

Background: Approved DNA demethylators do not directly inhibit Dnmt1, an oncogenic methyltransferase.
Results: Laccaic acid A (LCA) is a direct, DNA-competitive Dnmt1 inhibitor that reactivates genes silenced by DNA methylation in breast cancer cells synergistically with 5-azadC.
Conclusion: LCA is a natural product in a new class of Dnmt1-targeting small molecules.
Significance: By directly inhibiting Dnmt1, we may reveal and block specific carcinogenesis pathways.
Keywords: Cancer Therapy, Chemical Biology, DNA Methylation, DNA Methyltransferase, Drug Action, Drug Discovery, Enzyme Inhibitors, Fret, Gene Silencing, Microarray
Abstract
Methylation of cytosines in CpG dinucleotides is the predominant epigenetic mark on vertebrate DNA. DNA methylation is associated with transcriptional repression. The pattern of DNA methylation changes during development and with disease. Human DNA methyltransferase 1 (Dnmt1), a 1616-amino acid multidomain enzyme, is essential for maintenance of DNA methylation in proliferating cells and is considered an important cancer drug target. Using a fluorogenic, endonuclease-coupled DNA methylation assay with an activated form of Dnmt1 engineered to lack the replication foci targeting sequence domain, we discovered that laccaic acid A (LCA), a highly substituted anthraquinone natural product, is a direct inhibitor with a 310 nm Ki. LCA is competitive with the DNA substrate in in vitro methylation assays and alters the expression of methylated genes in MCF-7 breast cancer cells synergistically with 5-aza-2′-deoxycytidine. LCA represents a novel class of Dnmt-targeted molecular probes, with biochemical properties that allow it to distinguish between non DNA-bound and DNA-bound Dnmt1.
Introduction
Methylation of cytosine, which occurs predominantly in CpG2 dinucleotides, is an important epigenetic mark associated with gene repression. DNA methylation is crucial for development and differentiation, genome stability, genomic imprinting, X-chromosome inactivation, and silencing of retrotransposons (1). Three catalytically active DNA methyltransferases (Dnmts) are responsible for establishing and maintaining DNA methylation patterns in vertebrates. The Dnmt3 isoforms (Dnmt3a and Dnmt3b), termed de novo methyltransferases, are responsible for establishing DNA methylation marks during germ cell and embryonic development and thus act preferentially on nonmethylated DNA sequences (2). Dnmt1, the most abundant Dnmt, is responsible for maintaining DNA methylation patterns. Thus, hemimethylated DNA sequences are the preferred Dnmt1 substrates (3).
Disruption of normal DNA methylation patterns can lead to a variety of diseases including cancer. Indeed, cancer is the class of illness for which DNA methylation was first proposed as a therapeutic target (4). Increases in Dnmt1 activity and genomic changes in methylation patterns are observed in a variety of malignancies. Multiple common oncogenic pathways lead to the overexpression of Dnmt1 through transcriptional and post-translational mechanisms (5–8). This tends to produce global DNA hypomethylation with concomitant hypermethylation of promoter regions of tumor suppressor genes, enabling tumor-specific gene silencing (9, 10). In preclinical models, targeting of the Dnmt1 isozyme for cancer prevention has been validated genetically. Reducing Dnmt1 copy number and gene activity protects against tumor formation in the ApcMin mouse (11). In addition, targeting Dnmt1 with antisense oligonucleotides inhibits neoplasia in cell culture and in mouse tumor models (12, 13).
Gene-targeted experiments that result in the reduction or elimination of a protein can be used to distinguish among the functions of Dnmt1, Dnmt3a, and Dnmt3b. This is particularly important because Dnmt3a is inactivated in a high proportion of human tumors, such as acute myeloid leukemia (14). In contrast to the isozyme specificity of genetic experiments, most small molecule inhibitors work by mechanisms that are quite distinct from genetic knockouts and knockdowns. Indeed, small molecules that block enzyme activity or inhibit a specific protein-protein interaction would be expected to perform differently than genetic reagents that prevent expression of a protein. This is relevant for Dnmt1, which may be essential for the assembly of multiple protein complexes with different functions in different cell types.
The most commonly used small molecules for cellular and therapeutic DNA demethylation are neither isozyme-specific nor direct inhibitors of Dnmt1. Agents such as 5-azacytidine (5-azaC; see Fig. 1) and 5-aza-2′-deoxycytidine (5-azadC) effectively cause DNA demethylation and have been approved for use in myelodysplastic syndrome (15). 5-Aza nucleosides have complicated mechanisms of action. After cellular uptake, these compounds are converted to their triphosphate counterparts, which requires an additional ribonucleotide reductase-dependent step for 5-azaC. The resulting 5-azadCTP metabolite is then incorporated into DNA, albeit not specifically at CpG dinucleotides. Incorporated 5-azadCMP nucleotides induce a DNA damage response and covalent trapping of Dnmt isozymes, which is followed by proteolysis of Dnmt proteins, DNA demethylation, and reactivation of hypermethylated genes (15–17). The cytotoxicity of 5-aza nucleosides, lack of direct inhibition of Dnmts, and lack of Dnmt isozyme specificity suggests that there is room for improvement in molecular probes targeted against Dnmt1 enzyme activity. Such compounds might allow further characterization of the role of Dnmt1 and other Dnmt isozymes in carcinogenesis and could also serve as leads for new drug development.
FIGURE 1.

Structures of inhibitory compounds. 5-AzaC is a known demethylator in vivo and an approved drug for treatment of myelodysplastic syndrome (15). SGI-1027 is a previously reported Dnmt inhibitor (31). LCA is an insect-derived natural product and the focus of this work.
Human Dnmt1 is a 1616-amino acid multidomain protein with a conserved C-terminal catalytic domain and ∼1100 amino acids of N-terminal regulatory sequences (supplemental Fig. S1). Multiple globular conserved domains are found in the N-terminal regulatory region including the DMAP1 (DNA methyltransferase-associated protein 1) binding domain (18), the proliferating cell nuclear antigen binding domain (19), the replication focus targeting sequence (RFTS) domain (20), the CXXC domain (21), and two bromo-adjacent homology domains (22).
Recently several structures of Dnmt1 were reported (23–26). The first structures to be solved consisted of nonmethylated duplex DNA bound to a fragment of Dnmt1 that started with the CXXC domain (23). In these structures the linker between the CXXC domain and the first bromo-adjacent homology domain was positioned in the active site and proposed to serve an autoinhibitory role and occlude nonmethylated DNA binding. However, using a fluorescence-based DNA methylation assay, we showed that the RFTS domain of Dnmt1 (not present in the original crystal structure) is an endogenous DNA-competitive inhibitor of Dnmt1 activity, such that a truncation mutant lacking RFTS is ∼640-fold more active than a protein containing RFTS (24). Enzymatic assays and gel shift assays clearly showed that the RFTS domain inhibits association with DNA whether the DNA is hemimethylated or not. The kinetics of RFTS inhibition and the acidic nature of this domain prompted us to conclude that RFTS is the key autoinhibitory domain of Dnmt1 that must be removed from the DNA active site for catalysis to occur in the full-length enzyme (24). Consistent with our biochemical data and prediction, the crystal structure of a mouse Dnmt1 construct containing the RFTS domain confirmed that the RFTS domain binds in the substrate DNA binding pocket (25).
