

Background: Kruppel-like factor 4 (KLF4) is a zinc finger transcription factor that influences immunity.
Results: KLF4 binds to CACCC sequences in the IL-6 promoter, directly activates transcription, and impacts promoter acetylation.
Conclusion: KLF4 modulates IL-6 production by dendritic cells via both a direct transcriptional activation and epigenetic modification of the promoter.
Significance: KLF4 plays a significant and previously unrecognized role in regulation of IL-6.
Keywords: Cytokine, Dendritic Cells, Inflammation, Kruppel-like factor (KLF), Monocytes, IL-6, KLF4
Abstract
The initiation and maintenance of the immune response require a coordinated regulation of signal transduction pathways. Identifying the mechanisms by which these pathways are controlled and modulated is a significant goal of immunology. In the present report, we show a novel role for the zinc finger transcription factor Kruppel-like factor 4 (KLF4) in the modulation of the inflammatory immune response via its regulation of IL-6. We analyzed the role of KLF4 in the production of IL-6 by dendritic cells. Our data indicate that KLF4 can act in a dual function manner. It acts as a transcription factor in that it can bind to and activate the IL-6 promoter at specific binding sites. KLF4 also has a role in the chromatin remodeling of the IL-6 promoter in that cells deficient in KLF4 exhibited a relative hypoacetylation. These results indicate a molecular role for KLF4 in modulating the intensity of the inflammatory response and help to explain its pleiotropic role in different settings.
Introduction
Kruppel-like factor 4 (KLF4)2 is a member of the large Kruppel-like factor family, whose members are zinc finger transcription factors defined by having a composition of three Cys2/His2 zinc fingers that contain conserved residues. They are known to play significant roles in stem cell function, cell survival, proliferation, and differentiation (1–6). KLF4 regulates promoter activation via binding to its canonical site 5′-(G/A)(G/A)GG(C/T)G(C/T)-3′ (7) as well as to the CACCC motif (8). Depending on the setting, it has been shown to either activate or repress promoter activation. It may also act as part of a complex and in a context-related manner, can cooperate with, or compete with other factors as well (7).
KLF4 has been shown to significantly influence the immune system through different avenues. Previous work on this transcription factor has demonstrated its roles in both the development of inflammatory monocytes (1, 3) and the production of inflammatory cytokines from those monocytes that do develop (9). Alder et al. showed that mice genetically deficient in KLF4 had a notable absence of CD115+/Gr1hi monocytes, indicating a critical role for the factor in the differentiation of this cell type (1). When KLF4 was targeted in already mature cells, its requirement for the inflammatory molecule IFNγ was also demonstrated (9). These results suggest an important role for KLF4 in the development of an inflammatory response. In the present studies, we sought to elucidate the molecular mechanism by which KLF4 might contribute to the inflammatory process. To that end, we assessed its role in the transcriptional regulation and chromatin remodeling of another critical inflammatory molecule, IL-6.
IL-6 is a pleiotropic cytokine that has been studied in the context of a number of autoimmune and inflammatory settings (10–13). The contribution of IL-6 to the pathogenesis of disease has been investigated extensively, and has followed two broad pathways, one of which is the role of IL-6 in inducing and influencing the phenotypes of T cell responses (14) and the second is the T cell-independent effects by which IL-6 secretion leads to the recruitment and activation of other inflammatory cells. Its significance in one model system, experimental autoimmune encephalomyelitis (EAE), was supported by a number of studies. One of the most compelling being that IL-6 knock-out mice were resistant to EAE (11, 15) and had defects in the ability to activate antigen-specific T cells into effector status, despite having apparently normal T cell development (13). The IL-6 promoter contains both canonical KLF4 and CACCC binding sites, which led us to speculate that KLF4 might regulate the transcription of IL-6 and therefore have a downstream effect on production of IL-6. This possibility would indicate a further role for this molecule in the development of autoimmune disease.
The process of transcription requires the presence of at least one activation signal in a receptive environment. In order for a transcription factor to bind to a promoter, the chromatin must be in an unfolded, or relaxed state. In addition to its role as a transcription factor, KLF4 has been reported to function as a modulator of chromatin acetylation, which is one determinant of efficiency of transcription. The importance of histone acetylation in the process of gene activation was first described in 1964 (16), and since that time, numerous studies have expanded on its function and importance. Targets for acetylation include histones, activator proteins, and transcription factors themselves. In general, acetylation of histones is associated with an enhancement of access of transcription factors, leading ultimately to a more active state partly due to a weakened interaction of the histones with the DNA (reviewed recently in Ref. 17). A role for acetylation in the activity of KLF4 has previously been shown to occur via two mechanisms, in that KLF4 itself becomes acetylated by p300, and KLF4 can modulate the acetylation status of histone H4 (18). With these findings in mind, we assessed the role of KLF4 in the acetylation of the IL-6 promoter, and found that KLF4 itself increases the degree of acetylation in the proximal region of the promoter. These findings provide a new mechanism by which the level of expression of IL-6 may be modulated by KLF4.
