

Background: In animal cells, nine aminoacyl-tRNA synthetases form a supramolecular assembly.
Results: The low resolution solution structure of this complex is determined by SAXS.
Conclusion: This complex exhibits a non-compact, elongated, and multi-armed shape.
Significance: This type of structural organization provides new insights to understand the structure-function relationship and spatiotemporal regulation of the activity of its multiple components.
Keywords: Aminoacyl tRNA Synthetase, Protein Complexes, Protein Structure, Protein Synthesis, X-ray Scattering
Abstract
In animal cells, nine aminoacyl-tRNA synthetases are associated with the three auxiliary proteins p18, p38, and p43 to form a stable and conserved large multi-aminoacyl-tRNA synthetase complex (MARS), whose molecular mass has been proposed to be between 1.0 and 1.5 MDa. The complex acts as a molecular hub for coordinating protein synthesis and diverse regulatory signal pathways. Electron microscopy studies defined its low resolution molecular envelope as an overall rather compact, asymmetric triangular shape. Here, we have analyzed the composition and homogeneity of the native mammalian MARS isolated from rabbit liver and characterized its overall internal structure, size, and shape at low resolution by hydrodynamic methods and small-angle x-ray scattering in solution. Our data reveal that the MARS exhibits a much more elongated and multi-armed shape than expected from previous reports. The hydrodynamic and structural features of the MARS are large compared with other supramolecular assemblies involved in translation, including ribosome. The large dimensions and non-compact structural organization of MARS favor a large protein surface accessibility for all its components. This may be essential to allow structural rearrangements between the catalytic and cis-acting tRNA binding domains of the synthetases required for binding the bulky tRNA substrates. This non-compact architecture may also contribute to the spatiotemporal controlled release of some of its components, which participate in non-canonical functions after dissociation from the complex.
Introduction
Aminoacyl-tRNA synthetases are essential enzymes for genetic code decoding. They activate amino acids and transfer them on cognate tRNAs (1). Because the establishment of a univocal link between an amino acid and the nucleotide triplet present on the anticodon loop of the tRNA molecule is essential for the fidelity of translation, the process of tRNA aminoacylation has been extensively studied in the past 30 years. These enzymes are generally monomers (α), homodimers (α2), or tetramers (α4 or α2β2), and the three-dimensional structure of at least one member of each component of this family of 20 enzymes has been determined from bacteria, archaea, or lower eukaryotes. However, our knowledge of the structural arrangement of these enzymes in higher eukaryotes remains essentially unknown at the atomic level due to their organization into large supramolecular assemblies, which represents a challenge for high resolution structural studies.
Aminoacyl-tRNA synthetases from animal cells display several distinctive features (2, 3). First, polypeptide extensions are generally appended to the N- or C-terminal extremity of their bacterial-like core enzyme. There are tRNA binding domains (tRBD)4 that strengthen tRNA-enzyme interactions (4), or PBD (protein binding domains) involved in protein-protein associations (5). Second, examples of fused synthetases have been described, such as in the case of GluProRS where a long polypeptide of 1512 amino acid residues results from the fusion between bona fide GluRS and ProRS. Third, some of these enzymes are gathered within supramolecular assemblies. The emergence of stable multiprotein assemblies amenable to structural characterization has been essentially described in the bilaterian phylum of metazoans. The largest supramolecular complex of aminoacyl-tRNA synthetases described so far is the MARS complex (multi-aminoacyl-tRNA synthetase complex) containing the nine enzymes ArgRS, AspRS, GlnRS, GluRS, IleRS, LeuRS, LysRS, MetRS, and ProRS and the three nonsynthetase components p18, p38, and p43 (3, 6). The complexes isolated from arthropods (7) to human (8) have the same protein composition, but there is still some ambiguity about the stoichiometry and total number of components mainly because the native multi-protein assemblies are highly sensitive to uncontrolled proteolysis. In addition to its essential role in translation, MARS acts as a reservoir for aminoacyl-tRNA synthetases where, in response to cellular changes, synthetases are subsequently released from the complex to participate in non-canonical tasks (9). These include for example translational silencing of ceruloplasmin by GluProRS in inflammatory response (10) or regulation of gene expression by LysRS in the immune response (11). Released non-enzymatic components also play a role in diverse regulatory signal pathways. The auxiliary protein p43, which facilitates tRNA delivery in MARS (12, 13), is for example additionally involved in angiogenesis or displays cytokine activity after its release from MARS by caspase-7-mediated proteolysis (14, 15). The knowledge of the structural assembly of MARS complex, therefore, represents an essential step for understanding the spatiotemporal regulation of the activity of all its components.
The three-dimensional architecture of MARS has been mapped by different methods. Models of protein topology of this complex have been proposed based on two-hybrid approaches (16, 17) or in vitro cross-linking studies (18). By combining isolation by tandem-affinity purification of complexes assembled in cellulo in the absence of the p18, p38, or p43 components that were inactivated by siRNA silencing, it was proposed that MARS is made of two sub-complexes joined by the p38 scaffold protein (8). Sub-complex I contains GluProRS, IleRS, LeuRS, MetRS, and p18, whereas sub-complex II contains ArgRS, GlnRS, and p43. LysRS and AspRS would be directly linked to p38. A stable sub-complex containing p38, p43, ArgRS, and GlnRS could also be reconstituted in vitro after mixing individual recombinant proteins produced in bacteria or yeast (19). At the atomic level, the structures of only few individual components have been solved.
The crystal structure of the C-terminal moiety of p43 revealed a novel OB-fold based tRNA-binding site (12, 13). The p18 component that links MetRS to sub-complex I (8) adopts a glutathione S-transferase-like fold (20) similar to the N-terminal domain of the Arc1p protein that links MetRS to GluRS in yeast (21). The crystal structure of the human core LysRS also suggested how LysRS could interact with p38 (22, 23). The solution structure of one of the repeated WHEP domains located in the spacer region between the two synthetases in the fused GluProRS polypeptide revealed a helix-turn-helix-based tRNA binding domain (24, 25). Although these data shed some light on the structural organization of individual components of MARS, the detailed architecture of the whole particle is not yet deciphered. A structural working model of the particle has been proposed based on electron microscopy and three-dimensional reconstructions. These studies revealed an asymmetric three-domain Y-shaped arrangement of proteins showing large external dimensions ranging from 25 × 30 × 23 nm (26) to 19 × 16 × 10 nm (27), with a deep central cleft. Some of the components were tentatively located within the particle by labeling with antibodies or with specific tRNAs (28, 29).
To gain more insight into the functioning of the synthetases associated within supramolecular assemblies, it is necessary to get a deeper understanding of their structural organization. In this study we purified the native complex from rabbit liver, analyzed the structural homogeneity of the enzyme preparation, and characterized its overall size and shape at low resolution by hydrodynamic methods and small-angle x-ray scattering (SAXS) in solution. Our data reveal that the MARS complex has a much more elongated shape than expected from previous reports and displays the largest maximum particle size for a stable supramolecular assembly involved in translation, including the ribosome.