This activated form of Dnmt1 (residues 621–1600; supplemental Fig. S1) and the fluorogenic assay proved to be valuable tools to identify small molecule inhibitors of Dnmt1 activity in vitro. Here we show that laccaic acid A (LCA; Fig. 1), a highly substituted anthraquinone natural product, is a direct, DNA-competitive inhibitor of Dnmt1 with a submicromolar Ki. In addition, treatment of MCF-7 cells with LCA results in re-expression of genes known to be silenced by methylation, indicating that LCA is a useful lead compound for Dnmt1 inhibition. Significantly, LCA and 5-azadC synergized in activating expression of silenced tumor suppressor genes, thereby demonstrating the value of obtaining a molecular probe with a direct and complementary mechanism of action to that of 5-aza nucleosides.
EXPERIMENTAL PROCEDURES
Expression and Purification of Dnmts
Truncated versions of human Dnmt1 (amino acids 621–1600 and 351–1600; supplemental Fig. S1) were expressed and purified as previously described (24). The catalytic domain of human Dnmt3a (amino acids 590–912) was expressed as an N-terminal His-tagged maltose-binding protein fusion protein in Rosetta 2(DE3)pLysS competent cells (Novagen). Protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and cultures were grown at 30 °C for 4 h. After cell lysis, protein was purified via metal affinity using nickel-nitrilotriacetic acid resin (GE Healthcare). The fusion protein was cleaved with tobacco etch virus protease, and a second metal affinity column was used to remove the His-tagged protease and any uncleaved fusion protein. The C-terminal catalytic domain was then further purified using a Heparin HP Hi-Trap column (GE Healthcare). Bound protein was eluted using a linear gradient from 0.2 to 1.5 m NaCl in 20 mm NaPi, pH 7.5, 2 mm DTT buffer. Purified proteins were concentrated and stored at −80 °C in 50% glycerol. All proteins were quantified using A280 and calculated extinction coefficients.
DNA Methylation Assay
Dnmt activity was measured using a fluorescence-based DNA methylation assay (24). In short, hemimethylated hairpin oligonucleotide, termed 8006, with a 5′ fluorophore and a 3′ quencher was incubated with Dnmt and AdoMet in the presence of restriction enzyme Gla I, which cleaves the fully methylated product oligonucleotide and releases the fluorophore from the quencher, generating fluorescence in real-time (Fig. 2A). A control containing Gla I in the absence of methyltransferase was subtracted from each assay condition to account for the sum of background fluorescence of the internally quenched substrate and slow Gla I cleavage of the hemimethylated substrate oligonucleotide. Assays (0.1 ml) were conducted in 96-well black half-area plates in either a Wallac VICTOR2 or Biotek Synergy Neo plate reader at 37 °C in 10 mm Tris, pH 7.5, 100 mm potassium glutamate, 1 mm MgCl2, 1 mm DTT, 0.1 mg/ml BSA, and 5% glycerol. Assays contained varying amounts of oligonucleotide 8006 (5′-FAM-CCTATGCGmCATCAGTTTTCTGATGmCGmCATAGG-3′-Iowa Black, in which mC denotes 5-methyldeoxycytidylate residues (Integrated DNA Technologies, Coralville, IA), AdoMet (HPLC-purified, Sigma), and small molecule inhibitors (5-azaC, Sigma; SGI-1027, generous gift of Dr. Jian Jin, University of North Carolina; LCA, TCI America). Other anthraquinone compounds examined were purchased from ChemBridge Corp, San Diego, CA (UI1055 is N,N-diethyl-1-nitro-9,10-dioxo-9,10-dihydroanthracene-2-carboxamide; UI1060 is ethyl N-{[1-(butylthio)-9,10-dioxo-9,10-dihydroanthra-cen-2-yl] carbonyl}glycinate; UI1061 is 1-(butyl-sulfonyl)-9,10-dioxo-9,10-dihydroanthracene-2-carboxylicacid) and Sigma (anthraquinone and anthraquinone 2-carboxylic acid). All assays were conducted in triplicate and contained either human Dnmt1 (621–1600), human Dnmt1 (351–1600), human Dnmt3a (590–912), or M.SssI methyltransferase (New England Biolabs) and 0.8 units of Gla I (Sibenzyme, West Roxbury, MA), except for the Gla I control assay, which did not contain a methyltransferase. Data were fitted using Prism (GraphPad Software, Inc). Dose-response data were fit allowing the Hill slope to vary. In all cases, Hill slopes of −0.9 to −1.2 were obtained. Thus, all dose-response data were refitted using a constant Hill slope of −1.
FIGURE 2.

LCA inhibits Dnmt1 activity. A, shown is a schematic of the Gla I-coupled Dnmt assay. Oligonucleotide 8006 contains a single nonmethylated CpG dinucleotide (shown in red). The fully methylated product oligonucleotide is a substrate for the restriction endonuclease Gla I. Cleavage of the product oligonucleotide releases the 5′ fluorophore from the 3′ quencher and generates fluorescence. AdoHcy, S-adenosyl homocysteine. B, shown is time dependence of assays containing 10 nm oligonucleotide 8006, 10 μm AdoMet, and 1 nm RFTS-lacking Dnmt1 (amino acids 621–1600) in the presence of DMSO (black), 10 μm 5-azaC (green), 10 μm SGI-1027 (blue), and 10 μm LCA (red). The addition of SGI-1027 and LCA fully inhibit Dnmt1 activity, whereas 5-azaC has no effect on fluorescence generation. RFU, relative fluorescence units.
Gla I Activity Assay
Gla I activity was measured using a fluorescence-based assay. Assays (0.1 ml) were conducted in 96-well black half-area plates at 37 °C in a Wallac VICTOR2 using excitation and emission wavelengths of 485 and 535 nm, respectively. Each assay contained 10 mm Tris, pH 7.5, 100 mm potassium glutamate, 5 mm MgCl2, 1 mm DTT, 0.1 mg/ml BSA, 5% glycerol, 8 nm oligonucleotide 8007 (5′-FAM-CCTATGmCGmCATCAGTTTTCTGATGmCGmCATAGG-3′-IowaBlack, where mC denotes 5-methyldeoxycytidylate residues; Integrated DNA Technologies), 0.4 units Gla I, and 1% DMSO. Assays were conducted in triplicate in the presence and absence of 10 μm LCA. Cleavage of the oligonucleotide by Gla I releases the 5′-FAM fluorophore from the 3′-Iowa Black quencher and generates fluorescence. Data were fitted using Prism.
Differential Scanning Fluorometry (DSF)
DSF (28) was used to assess the ability of small molecules to bind directly to Dnmts in the absence of substrates and alter the observed melting temperature (Tm). Assays (25 μl) were conducted in triplicate in a Bio-Rad C1000 Thermal Cycler-CFX real-time system using the FRET channel and contained 100 mm Hepes, pH 7.5, 100 mm potassium glutamate, 5 mm MgCl2, 1–4 μm Dnmt, 5× Sypro Orange (Invitrogen), 50 or 100 μm concentrations of the compounds of interest, and 1% DMSO. The temperature was increased from 25 to 95 °C in 0.5 °C increments every minute. Fluorescence traces were exported and analyzed by fitting to the Boltzmann equation in Prism to determine the Tm.
