

Background: A hypoxia-inducible transcription factor (Hif2α) mediates induction of intestinal iron and copper transporters during iron deficiency.
Results: Specificity factor 1 (Sp1) is required for transcriptional induction of an intestinal copper transporter (Atp7a) by Hif2α.
Conclusion: Sp1 and Hif2α may synergistically mediate the genetic response to iron deficiency.
Significance: Understanding molecular mechanisms governing iron absorption may allow modulation of this process during disease states.
Keywords: Chromatin Immunoprecipitation (ChIP), Hypoxia-inducible Factor (HIF), Intestine, Iron, Transcription Coactivators, IEC-6 Cells, Cobalt Chloride
Abstract
Genes with G/C-rich promoters were up-regulated in the duodenal epithelium of iron-deficient rats including those encoding iron (e.g. Dmt1 and Dcytb) and copper (e.g. Atp7a and Mt1) metabolism-related proteins. It was shown previously that an intestinal copper transporter (Atp7a) was co-regulated with iron transport-related genes by a hypoxia-inducible transcription factor, Hif2α. In the current study, we sought to test the role of Sp1 in transcriptional regulation of Atp7a expression during iron deprivation/hypoxia. Initial studies in IEC-6 cells showed that mithramycin, an Sp1 inhibitor, reduced expression of Atp7a and iron transport-related genes (Dmt1, Dcytb, and Fpn1) and blocked their induction by CoCl2, a hypoxia mimetic. Consistent with this, overexpression of Sp1 increased endogenous Atp7a mRNA and protein expression and stimulated Atp7a, Dmt1, and Dcytb promoter activity. Site-directed mutagenesis and functional analysis of a basal Atp7a promoter construct revealed four functional Sp1 binding sites that were necessary for Hif2α-mediated induction of promoter activity. Furthermore, chromatin immunoprecipitation (ChIP) assays confirmed that Sp1 specifically interacts with the Atp7a promoter in IEC-6 cells and in rat duodenal enterocytes. This investigation has thus revealed a novel aspect of Atp7a gene regulation in which Sp1 may be necessary for the HIF-mediated induction of gene transcription during iron deficiency/hypoxia. Understanding regulation of Atp7a expression may help further clarify the physiological role of copper in the maintenance of iron homeostasis. Furthermore, this Sp1/Hif2α regulatory mechanism may have broader implications for understanding the genetic response of the intestinal epithelium to maintain whole-body iron homeostasis during states of deficiency.
Introduction
Iron is essential for life as it plays critical roles in biological systems including those related to mitochondrial electron transport and energy production, enzyme activity, oxygen transport, and regulation of gene expression (1). Systemic iron levels are maintained by intestinal absorption, which is precisely controlled as there is no active excretory mechanism in mammals. Iron absorption is enhanced during iron deprivation as reflected by increased expression of iron transport-related genes including divalent metal transporter 1 (Dmt12; an iron importer), duodenal cytochrome b (Dcytb; a brush-border membrane ferrireductase), and ferroportin 1 (Fpn1; an iron exporter) in duodenal enterocytes (2). Studies also found that the Menkes copper-transporting ATPase (Atp7a), an enterocyte copper exporter, was up-regulated in the rat duodenal epithelium during iron deficiency, consistent with noted increases in copper content of the intestinal mucosa, liver, and serum (2, 3). Similar perturbations in tissue copper levels have been noted in other mammalian species during states of iron deficiency (4–6). It has thus been hypothesized that copper plays a role in the maintenance of iron homeostasis (7). Importantly, two multicopper ferroxidases, one expressed in enterocytes of the small intestine (hephaestin) and one produced in liver and secreted into the blood (ceruloplasmin), provide key links between iron and copper homeostasis (8).
Depletion of body iron stores leads to decreased red blood cell hemoglobin levels causing tissue hypoxia. Low tissue oxygen tension in turn results in stabilization of trans-acting hypoxia-inducible factors (HIFs). The HIFs function as heterodimers, containing a constitutively expressed β subunit and a hypoxia-responsive α subunit (one of three known Hifα subunits). The increase in intestinal iron absorption when body iron stores are depleted has in fact been shown to be mediated via activation of Hif2α. This regulatory mechanism was revealed by two recent studies in which the α subunits (Hif1α and Hif2α) of the functional HIF protein complexes were specifically inactivated in the intestinal epithelium of mice (9, 10). Results showed that regulation of iron absorption was defective in mice lacking intestinal Hif2α, whereas induction of iron absorption during iron deprivation was maintained in mice lacking Hif1α. It was further shown that the Dmt1, Dcytb, and Fpn1 promoters contained functional hypoxia-responsive elements (HREs) that specifically interacted with Hif2α, explaining their induction during iron deficiency (and tissue hypoxia) (9–11). Hif2α is thus critical to maintain intestinal iron homeostasis in mice.
Interestingly, our previous studies in iron-deficient rats showed that Atp7a was up-regulated in the duodenal epithelium similarly to Dmt1, Dcytb, and Fpn1 (2), and we thus hypothesized that Atp7a was coordinately regulated with these iron transport-related genes. Subsequently, it was demonstrated that the Atp7a promoter was indeed a direct Hif2α target in rat intestinal epithelial (IEC-6) cells (12). Furthermore, in a previous investigation, it was noted that promoters of genes induced in the duodenal epithelium of iron-deficient rats contained a statistical overrepresentation of G/C-rich sequences (13). It was also shown that an abundance of genes up-regulated in differentiated Caco-2 cells (human intestinal adenocarcinoma cells) in response to iron chelation contained G/C-rich promoter sequences as well as putative HREs (14). Importantly, many of the iron and copper homeostasis-related genes induced in both models of intestinal iron transport contained G/C-rich promoters and putative HREs. These observations led us to hypothesize that a G/C-binding protein (e.g. specificity factor 1 (Sp1) or a related trans-acting factor) was important for the transcriptional response of the intestinal epithelium to iron deprivation (14). To test this hypothesis, in the current investigation, we performed a series of experiments to determine whether Sp1 is important for the Hif2α-mediated transactivation of Atp7a gene expression using an in vitro model of the intestinal epithelium (IEC-6 cells) and iron-deprived rats. Results of this investigation showed that Sp1 specifically interacts with cis-elements in the Atp7a promoter and furthermore that Sp1 binding is necessary for Hif2α-mediated induction of Atp7a transcription during hypoxia.
