

Background: Nuclear factor I (NFI) phosphorylation controls the expression of glial genes associated with migration.
Results: NFI is dephosphorylated and activated by a cleaved form of calcineurin in malignant glioma cells.
Conclusion: NFI transcriptional activity in malignant glioma is regulated in part by calcineurin.
Significance: Regulation of NFI by calcineurin provides a novel approach to control the expression of genes associated with migration/infiltration in malignant glioma.
Keywords: Brain Tumors, Calcineurin, Calpain, Cancer Biology, Phosphorylation, Nuclear Factor I, Gene Expression, Glial Fibrillary Acidic Protein, Transcription
Abstract
Malignant gliomas (MG), including grades III and IV astrocytomas, are the most common adult brain tumors. These tumors are highly aggressive with a median survival of less than 2 years. Nuclear factor I (NFI) is a family of transcription factors that regulates the expression of glial genes in the developing brain. We have previously shown that regulation of the brain fatty acid-binding protein (B-FABP; FABP7) and glial fibrillary acidic protein (GFAP) genes in MG cells is dependent on the phosphorylation state of NFI, with hypophosphorylation of NFI correlating with GFAP and B-FABP expression. Importantly, NFI phosphorylation is dependent on phosphatase activity that is enriched in GFAP/B-FABP+ve cells. Using chromatin immunoprecipitation, we show that NFI occupies the GFAP and B-FABP promoters in NFI-hypophosphorylated GFAP/B-FABP+ve MG cells. NFI occupancy, NFI-dependent transcriptional activity, and NFI phosphorylation are all modulated by the serine/threonine phosphatase calcineurin. Importantly, a cleaved form of calcineurin, associated with increased phosphatase activity, is specifically expressed in NFI-hypophosphorylated GFAP/B-FABP+ve MG cells. Calcineurin in GFAP/B-FABP+ve MG cells localizes to the nucleus. In contrast, calcineurin is primarily found in the cytoplasm of GFAP/B-FABP-ve cells, suggesting a dual mechanism for calcineurin activation in MG. Finally, our results demonstrate that calcineurin expression is up-regulated in areas of high infiltration/migration in grade IV astrocytoma tumor tissue. Our data suggest a critical role for calcineurin in NFI transcriptional regulation and in the determination of MG infiltrative properties.
Introduction
Malignant gliomas (MG),2 including grades III and IV astrocytomas, are the most common adult brain tumors. These tumors have a dismal prognosis with a median survival of less than 2 years (1). MGs are highly infiltrative, resulting in recurrence despite aggressive treatment, including surgical resection, radiotherapy, and chemotherapy (2). MGs have traditionally been hypothesized to arise from astrocytes as tumor cells express glial fibrillary acidic protein (GFAP), an intermediate filament protein expressed in differentiated astrocytes (3). More recent findings suggest that these tumors may arise from less differentiated glial cell types (4, 5). MG tumors express brain fatty acid-binding protein (B-FABP) (6), a marker of radial glial cells. Radial glial cells have been shown to have neural precursor cell properties as defined by the ability to self-renew and differentiate into glial and neuronal cells (7–11). B-FABP expression correlates with decreased survival in grade IV astrocytomas (12–14), and B-FABP expression increases MG cell migration and is associated with sites of infiltration in MG tumors (5, 15).
Expression of B-FABP and GFAP in MG cells is regulated by nuclear factor I (NFI) (16, 17). The NFI family of transcription factors consists of the four genes NFIA, NFIB, NFIC, and NFIX, all of which can bind to the consensus binding site 5′-TTGGCN5GCCAA-3′ as a homodimer or heterodimer to regulate target gene expression (18, 19). Although the N-terminal DNA binding domain is highly conserved in all four NFIs, the C-terminal domain shows divergence among family members (20). Our laboratory has demonstrated that specific NFIs have distinct effects on NFI-dependent promoter activity (16). Furthermore, NFIs can either activate or repress transcription from NFI-dependent promoters, and regulation by NFI is both tissue- and promoter context-dependent (16, 20).