EXPERIMENTAL PROCEDURES
Antibodies
The following antibodies were used: rabbit polyclonal against NF-κB p65 and pStat3 (Cell Signaling Technology, Danvers, MA) and rabbit polyclonal against Histone H3 (Biolegend, San Diego, CA), for Western blots; goat polyclonal against KLF4 for EMSA (R&D Systems, Minneapolis, MN). For immunoblots, the secondary antibody goat anti-rabbit IgG conjugated with HRP (Millipore, Billerica, MA) was used.
Plasmids
The following plasmids were obtained from Addgene (Cambridge, MA): pMXS-KLF4, and pMXS-gw (19, 20) The plasmid pcDNA-KLF4 was prepared by PCR amplification of the KLF4 cDNA from pMXS-KLF4 and ligation into pcDNA3.1 in a site created by endonuclease digestion with HindIII and EcoRV. The IL6 promoter-reporter plasmid pGL4-IL6 was generated by PCR amplification of the IL6 promoter from mouse genomic DNA using the following primers: IL6-FWD, CGCCTCGAGTGGATGTATGCTCCCGACTT; IL6 reverse, CGCAAGCTTGCTACAGACATCCCCAGTCTC. The resulting fragment was digested with XhoI and HindIII to generate overhangs and ligated into a site created by digestion with XhoI and HindIII. Transcription factor binding site mutations were generated by site-directed mutagenesis using the following primers: ΔNF-κB FWD, CACCCTCCAACAAAGATTTTTATCAAATGTCCCATTTTCCCATGAGTCTCAAAA; ΔNF-κB REV, TTTTGAGACTCATGGGAAAATGGGACATTTGATAAAAATCTTTGTTGGAGGGTG; ΔSTAT3 FWD, CCTTCAAGCCTCCTTGCATGATTTCCAAATGTTTTGGGGTGTCCTG; ΔSTAT3 REV, CAGGACACCCCAAAACATTTGGAAATCATGCAAGGAGGCTTGAAGG.
Cell Culture
The human cell line HEK 293T was cultured in Dulbecco's modified Eagle's Medium supplemented with 10% fetal bovine serum at 37 °C with 5% CO2 atmosphere and 95% humidity.
Mouse bone marrow-derived dendritic cells were generated from wild-type or KLF4−/− chimeric mice (1). The dendritic cells were expanded in complete RPMI supplemented with 10% fetal bovine serum, penicillin, streptomycin, 2 mm l-glutamine, 5 mm HEPES, non-essential amino acids, 1 mm sodium pyruvate, gentamicin, β-mercaptoethanol, and GM-CSF on non-tissue culture treated 100-mm dishes (Fisherbrand) at 37 °C under 5% CO2 and 95% humidity. Fresh medium was added after 3 days of culture and fresh medium was exchanged after 6 days in culture. Cells were typically used on days 7–10 of culture.
Luciferase Assay
293T cells were seeded in a 24-well tissue culture plate and 24 h later transfected in triplicate for each condition using Fugene6 transfection reagent (Promega, Madison, WI) according to manufacturer instructions with 60 ng pGL4 promoter reporter plasmid (Promega), with or without 150 ng of transcription factor plasmid, 1 ng SV40-RL control plasmid, and pMXS-gw empty vector plasmid to bring the total DNA per transfection to 300 ng. Transfected cells were harvested 48 h later. The cells were lysed and the Dual-Luciferase Reporter Assay (Promega) was performed and the resulting luciferase signal quantified on a Fluostar Omega plate reader (BMG Labtech, Ortenberg, Germany). Luminescence was measured for 6 s in 0.5-s intervals for each reaction. Promoter reporter luciferase activity was normalized to control Renilla luciferase activity.
Electrophoretic Mobility Shift Assay
DNA oligonucleotides labeled at the 5′-end with IRDye700 and unlabeled were obtained from Integrated DNA Technologies (Coralville, IA). Double stranded oligonucleotides were prepared according to Odyssey EMSA buffer kit instructions (Li-COR, Lincoln, NE). Transcription factors were expressed in HEK293T and whole cell lysates or nuclear fractions were used for binding experiments. Whole cell lysates were prepared in RIPA buffer supplemented with phosphatase and protease inhibitors and nuclear extracts were prepared as previously described (21).
DNA binding reactions were performed at room temperature using the Li-Cor EMSA Buffer Kit according to kit instructions. 5–10 μg of 293T nuclear extract or KLF4-expressing 293T nuclear extract were added to the binding reactions. Where noted, 1 μl of anti-KLF4 antibody was added to binding reactions to determine specificity of binding. Competition binding assays were performed with unlabeled competing oligos or unlabeled non-competing oligos. The binding reactions were resolved on Bio-Rad mini-protean 5% acrylamide TBE precast gels (Bio-Rad) and visualized using a Li-Cor Odyssey near-infrared imager and Odyssey Infrared Imaging System Software version 3.0.16.