EXPERIMENTAL PROCEDURES
Cell Culture
Rat intestinal epithelial (IEC-6) cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured as described previously (12, 15). For hypoxia experiments, IEC-6 cells at ∼85% confluence were cultured in a hypoxia chamber with 1% O2 and 5% CO2 (with the balance being nitrogen). To mimic hypoxia, 200 μm CoCl2 was added to the culture medium when the cells were ∼85% confluent, and cells were then cultured for 60 h. To interrupt Sp1 binding, fully differentiated IEC-6 cells (i.e. 7 days postconfluence) were treated with mithramycin (a G/C base pair-specific, DNA-binding antibiotic) (16, 17) at various concentrations for 24 h.
Animals and Diets
Weanling Sprague-Dawley rats (male) were purchased from Harlan (n = 12); raised in overhanging, wire mesh-bottomed cages in a room with 12-h light/dark cycles; and sacrificed at 10 a.m. Rats were fed custom AIN93G-based diets (Dyets, Bethlehem, PA) that varied only in iron content for 5 weeks; the control diet contained 198 ppm iron, whereas the iron-deficient diet contained 3 ppm iron. Animals were weighed weekly. Subsequently, rats were anesthetized by CO2 exposure and killed by cervical dislocation. Blood was collected by cardiac puncture, and hemoglobin and hematocrit were measured by routine methods (8). The duodenum was excised and inverted on a wooden stick after which enterocytes were isolated using a well established, previously published method (8, 18). Duodenal enterocytes were used for mRNA isolation, Western blot analysis, and chromatin immunoprecipitation experiments. All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Florida.
RNA Isolation and Real Time Quantitative RT-PCR
Total RNA was isolated from IEC-6 cells or duodenal enterocytes using TRIzol® reagent (Invitrogen) following a standard protocol. RNA was reverse transcribed using the iScript cDNA Synthesis kit (Bio-Rad), and the resulting cDNA was utilized for qRT-PCRs with SYBR Green PCR Master Mix (Bio-Rad). Primers (listed in supplemental Table S1) were designed to span large introns to avoid amplification from genomic DNA. Standard curve reactions and melt curves were routinely run to validate primer pairs and PCRs. Experimental genes were normalized to 18 S rRNA, and relative gene expression was quantified using routine methods.
Plasmid Construction
The rat Sp1 open reading frame (ORF) (GenBank accession number D12768) was cloned by PCR from cDNA derived from IEC-6 cells using Phusion® High Fidelity DNA Polymerase (Thermo Scientific, Pittsburgh, PA). The forward primer contained the translational start codon, and the reverse primer ended just 5′ of the stop codon. Primers were designed with overhanging KpnI (forward) and EcoRV (reverse) restriction enzyme cutting sites. The PCR-amplified Sp1 ORF amplicon and pcDNA3.1 expression vector (Invitrogen) were double digested with KpnI and EcoRV followed by column purification. Sp1 ORF was then subcloned into double digested pcDNA3.1 with the LigaFastTM Rapid DNA Ligation System (Promega, Madison, WI). An HA tag was inserted into the 3′-end of the Sp1 ORF by PCR amplifying the entire pcDNA-Sp1 plasmid with primers containing overhanging sequences containing an HA sequence tag and a stop codon. Primers were designed with the forward primer at the 3′-end of the Sp1 ORF and the reverse primer at the 5′-end of the EcoRV site on the pcDNA3.1 vector. Each primer was phosphorylated at the 5′-end, which allowed ligation of the PCR amplicons to reform the intact plasmid. Primer sequences are provided in supplemental Table S1.
Mutant Atp7a promoter constructs were prepared by PCR amplifying the entire wild-type (WT) promoter fragment (−224/+88) in the pGL4.18 vector (Promega) with the QuikChange® Lightning Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA). Primers contained mutations in putative Sp1 binding sites with amplification reactions proceeding in opposite directions. PCR products were digested with DpnI restriction enzyme (Agilent Technologies) to remove the template DNA (which was methylated during replication in bacteria). All DNA constructs were sequenced to confirm that promoter amplicons did not contain random mutations. Primer sequences are listed in supplemental Table S1.
Transfection and Luciferase Assays
WT or mutated Atp7a promoter constructs in the pGL4.18 vector (1 μg) were transiently transfected into IEC-6 cells at ∼60% confluence and cultured in 24-well plates. For Sp1 and Hif2α overexpression experiments, 1 μg of Atp7a promoter construct (WT or mutated) was co-transfected with 1 μg of either Sp1 (described above) or Hif2α overexpression vector (described previously) (12). Other constructs used were pGL4.18 plasmids containing ∼1-kb mouse Dcytb and Dmt1 promoter fragments (kindly provided by Dr. Yatrik Shah, University of Michigan). Co-transfected pRL-CMV plasmid (Invitrogen) expressing Renilla luciferase was used to normalize expression of firefly luciferase driven by experimental promoters. 36 h after transfection, luciferase activity was measured with the Dual-Luciferase® Reporter Assay System (Promega) according to the manufacturer's instructions.
Stable Sp1 Overexpression
IEC-6 cells were grown in 6-well plates and transfected with pcDNA3.1 (empty vector) or pcDNA-Sp1-HA vector with TurboFect® in Vitro Transfection kit (Thermo Scientific). 60 h after transfection, cells were treated with G418 (at a predetermined concentration) to kill non-transfected cells, allowing transfected cells expressing the neomycin gene in the pcDNA3.1 or pcDNA-Sp1-HA vectors to survive. IEC-6 cells with stable overexpression of Sp1 were used to analyze Atp7a and Sp1 mRNA expression using qRT-PCR and protein expression by Western blotting and to determine the effect of forced Sp1 expression on Atp7a promoter activity.
Protein Isolation and Western Blot Analysis
Cytosolic and nuclear proteins were isolated from IEC-6 cells cultured in 10-cm cell culture dishes using a kit from Active Motif as described previously (15). Protein concentrations were determined by BCA Protein Assay (Pierce). 30 μg of cytosolic or 50 μg of nuclear proteins were resolved by 7.5% SDS-PAGE followed by transfer to PVDF membranes. The membranes were blocked with 5% (w/v) nonfat milk and then incubated with one of the following primary antibodies: Atp7a (called 54-10) (3), Sp1 (catalog number 07-645, Millipore), phosphorylated Sp1 (catalog number ab37707, Abcam, Cambridge, MA), Hif1α (catalog number NB100-105, Novus Biologicals, Littleton, CO), or Hif2α (catalog number NB100-122, Novus Biologicals). Subsequently, membranes were incubated with an anti-rabbit IgG secondary antibody. Antibody binding was visualized using home-made ECL reagent (8) followed by exposure to x-ray film. For quantification, protein expression was normalized to total proteins on stained blots as this method does not rely on the expression level of any individual protein that may or may not be affected by various treatments (and as used extensively by us in the past).