EXPERIMENTAL PROCEDURES
Purification of MARS from Rabbit Liver
Livers (600 g) from 8 rabbits were recovered immediately after sacrifice of the animals and homogenized in a Waring blender (2 × 15 s) after the addition (1 ml/g) of extraction buffer (50 mm Tris-HCl, pH 7.5, 200 mm NaCl, 5 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, and 10% glycerol) containing protease inhibitors (10 mm PMSF, 10 mm benzamidine). Extract was cleared by centrifugation at 50,000 × g for 30 min and further subjected to high speed centrifugation at 230,000 × g for 90 min. The supernatant (500 ml) was applied on a 5600 ml (9 × 88 cm) Sephacryl S-400 HR column (GE Healthcare) equilibrated in buffer A (85 mm potassium phosphate buffer, pH 7.5, 1 mm EDTA, 10 mm 2-mercaptoethanol, and 10% glycerol) and developed at a flow rate of 5 ml/min. Fractions eluted between 2820 and 3600 ml were directly applied on a 170-ml (5 × 8.6 cm) tRNA-Sepharose column developed at a flow rate of 5 ml/min. After washing with buffer A, the complex was eluted with a linear gradient of potassium phosphate buffer (85–300 mm). After a 2.5-fold dilution with a solution containing 10 mm 2-mercaptoethanol and 10% glycerol, fractions were applied on a 4.6-ml Source 15Q column (GE Healthcare) equilibrated in buffer AQ (25 mm Tris-HCl, pH 7.5, 80 mm KCl, 1 mm DTT) and developed at 2 ml/min with a linear gradient of KCl from 80 to 200 mm. Fractions were concentrated by ultrafiltration on Vivaspin 50,000. Protein concentration was determined by using a calculated absorption coefficient of 0.931 A280 units·mg−1·cm2.
Analytical Gel Filtration
The apparent native molecular mass of MARS was determined by gel filtration on a Superose 6 HR 10/30 (GE Healthcare) column equilibrated in 20 mm Tris-HCl, pH 7.5, 200 mm NaCl, 10 mm 2-mercaeptoethanol, and developed at room temperature at a flow rate of 0.2 ml/min. All samples (50 μg of protein) were loaded in 0.2 ml. The calibration curve was established by using cytochrome c, ovalbumin, bovine serum albumin, and thyroglobulin as marker proteins. For a particular protein, its elution was described in term of the corresponding Kav value. Kav = (Ve − V0)/(Vt − V0), where Ve is the elution volume of the particular molecule, V0 is the void volume of the column, and Vt is the total bed volume. V0 and Vt were determined with dextran blue (>5 MDa) and vitamin B12 (1.35 kDa), respectively. The elution profile at 280 nm was fitted to a gaussian model following the equation y = a0 + ymaxexp[−(x − xmax)2/x1/22], where ymax is the value of A280 at the apex of the peak, xmax is the elution volume at ymax, x1/2 is the half-width of the peak at ymax/2, and a0 is the base line of A280.
Size Exclusion Chromatography Coupled to Multiangle Laser Light Scattering (SEC-MALLS)
For SEC-MALLS analysis, MARS (0.02 ml at 1 mg/ml) was loaded on a 300 × 4.6-mm BioSep-SEC-s4000 column (Phenomenex) equilibrated in 20 mm Hepes, pH 6.8, 200 mm NaCl, 2% glycerol, and 2 mm DTT and developed at room temperature on a Prominence HPLC system (Shimadzu) at a flow rate of 0.2 ml/min. Detection was performed using a three-detector static light scattering apparatus (MiniDAWN TREOS, Wyatt Technology, equipped with a quasi-elastic light scattering module) and a refractometer (Optilab rEX, Wyatt Technology). Molecular weight calculations were performed with the ASTRA V software (Wyatt Technology) using a dn/dc (Refractive Index Increment) value of 0.183 ml/g.
Dynamic Light Scattering
Hydrodynamic radius (Rh) measurements were made at 20 °C with a DynaPro DLS instrument (Protein Solutions Inc., Piscataway, NJ). Before measuring 90° light scattering intensity, samples were centrifuged at 20,000 × g for 10 min. MARS samples in different buffers were incubated at room temperature before measurements. Particle translational diffusion coefficients were calculated from decay curves of autocorrelation of light scattering data and converted to the Rh with the Stokes-Einstein equation using Dynamics data analysis software V5 from Protein Solutions Inc.
Sedimentation Velocity
Analytical ultracentrifugation (AUC) experiments were conducted in a Beckman Optima XL-A analytical ultracentrifuge using an An 60 Ti rotor and a double-sector cell of 12-mm path length. Sedimentation velocity measurements were performed at 25,000 rpm at 15 °C. Samples of MARS were extensively dialyzed against the buffers used for sedimentation analysis. Solvent densities were calculated using SEDNTERP software. Scans were analyzed to derive the distribution of sedimentation coefficients using SEDFIT (30).
Sedimentation Equilibrium
Experiments were conducted as described previously (31). Equilibrium was verified from the superimposition of duplicate scans recorded at 4-h intervals. The experimental sedimentation equilibrium data were fitted to a model for a single homogeneous species following the equation,
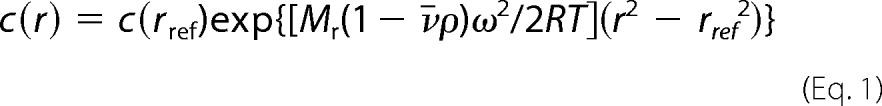 |
where c(r) is the protein concentration at radial position r, c(rref) is the concentration of the protein at an arbitrary reference radial distance rref, Mr is the molecular mass, ¯ν is the partial specific volume (0.733 at 4 °C for MARS) of the solute, ρ is the density of the solvent, ω is the angular velocity of the rotor, and R and T are the molar gas constant and the absolute temperature, respectively.
Small-angle X-ray Scattering
SAXS data were collected on the SWING beamline at SOLEIL Synchrotron using an Aviex CCD detector. The sample to detector distance was 1.84 m to cover a Q-range from 0.004 to 0.5 Å−1, with Q = 4πsinθ/λ, where 2θ is the scattering angle, and λ = 1.03 Å is the x-ray wavelength. Accurate SAXS measurements were performed on the MARS complex eluting from an analytical size-exclusion chromatography column with 20-MDa exclusion limit (Shodex KW405–4F 4.6 × 300 mm) directly into the SAXS flow-through capillary cell (32) at a flow rate of 0.05 ml/min in a buffer containing 20 mm Hepes, pH 7.5, 150 mm NaCl, 2 mm DTT. The volume and concentration of the MARS complex injected on the column were 0.1 ml and 8.5 mg/ml, respectively. The individual SAXS patterns of the frames displaying a constant radius of gyration (Rg) in the elution peak were averaged to provide a high statistics SAXS curve for the complex (32, 33). The individual SAXS patterns recorded before the column dead volume were also averaged to derive the buffer scattering. The data were processed and analyzed using Foxtrot, a SAXS software developed on the SWING beamline, and the program package ATSAS (v.2.5.0) (34) or the program foXS (35). The radius of gyration (Rg) and the intensity at the origin I(0) were derived from the distance distribution function P(r) of the particle using the program PRIMUS (36) as it was difficult to accurately derive the values from Guinier's approximation in the collected Q-range because of elongated shape and large dimensions of MARS particles (34). The three-dimensional envelopes of MARS complex were determined ab initio from the scattering curve using the program DAMMIF (37, 38), which generates a compact beads model fitting the experimental data to minimize the discrepancy,
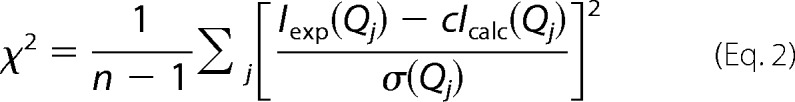 |
where n is the number of experimental points, Iexp(Qj) is the experimental intensity, Icalc(Qj) is the calculated intensity, σ(Qj) is the experimental error, and c is a scaling factor. Dummy atom models from repeated calculations were superimposed and compared using the program DAMAVER (39). EM density maps were converted into bead models in PDB-like formats using the program EM2DAM from ATSAS. Hydrodynamic parameters were calculated from bead models or crystal structures using HYDROPRO v.10 (40).