Chromatin Immunoprecipitation
Murine bone marrow-derived dendritic cells were cultured as described above. On day 8, chromatin immunoprecipitation was performed on dendritic cells using a MagnaChIP kit including the anti-acetyl H4-lysine antibody (Millipore, Billerica, MA), following the manufacturer's recommended protocol. The cell lysate was sonicated to shear the crosslinked DNA to 200–1000 base pair fragments (EpiShear, Active Motif, Carlsbad, CA). Following binding to antibody, collection of precipitated samples and elution, free DNA was purified with a spin filter and qPCR performed with primers spanning the region proximal to the start of the IL-6 promoter.
qPCR
For quantification of IL-6 message and for ChIP analysis, qPCR was conducted on a Bio-Rad iCycler, using the SYBR green system. Message was quantified using primers specific for IL-6 and actin as control for normalization, with cDNA as a template. For ChIP studies, the eluted DNA was used as a template, with primers for the proximal promoter region. Results were normalized to input DNA for analysis.
ELISA for Cytokine Production
DCs were cultured at a density of 1e6 cells/ml in DC medium and stimulated with 10 ng/ml LPS, 25 ng/ml LPS, or 10 μg/ml poly I:C. Supernatants were isolated, and cytokine concentrations were determined by ELISA according to the manufacturer's protocol (OptiEIA ELISA, BectonDickinson, San Jose, CA).
RESULTS
Transcription and Secretion of IL-6 Are Suppressed in KLF4−/− DCs
We assessed the level of production of IL-6 by DCs via two different methods to determine whether they were defective in IL-6 production and the mechanism behind any defect. First, we analyzed the production and secretion of IL-6 by ELISA to assess whether the loss of KLF4 affected protein levels. As shown in Fig. 1, the DCs from the KLF4−/− mice had a significant reduction in the amount of IL-6 secreted after stimulation with two different agents: LPS (A) or poly(I:C) (B) (p = .01). To determine whether this decrease of production was regulated at the level of transcription, we next compared the quantities of mRNA transcripts after stimulation with 25 ng/ml LPS. As shown in Fig. 1C, which depicts the relative fold change after stimulation in expression of mRNA for IL-6, the levels of mRNA paralleled the levels of protein, indicating that the deficiency occurred at the level of transcription (p = .039).
FIGURE 1.
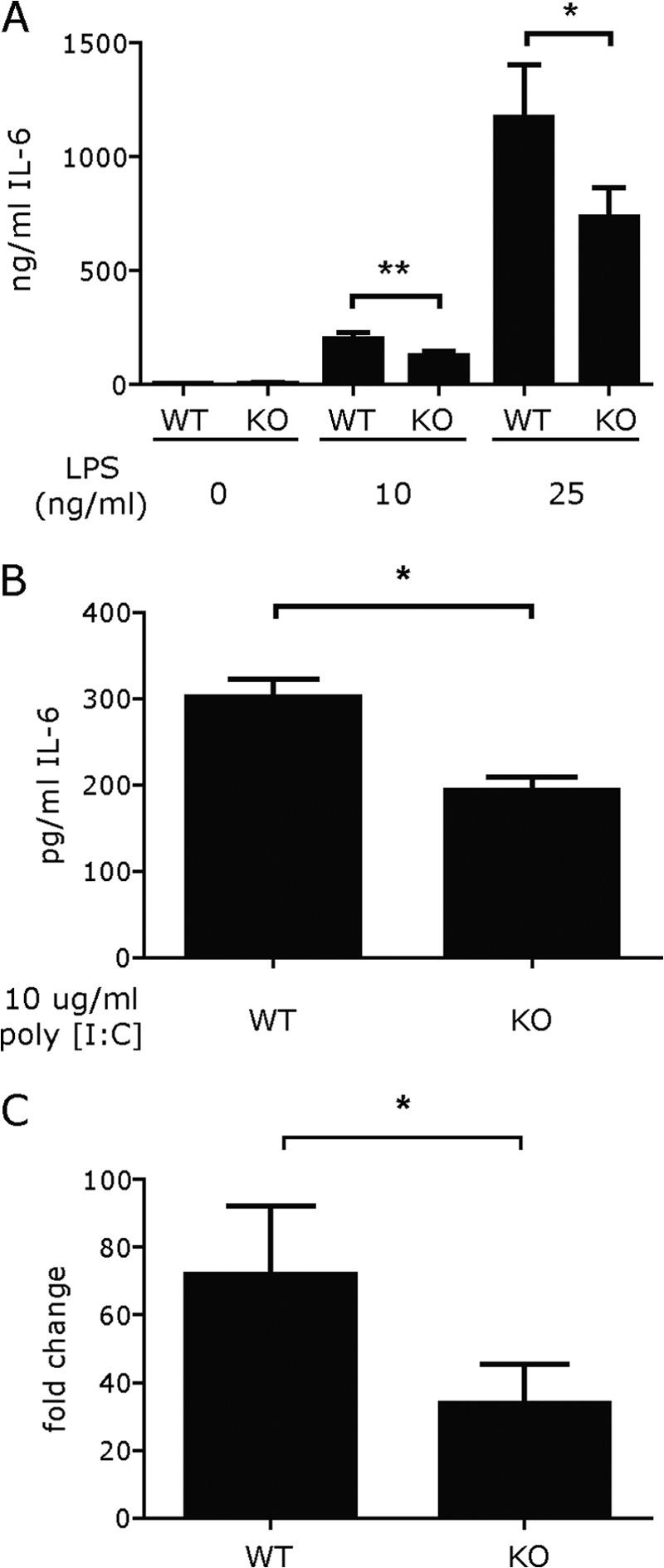
KLF4−/− DCs are deficient in IL-6 secretion and transcription. DCs from wild type and KLF4−/− transplanted mice were stimulated with LPS (10 or 25 ng/ml) overnight for supernatant collection (A), or poly(I:C), 10 μg/ml (B) and for 6 h for RNA collection (C). Supernatants were analyzed for cytokine production by ELISA and cDNA was prepared from the cells to quantify gene expression by qPCR. Bars represent the mean ± S.E. from at least four independent experiments. The production of IL-6 was significantly decreased for both RNA (p = .04) and protein (p = .01 for 10 ng/ml; .03 for 25 ng/ml LPS; p = .04 for poly(I:C).