Chromatin Immunoprecipitation (ChIP) Assay
Assays were performed as described previously (12). Briefly, IEC-6 cells or rat duodenal enterocytes were cross-linked with 1.1% (v/v) chloroform for 10 min followed by quenching with 0.3 m glycine. Cells were subsequently lysed with hypotonic buffer (Active Motif) and homogenized. Nuclei were collected and resuspended in nuclear lysis buffer followed by sonication with a BioRuptor (Diagenode, Liege, Belgium) for 30 cycles with 30 s on and 30 s off. Target DNA with bound protein was pulled down with anti-Sp1 (Millipore) or anti-Hif2α (Novus Biologicals) antibody. After removing cross-links, DNA samples were analyzed by PCR with primer sets listed in supplemental Table S1. Primers were designed to amplify regions of the Atp7a promoter containing putative Sp1 or Hif2α binding sites or other up- or downstream regions that did not contain predicted Sp- or HIF-like sites.
Statistical Analysis
One-way analysis of variance (with Tukey's post hoc test) and paired Student's t test (GraphPad, La Jolla, CA) were used to statistically compare data across groups. p < 0.05 was considered statistically significant.
RESULTS
Mithramycin Inhibits Expression of Iron and Copper Transport-related Genes
Expression of Atp7a and other genes was analyzed by qRT-PCR after mithramycin treatment of differentiated IEC-6 cells (Table 1). Mithramycin reduced expression of all experimental genes tested (Atp7a, Dmt1, Dcytb, and Fpn1) as well as positive control genes including ankyrin repeat domain 37 (Ankrd37), Hif2α, and Sp1. The inhibition was most significant for all tested genes with 500 nm mithramycin; higher concentrations were without additional effect (data not shown), although cellular toxicity was not noted with concentrations up to 1 μm. Ankrd37, which was strongly induced by iron deprivation (2), is a known Sp1 target gene (19) as is Hif2α (20). Interestingly, Sp1 is self-regulated via a positive feedback loop (21). Sp6 and transferrin receptor 1 (Tfr1) were selected as negative controls as neither gene is known to be regulated by Sp-like factors. Expression of Sp6 was unaffected by mithramycin treatment, whereas for unknown reasons, Tfr1 expression was induced.
TABLE 1.
Mithramycin inhibits mRNA expression of Sp1-regulated genes
Data are mean ± S.D. Significance between mithramycin-treated and control cells (Ctrl) (paired Student's t test) is indicated. n = 3 for all groups.
| Gene name | Ctrl | 100 nm | 300 nm | 500 nm |
|---|---|---|---|---|
| Ankrd37 | 1.00 | 0.50 ± 0.20a | 0.30 ± 0.20a | 0.10 ± 0.04a |
| Atp7a | 1.00 | 0.50 ± 0.06b | 0.40 ± 0.20c | 0.20 ± 0.10a |
| Dcytb | 1.00 | 0.50 ± 0.20b | 0.50 ± 0.30b | 0.30 ± 0.30c |
| Dmt1 | 1.00 | 0.80 ± 0.10 | 0.50 ± 0.30b | 0.30 ± 0.20a |
| Fpn1 | 1.00 | 0.70 ± 0.40 | 0.20 ± 0.10a | 0.30 ± 0.10a |
| Hif2α | 1.00 | 0.40 ± 0.20b | 0.30 ± 0.20a | 0.20 ± 0.20a |
| Sp1 | 1.00 | 0.40 ± 0.10a | 0.30 ± 0.20a | 0.20 ± 0.10a |
| Sp6 | 1.00 | 1.30 ± 0.40 | 1.30 ± 0.70 | 1.00 ± 0.30 |
| Tfr1 | 1.00 | 2.70 ± 1.20b | 2.90 ± 1.70b | 2.40 ± 1.00b |
a p < 0.005.
b p < 0.05.
c p < 0.01.
Inhibition of Sp1 Binding Blocks Hypoxia-mediated Gene Expression
Under normoxic conditions, the Hifα subunits are hydroxylated on conserved proline residues and subsequently targeted for proteasomal degradation. Hypoxia stabilizes the Hifα subunits by inhibiting the HIF prolyl hydroxylase enzymes that mediate this hydroxylation reaction (22). Hypoxia can be mimicked by treating cells with cobalt chloride, which effectively inhibits proteasomal degradation of the HIFα subunits under normoxic conditions (23, 24). Here, CoCl2 was utilized to mimic hypoxia in IEC-6 cells. Results showed that expression of experimental (Atp7a, Dcytb, Dmt1, and Fpn1) and positive control (Ankrd37 and vascular endothelial growth factor (Vegf)) genes was increased by CoCl2 exposure (Fig. 1). The Ankrd37 and Vegf genes are known Sp1 targets (19). Moreover, mithramycin decreased basal expression of all tested genes, and it inhibited the induction of Atp7a, Dcytb, Dmt1, and Fpn1 by CoCl2. Conversely, however, mithramycin did not affect the induction of Ankrd37 or Vegf expression by CoCl2.
FIGURE 1.

Effect of mithramycin on CoCl2-mediated transcriptional induction. Postconfluent IEC-6 cells were cultured for 60 h in the presence or absence (Ctrl) of 200 μm CoCl2. Mithramycin (Mith) (500 nm) was added to one set of culture dishes from each treatment group for the last 24 h. Gene expression levels were subsequently determined by qRT-PCR. Gene symbols are shown in each panel. Each bar represents the mean ± S.D. (n = 3). Different letters above each bar (a, b, and c) indicate significant differences between groups within each panel (p < 0.05; one-way analysis of variance).
Regulation of Atp7a Expression by Sp1
IEC-6 cells stably transfected with an Sp1 overexpression plasmid showed significant increases in Sp1 mRNA and protein expression as expected (Fig. 2). Sp1 overexpression also induced Atp7a mRNA and immunoreactive protein expression. Additionally, the Atp7a, Dcytb, and Dmt1 promoters were transactivated by Sp1 overexpression (Fig. 2).
FIGURE 2.