In addition to B-FABP and GFAP, NFI consensus binding sites have been identified in many brain-specific promoters (21), and NFIs have been shown to be regulators of glial cell differentiation (22–24). Nfia−/− and Nfib−/− mice exhibit agenesis of the corpus callosum, enlargement of lateral ventricles, and reduction of specific glial cell populations (25–27). In addition, Nfib−/− mice have defects in lung maturation (26, 28). Nfix−/− mice show enlargement of lateral ventricles and a host of skeletal defects (29). Unlike Nfia, Nfib, and Nfix knock-out mice, Nfic−/− mice have defects in tooth root development but no apparent brain defects (30, 31). In the developing spinal cord, NFIA and NFIB control glial fate specification (22). At early stages of development, both NFIA and NFIB are necessary for the maintenance of neural progenitor cells, including radial glial cells. At later stages of development, NFIA regulates the migration and differentiation of these precursor cells into astrocytes (22). NFIA has also been shown to be critical for astrocyte differentiation of neural precursor cells in the developing brain (23).
The B-FABP and GFAP promoters each contain three NFI consensus binding sites (16, 17, 32). Based on chromatin immunoprecipitation (ChIP) and electrophoretic mobility shift assays, NFI binds to all three NFI consensus sites in both genes. In addition, we have shown that modulation of NFI expression alters B-FABP and GFAP promoter activity, as well as endogenous expression of B-FABP and GFAP in MG cell lines (16). Our data indicate that NFI is differentially phosphorylated in different MG cell lines and that NFI phosphorylation state correlates with expression of B-FABP and GFAP, i.e. NFI is hyperphosphorylated in MG cell lines that do not express B-FABP or GFAP and is hypophosphorylated in MG cell lines that express B-FABP and GFAP (17). Intriguingly, this differential phosphorylation appears to be due to a phosphatase activity that is specifically present in MG cell lines with hypophosphorylated NFI (17). Thus, regulation of NFI dephosphorylation may be vital to the control of neural/glial gene expression in MG.
Calcineurin is a calcium-dependent serine/threonine phosphatase (33) composed of two subunits as follows: calcineurin A (CNA; PP2B), the catalytic subunit (33), and calcineurin B (CNB), a regulatory calcium-binding subunit (34). Calcineurin plays a wide variety of biological functions, linking calcium signaling to multiple outputs ranging from immediate cellular responses to long term alterations in gene expression (35, 36). In the brain, calcineurin is highly expressed and plays important roles in synaptic plasticity (37–39). In developing cerebellar granule neurons, calcineurin signaling activates NFAT binding to NFI target genes, blocking NFI occupancy. As these neurons mature, binding of NFAT is temporally down-regulated resulting in an increase in NFI binding to target genes (40). A more direct link between calcineurin and NFI comes from the observation that calcineurin is able to activate the transactivation domain of NFIC in fibroblasts (41).
Here, we investigate the regulation of NFI dephosphorylation and activity in MG cell lines. We show that calcineurin regulates NFI dephosphorylation and activity in MG cell lines. In addition, we identify a cleaved form of CNA that is specific to MG cell lines with hypophosphorylated NFI. A similar truncated form of CNA has previously been shown to have increased phosphatase activity, suggesting that NFI dephosphorylation and activation are regulated by activated calcineurin in MG. The discovery of a novel regulatory mechanism for controlling the expression of neural and glial genes in MG opens up new avenues for controlling the growth properties of MG.