Cell Culture
Human breast cancer MCF-7 cells (ATCC) were seeded at 6 × 104 per well in DMEM (Invitrogen) supplemented with 10% fetalplex (Gemini Bio-products) and grown overnight in 5% CO2 at 37 °C. The following day the cells were treated with 200 μm LCA or anthraquinone-2-carboxylic acid. In some experiments 0.5 μm 5-azadC (Sigma) was added. All compounds were dissolved in media. Each day for 5 days the media were replaced with fresh media plus the compounds, and then RNA and/or DNA were isolated from cells.
Microarray Analysis
Total RNA was isolated from 2 × 105 cells and used for microarray analysis (University of Iowa DNA Core Facility) in hybridization to Human Gene ST1.0 Array GeneChips (Affymetrix). Data were analyzed using Partek Genomics Suite Version 6.5. Pathway analysis was performed on the genes that changed expression in response to LCA using the WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) (29, 30).
Quantitative PCR
After drug treatment, total RNA was extracted from 2 × 105 cells using TRIzol (Invitrogen). RNA was treated with DNase I (Invitrogen), and cDNA was generated using the First strand cDNA synthesis kit (Invitrogen). Primers (supplemental Table 1) were designed using Primer3web software version 4.0.0, and qRT-PCR was performed using SYBR Green PCR master mix (Applied Biosystems). Reactions were performed in triplicate on three biologically independent samples using the Step One Plus Real-time PCR system (Applied Biosystems). Normalization was performed using cycle threshold (Ct) values for HPRT.
RESULTS
LCA Is a Direct, Potent Inhibitor of Dnmt1
Reversing epigenetic changes, such as aberrant DNA methylation, is a promising cancer therapy strategy (9). Because the link between Dnmt1 hyperactivity and cancer is well established (9, 10, 16), we sought to discover novel small molecule modulators of Dnmt1 function. We developed a high throughput assay to screen for compounds that inhibit activity of an activated form of Dnmt1 (621–1600). Deletion of sequences N-terminal of and including the RFTS domain results in an enzyme that has a kcat/Km of 1.9 × 106 m−1s−1 (DNA substrate), which is a 640-fold improvement over forms of the enzyme that include the RFTS domain (24). This catalytic power and the sensitivity of the fluorogenic assay were exploited to search for and characterize enzyme inhibitors. Using a miniaturized version of our Gla I-coupled DNA methylation assay (24), we screened the 2320 compound Spectrum library (Microsource, Gaylordsville, CT) for agents that inhibit Dnmt1 activity.3 In this screen, LCA was identified as a promising potential inhibitor with an unknown mechanism of action.
We employed multiple approaches to test whether LCA is an authentic and direct inhibitor of Dnmt1 activity. In the presence of 10 μm LCA, no fluorescence generating activity was observed in the coupled methylation assay using oligonucleotide 8006 (Fig. 2). As a positive control, we used SGI-1027 (Fig. 1), a quinoline compound reported to directly inhibit Dnmt isozymes (31). The addition of 10 μm SGI-1027 resulted in complete inhibition of the activated form of Dnmt1 in our assay (Fig. 2). However, 10 μm 5-azaC had no effect on Dnmt1 activity. This was anticipated because it is known that cytosine nucleoside inhibitors, such as 5-azaC, must be incorporated into DNA to cause cellular DNA demethylation (16). These data indicated that 5-azaC is not a direct inhibitor of Dnmt1 and suggested that LCA and SGI-1027 are either Dnmt1 inhibitors or cause interference with some aspect of the methylation assay.
The possibilities that LCA is an inhibitor of the restriction enzyme Gla I or a quencher of the 5′-FAM product were excluded by examining the effect of LCA on Gla I activity. The addition of 10 μm LCA to an assay containing oligonucleotide 8007, the fully methylated product oligonucleotide, had no effect on Gla I activity (supplemental Fig. S2). We then aimed to ensure that the inhibitory effects observed by the addition of LCA were structurally specific and could not be attributed to the anthracene or anthraquinone core. We, therefore, examined the effect of various anthraquinone compounds on Dnmt1 activity. No other anthraquinone compound examined inhibited Dnmt1 in the in vitro assay (supplemental Fig. S3 and Table 2). Thus, Dnmt1 inhibition cannot be attributed to nonspecific redox cycling or another generic characteristic of anthraquinone compounds.
Although LCA is not an inhibitor of Gla I, we aimed to exclude the possibility that the inhibitory activity of LCA stems from an interaction with the DNA substrate. To test if LCA interacts with RFTS-lacking Dnmt1 in the absence of DNA and AdoMet, we used DSF to determine the melting temperature (Tm) of Dnmt1 in the presence and absence of each candidate inhibitor. The addition of SGI-1027 resulted in a 2.0 ± 0.1 °C increase in the Tm (Table 1), indicating that the small molecule binds directly to Dnmt1 in the absence of substrates and stabilizes the enzyme against thermal denaturation. Because 5-azaC is a prodrug that does not directly inhibit Dnmt1, this compound did not alter the Tm. In contrast, the addition of LCA to the Dnmt1 DSF assay resulted in a 1.9 ± 0.1 °C increase in Tm from 43.8 °C in the DMSO control to 45.7 °C in the presence of saturating LCA. These data indicate that LCA binds directly to Dnmt1.
TABLE 1.
Changes in the observed melting temperature of Dnmt1 (621–1600) as determined by DSF
| Compound | ΔTm |
|---|---|
| °C | |
| 5-AzaC | 0 |
| SGI-1027 | 2.0 ± 0.1 |
| LCA | 1.9 ± 0.1 |
An authentic inhibitor should show concentration dependence. To calculate an IC50 value for each inhibitor, the concentrations of LCA and SGI-1027 were varied from 10 nm to 10 μm under standard assay conditions (10 nm oligonucleotide 8006 and 10 μm AdoMet). The percent activity observed was determined with respect to an uninhibited control. Both compounds showed concentration-dependent inhibition of Dnmt1 (621–1600). However, with an IC50 value of 650 ± 40 nm, LCA exhibited ∼2.5-fold higher potency at inhibiting Dnmt1 than SGI-1027 (Fig. 3).
FIGURE 3.

LCA is a more potent inhibitor of Dnmt1 than SGI-1027. The concentration dependence of inhibition by SGI-1027 (●) and LCA (■) was investigated at 10 nm oligonucleotide 8006 and 10 μm AdoMet using 1 nm RFTS-lacking Dnmt1 (amino acids 621–1600). The percent activity of each condition was determined by comparing to an uninhibited DMSO-containing control. Fitting the data using a Hill slope of −1 gives an IC50 of 1.6 ± 0.2 μm for SGI-1027 and 650 ± 40 nm for LCA.
We then investigated if LCA acts as a covalent inactivator. LCA was incubated with either Dnmt1 or Dnmt1 plus AdoMet for 30 min. Incubated enzymes were then buffer-exchanged into assay buffer that lacked LCA and used to initiate DNA methylation assays. In both cases, incubation with LCA had no effect on the observed activity of Dnmt1 (supplemental Fig. S4), indicating that LCA does not covalently inactivate the enzyme.
