

Background: The mechanism of repression of inflammasome caused by Francisella tularensis is not known.
Results: F. tularensis represses AIM2 and NLRP3 inflammasomes in a FTL_0325-dependent fashion.
Conclusion: Repression of inflammasome by F. tularensis results in fulminate infection.
Significance: This study advances the understanding of mechanisms of immune suppression caused by F. tularensis.
Keywords: Bacterial Pathogenesis, Cytokine, Immunosuppression, Inflammation, Macrophages, Francisella, Il-1b, Immune Subversion, Inflammasome
Abstract
Francisella tularensis is an important human pathogen responsible for causing tularemia. F. tularensis has long been developed as a biological weapon and is now classified as a category A agent by the Centers for Disease Control because of its possible use as a bioterror agent. F. tularensis represses inflammasome; a cytosolic multi-protein complex that activates caspase-1 to produce proinflammatory cytokines IL-1β and IL-18. However, the Francisella factors and the mechanisms through which F. tularensis mediates these suppressive effects remain relatively unknown. Utilizing a mutant of F. tularensis in FTL_0325 gene, this study investigated the mechanisms of inflammasome repression by F. tularensis. We demonstrate that muted IL-1β and IL-18 responses generated in macrophages infected with F. tularensis live vaccine strain (LVS) or the virulent SchuS4 strain are due to a predominant suppressive effect on TLR2-dependent signal 1. Our results also demonstrate that FTL_0325 of F. tularensis impacts proIL-1β expression as early as 2 h post-infection and delays activation of AIM2 and NLRP3-inflammasomes in a TLR2-dependent fashion. An enhanced activation of caspase-1 and IL-1β observed in FTL_0325 mutant-infected macrophages at 24 h post-infection was independent of both AIM2 and NLRP3. Furthermore, F. tularensis LVS delayed pyroptotic cell death of the infected macrophages in an FTL_0325-dependent manner during the early stages of infection. In vivo studies in mice revealed that suppression of IL-1β by FTL_0325 early during infection facilitates the establishment of a fulminate infection by F. tularensis. Collectively, this study provides evidence that F. tularensis LVS represses inflammasome activation and that F. tularensis-encoded FTL_0325 mediates this effect.
Introduction
Francisella tularensis is an important human pathogen responsible for causing tularemia in the northern hemisphere. F. tularensis has long been developed as a biological weapon and is now classified as a category A agent by the Centers for Disease Control because of its possible use as a bioterror agent (1–3). All virulent strains are classified under F. tularensis subsp. tularensis (type A) and F. tularensis subsp. holarctica (type B), whereas avirulent strains belong to F. tularensis subsp. novicida or Francisella mediasiatica. SchuS4 belongs to F. tularensis subsp. tularensis, whereas the live vaccine strain (LVS)2 is a derivative of F. tularensis subsp. holarctica developed in the United States (4). Being an intracellular pathogen, Francisella can survive and replicate in a variety of cell types including macrophages. F. tularensis has been shown to modulate macrophage functions and suppress proinflammatory cytokines (5–8). The virulence properties and mechanisms underlying innate immune subversion of F. tularensis are unique and remain largely undefined. F. tularensis lacks toxins or type III and IV secretion systems but possesses a type VI secretion system (9, 10). Macrophages are the primary sites of predilection for F. tularensis. The unique intramacrophage lifestyle of F. tularensis includes a transient phagosomal phase (lasting for ∼30–60 min) (11) that is associated with the silencing of NF-κB, mitogen-activated protein (MAPKs), and PI3K/AKT signaling (12). F. tularensis then escapes from the phagosomes into the cytosol where bacterial replication occurs.
Mammalian Toll-like receptors (TLRs) are members of the pattern-recognition receptors family of proteins implicated in host antimicrobial defenses and play a central role in the initiation of innate cellular immune responses and the subsequent adaptive immune responses to pathogens (13). TLRs are expressed on a number of immune effector cells including monocytes/macrophages, neutrophils, dendritic, epithelial, and endothelial cells. Recognition of microbial products by TLRs leads to the activation of NF-κB and MAPK, production of cytokines, and the induction of co-stimulatory molecules (14, 15). Of the 11 or so known human and mouse homologues, the two most well characterized are TLR2 and TLR4. It has been previously demonstrated that TLR4 does not contribute to resistance of mice to airborne type A F. tularensis infection (16). F. tularensis LPS is inert, lacks endotoxin-like inflammatory properties, and is not sensed by TLR4. TLR2 plays an essential role in innate immunity to F. tularensis LVS, and we have reported earlier that deficiency of TLR2 enhances murine susceptibility to infection (17). TLR2 forms heterodimers in association with TLR1 or TLR6 and plays a key role in the host defense against F. tularensis (18). In addition to TLRs, another family of pattern-recognition receptors, the Nod-like receptor (NLR) family, can detect bacterial products in the cytoplasm (19–21). Polymorphisms or mutations in the NLRs are associated with susceptibility to inflammatory disorders, suggesting their importance in inflammation and immunity (22). Because F. tularensis grows and replicates within the intracellular environment of several cell types, the role of cytosolic receptors against this pathogen assumes great significance. However, little is known regarding the role of NLR proteins and the composition of the inflammasome in host defense against the virulent strains of F. tularensis.
TLRs are key inducers of the pro-forms of IL-1β and IL-18 through NF-κB stimulation (23), which then perhaps primes the NLR-containing inflammasomes to respond to bacterial pathogen-associated molecular patterns (PAMPs). The activation of inflammasome requires two distinct signals. The first signal is provided by TLRs in the form of proIL-1β and NLR-pyrin domain-containing 3 (NLRP3) messages, whereas the second signal serves to activate the assembly of an inflammasome complex that results in the proteolytic cleavage, activation, and secretion of the mature forms of proinflammatory cytokines IL-1β and IL-18. Mice deficient for inflammasome components apoptosis-associated speck-like protein containing a CARD domain (ASC) or caspase-1 exhibit a susceptible phenotype accompanied by increased morbidity and mortality, higher bacterial burden, and pyroptotic cell death, demonstrating a key role of these molecules in innate immunity to F. novicida (24). There are reports demonstrating that F. tularensis LVS and F. novicida are sensed by NLRP3 and a non-NLR protein, absent in melanoma 2 (AIM2) in human and mouse macrophages, respectively (25, 26). AIM2 serves as a cytosolic sensor for bacterial DNA. It has been reported that stimulator of interferon genes (STING) and IRF3-dependent production of type I interferons primes the AIM2 inflammasome in F. novicida-infected macrophages (27). Identification of bacterial DNA as a ligand and the activation of AIM2-dependent inflammasome has been well established using F. novicida as a model (28).
Several bacterial pathogens have evolved sophisticated mechanisms to avoid their detection by cytosolic NLR sensors to prevent inflammasome activation and subvert innate immune responses. A type III secretion protein YopK promotes Yersinia pestis virulence by preventing the recognition of its type III secretory components by NLR sensors, NLRP3 and NLRC4, to suppress inflammasome activation (29, 30). Salmonella evades detection by NLRC4 by repressing flagellin expression and modifying type III secretory rod protein SsaI in the intracellular environment to inhibit inflammasome activation (31). Similarly, Mycobacterium tuberculosis prevents inflammasome activation and, consequently, IL-1β secretion by expressing a zinc protease Zmp1 (32), whereas expression of an exoenzyme, ExoU, by Pseudomonas aeruginosa results in the evasion of inflammasome activation in the infected host cells (33). The Francisella-encoded factors MviN and RipA were initially described as required for suppression of AIM2-mediated inflammasome responses (34, 35). However, a later study identified that the activation of AIM2 inflammasome in ripA or mviN mutants was due to enhanced intramacrophage lysis of the mutant bacteria leading to the release of DNA into the host cell cytosol and subsequent activation of AIM2 inflammasome (36). We earlier reported that F. tularensis LVS- and SchuS4-encoded factors FTL_0325 and FTT0831c, respectively, interfere with NF-κB signaling to limit the production of proinflammatory cytokines TNF-α and IL-6 (37). In this study we utilized the FTL_0325 mutant of F. tularensis to investigate the mechanism of inflammasome repression by F. tularensis. We report that F. tularensis represses both the AIM2 and NLRP3 inflammasomes and that FTL_0325 is required for this repression.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Cultures
Wild type F. tularensis LVS and SchuS4 strains obtained from BEI Resources (Manassas, VA) and the FTL_0325 mutant and transcomplemented strain of F. tularensis LVS described in a previous study (37) were used. F. novicida was obtained from the Microbiology Core at Albany Medical College. A frozen stock of bacteria was streaked out on Mueller-Hinton chocolate agar plates containing beef extract, an acid hydrolysate of casein, starch, agar, and hemoglobin, and IsoVitalex™ enrichment (BD Biosciences), and the plates were incubated for 48 h at 37 °C in the presence of 5% CO2. The bacterial cultures scraped off the plates were resuspended in growth medium, adjusted to an optical density 600 (A600) at 0.2, which represents ∼1 × 109 cfu/ml, diluted to achieve the required bacterial numbers and used in the experiments. All experiments with F. tularensis SchuS4 were performed in Centers for Disease Control-approved BSL3 facility at New York Medical College.
