN.
- Rb
- retinoblastoma
- PHD
- plant homeodomain
- SRA
- set- and ring-associated
- m.o.i.
- multiplicity of infection
- IP
- immunoprecipitation
- qPCR
- quantitative PCR
- nd
- non-degradable
- GO
- gene annotation
- MIA2
- melanoma inhibitory activity 2
- AZIN1
- antizyme inhibitor 1
- UHRF2
- ubiquitin-like with PHD and ring finger domains 2.
REFERENCES
- 1. Zhang H. S., Gavin M., Dahiya A., Postigo A. A., Ma D., Luo R. X., Harbour J. W., Dean D. C. (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 101, 79–89 [DOI] [PubMed] [Google Scholar]
- 2. Ogawa H., Ishiguro K., Gaubatz S., Livingston D. M., Nakatani Y. (2002) A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296, 1132–1136 [DOI] [PubMed] [Google Scholar]
- 3. Wu X., Levine A. J. (1994) p53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl. Acad. Sci. U.S.A. 91, 3602–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hallstrom T. C., Nevins J. R. (2003) Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc. Natl. Acad. Sci. U.S.A. 100, 10848–10853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bates S., Phillips A. C., Clark P. A., Stott F., Peters G., Ludwig R. L., Vousden K. H. (1998) p14ARF links the tumour suppressors RB and p53. Nature 395, 124–125 [DOI] [PubMed] [Google Scholar]
- 6. Irwin M., Marin M. C., Phillips A. C., Seelan R. S., Smith D. I., Liu W., Flores E. R., Tsai K. Y., Jacks T., Vousden K. H., Kaelin W. G., Jr. (2000) Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 407, 645–648 [DOI] [PubMed] [Google Scholar]
- 7. Stiewe T., Pützer B. M. (2000) Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat. Genet. 26, 464–469 [DOI] [PubMed] [Google Scholar]
- 8. Lissy N. A., Davis P. K., Irwin M., Kaelin W. G., Dowdy S. F. (2000) A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature 407, 642–645 [DOI] [PubMed] [Google Scholar]
- 9. Moroni M. C., Hickman E. S., Lazzerini Denchi E., Caprara G., Colli E., Cecconi F., Müller H., Helin K. (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 3, 552–558 [DOI] [PubMed] [Google Scholar]
- 10. Nahle Z., Polakoff J., Davuluri R. V., McCurrach M. E., Jacobson M. D., Narita M., Zhang M. Q., Lazebnik Y., Bar-Sagi D., Lowe S. W. (2002) Direct coupling of the cell cycle and cell death machinery by E2F. Nat. Cell Biol. 4, 859–864 [DOI] [PubMed] [Google Scholar]
- 11. Rogoff H. A., Pickering M. T., Debatis M. E., Jones S., Kowalik T. F. (2002) E2F1 induces phosphorylation of p53 that is coincident with p53 accumulation and apoptosis. Mol. Cell. Biol. 22, 5308–5318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Croxton R., Ma Y., Song L., Haura E. B., Cress W. D. (2002) Direct repression of the Mcl-1 promoter by E2F1. Oncogene 21, 1359–1369 [DOI] [PubMed] [Google Scholar]
- 13. Hershko T., Ginsberg D. (2004) Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 279, 8627–8634 [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez J. M., Glozak M. A., Ma Y., Cress W. D. (2006) Bok, Bcl-2-related ovarian killer, is cell cycle-regulated and sensitizes to stress-induced apoptosis. J. Biol. Chem. 281, 22729–22735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tracy K., Dibling B. C., Spike B. T., Knabb J. R., Schumacker P., Macleod K. F. (2007) BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell. Biol. 27, 6229–6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Y., Mori T., Hata H., Homma Y., Kochi H. (2004) NIRF induces G1 arrest and associates with Cdk2. Biochem. Biophys. Res. Commun. 319, 464–468 [DOI] [PubMed] [Google Scholar]