RESULTS
Purification and Subunit Composition of MARS
The components of MARS are multidomain proteins and are very sensitive to uncontrolled proteolysis that generates active, but truncated and free enzymes (41, 42). We previously established a purification strategy designated to provide protection against proteolysis (43). To obtain complexes suitable for structural analyses, the isolation scheme was slightly modified to exclude the use of PEG for fractional precipitation of the crude extract and to remove glycerol during the final step of the purification procedure. Approximately 5 mg of highly purified MARS could be reproducibly obtained using the 48-h protocol described under “Experimental Procedures.” The characteristic features of the eleven polypeptide components of MARS are summarized in Table 1. The polypeptide components range from 174 amino acid residues for p18, with a calculated Mr of 19,810 Da, to the bifunctional GluProRS made of 1512 residues, with a calculated Mr of 170,591 Da. According to the intensity of the polypeptides stained with Coomassie Blue after SDS-PAGE, determined by densitometric scanning (Ref. 42 and this study) and to active site titration of the synthetase components with aminoacyl adenylates (44), a relative stoichiometry of the 11 polypeptides is proposed (Table 1). These putative stoichiometries are consistent with the fact that ProRS, LysRS, AspRS, p43, and p38 are dimers in solution, whereas IleRS, LeuRS, MetRS, and GlnRS are monomers. Taking into account these values, the molecular mass of the complex is estimated to be equal to 1,505,993 Da. The dimer of GluProRS, which is a paradigm of a multidomain protein especially sensitive to proteolysis, accounts for ∼23% of the mass of the particle, whereas the two dimers of LysRS account for 18% (Fig. 1 and Table 1). The volume of one molecule of MARS was estimated taking into account an average partial specific volume of 0.73 cm3/g determined for soluble proteins (45). Its predicted volume of 1,822,252 Å3 suggests that MARS is the largest stable supramolecular assembly involved in translation, larger than the 50 S ribosomal subunit (1,374,538 Å3 (46)). This volume should be considered as a rough estimate, as the standard value of 0.73 cm3/g for protein partial specific volume may be inaccurate for multidomain non-globular proteins and their large molecular assemblies.
TABLE 1.
Summary of MARS composition
The following are indicated for each polypeptide component of MARS: number of amino acid residues per polypeptide chain, the corresponding calculated molecular mass, the molar ratio of each component per mole of complex (1 for a monomer, 2 for a dimer, and 1 + 1 or 2 + 2 for 2 monomers or 2 dimers per molecule of MARS), total mass for each component and the corresponding percentage of the total mass of the complex, and the total volume for each component calculated according to Voss and Gerstein (46).
| Component | Number of residues | Mr | Molar ratio | Mass | Mass | Volume |
|---|---|---|---|---|---|---|
| Da | Da | % | Å3 | |||
| GluProRS | 1512 | 170,591 | 2 | 341,182 | 22.7 | 412,830 |
| IleRS | 1266 | 144,941 | 1 | 144,941 | 9.6 | 175,379 |
| LeuRS | 1176 | 134,411 | 1 | 134,411 | 8.9 | 162,637 |
| MetRS | 900 | 101,116 | 1 | 101,116 | 6.7 | 122,350 |
| GlnRS | 775 | 87,799 | 1 | 87,799 | 5.8 | 106,237 |
| LysRS | 597 | 68,048 | 2 + 2 | 272,192 | 18.1 | 329,352 |
| ArgRS | 660 | 75,379 | 1 + 1 | 150,758 | 10.0 | 182,417 |
| AspRS | 501 | 57,192 | 2 | 114,384 | 7.6 | 138,405 |
| p43 | 312 | 34,352 | 2 | 68,704 | 4.6 | 83,132 |
| p38 | 320 | 35,348 | 2 | 70,696 | 4.7 | 85,542 |
| p18 | 174 | 19,810 | 1 | 19,810 | 1.3 | 23,970 |
| Total | 13,289a | 1,505,993a | 100 | 1,822,252a |
a Taking into account molar ratios indicated.
FIGURE 1.

Polypeptide composition of the MARS complex. The multi-aminoacyl-tRNA synthetase complex purified from rabbit liver was analyzed by SDS-PAGE, and visualized by Coomassie staining. The identity of the polypeptide subunits (left) and their molecular masses (right) are indicated according to Mirande et al. (62). The pie graph represents the percentage of the total mass contributed by each subunit of the complex taking into account the molar ratios reported in Table 1. GluProRS accounts for 22.7% of the mass of the complex and p18 accounts for only 1.3%.
Homogeneous MARS Diffuses as a Non-globular Protein
To evaluate the homogeneity of the MARS preparation and to get a first experimental estimate of the size of the molecule, MARS was analyzed by SEC on a Superose 6 column (Fig. 2A). The complex was eluted as a reasonably symmetrical peak, as assessed by the good correlation observed with the bell-shape curve obtained by fitting experimental data to a gaussian function. The deduced average partition coefficient Kav corresponds to that of a globular protein of >3,000 kDa (Fig. 2A, inset). Taking into account the calculated Mr of ∼1,500 kDa, these data suggest that MARS diffuses as a non-globular molecule. We also used SEC-MALLS to determine the mass of the complex (Fig. 2B). A mass of 1594 ± 170 kDa was obtained, with a Rh of 15.1 ± 1.1 nm, in good agreement with its theoretical mass (Table 1). The homogeneity of MARS was also appraised by dynamic light scattering (DLS), which gives an estimate of the size and distribution of molecules in solution. The Rh of MARS was evaluated in different buffers (HEPES/NaCl and potassium phosphate buffer) and at protein concentrations ranging from 0.2 to 2 mg/ml (Fig. 2C). The MARS sample showed a monomodal size distribution. At a low protein concentration of 0.6 mg/ml, the percentage of polydispersity was equal to 15.5%, a value that ascertains that, for a large molecule of the size of MARS, the protein sample is monodisperse in solution. When Rh was extrapolated to infinite dilution, a condition that allows minimizing the incidence of protein-protein interactions on the measure, Rh values of 14.7 ± 2.5 and 14.8 ± 2.5 nm could be determined for MARS in the HEPES/NaCl and potassium phosphate buffers, respectively. A spherical particle of 1.5 MDa is expected to display an Rh below 11 nm, whereas the Rh for an elongated particle would be higher (47), suggesting that MARS is an elongated molecule in solution.
FIGURE 2.

Structural homogeneity of the purified MARS complex. A, purified MARS was subjected to size exclusion chromatography on a Superose 6 column as described under “Experimental Procedures.” The experimental elution profile at 280 nm (red curve) was fitted to a Gaussian equation (black dotted line). The apparent molecular mass of MARS was deduced from its relative elution volume Kav of 0.147. B, for SEC-MALLS analysis, MARS was subjected to SEC on a BioSep-SEC-s4000 column. Elution of MARS was monitored at 254 nm (red curve) and molecular mass (right axis, blue curve) is displayed for chromatographic peak. C, the hydrodynamic radius Rh of MARS was determined by DLS. The left panel shows the distribution by mass of a sample of MARS analyzed at 0.6 mg/ml in 20 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm DTT. The right panel shows Rh as a function of the concentration of MARS in the sample when analyzed in 20 mm HEPES-NaOH, pH 7.5, 150 mm NaCl, 2 mm DTT (●) or in 250 mm potassium phosphate, pH 6.8, 2 mm DTT (■). Each value is the mean of three independent determinations.