The IL-6 Promoter Contains KLF4 Binding Sites
The minimal IL-6 promoter spans ∼2kb and contains a total of 2 canonical KLF4 binding sites and 7 CACCC sites as shown in Fig. 2 (each labeled as CAC and a sequential number). Thus, KLF4 could modulate IL-6 promoter activity by directly binding to target sequences within the promoter. As there were multiple sites for which KLF4 had an affinity, we generated nine different constructs; seven with mutated KLF4 sites, one with a mutated Stat3 site, and one with a mutated NF-κB site. The latter two were selected to assess whether KLF4 might function via an indirect activation. The strategy employed to dissect the importance of each potential site is shown as well, with sequences for each mutation that was analyzed.
FIGURE 2.
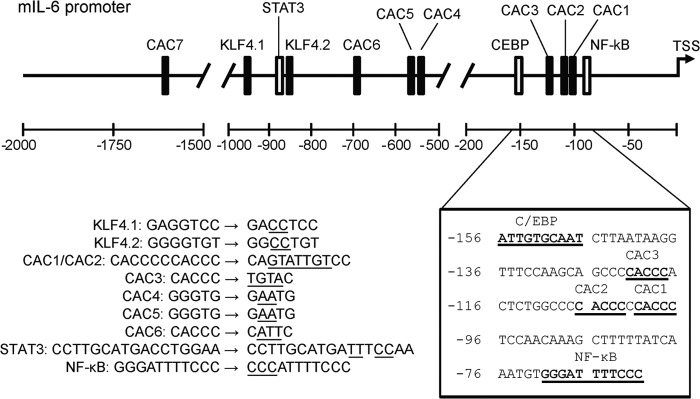
Illustration of relative positions of selected transcription factor binding sites in the mouse IL6 promoter. A sequence analysis of 2000 bp proximal to the IL6 transcriptional start site revealed two putative KLF4 binding sites, KLF4.1 and KLF4.2, based on the consensus binding sequence RRGGYGY and seven binding sites based on the CACCC motif (labeled CAC1-CAC7). The expanded sequence in the lower right shows the NF-κB - C/EBP activation complex positioned at −60 to −155 bp relative to the transcription start site with three CACCC binding motifs located within the complex. The more distal STAT3 response element and KLF4 consensus sites are clustered from −980 to −868 bp while the remaining CACCC sites are more widely distributed. The transcription factor binding sites that were mutated for this study are listed in the lower left. The underlined mutations were introduced by site-directed mutagenesis and the mutant promoter sequences analyzed by luciferase assay for KLF4 responsiveness.
KLF4 Activates the IL-6 Promoter by Binding to Specific Proximal CACCC Sites
We next sought to assess whether KLF4 activated the IL-6 promoter at a particular site or sites. Toward this end, we conducted a systematic promoter mutation analysis in which we point mutated each potential binding site and evaluated the impact on promoter activation of each individual mutation by a luciferase reporter assay. Proximal to the start site is a cluster of potential binding sites, which we initially targeted. Fig. 3A shows the resulting impact on luciferase expression of mutation of these proximal sites. As shown, the mutation of the three proximal CACCC sites led to a notable decrease in the activation of the promoter by KLF4, indicating that these were both active and important binding sites for KLF4. In contrast, mutation of the more distal CACCC sites did not result in a significant decrease in the activation of the promoter by KLF4, indicating that they were less critical sites for binding (Fig. 3B). The two canonical sequences for KLF4 binding are also located distal to the start site, and interestingly, mutation of those sites did not lead to a decrease in KLF4-mediated activation, (Fig. 3C), indicating that the proximal CACCC sequences were likely the sites at which KLF4 acts in this setting. Because of the critical nature of the CACCC sites in these results, we next sought to verify that KLF4 bound to this sequence. For these studies, we conducted electrophoretic mobility shift assays (EMSA) with oligos that contained either the two most proximal CACCC sites (which were not analyzed separately due to their proximity to each other) or the third proximal CACCC site. In the EMSA studies, we analyzed the specificity of the binding by determining whether the KLF4 protein binds to the target sequence, and then whether the addition of an antibody to the protein leads to a shift in the location of the band, supporting the specificity of the interaction. As shown in Fig. 3D, KLF4 binds to the oligonucleotide spanning the two most proximal CACCC sites, and the addition of antibody against KLF4, led to a shift of the band. Fig. 3E shows the results of the EMSA conducted with the KLF4 protein and an oligo spanning the third proximal CACCC site. As demonstrated, KLF4 also binds to the third CACCC sequence and is specific, as shown by the shift of the band after the addition of antibody to KLF4. Taken together, these results indicate that the activation of the IL-6 promoter by KLF4 results from the binding to the proximal CACCC sites rather than to the canonical KLF4 sequences, located distal to the promoter.