Effect of Sp1 overexpression on Atp7a expression and Atp7a, Dmt1, and Dcytb promoter activity. IEC-6 cells were transfected with HA-tagged Sp1 expression vector (Sp1) or empty expression vector (Ctrl; pcDNA3.1), and Atp7a (A and C) and Sp1 (B and D) mRNA and protein expression was determined. Western blots in C and D are representative of three experiments with similar results. C also shows quantitative data for Atp7a protein expression (*, p < 0.05). Atp7a (E), Dmt1 (F), and Dcytb (G) promoter constructs were co-transfected along with Sp1 overexpression vector into cells, and luciferase activity was measured as an indicator of promoter transactivation. Each bar represents the mean value ±S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.005 (paired Student's t test; n = 3–4 for all experiments presented in this figure).
Sp1-like cis-Elements Are Required for Basal Atp7a Promoter Activity
Phylogenetic footprinting analysis showed that multiple G/C-rich sequences in the basal Atp7a promoter region (−224/+88) were conserved among rats, mice, and humans (data not shown). Moreover, TFSEARCH was utilized to predict putative Sp1 binding sites; nine potential sites were identified. Initially, all nine sites were individually mutated in the basal Atp7a promoter, and promoter activity was assessed in IEC-6 cells. These experiments (n = 3) showed that four putative Sp1 binding sites had the most significant effects on basal promoter activity (data not shown), so these four sites (called S1–S4) were selected for further analysis. Mutation of each site individually significantly reduced basal promoter activity (Fig. 3). Combinatorial mutations (i.e. triple and quadruple mutations), however, had little additional effect on basal promoter activity. Additionally, to consider the functional role of the putative Sp1 binding sites, the effect of Sp1 overexpression on Atp7a promoter activity was assessed. Sp1 overexpression induced activity of the WT promoter (∼2.5-fold), whereas individual and combinatorial mutations had varying effects on promoter activity, with some mutations (e.g. S1–S3 and S2–S4) abolishing the increase caused by Sp1 overexpression (Fig. 3).
FIGURE 3.

Functional analysis of putative Sp1 binding sites on the Atp7a promoter. A shows a schematic representation of the Atp7a promoter (−224 to +88). The 5′-most transcriptional start site identified previously is marked as +1. Also shown are the previously identified HREs and the putative Sp1 binding sites (designated as S1–S4) with the mutated bases shown below the line. These putative Sp1 binding sites were mutated individually or in combination in the basal Atp7a promoter construct and subsequently transfected into IEC-6 cells. B shows the activity of mutated promoters in relation to the activity of the WT promoter. C shows the effect of forced Sp1 expression on WT and mutated Atp7a promoter activity. In B and C, each bar represents the mean value ±S.D. Different letters above bars indicate significant differences among groups (unpaired Student's t test; n = 3–4).
Sp1 Physically Interacts with the Atp7a Promoter
To assess potential interactions between Sp1 and the Atp7a promoter, ChIP assays were conducted. Chromosomal DNA containing cross-linked proteins was isolated from IEC-6 cells and sheared to ∼200 bp, and then DNA samples were pulled down with a ChIP-grade anti-Sp1 antibody. After reversing cross-links and purifying DNA, PCR analysis was utilized to determine whether specific regions of the Atp7a promoter were present in the immunoprecipitated samples. Results showed that all four putative Sp1 binding site regions were present, whereas up- or downstream Atp7a promoter regions lacking putative Sp1-like binding sites were not detected (Fig. 4). It was further shown that mithramycin significantly reduced the amount of Atp7a promoter DNA pulled down (containing all four putative Sp1 binding sites). In this experiment (and others), there was no apparent difference in the amount of input DNA among different reactions.
FIGURE 4.

ChIP analysis of Sp1 binding to the Atp7a promoter. Cross-linked chromosomal DNA was immunoprecipitated from IEC-6 cell nuclear extracts using a ChIP-grade Sp1 antibody. Subsequent PCR analysis was utilized to determine whether certain regions of the Atp7a promoter were pulled down by the antibody. A shows the typical size of DNA fragments after sonication. B depicts PCR analysis of recovered DNA. Results showed that all four putative Sp1 binding sites were present in the immunoprecipitated sample (Atp7a), but other regions of the promoter not containing putative Sp1 binding sites (−) were not detected. Also shown is amplification of the Sp1 binding site regions from the input DNA. ChIP analysis was also performed with nuclear extracts derived from control or mithramycin (Mith)-treated IEC-6 cells (C and D). C shows the effect of 500 nm mithramycin on the activity of the WT Atp7a promoter-transfected into cells. Each bar represents the mean value ±S.D. (n = 3; ***, p < 0.005; paired Student's t test). Mithramycin also decreased the amount of Atp7a promoter DNA containing the putative Sp1 binding sites detected by PCR after ChIP (Atp7a) (D). Amplification from input DNA samples was similar, indicating that equal amounts of starting material were used. Again, other unrelated promoter regions were not detected (−). ChIP experiments depicted here are typical of three independent experiments performed with similar results. Ctrl, control.
Sp1 Binding Is Required for Hif2α-mediated Up-regulation of Atp7a Expression
We next sought to determine whether the putative Sp1 binding sites were required for Hif2α-mediated induction of Atp7a promoter activity. As shown previously (12), Hif2α overexpression induced Atp7a promoter activity ∼5-fold in IEC-6 cells (Fig. 5). This induction was blunted by mutation of each Sp1 binding site individually, and combinatorial mutations abolished transactivation by Hif2α.
FIGURE 5.

Effect of Hif2α overexpression on Atp7a promoter activity. Hif2α expression vector was co-transfected into IEC-6 cells along with WT or mutant Atp7a promoter constructs, and luciferase assays were performed. The effect of Hif2α overexpression is shown relative to activity of the WT promoter without Hif2α overexpression. The specific Sp1 sites (S) mutated are indicated below each bar, and Hif2α overexpression is indicated further below. Each bar represents the mean value ±S.D. Different letters above bars indicate statistical differences between groups (p < 0.05; paired Student's t test; n = 3–4).