Cell Culture Assays
Immortalized bone marrow-derived macrophages (BMDMs) from C57BL/6 mice and primary BMDMs derived from mouse strains deficient in the inflammasome pathway components caspase-1, ASC, NLRP3, and AIM2 on a C57BL/6 background were used for experiments. The immortalized bone marrow-derived macrophages (IMCs) were a gift from Dr. Katherine Fitzgerald and were generated using gene trap insertions as described in previous publications (38). The macrophages were seeded at a concentration of 2 × 106 cells/well and infected with either the wild type (WT) F. tularensis LVS or the mutant at a multiplicity of infection (m.o.i.) of 100. Where indicated, macrophages were primed with Escherichia coli LPS (100 ng/ml) or recombinant IFN-β (100 units/ml). Cell lysates were analyzed at 12 and 24 h post-infection by Western blot analysis. RNA isolated from the infected cells 2 h post-infection was analyzed by quantitative real-time polymerase chain reaction (qRT-PCR).
RNA Isolation and qRT-PCR
Isolation of RNA from the macrophage cell lysates was carried out using the Invitrogen® RNA Isolation PureLink™ kit. Quantitative RT-PCR was carried out in a two step process. The cDNA was synthesized using the Bio-Rad® iScript™ cDNA kit, and then qRT-PCR was performed using the Bio-Rad® iQ™ SYBR® Green Supermix kit. For every cDNA reaction, 1 μg of isolated RNA was used in the reaction mixture with the 5× iScript Reaction Mix and iScript Reverse Transcriptase. The transcript levels of NLRP3, AIM2, caspase-1, proIL-1β, and proIL-18 were measured by running the reactions in a Bio-Rad MiniOpticon™ detector, CFD-3120, and MJ Mini™ Cycler, PTC-0148 qRT-PCR system. β-Actin was used as a normalization control. The primer sequences used are shown in Table 1.
TABLE 1.
List of primers used in the study
| Caspase-1 forward | 5′-GAA AGA ATT TGC TGC CTG CCC AGA-3′ |
| Caspase-1 reverse | 5′-GCT TGT CTT TCA AGC TTG GGC ACT-3′ |
| ProIL-1β forward | 5′-AAG GGC TGC TTC CAA ACC TTT GAC-3′ |
| ProIL-1β reverse | 5′-ATA CTG CCT GCC TGA AGC TCT TGT-3′ |
| ProIL-18 forward | 5′-TGC TGA AGA TGA CGA GTG TCC GTT-3′ |
| ProIL-18 reverse | 5′-TCT CGG GCG GGT AAT CTT CCA AAT-3′ |
| NLRP3 forward | 5′-GTT CAC TGG CTG CGG ATG GAA TTT-3′ |
| NLRP3 forward | 5′-ACT CCT GGA AAG TCA TGT GGC TGA-3′ |
| AIM2 forward | 5′-TCA CGG AGGAAG AAC TGA AAC GGT-3′ |
| AIM2 reverse | 5′-TGC ACT TTG AAT CAG GTG GTC AGC-3′ |
| β-Actin forward | 5′-ACG TCT GGC TCT CAT CAT CTG CAA-3′ |
| β-Actin reverse | 5′-ACG GCC AGC AAA CTG CAT TAA CTC-3′ |
SDS-PAGE and Western Blot Analysis
Cell lysates were prepared using Higgins cell (immunoprecipitation) lysis buffer (10 mm Tris-HCl, pH 7.5, 0.1% SDS, 0.5% w/v sodium deoxycholate, 0.5% w/v Triton-X, 1× Sigma protease inhibitor mixture, 1× Sigma phosphatase inhibitor mixture #2, 1× Sigma phosphatase inhibitor mixture #3, 0.5 mm EDTA, and 0.1 mm DTT). Protein concentrations in all samples were normalized using the Pierce® bicinchoninic acid protein assay kit. The proteins were resolved on 10% SDS-PAGE gels and transferred to polyvinylidene fluoride membranes (0.45 μm, Millipore, Immobilon-P) and probed against rabbit monoclonal antibody specific for mouse caspase-1 (Epitomics, 3345-1) and mouse monoclonal antibodies specific for mouse IL-1β (Santa-Cruz, sc-74133) and IL-18 (Santa-Cruz, sc-80048). For caspase-1, bovine-anti-rabbit monoclonal IgG-HRP antibody (Santa-Cruz, sc-2385), and for both IL-1β and IL-18, a bovine-anti-mouse IgG-HRP monoclonal antibody (Santa-Cruz, sc2386) were used. The blots were developed using Thermo Scientific SuperSignal West Pico Chemiluminescent Substrate and SuperSignal West Dura Extended Duration Substrate in a 1:1 ratio and imaged using the Bio-Rad Universal Hood II and Bio-Rad Image Lab™ Software Version 3.0. The blots were stripped and re-probed for β-actin to check for accuracy of protein loading. The band intensities on the blots were quantified utilizing the Bio-Rad Image Lab™ Software Version 3.0.
Lactate Dehydrogenase Release Assay and Propidium Iodide Staining
Macrophages (5 × 104/well) seeded in 96-well tissue culture plates were infected with F. tularensis LVS or the FTL_0325 mutant (m.o.i. 100), and cell death was quantified at the indicated times using a Cytotoxicity Detection kit (Roche Applied Science) according to the manufacturer's instructions. Analysis of the macrophages undergoing apoptotic cell death was done by staining infected macrophages with propidium iodide followed by flow cytometric assay to quantitate the hypodiploid cells as described earlier (39). Uninfected or macrophages infected with ΔiglC mutant of F. tularensis LVS were kept as controls.
In Vivo Studies
6–8-Week-old C57BL/6 mice were inoculated intranasally with 1 × 107 cfu of wild type F. tularensis LVS, the FTL_0325 mutant or the transcomplemented strain following a protocol described earlier (40). The mice were sacrificed at days 1 and 3 post-inoculation, and the lung homogenates were prepared for quantitation of bacterial burden and IL-1β levels as previously published (41). All the animal procedures were performed in accordance with the Institutional Animal Care and Use Committees protocol approved by the New York Medical College.
Cytokine Analysis
Measurement of IL-1β levels released into the cell culture supernatants or in the lung homogenates from the infected mice was carried out using the BD Biosciences Cytometric Bead Array (CBA) Flex Set kits and the BD Biosciences FACSVerse flow cytometer. The mean fluorescence intensity of each standard was determined by the FCAP software (BD Biosciences) creating the standard curve. The FCAP software determined the mean fluorescence intensity for each sample, and then using the standard curve, the concentration of IL-1β for each sample was calculated. IL-18 levels were quantified using a commercially available mouse IL-18 ELISA kit (eBioscience) following the manufacturer's protocol.
Statistical Analysis
Statistical analysis of all results was determined using GraphPad InStat® Version 3.05 program. Analysis was carried out using Student's t test or one-way ANOVA with the Tukey-Kramer post-test and a cut-off p value of 0.05 or less was considered significant.
RESULTS
Muted IL-1β and IL-18 Responses Are Observed in Naïve F. tularensis-infected Macrophages
We first examined the amounts of IL-1β and IL-18 produced by macrophages in response to infection with F. tularensis LVS and SchuS4 and compared with those obtained for F. novicida. Naïve macrophages were used in all the experiments, which were in contrast to previous studies that used macrophages primed with TLR agonists like LPS or Pam3CSK. Macrophages were infected with F. tularensis LVS, SchuS4, or F. novicida at 100 m.o.i., and the levels of IL-1β and IL-18 were determined in the culture supernatants 24 h later. A mutant of F. tularensis LVS in Francisella Pathogenicity Island (FPI) protein, IglC (ΔiglC), which cannot escape the phagosome, was used as a control. Very low levels of IL-1β and undetectable levels of IL-18 were observed in the culture supernatants from macrophages infected either with F. tularensis LVS or the SchuS4 strain, whereas very high levels of both these cytokines were observed in supernatants from the F. novicida-infected macrophages. Because phagosomal escape is required for induction of IL-1β and IL-18, no detectable levels of these cytokines were observed in macrophages infected with the ΔiglC mutant (Fig. 1, A and B). By performing a kinetic experiment, we next investigated if TLR2 and caspase-1 are required for induction of moderate amounts of IL-1β observed in macrophages infected with F. tularensis LVS. Macrophages derived from the WT C57BL/6, TLR2−/−, and caspase-1−/− mice were used to assess the impact of F. tularensis on IL-1β release. Similar to the results shown in Fig. 1A, a muted TLR2- and caspase-1-dependent IL-1β response was observed in the WT macrophages infected with F. tularensis LVS at 24 h post-infection (Fig. 1C). Similar to the results obtained in Fig. 1B, higher levels of IL-1β were observed in the culture supernatants of F. novicida-infected macrophages, which was also dependent on TLR2 and caspase-1(Fig. 1D). Collectively, these results demonstrate that muted IL-1β and IL-18 responses are generated in macrophages infected with F. tularensis LVS and SchuS4 as compared with F. novicida-infected macrophages, and both TLR2-dependent signal 1 and caspase-1-dependent signal 2 are required for secretion of mature IL-1β. These results also indicated that the muted IL-1β and IL-18 response in F. tularensis LVS- or SchuS4-infected macrophages may either be due to the suppressive effects or an inability of F. tularensis LVS or SchuS4 to induce inflammasome activation.