FIGURE 3.

KLF4 activates the IL-6 promoter via proximal CACCC sites. HEK293T cells were co-transfected in triplicate with the indicated IL-6 promoter-luciferase plasmids, pMXS-KLF4 expression plasmid or the pMXS-GW empty vector, and a SV40 promoter-Renilla luciferase control plasmid. Cells were lysed 48 h after transfection and promoter activity was measured by the Dual-Luciferase Assay. Mutation of CACCC sites proximal to the transcription start site of the IL-6 promoter reduced activation of the IL-6 promoter by KLF4 (A) while mutation of distal CACCC sites did not affect KLF4 activation (B). Mutation of the consensus KLF4 binding sites did not impair IL6 activation (C). The reporter luciferase activity was normalized to Renilla control in triplicate for each experiment. The data bars represent the mean of at least three independent experiments ± S.E. Electrophoretic mobility shift assays were performed with nuclear extracts of 293T cells or 293T cells transfected with a KLF4 expression plasmid and IRDye700-labeled DNA oligos containing CACCC sequences from the IL6 promoter at positions −121 (D) and −101 and −107 (E). Addition of an anti-KLF4 antibody generated a supershift in the presence of the KLF4 nuclear extract but not in the control extract. *, p < 0.05; **, p < 0.01; n.s., no significance (A); n.s., nonspecific (D and E).
KLF4−/− DCs Do Not Have Defects in Stat3 or NF-κB Signaling, and KLF4 Does Not Activate IL-6 via Their Promoter Consensus Binding Sites
Based on the critical nature of the CACCC binding sites and their proximity to binding sites for NF-κB and Stat3, which are known to be important regulators of IL-6 production, we next assessed whether KLF4 mediated its effects via either binding to their response elements or regulating their expression. As shown in Fig. 4A, mutation of either the NF-κB or the Stat3 response element did not affect the activation of the IL-6 promoter by KLF4. In addition, the absence of KLF4 did not decrease LPS-mediated translocation of p65 to the nuclei in DCs or phosphorylation of Stat3 (Fig. 4B), indicating that these pathways remained intact in KLF4−/− cells. These two pieces of data together indicate that KLF4 does not suppress either Stat3 or NF-κB activation nor does it bind to the response elements for these other two critical transcription factors.
FIGURE 4.
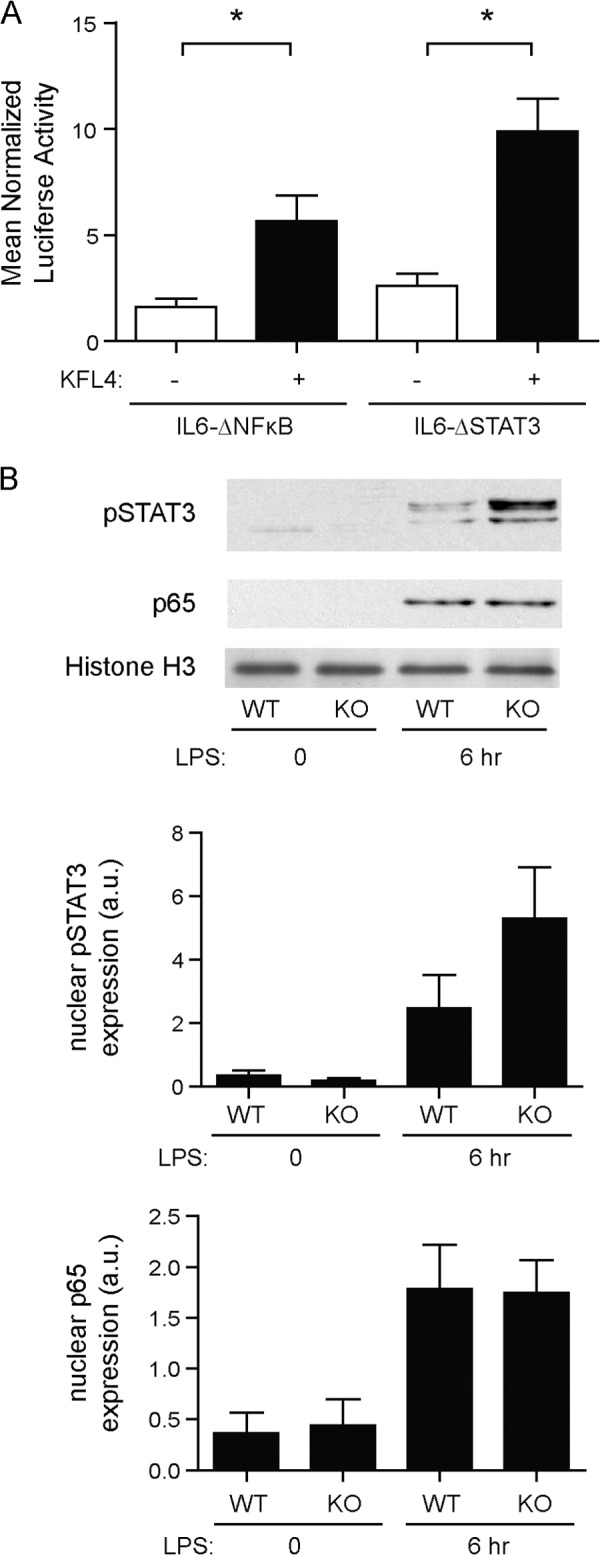
KLF4-induced activation of IL-6 is not mediated through NF-κB or STAT3. Plasmids containing the IL-6 promoter with mutated binding sites for NF-κB and STAT3 were co-transfected into HEK293T cells with an SV40 promoter-Renilla luciferase control plasmid and pMXS-KLF4 expression plasmid or pMXS-GW empty vector (A). Luciferase readings were performed in triplicate for each experiment. Data bars represent the mean ± S.E. (n > 3; *, p < 0.05). Protein expression of phospho-STAT3 was monitored by Western blot analysis of nuclear fractions from cultured BMDCs that were untreated or stimulated with LPS for 6 h (B). Western blot data were quantified by densitometric analysis and protein expression was normalized to histone H3 levels. Data bars represent mean ± S.E. of four independent experiments.