Analytical Ultracentrifugation Confirms an Elongated Shape of MARS
To get more insight into the homogeneity of the MARS preparation and into the shape of the MARS particle, we carried out analytical ultracentrifugation analysis. Sedimentation velocity analysis of MARS was conducted at an initial protein concentration of 1.55 mg/ml in 20 mm Tris-HCl, pH 7.5, 50 mm NaCl, 5 mm 2-mercaptoethanol in the presence or absence of 5% glycerol or in 25 mm potassium phosphate, pH 7.5, 5 mm 2-mercaptoethanol, 5% glycerol. Identical results were obtained, and only the analysis in the first of these buffers is shown (Fig. 3A). No rapidly sedimenting material was observed, ruling out the presence of large aggregates. The monomodal c(s) distribution observed in different buffer conditions is consistent with the presence of a single species in solution. Analysis of experimental data using SEDFIT (30) gave the best fit for a monodisperse solute with a sedimentation coefficient s20,w of 21.81 S. These data are compatible with a calculated frictional ratio f/f0 of 1.8 and a hydrodynamic radius Rh of 12.33 nm. The frictional ratio above 1.5, or the ratio Smax/S estimated between 1.8 and 2.0 (Table 2), further indicates that MARS particles display solution properties characteristic of proteins with a degree of asymmetry at the frontier between moderately and highly elongated proteins.
FIGURE 3.

Hydrodynamic properties of MARS determined by analytical ultracentrifugation. A, MARS (initial concentration of 1.55 mg/ml) was subjected to sedimentation velocity analysis at 25,000 rpm in 20 mm Tris-HCl, pH 7.5, 50 mm NaCl, 5 mm 2-mercaptoethanol, 5% glycerol at 15 °C. Radial scans were taken at 280 nm at 5-min intervals. Experimental values (open circles) were fitted (curve) to a single species model (only 10 scans are shown for clarity). Residuals are indicated. Sedimentation distribution c(s) versus s plot revealed the presence of a single species in solution. B, MARS (initial concentration of 0.24 mg/ml) was analyzed by equilibrium sedimentation at 5000 rpm for 72 h in 20 mm Tris-HCl, pH 7.5, 100 mm NaCl, and 5 mm 2-mercaptoethanol at 4 °C. Experimental values (closed circles) were fitted (curve) to a monodisperse solute of 1186 ± 102 kDa. The residuals are indicated.
TABLE 2.
Summary of the hydrodynamic properties and structural features determined for MARS particles in solution or estimated from bead models representing the ab initio SAXS and EM three-dimensional reconstructions of MARS molecular envelope
Mr, molecular mass; Rh/Rg = hydrodynamic/gyration radius; f/f0, frictional ratio; S, sedimentation coefficient; Sved, Svedberg units; Dmax, maximum particle size.
| Experimental or predicted values from | Mr | Rh | f/f0 | S | Rg | Dmax | Volume | Shape factorsa |
|
|---|---|---|---|---|---|---|---|---|---|
| Rg/Rh | Smax/S | ||||||||
| MDa | Å | Sved | Å | Å | 106 Å3 | ||||
| SDS-PAGE analysis + titrationb | ∼1.51 | ∼1.82c | ∼2.0 | ||||||
| SEC-MALLS | 1.59 ± 0.02 | 151 ± 11 | |||||||
| DLS | 147 ± 25 | 0.93–1.16 | |||||||
| AUC | ∼1.19 ± 0.1 | 123 | 1.8 | 21.81 | 1.13 | 1.85 ± 0.1 | |||
| SAXS | 139 ± 2 | 520 ± 25 | |||||||
| Selected SAXS bead models | 130–136 | 21–22f | 138–141 | 490–520 | 1–1.1 | 1.9–2.0 | |||
| EM volumes in negative staind | 80 | 35f | 67 | 218 | 1.5 | 0.84 | 1.2 | ||
| EM volumes in vitreous iced | 99 | 28f | 74 | 271 | 1.6 | 0.75 | 1.4 | ||
| Escherichia coli 50 S ribosomee | 1.55 | 99 | 50f | 73 | 271 | 1.7 | 0.74 | 1 | |
| E. coli 70 S ribosomee | 2.5 | 117 | 66f | 89 | 288 | 2.7 | 0.76 | 1 | |
a Rg/Rh is typically ∼0.77 and ∼1.4 for a globular and denatured protein, respectively (50), and Smax/S is in the ranges 1.2–1.3, 1.5–1.9, and 2.0–3.0 for globular, moderately elongated, and highly elongated proteins, respectively (47). Rg/Rh was determined using Rg obtained by SAXS or predicted from the bead models, and Smax/S was determined using Smax ∼ 0.00361(Mr)2/3 (47), Mr ∼ 1.2 MDa for all bead models as in Wolfe et al. (29), and S was obtained by AUC or predicted from the bead models.
b SDS-PAGE densitometric analysis combined with enzymatic titration.
c Sum of the standard atomic volumes of each component according to Voss and Gerstein (46) (Table I).
d Volumes were from EM images in negative stain and vitreous ice filtered to 30 and 36 Å, respectively, from Norcum and Boisset (27).
e PDB code 2AW4 (50 S); PDB codes 2AW4 + 2AVY for 70 S.
f Solution properties predicted from bead models or crystal structures with the program HYDROPRO (40).
To get an estimate of the mass of MARS that does not rely on the shape of the molecule, a sedimentation equilibrium analysis was performed (Fig. 3B). When MARS was subjected to centrifugation equilibrium at an initial protein concentration of 0.24 mg/ml, experimental data obtained after centrifugation for 72 h could be fitted to a single species with a molecular mass of 1186 ± 102 kDa. This value is significantly lower than the theoretical mass of the particle (1,505,993 Da; Table 1). The experimental mass of 1186 kDa was determined using a partial specific volume of 0.733, characteristic of globular proteins. A multidomain, non-compact, large supramolecular assembly may display a quite different ¯ν. A mass of ∼1.5 MDa was calculated when using a partial specific volume of 0.78 cm3/g, which would be more consistent with the data obtained by SAXS (see below). The origin of the discrepancy is not known but could also be related to the duration of an equilibrium sedimentation experiment of about 72 h.
The Low Resolution Solution Structure of the MARS Complex Obtained by SAXS Analysis Differs in Many Aspects from Previous Electron Microscopic Low Resolution Models
To determine more precisely the overall structure, size, and shape of MARS complex in solution, we undertook a low resolution structural analysis of this large molecular assembly by SAXS. Accurate SAXS measurements in solution were conducted near physiological ionic conditions on homogeneous and monodisperse particles of MARS eluted online from a size exclusion chromatography column (32). High quality SAXS data on MARS were thus obtained up to a momentum transfer (Qmax) of 0.25 Å−1 (Fig. 4A). The obtained experimental scattering pattern differs significantly from the scattering patterns computed from bead models representing the previous three-dimensional reconstructions of rabbit MARS particles from EM images of samples negatively stained or in vitreous ice and filtered to 30 and 36 Å resolution, respectively (27). The two EM envelopes have the same dimensions and rather compact, asymmetric Y-shape, but the molecular envelope obtained by cryoelectron microscopy displays additional openings and indentations on most sides of the three-domain Y-shaped structure, articulating potentially each main bulky Y-arm into several subdomains (Fig. 5B). The SAXS pattern derived from this latter model displays overall a slightly lower divergence with the experimental SAXS pattern of MARS in solution, but a very large discrepancy was observed with both EM models at small scattering angles (low Q values), indicating a strong divergence in the overall particle size (Fig. 4A).