EXPERIMENTAL PROCEDURES
Cell Lines, Constructs, Chemicals, and Transfections
The human MG cell lines have been previously described (6, 16). All cell lines were cultured in Dulbecco's modification of Eagle's minimum essential medium supplemented with 10% fetal calf serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). Cyclosporin A (CsA) was obtained from Sigma and ionomycin from Fisher. The pCAT/GFAP reporter construct contains −1708 to +8 bp of the GFAP promoter cloned into the pCAT basic vector. The pCAT/GFAP G-br1*, pCAT/GFAP G-br2*, pCAT/GFAP G-br3*, and pCAT/GFAP G-br1*/G-br2*/G-br3* reporter constructs contain mutations disrupting one or all three NFI-binding sites (16). The HA-tagged constitutively active CNA expression construct (CNA-CA) and catalytically inactive construct (CNA-IN) in pcDNA3 were obtained from Dr. R. Chen (School of Life Sciences, Xiamen University, China) and have previously been described (42). HA-DDX1 cloned into pcDNA3 (Invitrogen) was used as a transfection control. The calpastatin expression vector was obtained from Dr. D. Jay (Cross Cancer Institute, Edmonton, Canada) (43). U251 MG cells were transfected using polyethyleneimine (PEI) (Polysciences Inc). U87 MG cells were transfected by calcium phosphate-mediated DNA precipitation. Cells were harvested 60 h post-transfection. Where indicated, cells were treated 24 h post-transfection with drugs and harvested 24 h later. Chloramphenicol acetyltransferase (CAT) activity from pCAT (Promega) in lysates was measured following the manufacturer's protocol. Acetylated [14C]chloramphenicol was quantified in counts/min using a scintillation counter.
Chromatin Immunoprecipitation
ChIP was carried out as described previously (16, 44). Primers (Table 1) were designed to amplify regions of the GFAP and B-FABP promoters containing NFI-binding sites. The GAPDH promoter was used as a negative control. A pan-specific anti-NFI antibody sc-870 (Santa Cruz Biotechnology) and purified rabbit IgG (negative control) were used for immunoprecipitations.
TABLE 1.
Sequences of primers used for ChIP analysis
| Fragment | Forward sequence (5′–3′) | Reverse sequence (5′–3′) |
|---|---|---|
| G-br1 | GTC CTC TTG CTT CAG CGG | TGG GCT AGA CTG GCG ATG |
| G-br2/3 | CAG ACC TGG CAG CAT TGG | CTG CTC AAT GGG CTT CTC G |
| B-br1/2/3 | CGA ACC TGA AAG CCC TTC T | GCT CCT GCC TTC TTA TTT GG |
| GAPDH | GAA CCA GCA CCG ATC ACC | CCA GCC CAA GGT CTT GAG |
Western Blot Analysis and Phosphatase Treatment
Whole cell extracts were prepared by lysing cells on ice for 20 min in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 1× Complete protease inhibitor (Roche Applied Science), 1 mm PMSF. Nuclear extracts were prepared by lysing purified nuclei. Briefly, 3 × 106 cells were resuspended in 200 μl of nuclei isolation buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1.5 mm MgCl2, 0.2 mm CaCl2, 250 mm sucrose, 0.1% Nonidet P-40, 1× Complete protease inhibitor (Roche Applied Science), 1× PhoSTOP phosphatase inhibitor mixture (Roche Applied Science)) and incubated on ice for 10 min. Nuclei were pelleted by centrifugation at 3200 × g for 10 min at 4 °C, washed once in nuclei isolation buffer (45), and lysed in 200 μl of RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100) containing 1× Complete protease inhibitor and 1× PhoSTOP. Chromatin was digested by addition of micrococcal nuclease (New England Biolabs) and 1 μl of 1 m CaCl2. Protein extracts were electrophoresed in 10% polyacrylamide-SDS gels and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were immunostained with mouse anti-CNA (G182-1847; Pharmingen) (1:10,000), rabbit anti-CNAα (07-1492; Millipore) (1:1000), rabbit anti-CNAβ (07-1493; Millipore) (1:1000), rabbit anti-NFI (gift from Dr. N. Tanese, New York University Medical Center) (1:1000), rabbit anti-DDX1 (46) (1:5000), and mouse anti-α-tubulin (12G10; Developmental Studies Hybridoma Bank) (1:100,000) antibodies. Primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) using Immobilon (Millipore). For phosphatase treatment, nuclear extracts were prepared in the absence of 1× PhoSTOP and incubated for 1 h at 37 °C in λ-phosphatase buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 0.1 mm EGTA, 2 mm dithiothreitol, 0.01% Brij 35, 2 mm MnCl2) with or without 400 units of λ-phosphatase (New England Biolabs).