Sp1 and Hif2α Interact with the Atp7a Promoter in Vivo
As all experiments reported so far were from an in vitro model of the mammalian intestinal epithelium, it was important to confirm these observations in vivo. Accordingly, studies were performed in rats that were deprived of dietary iron for 5 weeks after weaning. The intent was to determine whether Sp1 and Hif2α bound to the Atp7a promoter in rat intestine and whether iron deprivation altered DNA-protein interactions. Iron-deprived rats had significantly decreased hemoglobin and hematocrit levels (both reduced >75%), which are indicative of iron deficiency anemia, consistent with previous observations (2, 8) (data not shown). Body weights were also lower (∼19%) in the rats fed the low iron diet. Dcytb, Dmt1, and Atp7a mRNA expression increased in duodenal enterocytes isolated from the iron-deficient rats as expected, and serum ceruloplasmin protein expression increased, consistent with previous observations (Fig. 6) (8). Moreover, Atp7a protein expression increased, and although inconsistent between animals, Hif2α protein levels were higher in the iron-deficient rats. Lack of Atp7a detection in one sample and Hif2α in two samples may relate to delays in sample processing (and subsequent partial protein degradation) because RNA purification was undertaken first. However, degradation was not observed upon visual inspection of the stained blots. Furthermore, Hif1α was undetectable under these conditions (but the antibody had been validated by us using other nuclear protein samples).
FIGURE 6.

Molecular analysis of control and iron-deficient rats. Weanling rats consumed control or low iron diets for 5 weeks and then were sacrificed. Expression of known iron-responsive genes was analyzed in isolated duodenal enterocytes by qRT-PCR (A–C). Each bar represents the mean value ±S.D. **, p < 0.01; ***, p < 0.005 (paired Student's t test; n = 6). Ceruloplasmin (Cp; in serum; D), Atp7a (in enterocytes; E), and Hif1/2α (in enterocytes; F) protein expression was assessed by Western blotting. Shown below the blots are total stained proteins exemplifying equal loading of the gels and efficient transfer of proteins to membranes. ChIP experiments were also performed using cross-linked chromosomal DNA isolated from duodenal enterocytes and ChIP-grade Hif2a (G) or Sp1 (H) antibodies. For Hif2a ChIP (G), primers that covered the region containing the three HREs on the Atp7a promoter were used. In H, the primers encompassed the Sp1 binding sites on the promoter. In G and H, (−) indicates the use of primers from unrelated up-or downstream sites within the Atp7a promoter, and “Input” indicates amplification from the DNA samples prior to immunoprecipitation. Ctrl, control; FeD, iron-deficient.
ChIP experiments confirmed that Sp1 and Hif2α specifically interacted with the Atp7a promoter in vivo (Fig. 6). There was a trend toward increased Hif2α binding in samples derived from iron-deficient rats, whereas conversely, no differences in Sp1 binding were noted among groups (data not shown). The detection of Hif2 binding in control rat samples was unexpected as the protein is normally degraded during normoxia. However, the intestinal epithelium exists in a natural state of mild hypoxia (25), particularly in epithelial cells at the villus tip, which is furthest from the blood supply (and where iron and copper transporters are expressed). This may explain the stabilization of the Hif2 protein in control duodenum.
Sp1 and Hif2α Synergistically Activate the Atp7a Promoter
Forced expression of Sp1 or Hif2α activates the Atp7a promoter in IEC-6 cells. To determine whether these two trans-acting factors can further enhance the promoter response when co-overexpressed, IEC-6 cells were transfected with Sp1 and Hif2α expression vectors individually or together along with the basal Atp7a promoter construct, and reporter gene assays were performed. Forced Sp1 expression increased activity ∼3-fold, whereas Hif2α overexpression increased activity ∼5-fold (Fig. 7). When both were overexpressed together, Atp7a promoter activity was further transactivated to ∼8-fold over control (empty vector-transfected) cells.
FIGURE 7.

Effect of Hif2α and/or Sp1 overexpression on Atp7a promoter activity. The basal Atp7a promoter construct (−224/+88) was co-transfected along with Hif2α and/or Sp1 expression plasmids into IEC-6 cells. Subsequent luciferase assays indicated promoter activity, which is shown in relation to promoter activity in the absence of Sp1 or Hif2α overexpression (Ctrl). Each bar represents the mean ± S.D. Different letters above each bar (a, b, c, and d) indicate significant differences (p < 0.05; one-way analysis of variance; n = 3–4).
CoCl2 and Low Oxygen Enhance Phosphorylation of Sp1
Sp1 transactivation properties are regulated by phosphorylation. We thus sought to determine whether the level of phospho-Sp1 was altered by hypoxia. Accordingly, IEC-6 cells were treated with CoCl2 to mimic hypoxia or grown in a hypoxia chamber (12), and Sp1/phospho-Sp1 proteins levels were determined by immunoblot analysis. Results showed significantly higher levels of phospho-Sp1 in treated cells, whereas total immunoreactive Sp1 levels were relatively constant when comparing the treatment groups with control (untreated) cells (Fig. 8).
FIGURE 8.
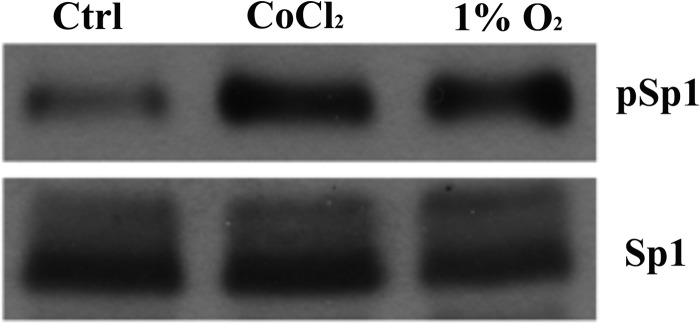
Immunoblot analysis of phosphorylated Sp1 protein expression. IEC-6 cells at 85% confluence were either untreated and grown under control conditions (Ctrl), treated with 200 μm CoCl2, or cultured in a hypoxia chamber (with 1% O2) for 60 h. Nuclear proteins were then isolated, and immunoblots were run for detection of Sp1 and phosphorylated Sp1 (pSp1). The pSp1 band was detected at ∼120 kDa, whereas the total Sp1 protein band was detected at ∼108 kDa. The blots shown are representative of three independent experiments with comparable results.
DISCUSSION
During iron deficiency, hemoglobin levels fall, decreasing oxygen delivery to tissues and cells, which leads to a hypoxic response. At the molecular level, this causes stabilization of the HIFα subunits that promotes nuclear localization and interaction with a constitutively expressed HIFβ subunit followed by DNA binding and activation of genes related to energy metabolism (glycolysis), angiogenesis, and iron homeostasis. In the intestinal mucosa, during iron deficiency/hypoxia, a Hif2α-specific transcriptional response enhances absorption of dietary iron by transactivating genes encoding proteins that mediate iron transport. Interestingly, Hif2α may also modulate intestinal copper absorption as reflected by induction of Atp7a and metallothionein in duodenal enterocytes during iron deprivation. The co-regulation of iron and copper transport during iron deficiency supports the concept that copper plays an important physiologic role in the maintenance of iron homeostasis. The current studies aimed to further evaluate mechanistic aspects of the Hif2α transcriptional response.