FIGURE 4.

The SAXS pattern analysis of MARS in solution suggests striking differences with previous EM three-dimensional reconstructions. A, the experimental SAXS pattern of monodisperse MARS particles (black) diverges significantly, especially at low Q values between 0 and 0.05 Å−1, from the scattering patterns computed from bead models filling the negative stain (green) and cryo (cyan) EM three-dimensional reconstructions filtered to 30 and 36 Å resolution, respectively, and shown in Fig. 5B. The red theoretical SAXS pattern computed from a typical ab initio elongated SAXS bead model generated by DAMMIF, as shown in Fig. 5A, displays a good fit with the experimental data with a χ2 of ∼1.5 up to Qmax = 0.25 Å−1. B, shown are dimensionless Kratky plots for MARS SAXS data (black) and the two EM models with the same color code as in A, suggesting an overall well-folded but non-compact conformation of MARS particles in solution (black). C, shown is distance distribution function P(r) derived from the intensity curves I(q) extrapolated to zero concentration with the same color codes. The values of Rg = 139 ± 2 Å and Dmax = 520 ± 25 Å for MARS particles derived from the black curve extend significantly beyond the values obtained from the two EM three-dimensional reconstructions (Rg/Dmax = 67/217 or 76/274 Å).
FIGURE 5.

Ab initio SAXS envelopes suggest an elongated and multi-armed shape for MARS particles in solution. A, shown are six independent ab initio low resolution bead models calculated using DAMMIF fitting the experimental SAXS pattern shown in Fig. 4A and the hydrodynamic properties obtained by DLS and AUC for MARS assemblies in solution. B, shown is superposition of 10 such bead SAXS models of MARS (red) and bead models from the negative stain (green) and cryo (cyan) EM three-dimensional reconstructions filtered to 30 and 36 Å resolution, respectively (27). The three low resolution envelopes are shown on the same scale in three different views. The radii of the beads in the superposed SAXS models are proportional to their frequency among individual models. C, for a scale comparison, MARS components for which structural data are known are shown: an 18-kDa C-terminal structural fragment of p43 (13), the 19-kDa dimeric p18 protein (green and blue protomers) (20), an N-terminally truncated, 58-kDa dimeric LysRS (KRS) (23), a 50-kDa dimeric AspRS (DRS) structural fragment (63). The bound structure of yeast tRNAAsp (64) is also shown in black sticks.
The degree of compactness or folding/unfolding of the complex was first assessed by a Kratky analysis of MARS SAXS data (48). A dimensionless Kratky plot was used to distinguish between a fully and partially unfolded protein assembly (49). In such representation a globular folded structure is expected to exhibit a bell-shaped profile between 0 < q.Rg < 3 with a single pronounced maximum at q.Rg ∼ 1.7 regardless of the size of the protein (49, 50). These main features are observed in the dimensionless Kratky plot derived from the calculated SAXS pattern of the bead model filling the rather compact EM Y-shape of negatively stained samples (27) (Fig. 4B, green curve). In contrast, the plot derived from MARS SAXS data exhibits a bell-shaped profile with two clear maxima at q·Rg values of 2.1 and 7.8. The presence of the first maximum combined with the absence of significant increase at higher scattering angles indicates no significant disorder and is consistent with the presence of folded domains within the protein assembly. The presence of the second strong peak suggests a structure either strongly symmetrical, which is unlikely here because MARS contains an array of proteins with different sizes, or with large scale density fluctuations, thus possibly containing several arms. In agreement with this hypothesis, second peaks appear in the Kratky plot (Fig. 4B, cyan curve) of the second EM model from frozen hydrated samples, containing small openings and indentations on most side of the triangular volume, whereas they are absent in the corresponding compact EM volume derived from negative staining (Fig. 4B, green curve). By analogy, large indentations can be expected on MARS molecular envelope obtained by SAXS.
The size of MARS particles was next derived in PRIMUS (36) from the probability distance distribution function P(r), which reflects the distribution of interatomic distances in macromolecules. The analysis further confirms that the MARS complex adopts an elongated averaged conformation in solution, in agreement with our hydrodynamic analyses. The P(r) function exhibits an asymmetric profile typical of an extended scattering object, with a peak at r ∼ 140 Å followed by a progressive decrease down to zero at the value of maximal extension Dmax (Fig. 4C). A shoulder at r ∼ 40 Å before the main peak and the limited height of the main peak are consistent with a rather broad distribution of the interatomic distances in several arms/subdomains. The radius of gyration (Rg) and the maximal diameter (Dmax) of the assembly derived from the P(r) are big for the MARS molecular weight with values of 139 ± 2 and 520 ± 25 Å, respectively. These values are much larger than the corresponding values expected for a spherical protein characterized by n = 13,290 residues (Table 1) (Rg ∼ 3n1/3 ∼ 71 Å and Dmax ∼ 2.6Rg ∼ 185 Å) or from the values derived from bead models filling the 30 and 36 Å resolution EM three-dimensional reconstructions (27) (Table 2). The P(r) function corresponding to the molecular envelope of negatively stained EM images exhibits in contrast a symmetric profile with a higher main maximum at r∼80 Å describing its rather compact Y-shaped structure characterized by an Rg and Dmax of 67 and 220 Å, respectively (Table 2). The close envelope obtained from cryoelectron micrographs shifts somewhat the P(r) function in the direction of the experimental P(r), i.e. toward a less symmetric profile and a slightly decreased maximum corresponding to the openings and small indentations found in the three main Y-arms (27) (Fig. 5B). In conclusion, all these structural analyses of the scattering pattern of entire and monodisperse MARS particles in solution suggest a protein assembly more extended and containing more broadly distributed arms/subdomains than in the previous low resolution structural analysis of EM images. This view is supported by the results of ab initio modeling based on the SAXS data.
To obtain three-dimensional models of the shape of MARS complex from scattering intensities, we used the ab initio bead model approach implemented in DAMMIF (38). Several independent ab initio shape determinations were performed starting within a spherical search volume with the diameter Dmax = 540 Å, no symmetry, and different packing radii r0 of dummy atoms. Different models fitting the SAXS experimental data with a low discrepancy χ2 between 1.4 and 1.9 were obtained with r0 = 6 Å (Fig. 4A, red curve). We further restrained among the possible shapes the models whose dimensions and predicted hydrodynamic values were the closest to those obtained experimentally by SEC-MALLS, DLS, and analytical ultracentrifugation. Thus, a final restrained set of bead models could be selected displaying structural and predicted hydrodynamic parameters within the following ranges: 490 ≤ Dmax ≤ 520 Å, 138 ≤ Rg ≤ 141 Å, 130 ≤ Rh ≤ 136 Å, and 21 ≤ s20,w ≤ 22 S. The overall appearance of this set of final models, displayed in Fig. 5A for six independent ab initio shape determinations, was quite similar; all of them displayed non-globular but elongated shapes containing several arms that differ from the previous EM three-dimensional reconstructions (27) (Fig. 5B). Given that the independent bead models yield nearly identical scattering patterns, the ambiguity illustrated in Fig. 5 had to be attributed to the loss of information in the scattering data due to spherical averaging. Thus, these different models represent possible elongated shapes for the whole MARS complex that are consistent with the hydrodynamic and low resolution structural characterizations obtained here in solutions near physiological ionic conditions.