Co-immunoprecipitations
Whole cell extracts were prepared as described above. For co-immunoprecipitations, 500 μg of U251 whole cell extracts were diluted in wash buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.25% sodium deoxycholate, 0.5% Nonidet P-40, 1× Complete protease inhibitor (Roche Applied Science)), incubated with 2 μl of α-CNA antibody or 2 μg of purified mouse IgG (negative control) for 3 h at 4 °C, and immunoprecipitated with protein G-Sepharose beads (GE Healthcare). Immunoprecipitates were washed three times and subjected to Western blot analysis.
Calcineurin Activity Assay
Whole cell lysates were prepared by lysing cells in hypotonic lysis buffer (50 mm Tris-HCl, pH 7.5, 0.1 mm EGTA, 1 mm EDTA, 0.5 mm DTT, 50 μg/ml PMSF, 50 μg/ml trypsin inhibitor, 10 μg/ml leupeptin, 10 μg/ml aprotinin) (47). Calcineurin activity in extracts was measured using γ-32P-RII peptide substrate as described previously (47).
Immunofluorescence Analysis
Cells growing on coverslips were treated for 1 h with 1 μm ionomycin or DMSO (control) and fixed with 1% (U251 cells) or 2% (U87 cells) paraformaldehyde in phosphate-buffered saline for 10 min, followed by permeabilization in 0.5% Triton X-100 for 5 min. Cells were immunostained with mouse α-CNA antibody (1:50–1:200), followed by Alexa 488-conjugated goat anti-mouse secondary antibody (1:200). Coverslips were mounted onto slides with polyvinyl alcohol containing 1 μg/ml 4′6-diamidino-2-phenylindole (DAPI). Images were acquired with a ×40/1.3 oil immersion lens on a Zeiss LSM 710 confocal microscope.
Immunohistochemical Analysis
Paraffin-embedded grade IV astrocytomas were obtained from the Brain Tumor Tissue Bank, London Health Sciences Centre, London (Canada). Tissues were de-waxed in xylene and rehydrated in 100% ethanol. Slides were microwaved for 10 min in citraconic anhydride epitope retrieval buffer, pH 7.4, and blocked with 0.5% fish gelatin in Tris-buffered saline with 0.1% Tween 20. Slides were incubated in anti-CNA antibody (1:500) overnight at 4 °C and then washed and incubated with anti-mouse DakoCytomation Envision+ System labeled polymer HRP (DakoCytomation, Denmark) for 1 h. CNA immunoreactivity was detected with Dako Liquid DAB+ Substrate Chromagen System and counterstained with hematoxylin.
RESULTS
In Vivo Occupancy of NFIs at Endogenous Promoters
B-FABP and GFAP expression correlates with NFI phosphorylation in MG cell lines. B-FABP and GFAP are expressed in MG cell lines that have hypophosphorylated NFIs but not in cell lines that have hyperphosphorylated NFIs (17). To compare NFI occupancy of the GFAP and B-FABP promoters in MG cells with hyperphosphorylated versus hypophosphorylated NFIs, we performed ChIP experiments using a pan-specific NFI antibody in B-FABP/GFAP+ve U251 (hypophosphorylated NFI) and B-FABP/GFAP-ve U87 (hyperphosphorylated NFI) MG cell lines (17). DNA cross-linked to NFI was PCR-amplified using primers flanking NFI-binding sites in the GFAP and B-FABP promoters (Table 1 and Fig. 1A). Bands corresponding to the NFI-binding sites in the GFAP (G-br1 and G-br2/3) and B-FABP (B-br1/2/3) promoters were detected in samples from U251 cells but not U87 cells (Fig. 1B). No bands were detected using primers to the GAPDH promoter or in the rabbit IgG lanes, which served as the negative controls for the ChIP experiments. Input represents sonicated genomic DNA and thus serves as a positive control for the PCRs. These results indicate that NFI occupies NFI-binding sites in U251 cells but not in U87 cells.
FIGURE 1.