It was noted previously that many genes induced by iron deprivation in the rat intestine and in Caco-2 cells have G/C-rich promoters (14), suggesting regulation by a trans-acting factor with an affinity for G-C base pairs. The classic example of such a transcription factor is Sp1. This widely expressed protein is a member of the Sp1-like and Krüppel-like factor family of DNA-binding proteins, which are integral parts of the transcriptional machinery of eukaryotic cells (26, 27). Sp1/Krüppel-like factors have highly conserved carboxyl-terminal DNA-binding domains containing three tandem zinc finger motifs. The amino-terminal regions are variable and contain transcription regulatory domains that interact with co-regulators. By regulating the expression of a large number of genes containing G/C-rich promoters, Sp1/Krüppel-like factor proteins are involved in many biological processes including cell proliferation, differentiation, apoptosis, and neoplastic transformation (26). Few studies to date have investigated Sp1-like factors in the regulation of iron homeostasis-related genes. One recent study suggested that the age-related decline in hepatic transferrin gene expression may relate in part to Sp1-like DNA binding activity (28). The hepatic Hfe gene, which is mutated in some types of hereditary hemochromatosis in humans, also has apparent Sp1-like binding sites (29). Sp1 and Sp3 were also shown to bind to an enhancer in the ferritin H gene and activate expression in fibroblast and liver cell lines (30). Additionally, it was reported previously that the Dmt1 gene has three predicted Sp1-like binding sites, but these were not experimentally verified (31). Moreover, although a role for Sp1-like factors in mediating the transcriptional response of intestinal epithelial cells to iron deprivation was postulated previously (13), this possibility has not been experimentally tested to date.
To elucidate a potential role for Sp1 in the Hif2α-mediated transcriptional response to iron deprivation, we performed initial studies on the Atp7a gene, which is coordinately regulated with iron transport-related genes during iron deprivation. Atp7a-mediated regulation of copper absorption may play an important physiologic role in the maintenance of intestinal iron transport possibly by enhancing activity of a multicopper ferroxidase (hephaestin), which couples iron oxidation to efflux via Fpn1 (32, 33). We previously evaluated the rat Atp7a promoter (12, 34) including mapping the transcriptional start site and defining the basal promoter region (−224/+88). The role of Sp1 in basal and Hif2α-stimulated Atp7a transcription, however, has not been examined. This investigation was thus undertaken to test the hypothesis that Sp1 (or an Sp1-like factor) is necessary for the Hif2α-mediated induction of gene expression in the duodenal mucosa during iron deficiency. Whenever possible, whether Atp7a-specific regulatory mechanisms were conserved among iron homeostasis-related genes (e.g. Dmt1, Dctyb, and Fpn1) was assessed to broaden the scope of this experimental analysis.
Initial experiments utilized a drug that blocks Sp1 binding to DNA (mithramycin) to assess a possible role for Sp1 in Atp7a gene transcription in IEC-6 cells. Mithramycin is a DNA-binding antibiotic that binds to the minor groove of G-C base pairs (35, 36). This interaction with DNA blocks trans-acting factor binding to G/C-rich regions. Although initial studies showed specific inhibition of Sp1 binding (16, 17, 37), mithramycin could theoretically block binding of any protein with an affinity for G-C base pairs. In the current study, mithramycin was utilized to show that Atp7a and other iron homeostasis-related genes were potentially regulated by Sp1 as mRNA expression was significantly inhibited. However, a G/C-binding protein was not absolutely required for basal transcriptional activation of these genes as expression was not abolished. These observations provide preliminary evidence that intestinal genes induced by Hif2α during iron deprivation/hypoxia may be regulated by Sp1.
CoCl2 chemically mimics hypoxia (under normoxic conditions) by stabilizing the HIFα subunits; Hif1α and Hif2α are both stabilized via inhibition of oxygen-dependent degradation. Expression of Atp7a and iron transport-related genes increased with CoCl2 treatment, consistent with their known regulation by Hif2α. Ankrd37 and Vegf were also up-regulated, likely reflecting regulation by Hif1α (19, 38). Interestingly, mithramycin had differing effects on the induction of mRNA expression by CoCl2; it blocked the increase of some genes, whereas other genes were unaffected. This exemplified two modes of regulation: one in which Sp1 (or a related G/C-binding protein) is necessary for the HIF response (e.g. for Atp7a and Dmt1) and another in which Sp1 is not required (e.g. for Ankrd37 and Vegf). These opposing regulatory mechanisms may relate to distinct transactivation properties of the different HIFα subunits. A trans-acting factor with affinity for G/C-rich DNA regions may thus be required for the Hif2α-mediated increase in gene expression, which ultimately promotes iron absorption during hypoxia.
Several experimental observations presented herein suggest that Atp7a gene transcription is regulated by Sp1 including the following. 1) Sp1 overexpression increased endogenous Atp7a mRNA and protein expression in IEC-6 cells and stimulated the exogenously expressed Atp7a promoter. 2) Putative Sp1 binding sites were shown to be required for full transactivation of Atp7a gene expression. 3) ChIP assays showed that Sp1 directly interacts with the Atp7a gene in IEC-6 cells and in rat duodenal enterocytes. 4) Mithramycin significantly decreased pulldown of Atp7a promoter DNA containing the putative Sp1 binding sites from IEC-6 cells, consistent with the documented decrease in Atp7a promoter activity in the presence of mithramycin. Furthermore, in the current investigation, the previously reported binding of Hif2α to the Atp7a promoter in IEC-6 cells (12) was confirmed in rat duodenal enterocytes. Atp7a is thus a bona fide Sp1 and Hif2α target gene.
A final series of experiments was designed to determine whether putative Sp1 binding sites were necessary for Hif2α-mediated induction of Atp7a promoter activity. Forced Hif2α expression increased promoter activity ∼5-fold, whereas individual Sp1 binding site mutations attenuated this increase to ∼3-fold. Combinatorial Sp1 site mutations abolished transactivation by Hif2α overexpression. Interestingly, Hif2α overexpression maintained basal Atp7a promoter activity at WT levels even when multiple Sp1 sites were mutated (in contrast to decreases in basal activity without forced Hif2α expression). Putative Sp1 binding sites are thus necessary for transactivation of the Atp7a gene by Hif2α.