LCA Is a DNA-competitive Inhibitor
The cytosine nucleoside DNA demethylating agents, such as 5-azaC, must be incorporated into DNA to exert their effects (15, 16). Several non-nucleoside inhibitors of DNA methylation have been reported in the literature (32). However, experimental information on the kinetics and mode of inhibition is lacking for many of these compounds. We set out to determine the mode of inhibition of LCA and SGI-1027 using the fluorescence-based DNA methylation assay. We titrated SGI-1027 in assays containing high concentrations of DNA (∼13 times Km,DNA) and varied concentrations of AdoMet to determine kinetic parameters. As suggested by previous results (31), SGI-1027 acts as an AdoMet competitor (Fig. 4A). The double reciprocal plot of the data clearly shows competitive inhibition with all lines intersecting on the y axis. Fitting the data to an AdoMet competitive model yields a Ki of 1.5 ± 0.3 μm. Surprisingly, LCA was not competitive with AdoMet. Instead, this compound exhibited mixed inhibition with respect to AdoMet (Fig. 4B and supplemental Fig. S5) and was competitive with the oligonucleotide substrate (Fig. 4C and supplemental Fig. S5). Titrating LCA into assays containing AdoMet at ∼15 times Km,AdoMet and varying concentrations of oligonucleotide 8006 yielded a double reciprocal plot in which all lines intersect on the y axis, indicating DNA competition. As the concentration of LCA was increased, the apparent Km,DNA was increased with no effect on the observed Vmax. Fitting these data by nonlinear regression, a Ki of 310 ± 80 nm was obtained (supplemental Fig. S5). Thus, in our in vitro DNA methylation assay, LCA is an ∼5-fold more potent inhibitor of Dnmt1 activity than SGI-1027 with a novel mechanism of action.
FIGURE 4.

LCA is a DNA-competitive inhibitor. A, shown are AdoMet-dependent SGI-1027 inhibition kinetics. SGI-1027 was used as an inhibitor in reactions containing 20 nm DNA (∼13× Km,DNA), and varying concentrations of AdoMet (1–25 μm) and initial velocities (relative fluorescence units/min) were determined. The double reciprocal plot of the data without SGI-1027 (●), 0.5 μm SGI-1027 (■), and 2 μm SGI-1027 (▴) shows that SGI-1027 is competitive with respect to AdoMet. Using nonlinear regression to fit the alteration in Kmapparent, a Ki of 1.5 ± 0.3 μm was determined. B, shown are AdoMet-dependent LCA inhibition kinetics. LCA was used as an inhibitor in reactions containing 20 nm DNA and varying concentrations of AdoMet (2–30 μm). The double reciprocal plot of the data without LCA (●), 0.4 μm LCA (■), 0.8 μm LCA (▴), and 1.5 μm LCA (♦) shows mixed inhibition, indicating that LCA binds both the free enzyme and the enzyme-AdoMet complex. Using nonlinear regression to fit the data (supplemental Fig. S5), a Ki,a of 640 ± 200 nm and a KI,b of 1.4 ± 0.4 μm was determined. C, DNA-dependent LCA inhibition kinetics are shown. Here, LCA was used as an inhibitor in reactions containing 30 μm AdoMet (∼15× Km,AdoMet) and varying concentrations of DNA (2–25 nm). The double reciprocal plot of the data without LCA (●), 0.4 μm LCA (■), 0.8 μm LCA (▴), and 1.5 μm LCA (♦) is indicative of competitive inhibition. Using nonlinear regression to fit the data (supplemental Fig. S5) gives a Ki of 310 ± 80 nm. In all cases, 1 nm RFTS-lacking Dnmt1 (621–1600) was used.
The activated form of Dnmt1 (621–1600) was used for probe discovery because it is ∼640-fold more active than forms of Dnmt1 with the RFTS domain. We have previously shown that RFTS functions as a DNA-competitive inhibitor of Dnmt1 (24). To test whether LCA would inhibit Dnmt1-containing RFTS, we first used DSF to test direct binding to Dnmt1 (351–1600). As observed for Dnmt1 (621–1600), the addition of 50 μm LCA shifts the melting temperature of Dnmt1 (351–1600) 2 °C to the right (Fig. 5A). The ability of LCA to stabilize against thermal denaturation of Dnmt1 (351–1600) indicates that the small molecule is capable of binding Dnmt1 even in the presence of the RFTS domain. We then investigated the ability of LCA to inhibit the methyltransferase activity of Dnmt1 (351–1600) under assay conditions suitable for this enzyme (100 nm oligonucleotide 8006 and 100 μm AdoMet). LCA concentration was varied from 2 to 100 μm. Under these conditions, LCA exhibited concentration-dependent inhibition of Dnmt1 with an IC50 value of 19.1 ± 1.7 μm (Fig. 5B). The higher IC50 for LCA inhibition of Dnmt1 (351–1600) than for Dnmt1 (621–1600) can be explained in two ways. First, the RFTS domain-containing enzyme must be assayed at a 10-fold higher concentration of DNA substrate. Second, because the RFTS domain is also DNA-competitive, a higher concentration of LCA would be required to displace the endogenous inhibitory protein domain. These findings suggest that LCA would be capable of inhibiting Dnmt1 in a cellular context, although at a higher concentration than used against Dnmt1 (621–1600).
FIGURE 5.

LCA is a direct inhibitor of RFTS-containing Dnmt1. A, DSF was used to determine the Tm of RFTS-containing Dnmt1 (351–1600) in the presence (●) and absence (■) of 50 μm LCA. Fitting the observed melting traces to the Boltzmann equation gives Tm values of 47.4 ± 0.1 °C and 49.3 ± 0.1 °C in the absence and presence of LCA, respectively. The nearly 2 °C shift in the observed Tm is indicative of direct binding. Fl, fluorescence. B, concentration-dependent inhibition by LCA was investigated at 100 nm oligonucleotide 8006 and 100 μm AdoMet using 10 nm RFTS-containing Dnmt1 (amino acids 351–1600). The percent activity of each condition was determined by comparison to an uninhibited DMSO-containing control. Fitting the data using a Hill slope of −1 gives an IC50 of 19.1 ± 1.7 μm.
LCA Specificity
We next investigated the specificity of LCA inhibition of cytosine DNA methyltransferases. Three catalytically active isoforms of Dnmt exist in vertebrates: Dnmt1, Dnmt3a, and Dnmt3b (1). To determine if LCA can interact directly with other Dnmts, we used DSF to detect changes in the observed melting temperature of the catalytic domain of Dnmt3a. In the presence of 100 μm LCA, the observed melting temperature is shifted more than 2 °C to the right from 43.0 ± 0.1 °C in the absence of LCA to 45.3 ± 0.1 °C, indicating a direct interaction between LCA and Dnmt3a. To further investigate specificity, IC50 values were determined for LCA inhibition of RFTS-containing Dnmt1 (351–1600), Dnmt3a (590–912), and the M.SssI methyltransferase from Spiroplasma sp. strain MQ1 under identical conditions (200 nm oligonucleotide 8006 and 0.25 mm AdoMet in standard assay buffer). LCA was varied from 5 to 100 μm, and the percent activity observed under each condition was determined with respect to an uninhibited control (Fig. 6). The IC50 values obtained under these conditions are 27.8 ± 1.8, 50.5 ± 4.3, and 53.6 ± 4.1 μm for RFTS-lacking Dnmt1, the catalytic domain of Dnmt3a, and M.SssI methyltransferase, respectively. Thus, LCA is capable of inhibiting all cytosine methyltransferases investigated. To directly compare potency, one must take into account the assay condition. Here, 200 nm DNA substrate was used. This is ∼13 times the reported Km,DNA for Dnmt1 (351–1600) (24). However, this DNA concentration is roughly equal to the Km,DNA for M.SssI methyltransferase (33) on a similar hairpin DNA substrate. The Km,DNA values reported for full-length Dnmt3a are 2.5 μm for 30-mer duplex non-methylated and hemi-methylated DNAs (34), 2.7 μm for poly(dIdC), and 3.5 μm for poly(dGdC) (35). All of these reported values are more than 10-fold larger than the DNA concentration used in the IC50 experiment. Yet, as can been seen in Fig. 6, LCA exhibited an ∼2-fold specificity for Dnmt1 under this condition. Because DNA substrates were presumably well below saturation for M.SssI methyltransferase and Dnmt3a, LCA appears to have substantial specificity for Dnmt1 vis à vis other methyltransferases.