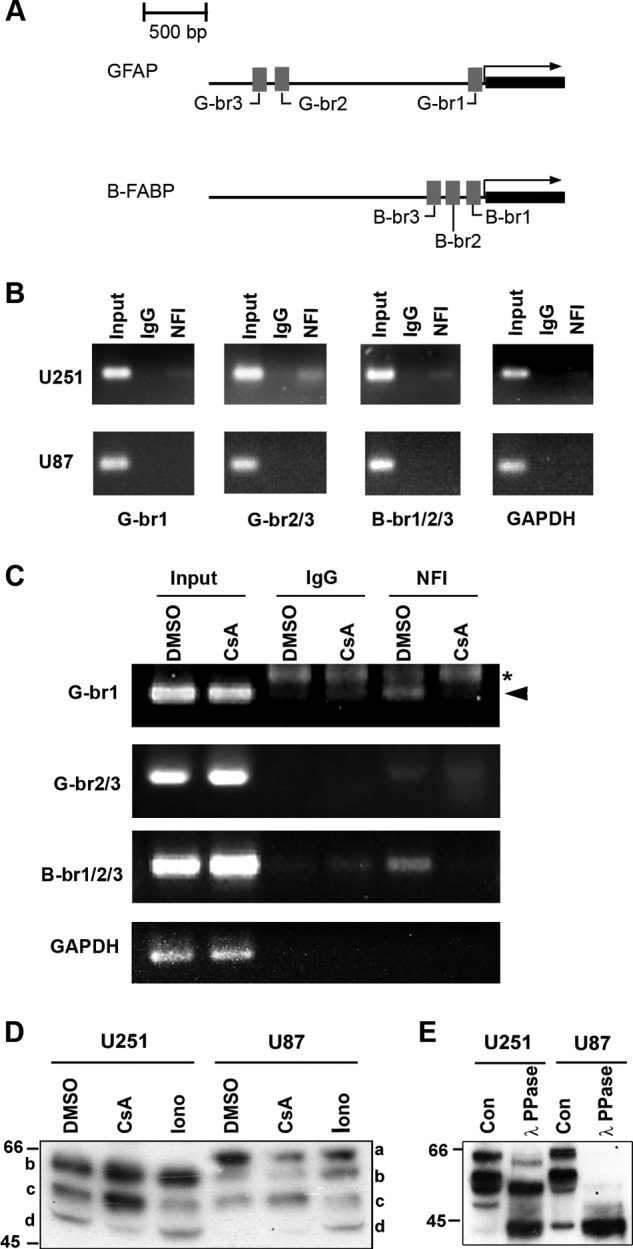
NFI-dependent promoter binding and activity. A, schematic diagram of the GFAP and B-FABP promoter regions showing the relative location of the three NFI-binding sites located in each promoter. B, ChIP analysis was carried out in U251 and U87 cells using either a pan-specific anti-NFI antibody or normal rabbit IgG as a negative control. Primers flanking the NFI-binding sites identified in the GFAP (G-br1 and G-br2/3) and B-FABP promoters (B-br1/2/3) were used for PCR amplification (Table 1). Primers flanking the proximal GAPDH promoter were used as a negative control. Input DNA represents DNA isolated from U251 or U87 cell lysates following sonication but prior to immunoprecipitation. Input DNA serves as the positive control for the PCR and reveals products of the expected sizes. C, U251 cells were treated for 1 h with 1 μm CsA or DMSO (control), followed by ChIP analysis as described in B. Asterisk denotes a nonspecific band, and arrowhead denotes a specific band. D, Western blot analysis of NFI in U251 and U87 cells treated with CsA and ionomycin (Iono). U251 and U87 cells were treated for 1 h with DMSO, 1 μm CsA, or 1 μm ionomycin and harvested using trypsin to dissociate the cells. Nuclear extracts were electrophoresed in a 10% SDS-polyacrylamide gel, transferred to a PVDF membrane, and immunostained with rabbit anti-NFI antibody. The primary antibody was detected with horseradish peroxidase-conjugated antibody, and the signal was detected with Immobilon reagent. E, λ-phosphatase treatment of U251 and U87 nuclear extracts. U251 and U87 nuclear extracts were prepared in the absence of phosphatase inhibitors and treated with or without λ-phosphatase (PPase) for 1 h at 30 °C. Following treatment, extracts were subjected to Western blot analysis as in D. Con, control.