Data presented here show that the HIF-mediated induction of Atp7a expression during iron deficiency/hypoxia involves Sp1. Sp1-dependent Hif2α transactivation of gene expression has not been reported in the scientific literature (to our knowledge), suggesting that this is a novel regulatory mechanism. Hif2α is preferentially stabilized during iron deprivation in the intestine of mice (9, 10) and rats (this study) and in Caco-2 cells (14). Hif2α protein levels likely increase due to tissue hypoxia in iron-deprived mice and rats and as a result of inhibition of the iron-dependent HIF prolyl hydroxylases in Caco-2 cells treated with deferoxamine (an iron chelator). What is not clear is the specific molecular mechanism by which Sp1 potentiates the HIF-mediated induction of Atp7a gene transcription. Sp1 is known to be regulated by phosphorylation (39–41), which alters its DNA binding affinity and/or transactivation capabilities. As such, we quantified Sp1/phospho-Sp1 levels in CoCl2-treated IEC-6 cells and in cells grown in 1% O2. Phosphorylation of Sp1 increased dramatically in treated cells, suggesting that posttranslational modification of the protein may play a role in induction of Atp7a expression during iron deprivation/hypoxia. Because ChIP assays showed no difference in the amount of Atp7a promoter DNA pulled down with Sp1 antibody from enterocytes isolated from control or iron-deficient rats, we speculate that Sp1 phosphorylation increases transactivation of Atp7a gene expression.
This investigation focused on the gene encoding the primary enterocyte copper exporter, Atp7a. Lack of fully functional Atp7a is the underlying cause of Menkes disease in humans, a Mendelian disorder in which inefficient absorption of dietary copper leads to systemic copper deficiency and the dire physiologic consequences of copper depletion (e.g. neurological damage, hypopigmentation, etc.) (42, 43). During iron deficiency/hypoxia, Atp7a expression increases dramatically, implicating copper in control of iron homeostasis. In fact, copper increases in tissues and cells important for homeostatic control of iron homeostasis (e.g. enterocytes and hepatocytes) during iron deficiency (3, 8). Given that Atp7a represents the rate-limiting step in acquisition of dietary copper, it may then play a key role in the compensatory response to iron deficiency. Thus, a detailed mechanistic understanding of Atp7a gene regulation may increase knowledge of regulatory aspects of whole-body iron homeostasis.
In summary, Sp1 binding is necessary for the hypoxia-mediated induction of Atp7a promoter activity in IEC-6 cells. Whether this mechanism is also true of in vivo regulation of Atp7a gene expression during iron deprivation is unknown, but we provide evidence that the Atp7a gene is a direct Hif2α and Sp1 target in rat duodenal enterocytes. Three lines of evidence suggest that these observations may have importance beyond understanding Atp7a gene regulation. 1) Atp7a is coordinately regulated by Hif2α along with genes encoding proteins required for iron absorption (Dcytb, Dmt1, and Fpn1). 2) Many genes up-regulated by iron deficiency in the mammalian duodenum have G/C-rich promoters and evolutionarily conserved HREs. 3) Hypoxia resulted in increased phosphorylation of Sp1, likely altering its transactivation properties. Sp1-dependent, Hif2α-mediated induction of gene expression may thus have broader implications for understanding additional mechanistic aspects of intestinal iron homeostasis.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01-DK074867 (to J. F. C.).

This article contains supplemental Table S1.
- Dmt1
- divalent metal transporter 1
- Ankrd37
- ankyrin repeat domain 37
- Dcytb
- duodenal cytochrome b
- Fpn1
- ferroportin 1
- HRE
- hypoxia-responsive element
- HIF
- hypoxia-inducible factor
- Sp
- specificity factor
- Tfr1
- transferrin receptor 1
- Atp7a
- Menkes copper-transporting ATPase
- qRT-PCR
- quantitative RT-PCR.
REFERENCES
- 1. Aisen P., Enns C., Wessling-Resnick M. (2001) Chemistry and biology of eukaryotic iron metabolism. Int. J. Biochem. Cell Biol. 33, 940–959 [DOI] [PubMed] [Google Scholar]
- 2. Collins J. F., Franck C. A., Kowdley K. V., Ghishan F. K. (2005) Identification of differentially expressed genes in response to dietary iron deprivation in rat duodenum. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G964–G971 [DOI] [PubMed] [Google Scholar]
- 3. Ravia J. J., Stephen R. M., Ghishan F. K., Collins J. F. (2005) Menkes copper ATPase (Atp7a) is a novel metal-responsive gene in rat duodenum, and immunoreactive protein is present on brush-border and basolateral membrane domains. J. Biol. Chem. 280, 36221–36227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee G. R., Nacht S., Lukens J. N., Cartwright G. E. (1968) Iron metabolism in copper-deficient swine. J. Clin. Investig. 47, 2058–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naveh Y., Hazani A., Berant M. (1981) Copper deficiency with cow's milk diet. Pediatrics 68, 397–400 [PubMed] [Google Scholar]
- 6. Prohaska J. R., Broderius M. (2006) Plasma peptidylglycine α-amidating monooxygenase (PAM) and ceruloplasmin are affected by age and copper status in rats and mice. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 143, 360–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gulec S., Collins J. F. (2013) Investigation of iron metabolism in mice expressing a mutant Menke's copper transporting ATPase (Atp7a) protein with diminished activity (Brindled; MoBr/y). PLoS One 8, e66010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ranganathan P. N., Lu Y., Jiang L., Kim C., Collins J. F. (2011) Serum ceruloplasmin protein expression and activity increases in iron-deficient rats and is further enhanced by higher dietary copper intake. Blood 118, 3146–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mastrogiannaki M., Matak P., Keith B., Simon M. C., Vaulont S., Peyssonnaux C. (2009) HIF-2α, but not HIF-1α, promotes iron absorption in mice. J. Clin. Investig. 