DISCUSSION
Here, we have characterized the solution low resolution structural properties of the whole rabbit MARS complex by combining hydrodynamic and small angle x-ray solution scattering studies. We have taken care to preserve the homogeneity and structural integrity of the whole complex during the sample preparation and to analyze its native structure and behavior in solution on monodisperse samples under near physiological ionic conditions. With a Rh, Rg, and Dmax above 120, 135, and 450 Å, respectively, MARS displays consequently large overall hydrodynamic and structural features for a supramolecular assembly involved in translation. These values are for example larger than those predicted from known crystal structures of prokaryotic large 50 S ribosomal subunits of ∼1.5 MDa or their 70 S assembly of ∼2.5 MDa, where Dmax values do not extend beyond 270 and 290 Å, respectively (Table 2).
All the determined hydrodynamic properties of MARS particles in solution and the SAXS analysis underline that the multiprotein assembly displays an elongated shape that is notably characterized by a large maximum intramolecular distance (Dmax) above 450 Å (Fig. 5). Such a maximum dimension within the multiprotein assembly is about twice larger than previously found in the rather compact, asymmetric triangular EM low resolution envelopes of rabbit (27) or human (29) MARS particles, where the Dmax fluctuates from 220 Å within the main asymmetric three-domain Y-shape to 280 Å including low resolution protruding densities (Fig. 5B). Both the predicted solution properties (Table 2) or scattering patterns (Fig. 4) computed from the asymmetric triangular volumes of rabbit MARS determined from EM images in negative stain or vitreous ice (27) suggest that the overall EM particle dimensions and shape are too globular to fit the solution or low resolution structural properties we have determined.
The apparently more compact shape of MARS particles observed in EM structural analysis could be the consequence of partial destabilization of MARS during sample preparation for EM analysis or of partial degradation of some of the MARS components. It is worth noting that in several of these studies, the cross-linking agent glutaraldehyde was used to stabilize the complex (26, 27, 51, 52), suggesting an intrinsic instability of MARS during sample preparation for EM analysis. In addition, GluProRS is one of the major components of MARS. It accounts for about one fourth of the mass of the complex (Fig. 1). It is noteworthy that this bifunctional protein, which results from the fusion of GluRS and ProRS on a single polypeptide (53), is especially sensitive to uncontrolled proteolysis, a process that leads to complexes with low content in GluProRS (54), or even to complexes where GluProRS is absent (55). The SDS-PAGE pattern of the MARS used in EM studies (29) reveals a strong deficit in GluProRS, compared with the complex used in our study (Fig. 1 and Table 1), which might contribute to the global size discrepancy. It is also conceivable that these two different approaches, EM, which requires adsorption of the complex on carbon films, and SAXS, which gives structural data of the complex in solution, could induce different shapes on the complexes. In this connection, a recent study emphasized the large structural changes undergone by the yeast GluRS·Arc1p·MetRS complex upon tRNA binding (56). A SAXS analysis revealed that the trimeric complex containing one copy of each component has an Rg of 97 Å, whereas the pentameric complex containing two molecules of tRNA bound to the GluRS·Arc1p·MetRS complex displays a more compact structure, with a Rg of 60 Å. Thus, we cannot exclude that the EM structure of MARS might correspond to a drastically compacted structure compared with the solution structure observed here in solution under nearly physiological conditions.
Ab initio shape determinations of monodisperse MARS particles from the SAXS data suggest possible average conformations of MARS in solution. However, ab initio structure determinations by SAXS cannot identify a unique functional assembly as different three-dimensional models can show identical SAXS one-dimensional profiles at a given resolution. We have further selected SAXS molecular envelopes whose predicted solution properties approach the experimental values we have determined (Fig. 5A). Average conformations of MARS in solution, which are consistent with our SAXS and hydrodynamic data, form elongated molecular envelopes with bulky protrusions or arms more evenly distributed along the largest dimension of the complex. The Kratky plot analysis of SAXS data does not reveal overall significant partial disorder/unfolding in MARS (Fig. 4B), but we cannot exclude that local flexibility exists in such an elongated and multi-armed shape. The elongated and multi-armed shapes of MARS eventually provide a larger surface of solvent accessibility to all its components than does a compact arrangement. This could be essential for the simultaneous catalytic activities of its nine aminoacyl-tRNA synthetases or for interactions with other factors involved in protein synthesis such as the EF1 complex. In the multiprotein assembly, the nine different bulky and negatively charged tRNA substrates have to move into and out of their cognate catalytic binding sites without inhibition due to electrostatic repulsion (57). The tRBD appended to eukaryotic aminoacyl-tRNA synthetases are positively charged and strengthen the interaction between tRNAs and enzymes. They may play the role of electrostatic insulators. In human MetRS, the C-terminal tRBD has a helix-turn-helix fold displaying five basic residues on one side of this protein domain (24). This helix-turn-helix motif is a cis-acting element for tRNA binding (58). A similar tRBD is also recovered in human GluProRS (three motifs in tandem), HisRS, GlyRS, or TrpRS. In the crystal structure of native, dimeric human TrpRS determined in the absence of tRNA, only one of the helix-turn-helix domains is visible in the electron density map and is related to the core enzyme by a disordered, flexible linker (59). In human LysRS, the N-terminal tRBD is a long α-helix (60, 61), but only an N-terminally truncated derivative of this enzyme could be crystallized (22). The multi-armed shape of MARS observed by SAXS could also at least be due in part to the flexibility of the tRBD appended to these synthetases. The non-compact structural organization of MARS would be especially suited to allow large structural rearrangements of the cis-acting polypeptide extensions of the synthetases upon tRNA binding. This shape is thus appropriate for functional constraints and may additionally favor efficient release of MARS subunits upon proteolysis/disassembly for their other non-canonical functions.
In conclusion, the present work represents a new stimulating low resolution structural template of MARS to understand the structure-function relationship and spatiotemporal regulation of the activity of its multiple components. A clear picture of the internal protein topography of MARS awaits further functional and structural studies. Similar solution structural studies of MARS bound to large binding partners such as the elongation factor complex or of stable sub-complexes of MARS will allow analysis of the potential plasticity and identification of the contribution of its different components in the overall molecular envelope.
Acknowledgments
We thank Karine Madiona (Laboratoire d'Enzymologie et Biochimie Structurales, Gif-sur-Yvette) and Christophe Velours (IMAGIF, Gif-sur-Yvette) for analytical ultracentrifugation and SEC-MALLS experiments and Eric Larquet (Laboratoire d'Enzymologie et Biochimie Structurales, Gif-sur-Yvette) for fruitful discussions. We are also indebted to Mona Norcum (University of Mississippi Medical Center, Jackson, MS) and to our late colleague and friend Nicolas Boisset (Université Pierre et Marie Curie, Paris) for kindly providing us the EM three-dimensional reconstruction maps of rabbit MARS.
This work was supported by grants from the Centre National de la Recherche Scientifique, the Association pour la Recherche sur le Cancer, and the Agence Nationale de Recherche sur le SIDA.
- tRBD
- tRNA binding domain
- MARS
- multi-aminoacyl-tRNA synthetase
- SAXS
- small-angle x-ray scattering
- SEC-MALLS
- size exclusion chromatography coupled to multiangle laser light scattering
- Rh
- hydrodynamic radius
- DLS
- dynamic light scattering
- Rg
- radius of gyration
- Dmax
- maximal diameter.