FIGURE 6.

Specificity of LCA inhibition. LCA inhibition of 10 nm RFTS-containing Dnmt1 (●), catalytic domain of Dnmt3a (■), and M.SssI methyltransferase (▴) was investigated at 200 nm oligonucleotide 8006 and 0.25 mm AdoMet using the Gla I-coupled DNA methylation assay. The percent activity of each condition (5–100 μm LCA) was determined by comparison to an uninhibited DMSO containing control. Fitting the data using a Hill slope of −1 gives IC50 values of 27.8 ± 1.8 μm for RFTS-containing Dnmt1, 50.5 ± 4.3 μm for Dnmt3a, and 53.6 ± 4.1 μm for M.SssI methyltransferase.
LCA Treatment Alters Gene Expression in MCF-7 Breast Cancer Cells
To determine whether LCA is cellularly available and capable of altering gene expression, we examined the gene expression consequences of LCA on MCF-7 breast cancer cells. Cells were treated with 5–1000 μm LCA in a five-day protocol, and the cells were harvested for RNA purification. qRT-PCR was performed with primers specific for genes known to be methylated in MCF-7 cells and that show increased expression upon 5-azaC treatment (36–41). Of the 10 genes tested, eight (CLDN6, DNMT1, FKBP4, GSTP1, KIF1A, PHD3, P16, and TFPI-2) showed no change in expression even at the highest concentrations of LCA. In contrast, VGF and MAL showed 3–5-fold up-regulation at 200 μm and higher concentrations (data not shown). Thus, specific methylated genes showed sensitivity to LCA treatment, indicating that LCA treatment is capable of alteration of gene expression in cell culture.
The selective activation of methylated genes prompted a genome-wide investigation of the effects of LCA. MCF-7 cells were treated for 5 days with 200 μm LCA or media alone as a control. Total RNA was isolated and used for microarray analysis (Affymetrix, Human Gene ST1.0 gene chip). Of the 21,014 human Refseq genes, 64 showed a 2-fold or greater change in expression with a p value of 0.05 or lower (supplemental Table 3 and accession number GSE45804, NCBI GEO database). Of these mRNAs, 35 were significantly up-regulated, and 29 were down-regulated. The majority of these genes (42/64) possess CpG islands within 1000 bp of their transcription start sites and are frequently methylated in cancer.
To determine whether the effects on gene expression are specific for LCA, cells were treated in parallel with a structurally related compound, anthraquinone-2-carboxylic acid, that had no effect on Dnmt1 activity in vitro (supplemental Fig. S3). Probes for 12 mRNAs were designed; 9 were up-regulated by LCA, 2 were down-regulated by LCA, and 1 was unmethylated in MCF-7 cells and was unaffected by LCA. qRT-PCR revealed that of the genes tested, only CYP1A showed greater than a 2-fold up-regulation in response to the control anthraquinone (Table 2). This finding is consistent with the reported expression of CYP1A in response to xenobiotic compounds (42, 43). CBX5, a gene that is unmethylated in MCF-7 cells (44), showed no change in expression with either anthraquinone (Table 2). The eight remaining up-regulated genes and two down-regulated genes examined were altered by LCA but not the control anthraquinone (Table 2), indicating that the observed gene expression changes are specific for LCA.
TABLE 2.
qPCR validation of LCA microarray analysis and changes in gene expression caused by treatment with control compound, anthraquinone-2-carboxylic acid
| Gene | Microarray | qPCR | Anthraquinone 2-carboxylic acid |
|---|---|---|---|
| ARRB1 | +2.29 | +2.37 ± 0.30 | +1.05 ± 0.33 |
| CEACAM5 | +3.24 | +4.97 ± 0.23 | +1.48 ± 0.51 |
| CYP1A1 | +13.47 | +48.77 ± 15.80 | +5.71 ± 2.31 |
| CYP1B1 | +2.26 | +2.79 ± 0.69 | +1.65 ± 0.30 |
| DHRS3 | +2.42 | +3.00 ± 0.20 | +1.55 ± 0.27 |
| GCNT1 | +2.33 | +2.96 ± 0.18 | +0.85 ± 0.12 |
| PPARG | +2.28 | +2.51 ± 0.13 | +1.21 ± 0.30 |
| RERG | +2.30 | +3.32 ± 0.59 | +0.85 ± 0.08 |
| RGS16 | +4.86 | +6.72 ± 0.81 | +1.17 ± 0.58 |
| SNAI2 | −3.27 | −8.26 ± 1.99 | +0.94 ± 0.13 |
| TGBF2 | −6.10 | −4.47 ± 0.59 | +1.40 ± 0.16 |
| CBX5 | +1.02 | 1.02 ± 0.10 | +0.90 ± 0.22 |
Given that LCA acts as a DNA-competitive inhibitor of Dnmt1, we reasoned that LCA would act synergistically with 5-azadC to re-activate methylated genes. In this treatment combination, incorporated 5-aza nucleotides would deplete Dnmt isozymes that are engaged with DNA, whereas LCA would directly inhibit newly synthesized Dnmt isozymes or Dnmt isozymes that are not DNA-associated, thereby preventing renewed DNA methylation. We were particularly interested in how mRNAs such as CEACAM5, DHRS3, and RGS16, which were strongly and specifically induced by LCA, would accumulate when treated with two agents of complementary mechanisms of action. To test this hypothesis, MCF-7 cells were treated for 5 days with 0.5 μm 5-azadC, 200 μm LCA, or both compounds. qRT-PCR of total RNA showed that each of the genes, CEACAM5, DHRS3, and RGS16, showed a synergistic response to the two compounds. The negative control, CBX5, showed no change in expression upon either treatment (Fig. 7A). These findings demonstrate the potential use of LCA to amplify the effects of 5-azadC on the reactivation of genes in cancer cells.
FIGURE 7.
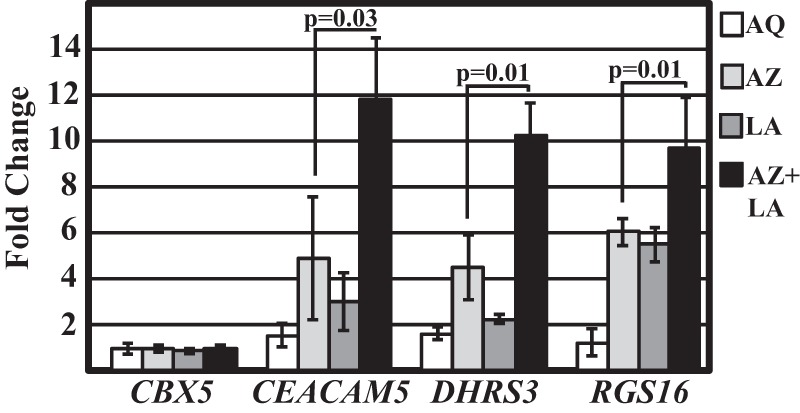
LCA and 5-azadC show synergistic effects on the activation of methylated genes. MCF-7 breast cancer cells were treated with the indicated compound, total RNA was isolated, and qRT-PCR was performed to examine the expression of 4 mRNAs. CEACAM5, DHRS3, and RGS16 possess CpG islands that are methylated in breast and other types of cancer. CBX5 possesses a CpG island that is hypomethylated in MCF-7 cells. Assays were performed on three biological replicates. Error bars represent the S.D. p values were calculated using Student's t test. AQ, 200 μm anthraquinone-2-carboxylic acid; AZ, 0.5 μm 5-azadC; LA, 200 μm LCA; AZ+LA, 0.5 μm 5-azadC plus 200 μm LCA.