FIGURE 1.

Muted IL-1β and IL-18 responses were observed in naïve F. tularensis (Ft)-infected macrophages. Murine macrophages were infected with 100 m.o.i. of ΔiglC mutant of F. tularensis LVS, wild type F. tularensis LVS, F. tularensis SchuS4, and F. novicida strains. Levels of IL-1β (A) and IL-18 (B) were measured in the cell culture supernatants 24 h post-infection using IL-1β flex-set and IL-18 ELISA kit. Macrophages isolated from WT, TLR2−/−, and caspase-1−/− C57BL/6 mice were infected with 100 m.o.i. of F. tularensis LVS or F. novicida. IL-1β levels in culture supernatants were analyzed in F. tularensis LVS-infected (C) or F. novicida (D)-infected macrophages. A representative of one of the three experiments conducted with similar results is shown. The data were analyzed by ANOVA with a Tukey-Kramer post-test, and a cut-off p value of 0.05 or less was considered significant. Comparisons are shown with F. novicida. *, p < 0.05; ***, p < 0.001. The levels of IL-18 in the ΔiglC mutant, F. tularensis LVS and SchuS4 infected macrophages were below the level of detection (25 pg/ml).
Differences in Magnitude of Inflammasome Activation in F. tularensis and F. novicida Are Due to Differential Activation of TLR2 Signaling Pathway
We next investigated if the difference in IL-1β response in F. tularensis LVS- and F. novicida-infected macrophages originates due to differences in the activation of TLR2-dependent signals. The expression of proIL-1β message was quantitated in macrophages infected either with F. tularensis LVS or F. novicida by performing a kinetic experiment at 6, 12, and 24 h post-infection. Significantly higher proIL-1β transcript levels were observed in F. tularensis LVS-infected macrophages as compared with the control uninfected cells at 6 and 12 h post-infection. However, these transcripts levels were significantly lower than those observed in F. novicida-infected WT macrophages at all the time points examined (Fig. 2A). Although the expression of proIL-1β in both the F. tularensis LVS- and F. novicida-infected macrophages was found to be dependent on TLR2 (Fig. 2A), the dependence on TLR2 did not appear to be as severe in F. novicida as compared with F. tularensis LVS-infected macrophages (Fig. 2A). We also examined the impact of TLR2 deficiency on the expression of NLR components NLRP3, procaspase-1, and a non-NLR component AIM2. TLR2-dependent expression of NLRP3 up-regulated as early as 2 h post-infection in F. tularensis LVS-infected macrophages and began to decline thereafter and returned to the base line values by 24 h post-infection (Fig. 2B). The elevated transcript levels of procaspase-1 were also dependent on TLR2; however, AIM2 levels remained unaltered in TLR2-deficient macrophages when examined at 24 h post-infection (Fig. 2C). Western blot analysis was performed to assess the role of TLR2 in inflammasome activation by visualizing the bioactive forms of caspase-1, IL-1β, and IL-18. Similar to transcription, activation of caspase-1, IL-1β, and IL-18 were also dependent on TLR2 as diminished levels of activated forms of these proteins were observed in TLR2−/− as compared with the WT macrophages infected with F. tularensis LVS at 24 h post-infection (Fig. 2D). Collectively, these results demonstrate that F. tularensis LVS has a suppressive effect on inflammasome activation either due to its failure to engage and activate TLR2 or because of inhibition of the TLR signaling pathway.
FIGURE 2.
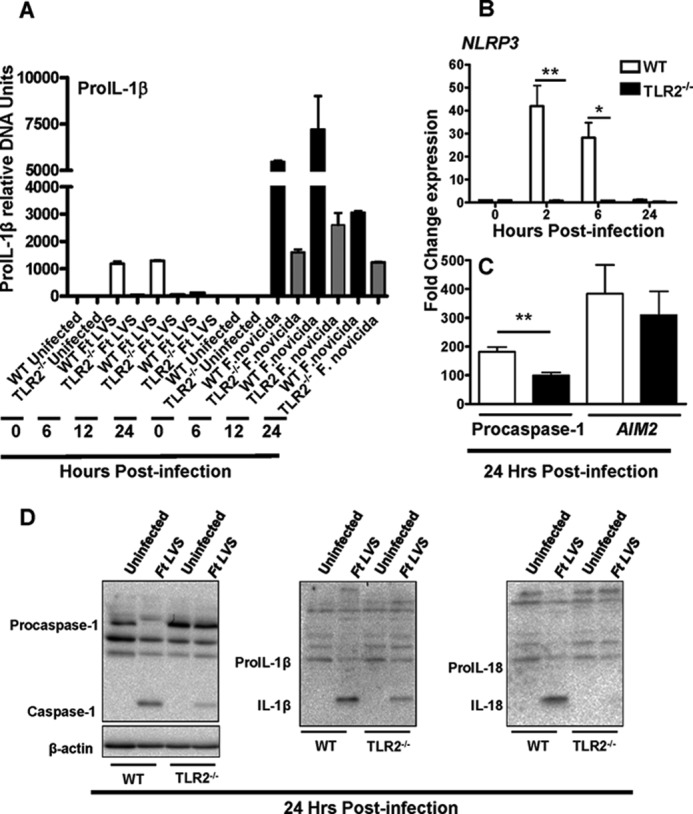
Differences in magnitude of inflammasome activation in F. tularensis (Ft) and F. novicida are due to differential activation of TLR2 signaling pathway. The proIL-1β (A) NLRP3 (B), and procaspase-1 and AIM2 (C) transcript levels were quantitated in macrophages infected with F. tularensis LVS and/or F. novicida at the indicated time points by qRT-PCR. The results are representative of at least two to three independent experiments. The data were analyzed by Student's t test, and a cut-off p value of 0.05 or less was considered significant. *, p < 0.05; **, p < 0.01. D, Western blot analysis shown is to visualize the activated forms of caspase-1, IL-1β, and IL-18 in the cell lysates from WT and TLR2−/− macrophages 24 h post-infection.
Stimulation through TLR4 but Not IFN-β Partially Restores IL-1β Suppression by F. tularensis LVS
Production of IL-1β requires two signals; a TLR-dependent signal 1 for the activation of transcription and translation of proIL-1β and an inflammasome-activating signal 2 for the proteolytic cleavage of proIL-1β into its active secreted form. Our results revealed that F. tularensis LVS induces very low amounts of IL-1β either due to failure to engage TLR2 or repression of TLR2 signaling. We investigated if an alternate TLR signal, specifically a potent TLR4 signal, when provided exogenously would induce higher IL-1β levels in F. tularensis LVS-infected macrophages. When macrophages were stimulated with E. coli LPS, a TLR4 ligand, the ability of TLR2−/− macrophages to secrete IL-1β was partially reconstituted. Significantly higher levels of IL-1β were observed in E. coli LPS-primed and F. tularensis LVS-infected WT and TLR2−/− macrophages as compared with the untreated controls 24 h post-infection (Fig. 3A); however, these levels remained significantly lower than those observed for F. novicida-infected macrophages (Fig. 3B). These results demonstrate that although there is a specific requirement for TLR-dependent signal 1 for IL-1β secretion, a TLR signal of sufficient strength does not fully restore IL-1β in F. tularensis LVS-infected macrophages, similar to levels observed for F. novicida. Collectively, these results indicate that F. tularensis LVS not only represses/fails to activate TLR2 but also interferes with E. coli LPS-induced TLR4 signals probably by acting downstream at a point common to both the TLR2 and TLR4.
FIGURE 3.

Stimulation through TLR4 but not IFN-β partially restores IL-1β suppression by F. tularensis (Ft) LVS. Macrophages were stimulated with E. coli LPS and infected with 100 m.o.i. of F. tularensis LVS (A) or F. novicida (B). The macrophages were treated with recombinant IFN-β and infected with F. tularensis LVS (C) or F. novicida (D). The IL-1β levels were measured in the culture supernatants at 6 and 24 h post-infection. The results are representative of at least two to three independent experiments. The data were analyzed by ANOVA with Tukey-Kramer post test, and a cut-off p value of 0.05 or less was considered significant. **, p < 0.01; ***, p < 0.001.