The IL-6 Promoter Is Hypoacetylated at the Proximal Region in KLF4−/− DCs
KLF4 has been reported to have dual functions in that it acts both as a specific transcription factor and also as an epigenetic modifier. We sought to determine whether KLF4 might affect the efficiency of transcription of IL-6 via its modification of chromatin structure. Toward this end, we analyzed the acetylation status of the IL-6 promoter in wild type and KLF4-deficient DCs. For these studies, BMDCs were generated, and subjected to chromatin immunoprecipitation analysis, using an antibody specific for lysine on histone H4. We then conducted qPCR on the immunoprecipitated DNA using primers targeting the IL-6 promoter at a region proximal to the transcriptional start site. As shown in Fig. 5, KLF4−/− DCs exhibited a relative hypoacetylation in this region, compared with their wild type counterparts. This result indicates a possible role for KLF4 in chromatin remodeling of promoters, and supports the notion that this mechanism may contribute to observed modulation of expression of multiple cytokines.
FIGURE 5.

KLF4 KO DCs are relatively deficient in histone acetylation. The relative degrees of histone acetylation in the IL-6 promoter were analyzed by ChIP. Sheared chromatin was prepared from formaldehyde cross-linked BMDCs. DNA from chromatin complexes was immunoprecipitated with an antibody specific for histone H4K27ac then purified and analyzed by qPCR using primers specific for the IL-6 promoter. A shows the relative abundance of acetylated H4 in KLF4−/− DCs compared with wild type controls (mean ± S.E., n = 4; **, p < 0.01).
DISCUSSION
Elucidation of the molecular mechanisms by which cytokine production is regulated is a critical step toward deciphering the immune response. The immune system depends on a tightly controlled and interactive series of events that lead to the generation of cytokines that activate and repress the mounting of a response, whether it creates either a beneficial or pathologic outcome. Each cytokine produced helps to form the nature of the response. IL-6 is a pleiotropic cytokine that has been implicated in a number of autoimmune diseases and whose regulation has not been completely understood. Our studies have furthered the elucidation through the identification of a previously unrecognized mechanism by which KLF4 can modulate the expression of IL-6.
Control of IL-6 expression is multifactorial, and a number of transcription factors have been shown to participate in its regulation. Among these are C/EBP (22), NF-κB (23), and Stat3 (24). Similarly, a number of different stimuli to monocytes and dendritic cells will elicit the production of this cytokine. These two features point to the complexity of the regulation and the potential for a multifaceted process. Our results indeed indicate that KLF4 can modulate the level of expression via both a direct transcriptional activation as well as though chromatin remodeling. The results of our studies showed that both mRNA and protein levels of IL-6 were significantly suppressed but not absent in DCs lacking KLF4. This finding indicated that KLF4 was required for optimal activation of this cytokine.
KLF4 has been reported to have a variety of functions, both in terms of development of specific cell types, and in the nature of the cells, as evidenced by a large number of publications, e.g.:(25–32). Because of its known role as a transcription factor, we sought to systematically determine how it interacted with the IL-6 promoter. The identification of multiple possible binding sites for KLF4 supported the likely possibility that KLF4 could directly modulate activation of the promoter. Our systematic dissection of the different potential binding sites showed that in luciferase reporter assays, KLF4 mediated its primary transcriptional effect through binding to consensus sequences containing the CACCC motif rather than through binding to its canonical sequences. The proximity of the CACCC sites to the start of transcription may influence the relative contributions of the two different types of sequences, in that only the most proximal CACCC sites appeared to be required for efficient activation.
Interestingly, the binding sites for NF-κB and C/EPB, other transcription factors known to contribute to IL-6 signaling, are also located in the same region as the proximal CACCC sites, and the Stat3 response element more distal. We considered the possibility that KLF4 might mediate some of its effects through binding to these other sites, possibly as part of a complex. Mutation of the binding sites for these additional transcription factors led to no significant decrease in the ability of KLF4 to activate the reporter, however, which suggests that KLF4 primarily acts through the proximal CACCC sites. This finding is of significance in that it shows that KLF4 can act on its own, that it is not restricted to binding through its canonical sequences, and that the location of binding sites can dramatically affect its efficiency of transcription.