DISCUSSION
Genetic lesions in cancer cells are not reversible and frequently result in the misregulation of gene expression. The plasticity of the epigenome provides an opportunity to correct gene expression. Epigenetic reprogramming could be expected to result in restoration of a more differentiated and less proliferative state and regression to a lower degree of drug resistance (45). Nucleoside demethylating agents, such as 5-azadC, are used in the treatment of myelodysplastic syndrome and acute myeloid leukemia (15, 46). Particularly at high doses, these compounds act non-specifically on the genome, causing the activation of many genes and potentially transposons, which can lead to genomic instability (47, 48). Furthermore, 5-azaC is incorporated into RNA and induces off-target effects (16). Thus, there is a need for the discovery of new compounds that would reduce side effects in patients. In addition, there is a need for research tools that will distinguish genes that require Dnmt1 enzymatic activity from those that require Dnmt1 physical interactions for gene silencing.
Here we show that LCA is a direct inhibitor of Dnmt1. LCA is the first of a new class of Dnmt inhibitors and suggests that specifically substituted anthraquinones may be a useful scaffold for Dnmt inhibitors. LCA, a component of lac dye, is a natural product from the red scales of the insect, Kerria lacca, and has been used as a red dye for millennia in food, cosmetics, paint, and fabrics. LCA has mild toxicity (49), unlike DNA demethylators such as 5-azaC, which are cytotoxic (15–17).
LCA is demonstrated to have a DNA-competitive mechanism of action and to inhibit Dnmt3a and M.SssI methyltransferase with moderate selectivity for Dnmt1. Recently, 3-nitroflavanones were identified by screening against Dnmt3a and suggested by molecular docking to compete for DNA binding (50). We suggest that specifically substituted flavones, anthraquinones, and related compounds may produce multiple hits against Dnmts. Potentially by maximizing interactions with the target recognition sequences, which are unique to each isozyme (51), inhibitors of this class may be identified with strong isozyme selectivity.
Because LCA is DNA-competitive and Dnmt1 is reported to be highly processive (52, 53), the population of Dnmt1 that is tightly DNA associated should by definition be resistant to inhibition. In contrast, non-DNA-bound enzyme should be sensitive to this class of compound. The set of genes activated by LCA, which appear to be a subset of those silenced by DNA methylation, may serve to be highly revealing of cellular mechanisms of gene silencing. We suggest that the genes that are resistant to LCA but sensitive to 5-azadC are less actively demethylated or more tightly associated with Dnmt proteins than those that are sensitive to LCA. According to this view, LCA-sensitive genes would be those for which Dnmt enzyme activity rather than Dnmt protein complex occupancy is determinative for gene silencing. This can be tested by next generation methods including methylation sequencing and Dnmt-targeted chIPseq.
Interestingly, the genes that changed expression in response to LCA treatment were enriched in 10 common biological pathways, many of which are involved in cell adhesion and signaling (Table 3). These pathways contain shared genes that play roles in breast cancer progression. LAMA3 (encoding a subunit of laminin 322/laminin 5), up-regulated by LCA treatment, has tumor suppressor functions and aberrant promoter DNA methylation in breast tumors (29, 30). PPARG (peroxisome proliferator-activated receptor γ), up-regulated by LCA treatment, induces terminal differentiation of breast cancer cells, providing a natural therapeutic block of proliferation (54). Up-regulation of PPARG may provide additional benefits because pioglitazone, a PPARγ agonist, inhibits the expression of aromatase, which mediates estrogen synthesis in adipocytes (55). Thus, knowledge of which genes change expression upon LCA treatment, coupled with genomic profiling of patients, could help to personalize treatment.
TABLE 3.
Pathways regulated by LCA in MCF-7 breast cancer cells
TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; EGFR, EGF receptor.
| Pathway | # Genes | p value |
|---|---|---|
| α9β1 integrin signaling | 13 | 3.78e-07 |
| Integrin-linked kinase signaling | 10 | 3.78e-07 |
| IL3-mediated signaling | 13 | 3.78e-07 |
| AP-1 transcription factor network | 10 | 3.78e-07 |
| Nectin adhesion pathway | 13 | 3.78e-07 |
| Integrin family cell surface interactions | 14 | 3.78e-07 |
| TRAIL signaling pathway | 13 | 3.89e-07 |
| β1 integrin cell surface interactions | 13 | 4.16e-07 |
| Regulation of CDC42 activity | 10 | 4.96e-07 |
| EGFR-dependent endothelin signaling | 12 | 4.96e-07 |
We predicted that LCA would potentiate the gene activation effects of 5-azadC at LCA-sensitive genes in MCF-7 cells. The target genes examined, CEACAM5, DHRS3, RGS16, are silenced by methylation in breast and other malignancies (46, 56–58), and in all cases, combined treatment resulted in enhanced gene expression as compared with either LCA or 5-azadC alone. This is potentially clinically important because the current goal of expanding the utility of 5-aza nucleosides to solid tumors depends on using low doses that are not cytotoxic (45).
Further development of LCA as a molecular probe or lead compound is expected to depend on structural characterization and cellular comparisons between LCA and control anthraquinones, such as UI1055 and anthraquinone-2-carboxylic acid, which are inert with respect to Dnmt1. We have been particularly eager to test LCA in cancer models that are driven by Dnmt1. Because Rgs6 functions as a negative regulator of Dnmt1 (27), we tested whether LCA can inhibit Ras-induced proliferative and apoptotic phenotypes in a rgs6 knock-out model. In these studies, Dnmt1 was strongly overexpressed and drove cellular phenotypes that were effectively reversed by LCA but not by the control anthraquinone (59). These data indicate that LCA is a novel DNA-competitive inhibitor of Dnmt1 with the potential to improve the understanding of biology of DNA methylation and the enzymology of Dnmt isozymes and serve as a lead compound for combination therapy agents.
Supplementary Material
Acknowledgment
We thank Dr. Jian Jin from the University of North Carolina for the generous gift of SGI-1027.
This work was supported by National Institutes of Health Grant R01CA075954, Contract HHSN261200433000C from the National Cancer Institute and a generous gift from the Roy J. Carver Charitable Trust (to C. B.). This work was also supported by Oberley Seed Grants from the University of Iowa Holden Comprehensive Cancer Center (to C. B. and L. L. W.), and an American Cancer Society postdoctoral fellowship (to R. L. F.).
The Gene Expression Omnibus (GEO) accession number GSE45804 reported in this work can be accessed through the NCBI GEO database.

This article contains supplemental Tables 1–3 and Figs. S1–S4.