It has been shown for F. novicida infection that type I IFN, especially IFN-β ,provides signal 2 and is required for priming of the AIM2 inflammasome and IL-1β secretion (42). We asked the question if providing a second signal by exogenous addition of IFN-β would restore the activation of IL-1β to a higher extent in F. tularensis LVS-infected macrophages. As shown in Fig. 3C, it was observed that the addition of IFN-β did not significantly alter the levels of secreted IL-1β either in the WT, TLR2−/−, or caspase-1−/− macrophages infected with F. tularensis LVS, and the amounts of IL-1β remained similar to those observed for the untreated controls. Similarly, the addition of IFN-β to F. novicida-infected macrophages did not further enhance the existing elevated levels of IL-1β observed in untreated but F. novicida-infected macrophages (Fig. 3D). Collectively, these results confirm an absolute requirement for TLR and caspase-1-dependent signaling for IL-1β production in both the F. tularensis LVS and F. novicida-infected macrophages. Furthermore, these results also indicate that due to a predominant suppressive effect of F. tularensis LVS on signal 1, even if a signal 2 of sufficient strength is available, the inflammasome activation and enhanced IL-1β secretion does not occur.
FTL_0325 of F. tularensis Represses the Expression of ProIL-1β
Because our results indicated that F. tularensis probably acts downstream of TLR signaling to suppress IL-1β production, we further investigated this notion. We reported earlier that F. tularensis LVS- and SchuS4-encoded factors FTL_0325 and FTT0831c, respectively, interfere with NF-κB signaling to limit the production of proinflammatory cytokines TNF-α and IL-6 (37). We utilized the FTL_0325 mutant of F. tularensis to investigate if FTL_0325 represses expression of proIL-1β, proIL-18, NLR components, and AIM2. Macrophages were infected either with F. tularensis LVS or the FTL_0325 mutant at 100 m.o.i., and RNA was isolated at 2 h post-infection and quantitated for the expression of NLRP3, AIM2, procaspase-1, proIL-1β, and proIL-18 by qRT-PCR. Modest increases in the expression of NLRP3, AIM2, and proIL-18 transcript levels were observed in macrophages infected with the FTL_0325 mutant as compared with the LVS-infected macrophages; however, these differences did not achieve statistical significance. On the other hand, significantly elevated proIL-1β transcripts levels were observed in macrophages infected with the FTL_0325 mutant as compared with F. tularensis LVS-infected macrophages (Fig. 4). These results demonstrate that FTL_0325 of F. tularensis impacts proIL-1β expression as early as 2 h post-infection.
FIGURE 4.
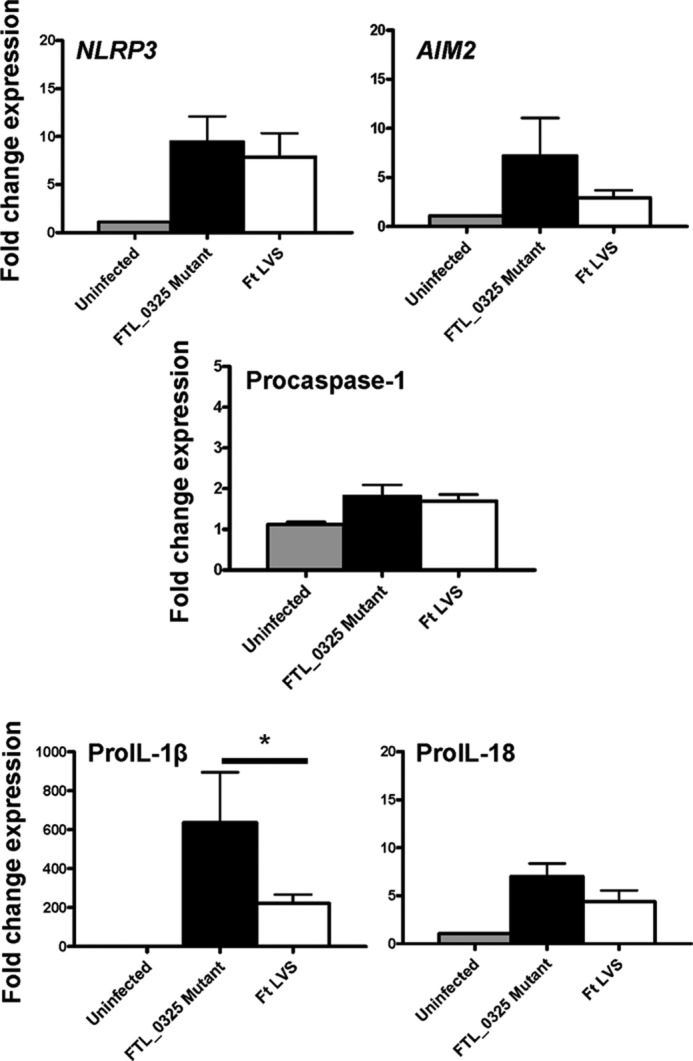
FTL_0325 of F. tularensis (Ft) represses the expression of proIL-1β. Shown are Transcript levels of NLRP3, AIM2, procaspase-1, proIL-1β, and proIL-18 in IMCs derived from C57BL/6 mice. The IMCs were infected with either the FTL_0325 mutant or F. tularensis LVS at an m.o.i. of 100 for 2 h, and transcript levels were quantitated by qRT-PCR. The results are expressed as -fold change expression and are representative of three independent experiments performed using duplicate samples. The data were analyzed by ANOVA with Tukey-Kramer post-test, and a cut-off p value of 0.05 or less was considered significant. *, p < 0.05.
FTL_0325 Mutant of F. tularensis LVS Induces an Early Activation of Inflammasome in a TLR2-dependent Fashion
We observed that F. tularensis activates inflammasome later during infection at 24 h post-infection (Fig. 2D). We further examined if FTL_0325 of F. tularensis is responsible for this delayed response. The bioactive forms of caspase-1, IL-1β, and IL-18 in uninfected or the F. tularensis LVS- or FTL_0325 mutant-infected macrophages were determined at 12 h post-infection by Western blot analysis. Significantly elevated bioactive forms of caspase-1, IL-1β, and IL-18 were detected in macrophages infected with the FTL_0325 mutant as compared with the LVS-infected macrophages at 12 h post-infection (Fig. 5, A, B, and C). The inflammasome-dependent responses of LVS-infected macrophages were muted at this early time point in infection. These results suggested that FTL_0325 delays the activation of inflammasome in F. tularensis-infected macrophages. Next, we investigated the requirement for TLR2 in this early induction of inflammasome response in the FTL_0325 mutant-infected macrophages. It was found that the increased activation of caspase-1, IL-1β, and IL-18 in the FTL_0325 mutant-infected macrophages was dependent on TLR2 (Fig. 5, D, E, and F). Collectively, these results demonstrate that FTL_0325 of F. tularensis LVS delays the activation of inflammasome in a TLR2-dependent fashion.
FIGURE 5.

FTL_0325 mutant of F. tularensis LVS induces an early activation of inflammasome in a TLR2-dependent fashion. Western blot analysis visualizes activated caspase-1 (A), IL-1β (B), and IL-18 (C). IMCs derived from WT C57BL/6 mice were infected with either the FTL_0325 mutant or F. tularensis LVS at an m.o.i. of 100. The cells were lysed 12 h post-infection and analyzed by Western blot analysis. The blots were stripped and re-probed for β-actin, which was used as a loading control. The Western blot images are representative of at least two-three independent experiments. The relative quantification of the bands from two-three blots is shown and expressed as relative intensity units (RIU). The data were analyzed by ANOVA with Tukey-Kramer post-test, and a cut-off p value of 0.05 or less was considered significant. **, p < 0.01; ***, p < 0.001. D, E, and F, shown is Western blot analysis using wild type and TLR2−/− macrophages infected with the FTL_0325 mutant or F. tularensis LVS at 12 h post-infection.
Early Activation of Inflammasome in the FTL_0325 Mutant-infected Macrophages Is Partially Dependent on AIM2 and NLRP3
It has been reported that both the AIM2 and NLRP3 inflammasomes can sense F. tularensis LVS (25). We determined the relative contributions of AIM2 and NLRP3 in the activation of caspase-1, IL-1β, and IL-18 in the FTL_0325 mutant-infected macrophages. The results indicated that the activation of caspase-1, IL-1β, and IL-18 in the FTL_0325 mutant-infected WT macrophages was dependent on AIM2, as their levels reduced drastically in AIM2−/− macrophages (Fig. 6). The results from NLRP3−/− macrophages revealed a partial dependence on NLRP3 for the activation of IL-1β in macrophages infected with the FTL_0325 mutant, as the levels of bioactive IL-1β reduced significantly but were not completely obliterated in the NLRP3−/− macrophages (Fig. 7). In contrast to the IL-1β results, the processing of caspase-1 and IL-18 was independent of NLRP3 (Fig. 7). As observed previously, LVS suppressed the activation of caspase-1, IL-1β, and IL-18 in infected macrophages during the early stage of infection (Figs. 6 and 7). Collectively, these results demonstrate that F. tularensis LVS delays the activation of both the AIM2 and NLRP3 inflammasomes in an FTL_0325-dependent fashion. These results also suggest that enhancement of proIL-1β signal due to the loss of FTL_0325 also results in an enhanced AIM2- and NLRP3-dependent processing of IL-1β in murine macrophages.
FIGURE 6.