- 17. Bronner C., Achour M., Arima Y., Chataigneau T., Saya H., Schini-Kerth V. B. (2007) The UHRF family. Oncogenes that are drugable targets for cancer therapy in the near future? Pharmacol. Ther. 115, 419–434 [DOI] [PubMed] [Google Scholar]
- 18. Pichler G., Wolf P., Schmidt C. S., Meilinger D., Schneider K., Frauer C., Fellinger K., Rottach A., Leonhardt H. (2011) Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J. Cell Biochem. 112, 2585–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J., Gao Q., Li P., Liu X., Jia Y., Wu W., Li J., Dong S., Koseki H., Wong J. (2011) S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 21, 1723–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jeanblanc M., Mousli M., Hopfner R., Bathami K., Martinet N., Abbady A. Q., Siffert J. C., Mathieu E., Muller C. D., Bronner C. (2005) The retinoblastoma gene and its product are targeted by ICBP90. A key mechanism in the G1/S transition during the cell cycle. Oncogene 24, 7337–7345 [DOI] [PubMed] [Google Scholar]
- 21. Mori T., Ikeda D. D., Fukushima T., Takenoshita S., Kochi H. (2011) NIRF constitutes a nodal point in the cell cycle network and is a candidate tumor suppressor. Cell Cycle 10, 3284–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morita S., Kojima T., Kitamura T. (2000) Plat-E. An efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 23. Silva J. M., Li M. Z., Chang K., Ge W., Golding M. C., Rickles R. J., Siolas D., Hu G., Paddison P. J., Schlabach M. R., Sheth N., Bradshaw J., Burchard J., Kulkarni A., Cavet G., Sachidanandam R., McCombie W. R., Cleary M. A., Elledge S. J., Hannon G. J. (2005) Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 37, 1281–1288 [DOI] [PubMed] [Google Scholar]
- 24. Nelson J. D., Denisenko O., Bomsztyk K. (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1, 179–185 [DOI] [PubMed] [Google Scholar]
- 25. Mousli M., Hopfner R., Abbady A. Q., Monté D., Jeanblanc M., Oudet P., Louis B., Bronner C. (2003) ICBP90 belongs to a new family of proteins with an expression that is deregulated in cancer cells. Br. J. Cancer 89, 120–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hellerbrand C., Amann T., Schlegel J., Wild P., Bataille F., Spruss T., Hartmann A., Bosserhoff A. K. (2008) The novel gene MIA2 acts as a tumour suppressor in hepatocellular carcinoma. Gut 57, 243–251 [DOI] [PubMed] [Google Scholar]
- 27. Filimonenko M., Isakson P., Finley K. D., Anderson M., Jeong H., Melia T. J., Bartlett B. J., Myers K. M., Birkeland H. C., Lamark T., Krainc D., Brech A., Stenmark H., Simonsen A., Yamamoto A. (2010) The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 38, 265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mudryj M., Devoto S. H., Hiebert S. W., Hunter T., Pines J., Nevins J. R. (1991) Cell cycle regulation of the E2F transcription factor involves an interaction with cyclin A. Cell 65, 1243–1253 [DOI] [PubMed] [Google Scholar]
- 29. Bandara L. R., Adamczewski J. P., Hunt T., La Thangue N. B. (1991) Cyclin A and the retinoblastoma gene product complex with a common transcription factor. Nature 352, 249–251 [DOI] [PubMed] [Google Scholar]
- 30. Koguchi K., Kobayashi S., Hayashi T., Matsufuji S., Murakami Y., Hayashi S. (1997) Cloning and sequencing of a human cDNA encoding ornithine decarboxylase antizyme inhibitor. Biochim. Biophys. Acta 1353, 209–216 [DOI] [PubMed] [Google Scholar]
- 31. Zender L., Xue W., Zuber J., Semighini C. P., Krasnitz A., Ma B., Zender P., Kubicka S., Luk J. M., Schirmacher P., McCombie W. R., Wigler M., Hicks J., Hannon G. J., Powers S., Lowe S. W. (2008) An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 135, 852–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clement P. M., Henderson C. A., Jenkins Z. A., Smit-McBride Z., Wolff E. C., Hershey J. W., Park M. H., Johansson H. E. (2003) Identification and characterization of eukaryotic initiation factor 5A-2. Eur. J. Biochem. 270, 4254–4263 [DOI] [PubMed] [Google Scholar]