Initial investigations on KLF4 revealed that it serves a vital physiologic role as mice that were pan-genetically deficient in KLF4 died shortly after birth due to an epithelial defect (4). This result demonstrated the high level of importance of KLF4 in cellular functioning. As described, our and others' works have bypassed this limitation via selective deletion in immune cells via bone marrow chimeras (1). This system has allowed us to query the role of KLF4 in both the development of immune cells and in the production of cytokines by cells that develop in its absence, without affecting the stromal population.
An interesting feature of KLF4 is its pleiotropic nature, as demonstrated by its ability to either activate or repress transcription, depending on the context (9, 18, 31, 33). Our data support a previous contention that this characteristic may be at least partially explained by its multifunctional role as a promoter binding transcription factor and as a chromatin remodeler. During the process of transcription, a number of events must occur. In a resting cell, expression of many genes is limited, and control of the latent state occurs on numerous levels. Activating signals to generate either production or translocation of transcription factors often occurs to initiate the process. In addition, the nucleosome structure itself is maintained in a closed and relatively inactive state for many promoters. Thus, the chromatin structure of the promoter often needs to be modified to generate a more permissive state. Acetylation of promoters at lysine residues is one well recognized method by which this remodeling process can occur. KLF4 has previously been shown to participate in acetylation of promoters and interestingly is a target for acetylation itself. Results showed that KLF4 was a target for acetylation by p300, and that this process regulated its transcactivation potential. Interestingly, while acetylation was required for activation, it was not required for repression (18). These results suggested a possible explanation for KLF4's diverse roles in that in order for KLF4 to activate transcription, it requires recruitment of a cofactor and histone acetylation. In the absence of an appropriate cofactor/acetylation status, repression may ensue.
Our results are consistent with this model and are of significance in that the finding that KLF4 modulates the level of expression of IL-6 via both direct promoter activation and acetylation shows that optimal expression levels of critical cytokines may depend on multiple transcription factors and on chromatin remodeling as well as promoter activation by these factors. These results indicate the importance of considering the milieu in which KLF4 is expressed and may help to explain how it can exert its varied effects. This requirement for a permissive and collaborative environment both highlights the importance of understanding the full context in which KLF4 is expressed and the considerations in manipulations of its expression.
This work was supported in part by grants from the National Multiple Sclerosis Society, R01 NS41435, and the Silverman Foundation.
- KLF4
- Kruppel-like factor 4
- EAE
- experimental autoimmune encephalomyelitis
- DC
- dendritic cell
- BMDC
- bone marrow dendritic cell
- EMSA
- electromobility shift assay.
REFERENCES
- 1. Alder J. K., Georgantas R. W., 3rd, Hildreth R. L., Kaplan I. M., Morisot S., Yu X., McDevitt M., Civin C. I. (2008) Kruppel-like factor 4 is essential for inflammatory monocyte differentiation in vivo. J. Immunol. 180, 5645–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buckley A. F., Kuo C. T., Leiden J. M. (2001) Transcription factor LKLF is sufficient to program T cell quiescence via a c-Myc-dependent pathway. Nature Immunol. 2, 698–704 [DOI] [PubMed] [Google Scholar]
- 3. Feinberg M. W., Wara A. K., Cao Z., Lebedeva M. A., Rosenbauer F., Iwasaki H., Hirai H., Katz J. P., Haspel R. L., Gray S., Akashi K., Segre J., Kaestner K. H., Tenen D. G., Jain M. K. (2007) The Kruppel-like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J. 26, 4138–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garrett-Sinha L. A., Eberspaecher H., Seldin M. F., de Crombrugghe B. (1996) A gene for a novel zinc-finger protein expressed in differentiated epithelial cells and transiently in certain mesenchymal cell. J. Biol. Chem. 271, 31384–31390 [DOI] [PubMed] [Google Scholar]
- 5. Kuo C. T., Veselits M. L., Barton K. P., Lu M. M., Clendenin C., Leiden J. M. (1997) The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 11, 2996–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shields J. M., Christy R. J., Yang V. W. (1996) Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J. Biol. Chem. 271, 20009–20017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shields J. M., Yang V. W. (1998) Identification of the DNA sequence that interacts with the gut-enriched Kruppel-like factor. Nucleic Acids Res. 26, 796–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miller I. J., Bieker J. J. (1993) A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol. Cell. Biol. 13, 2776–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feinberg M. W., Cao Z., Wara A. K., Lebedeva M. A., Senbanerjee S., Jain M. K. (2005) Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J. Biol. Chem. 280, 38247–38258 [DOI] [PubMed] [Google Scholar]