119, 1159–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shah Y. M., Matsubara T., Ito S., Yim S. H., Gonzalez F. J. (2009) Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 9, 152–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taylor M., Qu A., Anderson E. R., Matsubara T., Martin A., Gonzalez F. J., Shah Y. M. (2011) Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 140, 2044–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xie L., Collins J. F. (2011) Transcriptional regulation of the Menkes copper ATPase (Atp7a) gene by hypoxia-inducible factor (HIF2α) in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 300, C1298–C1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Collins J. F., Hu Z. (2007) Promoter analysis of intestinal genes induced during iron-deprivation reveals enrichment of conserved SP1-like binding sites. BMC Genomics 8, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Z., Gulec S., Collins J. F. (2010) Cross-species comparison of genomewide gene expression profiles reveals induction of hypoxia-inducible factor-responsive genes in iron-deprived intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 299, C930–C938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie L., Collins J. F. (2013) Copper stabilizes the Menkes copper-transporting ATPase (Atp7a) protein expressed in rat intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 304, C257–C262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blume S. W., Snyder R. C., Ray R., Thomas S., Koller C. A., Miller D. M. (1991) Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J. Clin. Investig. 88, 1613–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ray R., Snyder R. C., Thomas S., Koller C. A., Miller D. M. (1989) Mithramycin blocks protein binding and function of the SV40 early promoter. J. Clin. Investig. 83, 2003–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang L., Ranganathan P., Lu Y., Kim C., Collins J. F. (2011) Exploration of the copper related compensatory response in the Belgrade rat model of genetic iron deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G877–G886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benita Y., Kikuchi H., Smith A. D., Zhang M. Q., Chung D. C., Xavier R. J. (2009) An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 37, 4587–4602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wada T., Shimba S., Tezuka M. (2006) Transcriptional regulation of the hypoxia inducible factor-2α (HIF-2α) gene during adipose differentiation in 3T3-L1 cells. Biol. Pharm. Bull. 29, 49–54 [DOI] [PubMed] [Google Scholar]
- 21. Liang Z. D., Tsai W. B., Lee M. Y., Savaraj N., Kuo M. T. (2012) Specificity protein 1 (Sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol. Pharmacol. 81, 455–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salceda S., Caro J. (1997) Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 272, 22642–22647 [DOI] [PubMed] [Google Scholar]
- 23. Yuan Y., Beitner-Johnson D., Millhorn D. E. (2001) Hypoxia-inducible factor 2α binds to cobalt in vitro. Biochem. Biophys. Res. Commun. 288, 849–854 [DOI] [PubMed] [Google Scholar]
- 24. Yuan Y., Hilliard G., Ferguson T., Millhorn D. E. (2003) Cobalt inhibits the interaction between hypoxia-inducible factor-α and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-α. J. Biol. Chem. 278, 15911–15916 [DOI] [PubMed] [Google Scholar]
- 25. Taylor C. T., Colgan S. P. (2007) Hypoxia and gastrointestinal disease. J. Mol. Med. 85, 1295–1300 [DOI] [PubMed] [Google Scholar]
- 26. Kaczynski J., Cook T., Urrutia R. (2003) Sp1- and Kruppel-like transcription factors. Genome Biol. 4, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Black A. R., Black J. D., Azizkhan-Clifford J. (2001) Sp1 and Kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell. Physiol. 188, 143–160 [DOI] [PubMed] [Google Scholar]
- 28. Adrian G. S., Seto E., Fischbach K. S., Rivera E. V., Adrian E. K., Herbert D. C., Walter C. A., Weaker F. J., Bowman B. H. (1996) YY1 and Sp1 transcription factors bind the human transferrin gene in an age-related manner. J. Gerentol. A Biol. Sci. Med. Sci. 51, B66–B75 [DOI] [PubMed] [Google Scholar]
- 29. Mura C., Le Gac G., Jacolot S., Férec C. (2004) Transcriptional regulation of the human HFE gene indicates high liver expression and erythropoiesis coregulation. FASEB J. 18, 1922–1924 [DOI] [PubMed] [Google Scholar]
- 30. Tsuji Y., Torti S. V., Torti F. M. (1998) Activation of the ferritin H enhancer, FER-1, by the cooperative action of members of the AP1 and Sp1 transcription factor families. J. Biol. Chem. 273, 2984–2992 [DOI] [PubMed] [Google Scholar]
- 31. Lee P. L., Gelbart T., West C., Halloran C., Beutler E. (1998) The human Nramp2 gene: characterization of the gene structure, alternative splicing, promoter region and polymorphisms. Blood Cells Mol. Dis. 24, 199–215 [DOI] [PubMed] [Google Scholar]
- 32. Fuqua B. K., Vulpe C. D., Anderson G. J. (2012) Intestinal iron absorption. J. Trace Elem. Med. Biol. 26, 115–119 [DOI] [PubMed] [Google Scholar]
- 33. Vulpe C. D., Kuo Y. M., Murphy T. L., Cowley L., Askwith C., Libina N., Gitschier J., Anderson G. J. (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 21, 195–199 [DOI] [PubMed] [Google Scholar]
- 34. Collins J. F., Hua P., Lu Y., Ranganathan P. N. (2009) Alternative splicing of the Menkes copper ATPase (Atp7a) transcript in the rat intestinal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G695–G707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schnedl W., Breitenbach M., Stranzinger G. (1977) Mithramycin and DIPI: a pair of fluorochromes specific for GC-and AT-rich DNA respectively. Hum. Genet. 36, 299–305 [DOI] [PubMed] [Google Scholar]
- 36. Ward D. C., Reich E., Goldberg I. H. (1965) Base specificity in the interaction of polynucleotides with antibiotic drugs. Science 149, 1259–1263 [DOI] [PubMed] [Google Scholar]
- 37. Snyder R. C., Ray R., Blume S., Miller D. M. (1991) Mithramycin blocks transcriptional initiation of the c-myc P1 and P2 promoters. Biochemistry 30, 4290–4297 [DOI] [PubMed] [Google Scholar]
- 38. Dvorak H. F. (2002) Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 20, 4368–4380 [DOI] [PubMed] [Google Scholar]
- 39. Daniel S., Zhang S., DePaoli-Roach A. A., Kim K. H. (1996) Dephosphorylation of Sp1 by protein phosphatase 1 is involved in the glucose-mediated activation of the acetyl-CoA carboxylase gene. J. Biol. Chem. 271, 14692–14697 [DOI] [PubMed] [Google Scholar]
- 40. Rohlff C., Glazer R. I. (1998) Regulation of the MDR1 promoter by cyclic AMP-dependent protein kinase and transcription factor Sp1. Int. J. Oncol. 12, 383–386 [DOI] [PubMed] [Google Scholar]
- 41. Rohlff C., Ahmad S., Borellini F., Lei J., Glazer R. I. (1997) Modulation of transcription factor Sp1 by cAMP-dependent protein kinase. J. Biol. Chem. 272, 21137–21141 [DOI] [PubMed] [Google Scholar]
- 42. Tümer Z., Møller L. B. (2010) Menkes disease. Eur. J. Hum. Genet. 18, 511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kaler S. G. (1994) Menkes disease. Adv. Pediatr. 41, 263–304 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.