R. L. Fagan, M. Wu, and C. Brenner, submitted for publication.
- CpG
- cytosine-phosphate-guanosine dinucleotide
- Dnmt
- DNA methyltransferase
- 5-azaC
- 5-aza-cytidine
- 5-azadC
- 5-aza-2′-deoxycytidine
- RFTS
- replication foci targeting sequence
- LCA
- laccaic acid A
- AdoMet
- S-adenosyl methionine
- DSF
- differential scanning fluorometry
- Tm
- melting temperature
- FAM
- 6-carboxyfluorescein
- qRT
- quantitative real-time.
REFERENCES
- 1. Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- 2. Yokochi T., Robertson K. D. (2002) Preferential methylation of unmethylated DNA by Mammalian de novo DNA methyltransferase Dnmt3a. J. Biol. Chem. 277, 11735–11745 [DOI] [PubMed] [Google Scholar]
- 3. Yoder J. A., Soman N. S., Verdine G. L., Bestor T. H. (1997) DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J. Mol. Biol. 270, 385–395 [DOI] [PubMed] [Google Scholar]
- 4. Szyf M. (1994) DNA methylation properties. Consequences for pharmacology. Trends Pharmacol. Sci. 15, 233–238 [DOI] [PubMed] [Google Scholar]
- 5. MacLeod A. R., Rouleau J., Szyf M. (1995) Regulation of DNA methylation by the Ras signaling pathway. J. Biol. Chem. 270, 11327–11337 [DOI] [PubMed] [Google Scholar]
- 6. Slack A., Cervoni N., Pinard M., Szyf M. (1999) DNA methyltransferase is a downstream effector of cellular transformation triggered by simian virus 40 large T antigen. J. Biol. Chem. 274, 10105–10112 [DOI] [PubMed] [Google Scholar]
- 7. Bigey P., Ramchandani S., Theberge J., Araujo F. D., Szyf M. (2000) Transcriptional regulation of the human DNA Methyltransferase (dnmt1) gene. Gene 242, 407–418 [DOI] [PubMed] [Google Scholar]
- 8. Detich N., Ramchandani S., Szyf M. (2001) A conserved 3′-untranslated element mediates growth regulation of DNA methyltransferase 1 and inhibits its transforming activity. J. Biol. Chem. 276, 24881–24890 [DOI] [PubMed] [Google Scholar]
- 9. Daniel F. I., Cherubini K., Yurgel L. S., de Figueiredo M. A., Salum F. G. (2011) The role of epigenetic transcription repression and DNA methyltransferases in cancer. Cancer 117, 677–687 [DOI] [PubMed] [Google Scholar]
- 10. Teodoridis J. M., Hardie C., Brown R. (2008) CpG island methylator phenotype (CIMP) in cancer. Causes and implications. Cancer Lett. 268, 177–186 [DOI] [PubMed] [Google Scholar]
- 11. Eads C. A., Nickel A. E., Laird P. W. (2002) Complete genetic suppression of polyp formation and reduction of CpG-island hypermethylation in Apc(Min/+) Dnmt1-hypomorphic Mice. Cancer Res. 62, 1296–1299 [PubMed] [Google Scholar]
- 12. MacLeod A. R., Szyf M. (1995) Expression of antisense to DNA methyltransferase mRNA induces DNA demethylation and inhibits tumorigenesis. J. Biol. Chem. 270, 8037–8043 [DOI] [PubMed] [Google Scholar]
- 13. Ramchandani S., MacLeod A. R., Pinard M., von Hofe E., Szyf M. (1997) Inhibition of tumorigenesis by a cytosine-DNA, methyltransferase, antisense oligodeoxynucleotide. Proc. Natl. Acad. Sci. U.S.A. 94, 684–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ley T. J., Ding L., Walter M. J., McLellan M. D., Lamprecht T., Larson D. E., Kandoth C., Payton J. E., Baty J., Welch J., Harris C. C., Lichti C. F., Townsend R. R., Fulton R. S., Dooling D. J., Koboldt D. C., Schmidt H., Zhang Q., Osborne J. R., Lin L., O'Laughlin M., McMichael J. F., Delehaunty K. D., McGrath S. D., Fulton L. A., Magrini V. J., Vickery T. L., Hundal J., Cook L. L., Conyers J. J., Swift G. W., Reed J. P., Alldredge P. A., Wylie T., Walker J., Kalicki J., Watson M. A., Heath S., Shannon W. D., Varghese N., Nagarajan R., Westervelt P., Tomasson M. H., Link D. C., Graubert T. A., DiPersio J. F., Mardis E. R., Wilson R. K. (2010) DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoo C. B., Jones P. A. (2006) Epigenetic therapy of cancer. Past, present, and future. Nat. Rev. Drug Discov. 5, 37–50 [DOI] [PubMed] [Google Scholar]
- 16. Stresemann C., Lyko F. (2008) Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 123, 8–13 [DOI] [PubMed] [Google Scholar]
- 17. Taberlay P. C., Jones P. A. (2011) DNA methylation and cancer. Prog. Drug. Res. 67, 1–23 [DOI] [PubMed] [Google Scholar]
- 18. Rountree M. R., Bachman K. E., Baylin S. B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277 [DOI] [PubMed] [Google Scholar]
- 19. Chuang L. S., Ian H. I., Koh T. W., Ng H. H., Xu G., Li B. F. (1997) Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277, 1996–2000 [DOI] [PubMed] [Google Scholar]
- 20. Leonhardt H., Page A. W., Weier H. U., Bestor T. H. (1992) A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71, 865–873 [DOI] [PubMed] [Google Scholar]
- 21. Bestor T. H. (1992) Activation of mammalian DNA methyltransferase by cleavage of a zinc binding regulatory domain. EMBO J. 11, 2611–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Callebaut I., Courvalin J. C., Mornon J. P. (1999) The BAH (bromo-adjacent homology) domain. A link between DNA methylation, replication and transcriptional regulation. FEBS Lett. 446, 189–193 [DOI] [PubMed] [Google Scholar]
- 23. Song J., Rechkoblit O., Bestor T. H., Patel D. J. (2011) Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Syeda F., Fagan R. L., Wean M., Avvakumov G. V., Walker J. R., Xue S., Dhe-Paganon S., Brenner C. (2011) The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J. Biol. Chem. 286, 15344–15351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takeshita K., Suetake I., Yamashita E., Suga M., Narita H., Nakagawa A., Tajima S. (2011) Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc. Natl. Acad. Sci. U.S.A. 108, 9055–9059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Song J., Teplova M., Ishibe-Murakami S., Patel D. J. (2012) Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 335, 709–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Z., Fisher R. A. (2004) RGS6 interacts with DMAP1 and DNMT1 and inhibits DMAP1 transcriptional repressor activity. J. Biol. Chem. 279, 14120–14128 [DOI] [PubMed] [Google Scholar]
- 28. Niesen F. H., Berglund H., Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 [DOI] [PubMed] [Google Scholar]
- 29. Wang J., Duncan D., Shi Z., Zhang B. (2013) WEB-based GEne SeT AnaLysis Toolkit (WebGestalt). Update 2013. Nucleic Acids Res. 41, W77–W83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang B., Kirov S., Snoddy J. (2005) WebGestalt. An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33, W741--W748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Datta J., Ghoshal K., Denny W. A., Gamage S. A., Brooke D. G., Phiasivongsa P., Redkar S., Jacob S. T. (2009) A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 69, 4277–4285 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Gros C., Fahy J., Halby L., Dufau I., Erdmann A., Gregoire J. M., Ausseil F., Vispé S., Arimondo P. B. (2012) DNA methylation inhibitors in cancer. Recent and future approaches. Biochimie 94, 2280–2296 [DOI] [PubMed] [Google Scholar]