- 10. Eriksson U., Kurrer M. O., Schmitz N., Marsch S. C., Fontana A., Eugster H. P., Kopf M. (2003) Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation 107, 320–325 [DOI] [PubMed] [Google Scholar]
- 11. Eugster H. P., Frei K., Kopf M., Lassmann H., Fontana A. (1998) IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur. J. Immunol. 28, 2178–2187 [DOI] [PubMed] [Google Scholar]
- 12. Gijbels K., Brocke S., Abrams J. S., Steinman L. (1995) Administration of neutralizing antibodies to interleukin-6 (IL-6) reduces experimental autoimmune encephalomyelitis and is associated with elevated levels of IL-6 bioactivity in central nervous system and circulation. Mol. Med. 1, 795–805 [PMC free article] [PubMed] [Google Scholar]
- 13. Samoilova E. B., Horton J. L., Hilliard B., Liu T. S., Chen Y. (1998) IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J. Immunol. 161, 6480–6486 [PubMed] [Google Scholar]
- 14. Serada S., Fujimoto M., Mihara M., Koike N., Ohsugi Y., Nomura S., Yoshida H., Nishikawa T., Terabe F., Ohkawara T., Takahashi T., Ripley B., Kimura A., Kishimoto T., Naka T. (2008) IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A. 105, 9041–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okuda Y., Sakoda S., Bernard C. C., Fujimura H., Saeki Y., Kishimoto T., Yanagihara T. (1998) IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 10, 703–708 [DOI] [PubMed] [Google Scholar]
- 16. Allfrey V. G., Faulkner R., Mirsky A. E. (1964) Acetylation and Methylation of Histones and Their Possible Role in the Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. U. S. A. 51, 786–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Musselman C. A., Lalonde M. E., Cote J., Kutateladze T. G. Perceiving the epigenetic landscape through histone readers. Nature Struct. Mol. Biol. 19, 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Evans P. M., Zhang W., Chen X., Yang J., Bhakat K. K., Liu C. (2007) Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J. Biol. Chem. 282, 33994–34002 [DOI] [PubMed] [Google Scholar]
- 19. Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. (2003) Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 31, 1007–1014 [PubMed] [Google Scholar]
- 20. Takahashi K., Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- 21. Oelgeschläger M., Nuchprayoon I., Lüscher B., Friedman A. D. (1996) C/EBP, c-Myb, and PU. 1 cooperate to regulate the neutrophil elastase promoter. Mol. Cell. Biol. 16, 4717–4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu H. M., Tian Q., Baer M., Spooner C. J., Williams S. C., Johnson P. F., Schwartz R. C. (2000) The C/EBP bZIP domain can mediate lipopolysaccharide induction of the proinflammatory cytokines interleukin-6 and monocyte chemoattractant protein-1. J. Biol. Chem. 275, 16373–16381 [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y., Broser M., Rom W. N. (1994) Activation of the interleukin 6 gene by Mycobacterium tuberculosis or lipopolysaccharide is mediated by nuclear factors NF-IL6 and NF-κB. Proc. Natl. Acad. Sci. U. S. A. 91, 2225–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park S. J., Nakagawa T., Kitamura H., Atsumi T., Kamon H., Sawa S., Kamimura D., Ueda N., Iwakura Y., Ishihara K., Murakami M., Hirano T. (2004) IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 173, 3844–3854 [DOI] [PubMed] [Google Scholar]
- 25. Kharas M. G., Yusuf I., Scarfone V. M., Yang V. W., Segre J. A., Huettner C. S., Fruman D. A. (2007) KLF4 suppresses transformation of pre-B cells by ABL oncogenes. Blood 109, 747–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patel N. V., Ghaleb A. M., Nandan M. O., Yang V. W. Expression of the tumor suppressor Kruppel-like factor 4 as a prognostic predictor for colon cancer. Cancer Epidemiol. Biomarkers Prev. 19, 2631–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yusuf I., Kharas M. G., Chen J., Peralta R. Q., Maruniak A., Sareen P., Yang V. W., Kaestner K. H., Fruman D. A. (2008) KLF4 is a FOXO target gene that suppresses B cell proliferation. Int. Immunol. 20, 671–681 [DOI] [PubMed] [Google Scholar]
- 28. Chen X., Johns D. C., Geiman D. E., Marban E., Dang D. T., Hamlin G., Sun R., Yang V. W. (2001) Kruppel-like factor 4 (gut-enriched Kruppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J. Biol. Chem. 276, 30423–30428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Z. Y., Wang X., Zhou Y., Offner G., Tseng C. C. (2005) Destabilization of Kruppel-like factor 4 protein in response to serum stimulation involves the ubiquitin-proteasome pathway. Cancer Res. 65, 10394–10400 [DOI] [PubMed] [Google Scholar]
- 30. Katz J. P., Perreault N., Goldstein B. G., Actman L., McNally S. R., Silberg D. G., Furth E. E., Kaestner K. H. (2005) Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology 128, 935–945 [DOI] [PubMed] [Google Scholar]
- 31. Rowland B. D., Peeper D. S. (2006) KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 6, 11–23 [DOI] [PubMed] [Google Scholar]
- 32. Shie J. L., Chen Z. Y., Fu M., Pestell R. G., Tseng C. C. (2000) Gut-enriched Kruppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res. 28, 2969–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rowland B. D., Bernards R., Peeper D. S. (2005) The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nature Cell Biol. 7, 1074–1082 [DOI] [PubMed] [Google Scholar]