REFERENCES
- 1. Ibba M., Soll D. (2000) Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 69, 617–650 [DOI] [PubMed] [Google Scholar]
- 2. Guo M., Schimmel P., Yang X. L. (2010) Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Lett. 584, 434–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mirande M. (2005) The Aminoacyl-tRNA Synthetases (Ibba M., Francklyn C., Cusack S., eds.) pp. 298–308, Landes Bioscience, Georgetown, TX [Google Scholar]
- 4. Mirande M. (2010) Processivity of translation in the eukaryote cell. Role of aminoacyl-tRNA synthetases. FEBS Lett. 584, 443–447 [DOI] [PubMed] [Google Scholar]
- 5. Mirande M. (1991) Aminoacyl-tRNA synthetase family from prokaryotes and eukaryotes. Structural domains and their implications. Prog. Nucleic Acid Res. Mol. Biol. 40, 95–142 [DOI] [PubMed] [Google Scholar]
- 6. Lee S. W., Cho B. H., Park S. G., Kim S. (2004) Aminoacyl-tRNA synthetase complexes. Beyond translation. J. Cell Sci. 117, 3725–3734 [DOI] [PubMed] [Google Scholar]
- 7. Kerjan P., Cerini C., Sémériva M., Mirande M. (1994) The multienzyme complex containing nine aminoacyl-tRNA synthetases is ubiquitous from Drosophila to mammals. Biochim. Biophys. Acta 1199, 293–297 [DOI] [PubMed] [Google Scholar]
- 8. Kaminska M., Havrylenko S., Decottignies P., Gillet S., Le Maréchal P., Negrutskii B., Mirande M. (2009) Dissection of the structural organization of the aminoacyl-tRNA synthetase complex. J. Biol. Chem. 284, 6053–6060 [DOI] [PubMed] [Google Scholar]
- 9. Ray P. S., Arif A., Fox P. L. (2007) Macromolecular complexes as depots for releasable regulatory proteins. Trends Biochem. Sci. 32, 158–164 [DOI] [PubMed] [Google Scholar]
- 10. Arif A., Jia J., Moodt R. A., DiCorleto P. E., Fox P. L. (2011) Phosphorylation of glutamyl-prolyl tRNA synthetase by cyclin-dependent kinase 5 dictates transcript-selective translational control. Proc. Natl. Acad. Sci. U.S.A. 108, 1415–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yannay-Cohen N., Carmi-Levy I., Kay G., Yang C. M., Han J. M., Kemeny D. M., Kim S., Nechushtan H., Razin E. (2009) LysRS serves as a key signaling molecule in the immune response by regulating gene expression. Mol. Cell 34, 603–611 [DOI] [PubMed] [Google Scholar]
- 12. Kim Y., Shin J., Li R., Cheong C., Kim K., Kim S. (2000) A novel anti-tumor cytokine contains an RNA binding motif present in aminoacyl-tRNA synthetases. J. Biol. Chem. 275, 27062–27068 [DOI] [PubMed] [Google Scholar]
- 13. Renault L., Kerjan P., Pasqualato S., Ménétrey J., Robinson J. C., Kawaguchi S., Vassylyev D. G., Yokoyama S., Mirande M., Cherfils J. (2001) Structure of the EMAPII domain of human aminoacyl-tRNA synthetase complex reveals evolutionary dimer mimicry. EMBO J. 20, 570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park S. G., Choi E. C., Kim S. (2010) Aminoacyl-tRNA synthetase-interacting multifunctional proteins (AIMPs). A triad for cellular homeostasis. IUBMB Life 62, 296–302 [DOI] [PubMed] [Google Scholar]
- 15. Shalak V., Guigou L., Kaminska M., Wautier M. P., Wautier J. L., Mirande M. (2007) Characterization of p43(ARF), a derivative of the p43 component of multiaminoacyl-tRNA synthetase complex released during apoptosis. J. Biol. Chem. 282, 10935–10943 [DOI] [PubMed] [Google Scholar]
- 16. Quevillon S., Robinson J. C., Berthonneau E., Siatecka M., Mirande M. (1999) Macromolecular assemblage of aminoacyl-tRNA synthetases. Identification of protein-protein interactions and characterization of a core protein. J. Mol. Biol. 285, 183–195 [DOI] [PubMed] [Google Scholar]
- 17. Rho S. B., Kim M. J., Lee J. S., Seol W., Motegi H., Kim S., Shiba K. (1999) Genetic dissection of protein-protein interactions in multi-tRNA synthetase complex. Proc. Natl. Acad. Sci. U.S.A. 96, 4488–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Norcum M. T., Warrington J. A. (1998) Structural analysis of the multienzyme aminoacyl-tRNA synthetase complex. A three-domain model based on reversible chemical cross-linking. Protein Sci. 7, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson J. C., Kerjan P., Mirande M. (2000) Macromolecular assemblage of aminoacyl-tRNA synthetases. Quantitative analysis of protein-protein interactions and mechanism of complex assembly. J. Mol. Biol. 304, 983–994 [DOI] [PubMed] [Google Scholar]
- 20. Kim K. J., Park M. C., Choi S. J., Oh Y. S., Choi E. C., Cho H. J., Kim M. H., Kim S. H., Kim D. W., Kim S., Kang B. S. (2008) Determination of three-dimensional structure and residues of the novel tumor suppressor AIMP3/p18 required for the interaction with ATM. J. Biol. Chem. 283, 14032–14040 [DOI] [PubMed] [Google Scholar]
- 21. Simader H., Hothorn M., Köhler C., Basquin J., Simos G., Suck D. (2006) Structural basis of yeast aminoacyl-tRNA synthetase complex formation revealed by crystal structures of two binary sub-complexes. Nucleic Acids Res. 34, 3968–3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo M., Ignatov M., Musier-Forsyth K., Schimmel P., Yang X. L. (2008) Crystal structure of tetrameric form of human lysyl-tRNA synthetase. Implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. U.S.A. 105, 2331–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ofir-Birin Y., Fang P., Bennett S. P., Zhang H. M., Wang J., Rachmin I., Shapiro R., Song J., Dagan A., Pozo J., Kim S., Marshall A. G., Schimmel P., Yang X. L., Nechushtan H., Razin E., Guo M. (2013) Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol. Cell 49, 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cahuzac B., Berthonneau E., Birlirakis N., Guittet E., Mirande M. (2000) A recurrent RNA-binding domain is appended to eukaryotic aminoacyl-tRNA synthetases. EMBO J. 19, 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jeong E. J., Hwang G. S., Kim K. H., Kim M. J., Kim S., Kim K. S. (2000) Structural analysis of multifunctional peptide motifs in human bifunctional tRNA synthetase. Identification of RNA-binding residues and functional implications for tandem repeats. Biochemistry 39, 15775–15782 [DOI] [PubMed] [Google Scholar]
- 26. Norcum M. T. (1999) Ultrastructure of the eukaryotic aminoacyl-tRNA synthetase complex derived from two-dimensional averaging and classification of negatively stained electron microscopic images. FEBS Lett. 447, 217–222 [DOI] [PubMed] [Google Scholar]
- 27. Norcum M. T., Boisset N. (2002) Three-dimensional architecture of the eukaryotic multisynthetase complex determined from negatively stained and cryoelectron micrographs. FEBS Lett. 512, 298–302 [DOI] [PubMed] [Google Scholar]
- 28. Norcum M. T., Dignam J. D. (1999) Immunoelectron microscopic localization of glutamyl-/prolyl-tRNA synthetase within the eukaryotic multisynthetase complex. J. Biol. Chem. 274, 12205–12208 [DOI] [PubMed] [Google Scholar]
- 29. Wolfe C. L., Warrington J. A., Treadwell L., Norcum M. T. (2005) A three-dimensional working model of the multienzyme complex of aminoacyl-tRNA synthetases based on electron microscopic placements of tRNA and proteins. J. Biol. Chem. 280, 38870–38878 [DOI] [PubMed] [Google Scholar]