Early activation of inflammasome in the FTL_0325 mutant-infected macrophages is partially dependent on AIM2. WT or AIM2−/− macrophages were infected with either the FTL_0325 mutant or F. tularensis (Ft) LVS at an m.o.i. of 100. The cells were lysed 12 h post-infection and analyzed by Western blot analysis for visualization of active caspase-1, IL-1β, and IL-18. The blots were stripped and re-probed for β-actin, which was used as a loading control. The Western blot images are representative of at least two-three independent experiments. The relative quantification of the bands from two-three blots is shown and expressed as relative intensity units (RIU). The data were analyzed by ANOVA with Tukey-Kramer post-test, and a cut-off p value of 0.05 or less was considered significant. **, p < 0.01; ***, p < 0.001.
FIGURE 7.

Early activation of inflammasome in the FTL_0325 mutant-infected macrophages is partially dependent on NLRP3. Ft, F. tularensis WT or NLRP3−/− macrophages were infected with either the FTL_0325 mutant or Ft LVS at an m.o.i. of 100. The cells were lysed 12 h post-infection and analyzed by Western blot analysis for visualization of active caspase-1, IL-1β, and IL-18. The blots were stripped and re-probed for β-actin, which was used as a loading control. The Western blot images are representative of at least two-three independent experiments. The relative quantification of the bands from two-three blots is shown and expressed as relative intensity units (RIU). The data were analyzed by ANOVA with Tukey-Kramer post-test, and a cut-off p value of 0.05 or less was considered significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Activation of Inflammasome in the FTL_0325 Mutant-infected Macrophages Is Independent of Both AIM2 and NLRP3 during the Later Stages of Infection
It has been previously reported that F. tularensis LVS induces the activation of AIM2-dependent inflammasome in unprimed (34), LPS-primed (42), or thioglycollate-elicited peritoneal macrophages (38) at 16–24 h post-infection. To determine the relative contribution of AIM2 and NLRP3 on the activation of inflammasome components during the later stages of infection, WT, AIM2−/−, and NLRP3−/− macrophages were infected with F. tularensis LVS or the FTL_0325 mutant, lysed, and analyzed by Western blot analysis at 24 h post-infection. Similar to earlier published reports (34), F. tularensis LVS-infected macrophages were found to be capable of activating caspase-1, IL-1β, and IL-18 at levels similar to FTL_0325-infected macrophages in an AIM2-dependent (Fig. 8) but NLRP3-independent manner (Fig. 9) 24 h post-infection. Interestingly, an enhanced activation of all these inflammasome components observed in the FTL_0325 mutant-infected macrophages at 24 h post-infection was independent of both the AIM2 and NLRP3 (Figs. 8 and 9).
FIGURE 8.

Activation of inflammasome in the FTL_0325 mutant-infected macrophages is independent of AIM2 during the later stages of infection. WT or AIM2−/− macrophages were infected with either the FTL_0325 mutant or F. tularensis (Ft) LVS at an m.o.i. of 100. The cells were lysed 24 h post-infection and analyzed by Western blot analysis for visualization of active caspase-1, IL-1β, and IL-18. The blots were stripped and re-probed for β-actin, which was used as a loading control. The Western blot images are representative of at least two-three independent experiments. The relative quantification of the bands from two-three blots is shown and expressed as relative intensity units (RIU). The data were analyzed by ANOVA with Tukey-Kramer post-test and a cut-off p value of 0.05 or less was considered significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 9.

Activation of inflammasome in the FTL_0325 mutant-infected macrophages is independent of NLRP3 during the later stages of infection. WT or NLRP3−/− macrophages were infected with either the FTL_0325 mutant or F. tularensis (Ft) LVS at an m.o.i. of 100. The cells were lysed 24 h post-infection and analyzed by Western blot analysis for visualization of active caspase-1, IL-1β, and IL-18. The blots were stripped and re-probed for β-actin, which was used as a loading control. The Western blot images are representative of at least two-three independent experiments. The relative quantification of the bands from two-three blots is shown and expressed as relative intensity units (RIU). The data were analyzed by ANOVA with Tukey-Kramer post-test, and a cut-off p value of 0.05 or less was considered significant. *, p < 0.05.
Early Activation of Inflammasome in FTL_0325 Mutant-infected Macrophages Results in Early Induction of Cell Death
F. tularensis delays cell death in infected macrophages. It has also been reported that macrophages infected with F. novicida and F. tularensis LVS undergo a caspase-1-, ASC- and AIM2- dependent pyroptotic cell death (25, 28, 38). To determine the impact of delayed inflammasome activation on cell death, we used propidium iodide staining to determine the loss of plasma cell integrity followed by flow cytometry. It was observed that a significantly higher percentage of cells infected with the FTL_0325 mutant (13.4%) as compared with the F. tularensis LVS-infected macrophages (5.25%) underwent cell death at 12 h post-infection. The cytotoxicity in macrophages infected with the ΔiglC mutant was similar to that observed for the uninfected control at this time point (Fig. 10, A–D). To determine the type of cell death induced by F. tularensis, we employed an lactate dehydrogenase release assay to measure pyroptotic (caspase-1-dependent) and pyronecrotic (NLRP3-dependent, caspase-1 independent) cell death. WT, TLR2−/−, ASC−/−, caspase-1−/−, NLRP3−/−, and AIM2−/− macrophages were infected either with F. tularensis LVS or the FTL_0325 mutant, and cell cytotoxicity was monitored at 12 h post-infection by lactate dehydrogenase release assay. A significantly higher percentage of macrophages infected with the FTL_0325 mutant underwent ASC-, caspase-1-, and AIM2-dependent pyroptotic cell death as opposed to NLRP3-dependent pyronecrosis (Fig. 10E). These results indicate that F. tularensis LVS delays pyroptotic cell death of the infected macrophages in a FTL_0325-dependent manner during the early stages of infection.
FIGURE 10.
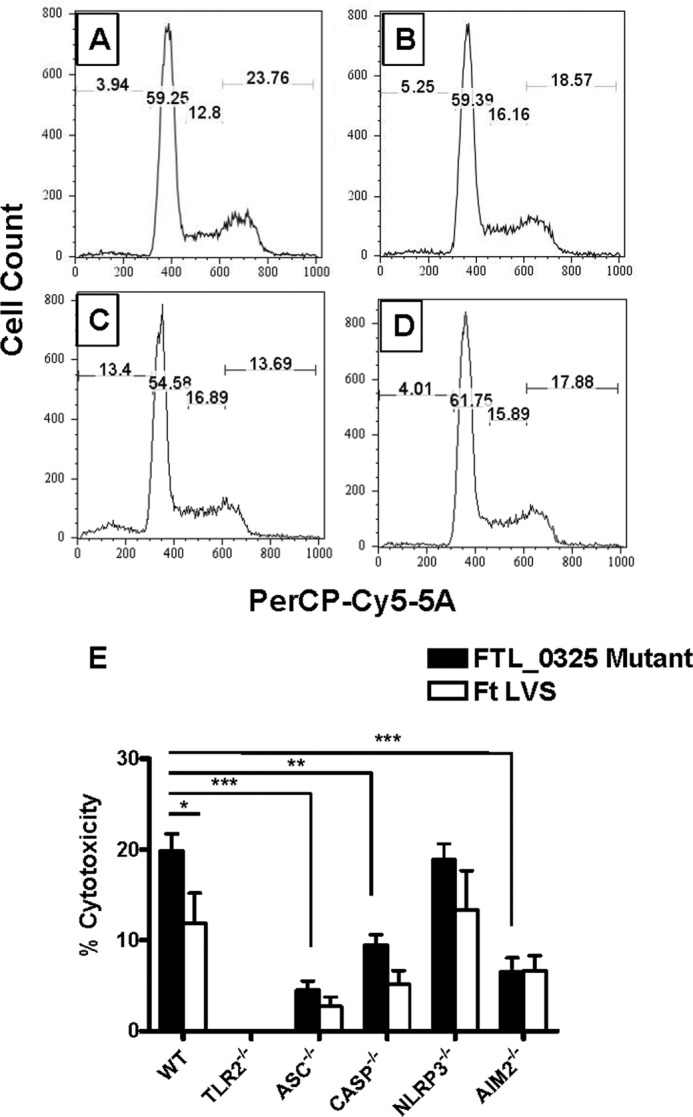
FTL_0325 delays pyroptotic cell death in infected macrophages. Wild type uninfected macrophages (A) or infected with F. tularensis LVS (B), the FTL_0325 mutant (C), or the ΔiglC mutant (D) were stained with propidium iodide and analyzed by flow cytometry 12 h post-infection. E, WT, TLR2−/−, ASC−/−, caspase-1−/−, NLRP3−/−, and AIM2−/− IMCs of C57BL/6 origin were infected with either the FTL_0325 mutant or F. tularensis (Ft) LVS at an m.o.i. of 100. At 12 h post-infection the lactate dehydrogenase release assay was performed using the Roche Applied Science cytotoxicity detection kit. The absorbance of the samples was read at 492 nm and adjusted using a background control before calculating the % cytotoxicity. The data are representative of three independent experiments, each conducted with three replicate samples. The data were analyzed by ANOVA with a Tukey-Kramer post-test, and a cut-off p value of 0.05 or less was considered significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FTL_0325-dependent Early Suppression of IL-1β Facilitates the Establishment of a Fulminate Infection by F. tularensis
Last, we examined the impact of delayed inflammasome activation and a muted IL-1β response on F. tularensis clearance using mouse model of tularemia. C57BL/6 mice were infected with F. tularensis LVS, the FTL_0325 mutant, or the transcomplemented strain (FTL_0325 mutant + FTL_0325) by intranasal route. The mice were sacrificed at days 1 and 3 post-infection, and IL-1β levels and bacterial burden were determined in the lungs of the infected mice. Significantly elevated levels of IL-1β were observed in the lung homogenates of mice infected with the FTL_0325 mutant as compared with the F. tularensis LVS or the transcomplemented strain as early as day 1 post-infection. These results corroborate our in vitro findings that F. tularensis suppresses inflammasome activation and IL-1β production in an FTL_0325-dependent manner. Similarly, in accordance with the delayed activation of inflammasome observed during the later stages of infection in F. tularensis-infected macrophages, significantly elevated levels of IL-1β were observed in F. tularensis LVS as compared with the FTL_0325 mutant-infected mice at day 3 post-infection (Fig. 11A). We also determined bacterial burden in the infected mice to investigate that the increased IL-1β in the lung homogenates of FTL_0325 mutant-infected mice at day 1 post-infection was not simply due to the differences in bacterial numbers. No differences were observed in the bacterial numbers recovered from the lungs of mice infected with all the three Francisella strains at day 1 post-infection, suggesting that the observed differences in the IL-1β are due to the ability of FTL_0325 to suppress this response. Furthermore, an early IL-1β response observed in the lungs of the FTL_0325 mutant-infected mice also resulted in the rapid clearance of the FTL_0325 mutant at day 3 post-infection (Fig. 11B). Collectively, these results demonstrate that suppression of IL-1β in the early stages of infection facilitates F. tularensis to establish a fulminate infection.