- 33. Wood R. J., McKelvie J. C., Maynard-Smith M. D., Roach P. L. (2010) A real-time assay for CpG-specific cytosine-C5 methyltransferase activity. Nucleic Acids Res. 38, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gowher H., Jeltsch A. (2001) Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse. The enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 309, 1201–1208 [DOI] [PubMed] [Google Scholar]
- 35. Aoki A., Suetake I., Miyagawa J., Fujio T., Chijiwa T., Sasaki H., Tajima S. (2001) Enzymatic properties of de novo-type mouse DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 29, 3506–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rao C. N., Segawa T., Navari J. R., Xu L., Srivastava S., Moul J. W., Phillips B. (2003) Methylation of TFPI-2 gene is not the sole cause of its silencing. Int. J. Oncol. 22, 843–848 [PubMed] [Google Scholar]
- 37. Segura-Pacheco B., Trejo-Becerril C., Perez-Cardenas E., Taja-Chayeb L., Mariscal I., Chavez A., Acuña C., Salazar A. M., Lizano M., Dueñas-Gonzalez A. (2003) Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin. Cancer Res. 9, 1596–1603 [PubMed] [Google Scholar]
- 38. Ostrow K. L., Park H. L., Hoque M. O., Kim M. S., Liu J., Argani P., Westra W., Van Criekinge W., Sidransky D. (2009) Pharmacologic unmasking of epigenetically silenced genes in breast cancer. Clin. Cancer Res. 15, 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paluszczak J., Krajka-Kúlzniak V., Baer-Dubowska W. (2010) The effect of dietary polyphenols on the epigenetic regulation of gene expression in MCF7 breast cancer cells. Toxicol. Lett. 192, 119–125 [DOI] [PubMed] [Google Scholar]
- 40. Place T. L., Fitzgerald M. P., Venkataraman S., Vorrink S. U., Case A. J., Teoh M. L., Domann F. E. (2011) Aberrant promoter CpG methylation is a mechanism for impaired PHD3 expression in a diverse set of malignant cells. PLoS ONE 6, e14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tao Y., Liu S., Briones V., Geiman T. M., Muegge K. (2011) Treatment of breast cancer cells with DNA demethylating agents leads to a release of Pol II stalling at genes with DNA-hypermethylated regions upstream of TSS. Nucleic Acids Res. 39, 9508–9520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohno M., Ikenaka Y., Ishizuka M. (2012) Sudan III dye strongly induces CYP1A1 mRNA expression in HepG2 cells. J. Biochem. Mol. Toxicol. 26, 16–22 [DOI] [PubMed] [Google Scholar]
- 43. Oleaga C., García M., Solé A., Ciudad C. J., Izquierdo-Pulido M., Noé V. (2012) CYP1A1 is overexpressed upon incubation of breast cancer cells with a polyphenolic cocoa extract. Eur. J. Nutr. 51, 465–476 [DOI] [PubMed] [Google Scholar]
- 44. Norwood L. E., Grade S. K., Cryderman D. E., Hines K. A., Furiasse N., Toro R., Li Y., Dhasarathy A., Kladde M. P., Hendrix M. J., Kirschmann D. A., Wallrath L. L. (2004) Conserved properties of HP1(Hsα). Gene 336, 37–46 [DOI] [PubMed] [Google Scholar]
- 45. Azad N., Zahnow C. A., Rudin C. M., Baylin S. B. (2013) The future of epigenetic therapy in solid tumours-lessons from the past. Nat. Rev. Clin. Oncol. 10, 256–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karahoca M., Momparler R. L. (2013) Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin. Epigenetics 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robertson K. D. (2001) DNA methylation, methyltransferases, and cancer. Oncogene 20, 3139–3155 [DOI] [PubMed] [Google Scholar]
- 48. Khan S. I., Aumsuwan P., Khan I. A., Walker L. A., Dasmahapatra A. K. (2012) Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem. Res. Toxicol. 25, 61–73 [DOI] [PubMed] [Google Scholar]
- 49. Tanaka T. (1997) Reproductive and neurobehavioural effects of lac dye administered in the diet to mice. Food Addit. Contam. 14, 373–380 [DOI] [PubMed] [Google Scholar]
- 50. Ceccaldi A., Rajavelu A., Champion C., Rampon C., Jurkowska R., Jankevicius G., Sénamaud-Beaufort C., Ponger L., Gagey N., Ali H. D., Tost J., Vriz S., Ros S., Dauzonne D., Jeltsch A., Guianvarc'h D., Arimondo P. B. (2011) C5-DNA methyltransferase inhibitors. From screening to effects on zebrafish embryo development. Chembiochem. 12, 1337–1345 [DOI] [PubMed] [Google Scholar]
- 51. Jurkowska R. Z., Jurkowski T. P., Jeltsch A. (2011) Structure and function of mammalian DNA methyltransferases. Chembiochem 12, 206–222 [DOI] [PubMed] [Google Scholar]
- 52. Vilkaitis G., Suetake I., Klimasauskas S., Tajima S. (2005) Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J. Biol. Chem. 280, 64–72 [DOI] [PubMed] [Google Scholar]
- 53. Goyal R., Reinhardt R., Jeltsch A. (2006) Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res. 34, 1182–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mueller E., Sarraf P., Tontonoz P., Evans R. M., Martin K. J., Zhang M., Fletcher C., Singer S., Spiegelman B. M. (1998) Terminal differentiation of human breast cancer through PPAR γ. Mol. Cell 1, 465–470 [DOI] [PubMed] [Google Scholar]
- 55. Subbaramaiah K., Howe L. R., Zhou X. K., Yang P., Hudis C. A., Kopelovich L., Dannenberg A. J. (2012) Pioglitazone, a PPARγ agonist, suppresses CYP19 transcription. evidence for involvement of 15-hydroxyprostaglandin dehydrogenase and BRCA1. Cancer Prev. Res. (Phila) 5, 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56. Furuta J., Nobeyama Y., Umebayashi Y., Otsuka F., Kikuchi K., Ushijima T. (2006) Silencing of peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 66, 6080–6086 [DOI] [PubMed] [Google Scholar]
- 57. Rivenbark A. G., Jones W. D., Risher J. D., Coleman W. B. (2006) DNA methylation-dependent epigenetic regulation of gene expression in MCF-7 breast cancer cells. Epigenetics 1, 32–44 [DOI] [PubMed] [Google Scholar]
- 58. Wiechec E., Overgaard J., Hansen L. L. (2008) A fragile site within the HPC1 region at 1q25.3 affecting RGS16, RGSL1, and RGSL2 in human breast carcinomas. Genes Chromosomes Cancer 47, 766–780 [DOI] [PubMed] [Google Scholar]
- 59. Huang J., Stewart A., Maity B., Hagen J., Fagan R. L., Yang J., Quelle D. E., Brenner C., Fisher R. A. (2013) RGS6 supresses Ras-induced cellular transformation by facilitating Tip60-mediated Dnmt1 degradation and promoting apoptosis. Oncogene, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.