- 30. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agou F., Waller J. P., Mirande M. (1996) Expression of rat aspartyl-tRNA synthetase in Saccharomyces cerevisiae. Role of the NH2-terminal polypeptide extension on enzyme activity and stability. J. Biol. Chem. 271, 29295–29303 [DOI] [PubMed] [Google Scholar]
- 32. David G., Pérez J. (2009) Combined sampler robot and high-performance liquid chromatography. A fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 42, 892–900 [Google Scholar]
- 33. Pérez J., Nishino Y. (2012) Advances in X-ray scattering. From solution SAXS to achievements with coherent beams. Curr. Opin. Struct. Biol. 22, 670–678 [DOI] [PubMed] [Google Scholar]
- 34. Konarev P. V., Volkov V. V., Petoukhov M. V., Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schneidman-Duhovny D., Hammel M., Sali A. (2010) FoXS. A web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS. A Windows-PC based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 37. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ortega A., Amorós D., García de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cirakoglu B., Waller J. P. (1985) Leucyl-tRNA and lysyl-tRNA synthetases, derived from the high-Mr complex of sheep liver, are hydrophobic proteins. Eur. J. Biochem. 151, 101–110 [DOI] [PubMed] [Google Scholar]
- 42. Mirande M., Kellermann O., Waller J. P. (1982) Macromolecular complexes from sheep and rabbit containing seven aminoacyl-tRNA synthetases. II. Structural characterization of the polypeptide components and immunological identification of the methionyl-tRNA synthetase subunit. J. Biol. Chem. 257, 11049–11055 [PubMed] [Google Scholar]
- 43. Mirande M., Le Corre D., Waller J. P. (1985) A complex from cultured Chinese hamster ovary cells containing nine aminoacyl-tRNA synthetases. Thermolabile leucyl-tRNA synthetase from the tsH1 mutant cell line is an integral component of this complex. Eur. J. Biochem. 147, 281–289 [DOI] [PubMed] [Google Scholar]
- 44. Johnson D. L., Yang D. C. (1981) Stoichiometry and composition of an aminoacyl-tRNA synthetase complex from rat liver. Proc. Natl. Acad. Sci. U.S.A. 78, 4059–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harpaz Y., Gerstein M., Chothia C. (1994) Volume changes on protein folding. Structure 2, 641–649 [DOI] [PubMed] [Google Scholar]
- 46. Voss N. R., Gerstein M. (2005) Calculation of standard atomic volumes for RNA and comparison with proteins. RNA is packed more tightly. J. Mol. Biol. 346, 477–492 [DOI] [PubMed] [Google Scholar]
- 47. Erickson H. P. (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Putnam C. D., Hammel M., Hura G. L., Tainer J. A. (2007) X-ray solution scattering (SAXS) combined with crystallography and computation. Defining accurate macromolecular structures, conformations, and assemblies in solution. Q. Rev. Biophys. 40, 191–285 [DOI] [PubMed] [Google Scholar]
- 49. Durand D., Vivès C., Cannella D., Pérez J., Pebay-Peyroula E., Vachette P., Fieschi F. (2010) NADPH oxidase activator p67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J. Struct. Biol. 169, 45–53 [DOI] [PubMed] [Google Scholar]
- 50. Receveur-Brechot V., Durand D. (2012) How random are intrinsically disordered proteins? A small angle scattering perspective. Curr. Protein Pept. Sci. 13, 55–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gulik A., Orsini G. (1984) Electron microscopy study of the aminoacyl-tRNA synthetase multienzymatic complex purified from rabbit reticulocytes. Mol. Biol. Rep. 10, 23–30 [DOI] [PubMed] [Google Scholar]
- 52. Norcum M. T. (1989) Isolation and electron microscopic characterization of the high molecular mass aminoacyl-tRNA synthetase complex from murine erythroleukemia cells. J. Biol. Chem. 264, 15043–15051 [PubMed] [Google Scholar]
- 53. Cerini C., Kerjan P., Astier M., Gratecos D., Mirande M., Sémériva M. (1991) A component of the multisynthetase complex is a multifunctional aminoacyl-tRNA synthetase. EMBO J. 10, 4267–4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kerjan P., Triconnet M., Waller J. P. (1992) Mammalian prolyl-tRNA synthetase corresponds to the 150-kDa subunit of the high Mr aminoacyl-tRNA synthetase complex. Biochimie 74, 195–205 [DOI] [PubMed] [Google Scholar]
- 55. Kellermann O., Tonetti H., Brevet A., Mirande M., Pailliez J. P., Waller J. P. (1982) Macromolecular complexes from sheep and rabbit containing seven aminoacyl-tRNA synthetases. I. Species specificity of the polypeptide composition. J. Biol. Chem. 257, 11041–11048 [PubMed] [Google Scholar]
- 56. Koehler C., Round A., Simader H., Suck D., Svergun D. (2013) Quaternary structure of the yeast Arc1p-aminoacyl-tRNA synthetase complex in solution and its compaction upon binding of tRNAs. Nucleic Acids Res. 41, 667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mirande M., Cirakoğlu B., Waller J. P. (1983) Seven mammalian aminoacyl-tRNA synthetases associated within the same complex are functionally independent. Eur. J. Biochem. 131, 163–170 [DOI] [PubMed] [Google Scholar]
- 58. Kaminska M., Shalak V., Mirande M. (2001) The appended C-domain of human methionyl-tRNA synthetase has a tRNA-sequestering function. Biochemistry 40, 14309–14316 [DOI] [PubMed] [Google Scholar]
- 59. Yang X. L., Otero F. J., Skene R. J., McRee D. E., Schimmel P., Ribas de Pouplana L. (2003) Crystal structures that suggest late development of genetic code components for differentiating aromatic side chains. Proc. Natl. Acad. Sci. U.S.A. 100, 15376–15380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Francin M., Kaminska M., Kerjan P., Mirande M. (2002) The N-terminal domain of mammalian lysyl-tRNA synthetase is a functional tRNA binding domain. J. Biol. Chem. 277, 1762–1769 [DOI] [PubMed] [Google Scholar]
- 61. Francin M., Mirande M. (2003) Functional dissection of the eukaryotic-specific tRNA-interacting factor of lysyl-tRNA synthetase. J. Biol. Chem. 278, 1472–1479 [DOI] [PubMed] [Google Scholar]
- 62. Mirande M., Cirakoğlu B., Waller J. P. (1982) Macromolecular complexes from sheep and rabbit containing seven aminoacyl-tRNA synthetases. III. Assignment of aminoacyl-tRNA synthetase activities to the polypeptide components of the complexes. J. Biol. Chem. 257, 11056–11063 [PubMed] [Google Scholar]
- 63. Kim K. R., Park S. H., Kim H. S., Rhee K. H., Kim B. G., Kim D. G., Park M. S., Kim H. J., Kim S., Han B. W. (2013) Crystal structure of human cytosolic aspartyl-tRNA synthetase, a component of multi-tRNA synthetase complex. Proteins 10.1002/prot.24306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cavarelli J., Eriani G., Rees B., Ruff M., Boeglin M., Mitschler A., Martin F., Gangloff J., Thierry J. C., Moras D. (1994) The active site of yeast aspartyl-tRNA synthetase. Structural and functional aspects of the aminoacylation reaction. EMBO J. 13, 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]