FIGURE 11.
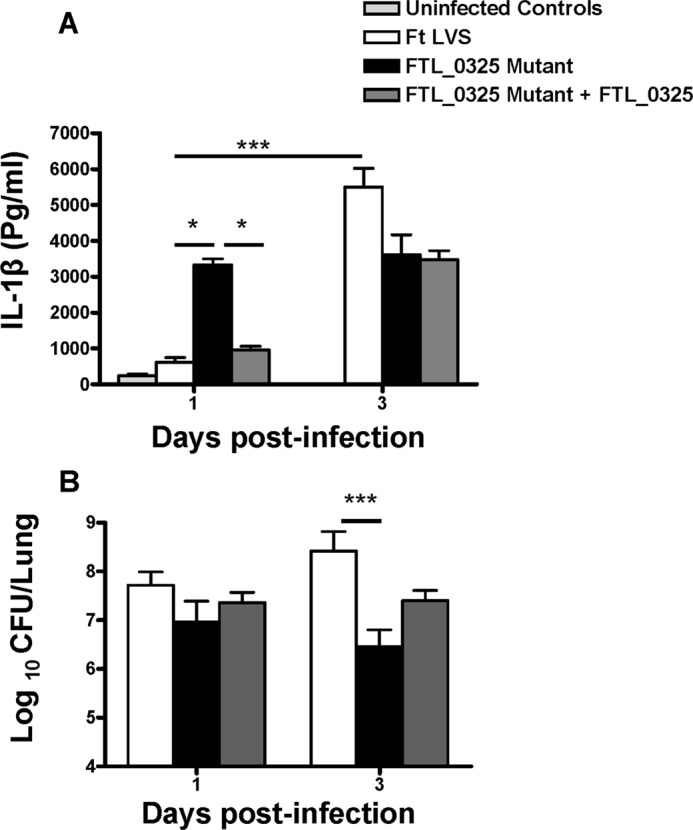
FTL_0325-dependent early suppression of IL-1β facilitates the establishment of a fulminate infection by F. tularensis (Ft). C57BL/6 mice were inoculated intranasally with 1 × 107 cfu of F. tularensis LVS, the FTL_0325 mutant, or the transcomplemented strain. The mice were sacrificed at days 1 and 3 post-infection. The IL-1β levels (A) and bacterial numbers (B) were determined at the indicated times in the lung homogenates. The data are cumulative of two independent experiments each conducted with three mice each (total n = 6). The data were analyzed by ANOVA with Tukey-Kramer post-test, and a cut-off p value of 0.05 or less was considered significant. *, p < 0.05; ***, p < 0.001.
DISCUSSION
This study provides evidence that F. tularensis LVS represses inflammasome activation and that F. tularensis encoded FTL_0325 mediates this effect. Several bacterial pathogens have evolved sophisticated mechanisms to suppress inflammasome activation. Effector proteins secreted by type III secretion systems of Pseudomonas and Yersinia diminish inflammasome responses. M. tuberculosis inhibits inflammasome activation via zinc metalloproteases, whereas pneumolysin of Streptococcus pneumoniae prevents caspase-1 activation and IL-1β secretion not by direct inhibition of inflammasome but by interfering with NF-κB and MAPK signaling pathways. Francisella is different from these pathogens as it lacks type III or type IV secretion systems and exotoxins. DotU and VgrG have been identified as the core components of the type VI secretion system in F. tularensis, and null mutation of these two genes fail to activate the inflammasome (9). However, these results may apparently be due to the inability of both the dotU and vgrG mutants to escape phagosomes rather than their inhibitory effects on assembly and activation of the inflammasome components. A number of F. tularensis LVS and F. novicida genes that were identified earlier to have inflammasome inhibitory properties turned out to be a consequence of compromised structural integrity of the mutants lacking these genes. The activation of AIM2-dependent inflammasome in macrophages infected with these mutants was later reported to be due to enhanced intramacrophage lysis, and it was inferred that Francisella does not actively suppress or modulate the inflammasome. Moreover, studies conducted with F. novicida have shown an AIM2-dependent activation of inflammasome, caspase-1 activation, and induction of the mature form of IL-1β, suggesting that the infected macrophages respond to and induce a protective immune response against F. novicida. Using F. novicida as a model, it has also been reported that TLR2 signaling is required for rapid inflammasome activation highlighting the importance of priming signal 1 provided by the TLRs (43).
Much of the previous work done to understand the role of inflammasome after F. novicida and F. tularensis LVS infection has used macrophages activated either by strong TLR agonists such as E. coli LPS, heat-killed bacteria, or IFN-γ. However, such model systems failed to explain the significance of priming signal provided by TLRs in the context of naïve macrophages infected with F. tularensis. In this study unprimed naïve macrophages were used to investigate inflammasome-mediated response after infection with both the F. tularensis LVS and F. novicida. Additionally, various Francisella strains differ in their virulence and pathogenicity in mouse and humans; however, very few studies comparing the intrinsic differences between strains that impact their ability to elicit innate immune responses have been conducted. We, therefore, examined differences in inflammasome responses after infection with F. novicida and F. tularensis LVS. As reported earlier, F. novicida induced a vigorous IL-1β response (44). By comparison, IL-1β responses in the F. tularensis LVS- and SchuS4-infected macrophages were quite modest and observed only after 24 h of infection but were nevertheless dependent upon TLR2 and caspase-1. Furthermore, unlike F. novicida-infected macrophages, priming of TLR2−/− macrophages with LPS only partially restored IL-1β production after F. tularensis LVS infection. These results point to the fact the F. tularensis exerts a suppressive effect common to both TLR2 and TLR4 signaling probably at the level of NF-κB. This notion is supported by previous observations that LPS-primed and F. tularensis-infected macrophages fail to induce a secondary response (5).
Recently, we established the role of FTL_0325 protein of F. tularensis LVS and FTT0831c, an ortholog of FTL_0325, in the virulent F. tularensis SchuS4 strain (37). The FTL_0325 gene encodes for a 45.75-kDa protein that is annotated as an OmpA-like protein due to its conserved OmpA domain. However, the remainder of the protein does not show any significant homology to other bacterial proteins, indicating its structural uniqueness. Bioinformatics analysis of FTL_0325 using ExPASy, DAS, and TMpred software predicted a single transmembrane segment (residues 3–23) and the signature 17LXXC20 signal peptide cleavage site at the N terminus, suggesting that this protein may not span the bacterial membrane several times and surface-exposed residues may exist. Likewise, we have demonstrated that this protein is surface-localized with surface-exposed regions (37). A motif scan using PROSITE (45) revealed a periplasmic peptidoglycan-associating motif and three other major motifs, KID repeat (amino acid residues 187–197), a possible RNA binding repeat profile, and a low score resemblance to the bacterial Ig-like domain (Big-1) (amino acid residues 398–417). The region from amino acid residues 184–200 showed homology to leucine-rich repeats of internalin B protein of Listeria monocytogenes. The leucine-rich repeats of internalin B has been shown to be required for inducing phagocytosis in non-phagocytic cells such as hepatocytes and endothelial and epithelial cells (46, 47). We reported that FTL_0325 suppresses proinflammatory cytokines primarily by interfering with NF-κB signaling (37). We have also reported that FTL_0325 is required for both in vitro and in vivo growth, virulence in mice, and has a unique ability to induce an early production of TNF-α and IL-6 as early as day 1 post-infection in mice (41). In the present study we took advantage of this mutant to understand how F. tularensis regulates inflammasome repression.