- 33. Hu J., Yuan W., Tang M., Wang Y., Fan X., Mo X., Li Y., Ying Z., Wan Y., Ocorr K., Bodmer R., Deng Y., Wu X. (2010) KBTBD7, a novel human BTB-kelch protein, activates transcriptional activities of SRE and AP-1. BMB Rep. 43, 17–22 [DOI] [PubMed] [Google Scholar]
- 34. Mori T., Li Y., Hata H., Ono K., Kochi H. (2002) NIRF, a novel RING finger protein, is involved in cell-cycle regulation. Biochem. Biophys. Res. Commun. 296, 530–536 [DOI] [PubMed] [Google Scholar]
- 35. Rothbart S. B., Krajewski K., Nady N., Tempel W., Xue S., Badeaux A. I., Barsyte-Lovejoy D., Martinez J. Y., Bedford M. T., Fuchs S. M., Arrowsmith C. H., Strahl B. D. (2012) Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 19, 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Unoki M., Brunet J., Mousli M. (2009) Drug discovery targeting epigenetic codes: the great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochem. Pharmacol. 78, 1279–1288 [DOI] [PubMed] [Google Scholar]
- 37. Bostick M., Kim J. K., Estève P. O., Clark A., Pradhan S., Jacobsen S. E. (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317, 1760–1764 [DOI] [PubMed] [Google Scholar]
- 38. Sharif J., Muto M., Takebayashi S., Suetake I., Iwamatsu A., Endo T. A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K., Tajima S., Mitsuya K., Okano M., Koseki H. (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912 [DOI] [PubMed] [Google Scholar]
- 39. Hashimoto H., Horton J. R., Zhang X., Bostick M., Jacobsen S. E., Cheng X. (2008) The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature 455, 826–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Avvakumov G. V., Walker J. R., Xue S., Li Y., Duan S., Bronner C., Arrowsmith C. H., Dhe-Paganon S. (2008) Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 455, 822–825 [DOI] [PubMed] [Google Scholar]
- 41. Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. (2008) Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature 455, 818–821 [DOI] [PubMed] [Google Scholar]
- 42. Muto M., Kanari Y., Kubo E., Takabe T., Kurihara T., Fujimori A., Tatsumi K. (2002) Targeted disruption of Np95 gene renders murine embryonic stem cells hypersensitive to DNA damaging agents and DNA replication blocks. J. Biol. Chem. 277, 34549–34555 [DOI] [PubMed] [Google Scholar]
- 43. Hervouet E., Lalier L., Debien E., Cheray M., Geairon A., Rogniaux H., Loussouarn D., Martin S. A., Vallette F. M., Cartron P. F. (2010) Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis from human and mice glial cells. PLoS One. 5, e11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jenkins Y., Markovtsov V., Lang W., Sharma P., Pearsall D., Warner J., Franci C., Huang B., Huang J., Yam G. C., Vistan J. P., Pali E., Vialard J., Janicot M., Lorens J. B., Payan D. G., Hitoshi Y. (2005) Critical role of the ubiquitin ligase activity of UHRF1, a nuclear RING finger protein, in tumor cell growth. Mol. Biol. Cell 16, 5621–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Unoki M., Nishidate T., Nakamura Y. (2004) ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene 23, 7601–7610 [DOI] [PubMed] [Google Scholar]
- 46. Mori T., Li Y., Hata H., Kochi H. (2004) NIRF is a ubiquitin ligase that is capable of ubiquitinating PCNP, a PEST-containing nuclear protein. FEBS Lett. 557, 209–214 [DOI] [PubMed] [Google Scholar]