The present study reaffirms our previous observations and indicates that in addition to TNF-α and IL-6 production, FTL_0325 also impacts IL-1β production by suppressing NF-κB-dependent transcription of proIL-1β. We demonstrate that up-regulated expression of proIL-1β and NLRP3 in macrophages infected with the FTL_0325 mutant as early as 2 h post-infection is associated with an early activation of inflammasome. A phenotype similar to the FTL_0325 has also been reported for the ΔripA and mviN mutant of F. tularensis LVS. RipA suppresses the MAPK signaling pathway to down-regulate proIL-1β synthesis and IL-1β production and that activation of IL-1β in ΔripA mutant is mediated by an AIM2-dependent inflammasome (34). However, Peng et al. (36) reported that the hyperinflammatory and hypercytotoxic nature of the ΔripA mutant was due to altered membrane integrity and intramacrophage lysis of the mutant, resulting in the release of excessive bacterial DNA and activation of AIM2 inflammasome. Using the assay described by Peng et al. (36), we monitored the intramacrophage bacterial lysis and have demonstrated that although the FTL_0325 mutant undergoes intramacrophage lysis 24 h post-infection, the extent of lysis is similar to that observed for the parental F. tularensis LVS strain (37). Additionally, we have also shown that unlike ΔripA and mviN mutants, the FTL_0325 mutant does not exhibit compromised structural integrity or growth defects when grown under acellular growth conditions. Furthermore, the inability of FTL_0325 mutant to survive in macrophages or mice indicates that generation of an early and enhanced innate immune response is in fact responsible for in vitro and in vivo killing of this mutant (37, 41).
Results from this study demonstrate that F. tularensis represses an early activation of inflammasome. The support for these observations comes from the fact that activation of inflammasome was not observed in F. tularensis LVS-infected macrophages at 12 h post-infection; however, an enhanced activation was observed at 24 h. It has been speculated that limited intramacrophage lysis and release of bacterial ligands, especially DNA by F. tularensis LVS and SchuS4, prevents the priming of AIM2-dependent inflammasome. Furthermore, it is also possible that endogenous AIM2 levels are not adequate to sense small quantities of DNA released by these two strains in the macrophage cytosol. Rather, our results demonstrate that repression of AIM2 inflammasome early in infection may be due to inadequate proIL-1β levels resulting primarily from suppression of TLR2-NF-κB signaling by F. tularensis LVS. When this repression is relieved and sufficient proIL-1β transcripts are available as observed in the FTL_0325 mutant-infected macrophages, the endogenous levels of AIM2 are sufficient to prime the inflammasome and activate caspase-1, IL-1β, and IL-18. Furthermore, the lowered transcription of these precursors, as observed in F. tularensis LVS-infected macrophages, does not result in enhanced IL-1β production upon exogenous addition of IFN-β that primes the AIM2-dependent inflammasome as reported earlier (42). These results highlight the significance of a TLR2-NF-κB signaling axis in regulation of inflammasome activation and IL-1β production in F. tularensis LVS-infected macrophages. Unlike AIM2, the role of NLRP3-dependent inflammasome is not completely understood in F. tularensis-infected macrophages. Our previous work employing reconstitution and knockdown approaches has shown that F. tularensis LVS and SchuS4 can be sensed by the NLRP3 inflammasome. In this study we observed an early activation of NLRP3-dependent IL-1β in murine macrophages in the FTL_0325 mutant but not in F. tularensis LVS-infected macrophages at 12 h post-infection, suggesting a repression of NLRP3-mediated response by F. tularensis early in infection. However, as reported in previous studies, we did not observe this NLRP3 dependence at 24 h post-infection either in F. tularensis LVS or the FTL_0325 mutant-infected macrophages. Similar to several previous reports, our results also demonstrate an AIM2-dependent but NLRP3-independent inflammasome activation at 16–24 h post-infection in F. tularensis LVS-infected macrophages (34, 35, 48).
Interestingly, the activation of inflammasome components caspase-1, IL-1β, and IL-18, which remained elevated in the FTL_0325 mutant-infected macrophages at 24 h post-infection, was independent of both AIM2 and NLRP3. In fact, the levels of activated caspase-1, IL-1β, and IL-18 were significantly higher in the AIM2−/− macrophages than their wild type counterparts in the FTL_0325 mutant-infected macrophages at 24 h post-infection. These results are intriguing and have led us to speculate that loss of AIM2/NLRP3 may lead to a compensatory mechanism or activation of an unknown NLR that might be at play during the later stages of infection with the FTL_0325 mutant. It is also possible that another DNA sensor such as IFI16 (also known as p204) is activated during the later stages of infection. IFI16, similar to AIM2, is a member of the PYHIN family, and it has been reported that like AIM2, IFI16 can bind to DNA in a non-sequence-specific manner and interact with STING (stimulator of interferon gene) to activate Tank-binding kinase 1 (TBK)-dependent IFN-β production, which results in enhanced transcriptional activation of NF-κB genes such as TNF-α and proIL-1β and caspase-1 activation through an unknown mechanism (49). These possibilities are currently being investigated in our laboratory.
Our in vivo findings in mice infected intranasally with F. tularensis LVS corroborate our in vitro findings that F. tularensis suppresses early inflammasome activation and IL-1β production in an FTL_0325-dependent manner. Delayed IL-1β production in F. tularensis LVS-infected mice was associated with enhanced bacterial burden in the lungs. Contrary to F. tularensis LVS-infected mice, significantly elevated IL-1β levels were observed in the lung homogenates of mice infected with the FTL_0325 mutant as early as day 1 post-infection. Furthermore, an early IL-1β response observed in the lungs of the FTL_0325 mutant-infected mice also resulted in rapid clearance of the FTL_0325 mutant at day 3 post-infection (Fig. 11B). Collectively, these findings are consistent with the biphasic nature of innate immune response generated by F. tularensis, which is associated with an early repression followed by an exaggerated response during the later stages of infection (50).
To conclude, this study investigated the mechanism of repression of inflammasome by F. tularensis. We provide evidence that F. tularensis LVS represses inflammasome activation and that F. tularensis-encoded FTL_0325 mediates these effects.
This work was supported, in whole or in part, by National Institutes of Health Grants R21AI075250-01A2 (to M. M.), 2P01AI056320, and R56AI090072-02 (to C. S. B.). This work was also supported by startup funds from the Albany College of Pharmacy and Health Sciences (to M. M.) and New York Medical College (to C. S. B.).
- LVS
- live vaccine strain
- TLR
- Toll-like receptor
- NLR
- Nod-like receptor
- NLRP3
- NLR-pyrin domain-containing 3
- IMC
- immortalized macrophage
- qRT
- quantitative real-time
- AIM2
- absent in melanoma 2
- m.o.i.
- multiplicity of infection
- ANOVA
- analysis of variance
- ASC
- apoptosis-associated speck-like protein containing a CARD domain.
REFERENCES
- 1.(2001) Tularemia could be bioweapons threat. J. Environ. Health 64, 47–48 [PubMed] [Google Scholar]
- 2. Bossi P., Bricaire F. (2003) Tularemia, a potential bioterrorism weapon. Presse. Med. 32, 1126–1130 [PubMed] [Google Scholar]
- 3. Dennis D. T., Inglesby T. V., Henderson D. A., Bartlett J. G., Ascher M. S., Eitzen E., Fine A. D., Friedlander A. M., Hauer J., Layton M., Lillibridge S. R., McDade J. E., Osterholm M. T., O'Toole T., Parker G., Perl T. M., Russell P. K., Tonat K. (2001) Tularemia as a biological weapon. Medical and public health management. JAMA 285, 2763–2773 [DOI] [PubMed] [Google Scholar]
- 4. Wayne Conlan J., Oyston P. C. (2007) Vaccines against Francisella tularensis. Ann. N.Y. Acad. Sci. 1105, 325–350 [DOI] [PubMed] [Google Scholar]
- 5. Bosio C. M., Bielefeldt-Ohmann H., Belisle J. T. (2007) Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J. Immunol. 178, 4538–4547 [DOI] [PubMed] [Google Scholar]
- 6. Chase J. C., Celli J., Bosio C. M. (2009) Direct and indirect impairment of human dendritic cell function by virulent Francisella tularensis Schu S4. Infect. Immun. 77, 180–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bosio C. M., Dow S. W. (2005) Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 175, 6792–6801 [DOI] [PubMed] [Google Scholar]
- 8. Butchar J. P., Cremer T. J., Clay C. D., Gavrilin M. A., Wewers M. D., Marsh C. B., Schlesinger L. S., Tridandapani S. (2008) Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS ONE 3, e2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bröms J. E., Meyer L., Lavander M., Larsson P., Sjöstedt A. (2012) DotU and VgrG, core components of type VI secretion systems, are essential for Francisella LVS pathogenicity. PLoS ONE 7, e34639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bröms J. E., Sjöstedt A., Lavander M. (2010) The role of the Francisella tularensis pathogenicity island in type VI secretion, intracellular survival, and modulation of host cell signaling. Front. Microbiol. 1, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santic M., Molmeret M., Klose K. E., Abu Kwaik Y. (2006) Francisella tularensis travels a novel, twisted road within macrophages. Trends Microbiol. 14, 37–44 [DOI] [PubMed] [Google Scholar]
- 12. Rajaram M. V., Ganesan L. P., Parsa K. V., Butchar J. P., Gunn J. S., Tridandapani S. (2006) Akt/Protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J. Immunol. 177, 6317–6324 [DOI] [PubMed] [Google Scholar]
- 13. Pasare C., Medzhitov R. (2004) Toll-like receptors. linking innate and adaptive immunity. Microbes Infect. 6, 1382–1387 [DOI] [PubMed] [Google Scholar]
- 14. Re F., Strominger J. L. (2001) Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J. Biol. Chem. 276, 37692–37699 [DOI] [PubMed] [Google Scholar]
- 15. Medzhitov R., Preston-Hurlburt P., Janeway C. A. (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 [DOI] [PubMed] [Google Scholar]
- 16. Chen W., KuoLee R., Shen H., Bùsa M., Conlan J. W. (2004) Toll-like receptor 4 (TLR4) does not confer a resistance advantage on mice against low-dose aerosol infection with virulent type A Francisella tularensis. Microb. Pathog. 37, 185–191 [DOI] [PubMed] [Google Scholar]
- 17. Malik M., Bakshi C. S., Sahay B., Shah A., Lotz S. A., Sellati T. J. (2006) Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect. Immun. 74, 3657–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H., Nookala S., Bina X. R., Bina J. E., Re F. (2006) Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. J. Leukoc. Biol. 80, 766–773 [DOI] [PubMed] [Google Scholar]
- 19. Harton J. A., Linhoff M. W., Zhang J., Ting J. P. (2002) Cutting edge. CATERPILLER, a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J. Immunol. 169, 4088–4093 [DOI] [PubMed] [Google Scholar]
- 20. Inohara, Chamaillard, McDonald C., Nuñez G. (2005) NOD-LRR proteins. Role in host-microbial interactions and inflammatory disease. Ann. Rev. Biochem. 74, 355–383 [DOI] [PubMed] [Google Scholar]
- 21. Inohara N., Nuñez G. (2003) NODs. Intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3, 371–382 [DOI] [PubMed] [Google Scholar]
- 22. Shoham N. G., Centola M., Mansfield E., Hull K. M., Wood G., Wise C. A., Kastner D. L. (2003) Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. U.S.A. 100, 13501–13506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kahlenberg J. M., Lundberg K. C., Kertesy S. B., Qu Y., Dubyak G. R. (2005) Potentiation of caspase-1 activation by the P2X7 receptor is dependent on TLR signals and requires NF-κB-driven protein synthesis. J. Immunol. 175, 7611–7622 [DOI] [PubMed] [Google Scholar]
- 24. Mariathasan S., Weiss D. S., Dixit V. M., Monack D. M. (2005) Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Atianand M. K., Duffy E. B., Shah A., Kar S., Malik M., Harton J. A. (2011) Francisella tularensis reveals a disparity between human and mouse NLRP3 inflammasome activation. J. Biol. Chem. 286, 39033–39042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fernandes-Alnemri T., Yu J. W., Juliana C., Solorzano L., Kang S., Wu J., Datta P., McCormick M., Huang L., McDermott E., Eisenlohr L., Landel C. P., Alnemri E. S. (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Broz P., von Moltke J., Jones J. W., Vance R. E., Monack D. M. (2010) Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host. Microbe 8, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones J. W., Kayagaki N., Broz P., Henry T., Newton K., O'Rourke K., Chan S., Dong J., Qu Y., Roose-Girma M., Dixit V. M., Monack D. M. (2010) Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brodsky I. E., Palm N. W., Sadanand S., Ryndak M. B., Sutterwala F. S., Flavell R. A., Bliska J. B., Medzhitov R. (2010) A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host. Microbe 7, 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng Y., Lilo S., Brodsky I. E., Zhang Y., Medzhitov R., Marcu K. B., Bliska J. B. (2011) A Yersinia effector with enhanced inhibitory activity on the NF-κB pathway activates the NLRP3/ASC/caspase-1 inflammasome in macrophages. PLoS Pathog. 7, e1002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miao E. A., Rajan J. V. (2011) Salmonella and caspase-1. A complex interplay of detection and evasion. Front. Microbiol. 2, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Master S. S., Rampini S. K., Davis A. S., Keller C., Ehlers S., Springer B., Timmins G. S., Sander P., Deretic V. (2008) Mycobacterium tuberculosis prevents inflammasome activation. Cell Host. Microbe 3, 224–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sutterwala F. S., Mijares L. A., Li L., Ogura Y., Kazmierczak B. I., Flavell R. A. (2007) Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang M. T., Mortensen B. L., Taxman D. J., Craven R. R., Taft-Benz S., Kijek T. M., Fuller J. R., Davis B. K., Allen I. C., Brickey W. J., Gris D., Wen H., Kawula T. H., Ting J. P. (2010) Deletion of ripA alleviates suppression of the inflammasome and MAPK by Francisella tularensis. J. Immunol. 185, 5476–5485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ulland T. K., Buchan B. W., Ketterer M. R., Fernandes-Alnemri T., Meyerholz D. K., Apicella M. A., Alnemri E. S., Jones B. D., Nauseef W. M., Sutterwala F. S. (2010) Cutting edge. Mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J. Immunol. 185, 2670–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peng K., Broz P., Jones J., Joubert L. M., Monack D. (2011) Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell. Microbiol. 13, 1586–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mahawar M., Atianand M. K., Dotson R. J., Mora V., Rabadi S. M., Metzger D. W., Huntley J. F., Harton J. A., Malik M., Bakshi C. S. (2012) Identification of a novel Francisella tularensis factor required for intramacrophage survival and subversion of innate immune response. J. Biol. Chem. 287, 25216–25229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rathinam V. A., Jiang Z., Waggoner S. N., Sharma S., Cole L. E., Waggoner L., Vanaja S. K., Monks B. G., Ganesan S., Latz E., Hornung V., Vogel S. N., Szomolanyi-Tsuda E., Fitzgerald K. A. (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riccardi C., Nicoletti I. (2006) Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 1, 1458–1461 [DOI] [PubMed] [Google Scholar]
- 40. Bakshi C. S., Malik M., Mahawar M., Kirimanjeswara G. S., Hazlett K. R., Palmer L. E., Furie M. B., Singh R., Melendez J. A., Sellati T. J., Metzger D. W. (2008) An improved vaccine for prevention of respiratory tularemia caused by Francisella tularensis SchuS4 strain. Vaccine 26, 5276–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mahawar M., Rabadi S. M., Banik S., Catlett S. V., Metzger D. W., Malik M., Bakshi C. S. (2013) Identification of a live attenuated vaccine candidate for tularemia prophylaxis. PLoS ONE 8, e61539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Henry T., Brotcke A., Weiss D. S., Thompson L. J., Monack D. M. (2007) Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jones C. L., Weiss D. S. (2011) TLR2 signaling contributes to rapid inflammasome activation during F. novicida infection. PLoS ONE 6, e20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weiss D. S., Henry T., Monack D. M. (2007) Francisella tularensis. Activation of the inflammasome. Ann. N.Y. Acad. Sci. 1105, 219–237 [DOI] [PubMed] [Google Scholar]
- 45. Sigrist C. J., Cerutti L., de Castro E., Langendijk-Genevaux P. S., Bulliard V., Bairoch A., Hulo N. (2010) PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 38, D161--D166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chiba S., Nagai T., Hayashi T., Baba Y., Nagai S., Koyasu S. (2011) Listerial invasion protein internalin B promotes entry into ileal Peyer's patches in vivo. Microbiol. Immunol. 55, 123–129 [DOI] [PubMed] [Google Scholar]
- 47. Parida S. K., Domann E., Rohde M., Müller S., Darji A., Hain T., Wehland J., Chakraborty T. (1998) Internalin B is essential for adhesion and mediates the invasion of Listeria monocytogenes into human endothelial cells. Mol. Microbiol. 28, 81–93 [DOI] [PubMed] [Google Scholar]
- 48. Ulland T. K., Janowski A. M., Buchan B. W., Faron M., Cassel S. L., Jones B. D., Sutterwala F. S. (2013) Francisella tularensis live vaccine strain folate metabolism and pseudouridine synthase gene mutants modulate macrophage caspase-1 activation. Infect. Immun. 81, 201–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Atianand M. K., Fitzgerald K. A. (2013) Molecular basis of DNA recognition in the immune system. J. Immunol. 190, 1911–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chiavolini D., Alroy J., King C. A., Jorth P., Weir S., Madico G., Murphy J. R., Wetzler L. M. (2008) Identification of immunologic and pathologic parameters of death versus survival in respiratory tularemia. Infect. Immun. 76, 486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]