CsA Regulates NFI Promoter Binding and Dephosphorylation
Our laboratory has previously shown by gel shifts and phosphatase inhibition experiments that there is phosphatase activity in NFI-hypophosphorylated MG cells that is absent in NFI-hyperphosphorylated MG cells (17). The serine/threonine phosphatase calcineurin is expressed in neurons and reactive astrocytes (48, 49) and has previously been associated with NFIC transactivation in NIH3T3 cells (41). We performed ChIP on U251 MG cells treated with the calcineurin inhibitor CsA to determine whether there was any effect on NFI occupancy of endogenous promoters. U251 cells were incubated with 1 μm CsA or DMSO (control) for 1 h followed by ChIP analysis as described above. Band intensity was markedly decreased in the presence of CsA compared with the DMSO control in the NFI immunoprecipitation lanes using primers flanking G-br1 and B-br1/2/3 (Fig. 1C) indicating a decrease in immunoprecipitated DNA. No change was observed using primers flanking G-br2/3 (Fig. 1C, compare IgG and CsA lanes), and no signal was detected using primers to the GAPDH promoter. These results indicate that inhibition of calcineurin in U251 cells decreases NFI binding at endogenous consensus binding sites.
To determine whether the decrease in B-FABP and GFAP promoter binding is due to changes in NFI phosphorylation, we treated U251 and U87 MG cells with 1 μm CsA, isolated nuclear extracts, and analyzed NFI phosphorylation by Western blotting. The banding patterns of NFI were distinctly different in U251 and U87 cells (Fig. 1D), with a slower migrating band (band a) specific to U87 cells observed in the DMSO lane. Furthermore, the fastest migrating band (Fig. 1D, band d) in the U251 control (DMSO) lane was absent in the U87 control (DMSO) lane. This recapitulates the increased phosphorylation of NFI reported in U87 compared with U251 MG cells (17). Upon inhibition of calcineurin with CsA, there was a shift up toward the slower migrating bands (Fig. 1D, bands b and c) in U251 cells (CsA lane) with a decrease in the intensity of the fastest migrating band (band d). CsA treatment did not alter the intensity of the slower migrating bands (Fig. 1D, bands b and c) in U87 cells.
Calcineurin is a calcium-dependent phosphatase. We therefore used the calcium ionophore ionomycin to increase the level of intracellular calcium in MG cells to determine whether NFI phosphorylation is calcium-sensitive. Western blot analysis of NFI following exposure of U87 cells to 1 μm ionomycin for 1 h revealed a change in NFI phosphorylation. In particular, we observed a faster migrating band in U87 cells that corresponds in size to the fastest migrating band in U251 (Fig. 1D, 6th lane, band d). There was no significant change in the NFI phosphorylation status of U251 cells treated with ionomycin.
To verify that changes in NFI mobility were due to phosphorylation, U251 and U87 nuclear extracts were prepared in the absence of phosphatase inhibitors and treated with λ-phosphatase (Fig. 1E). A shift to faster migrating bands was observed in the presence of λ-phosphatase in U251 cells. In comparison, all bands were shifted to the fastest migrating form when U87 cell lysates were incubated with λ-phosphatase. The persistence of slower migrating bands in phosphatase-treated U251 cell lysates suggests the presence of a different population of NFI family members.
Calcineurin Regulates NFI Activity
To study the effect of calcineurin modulation on NFI-dependent transcriptional activity, U251 cells were transfected with a CAT reporter gene under the control of the GFAP promoter (pCAT/GFAP) containing three well characterized NFI-binding sites and were treated with CsA or DMSO (control). Following treatment with CsA, CAT activity decreased to 74% of control levels (p < 0.001) (Fig. 2A). U251 cells were also transfected with the CAT reporter gene under the control of the GFAP promoter with each of the three NFI-binding sites mutated singly (pCAT/GFAP G-br1*, pCAT/GFAP G-br2*, and pCAT/GFAP G-br3*) and in combination (pCAT/GFAP G-br1*/2*/3*). As reported previously, mutation of G-br1 had the most striking effect on promoter activity, with no further decreases observed upon mutation of all three NFI-binding sites (16). These results are consistent with our ChIP results showing the strongest binding of NFI to G-br1. CsA treatment had no effect on the CAT activity of the mutated constructs.
FIGURE 2.
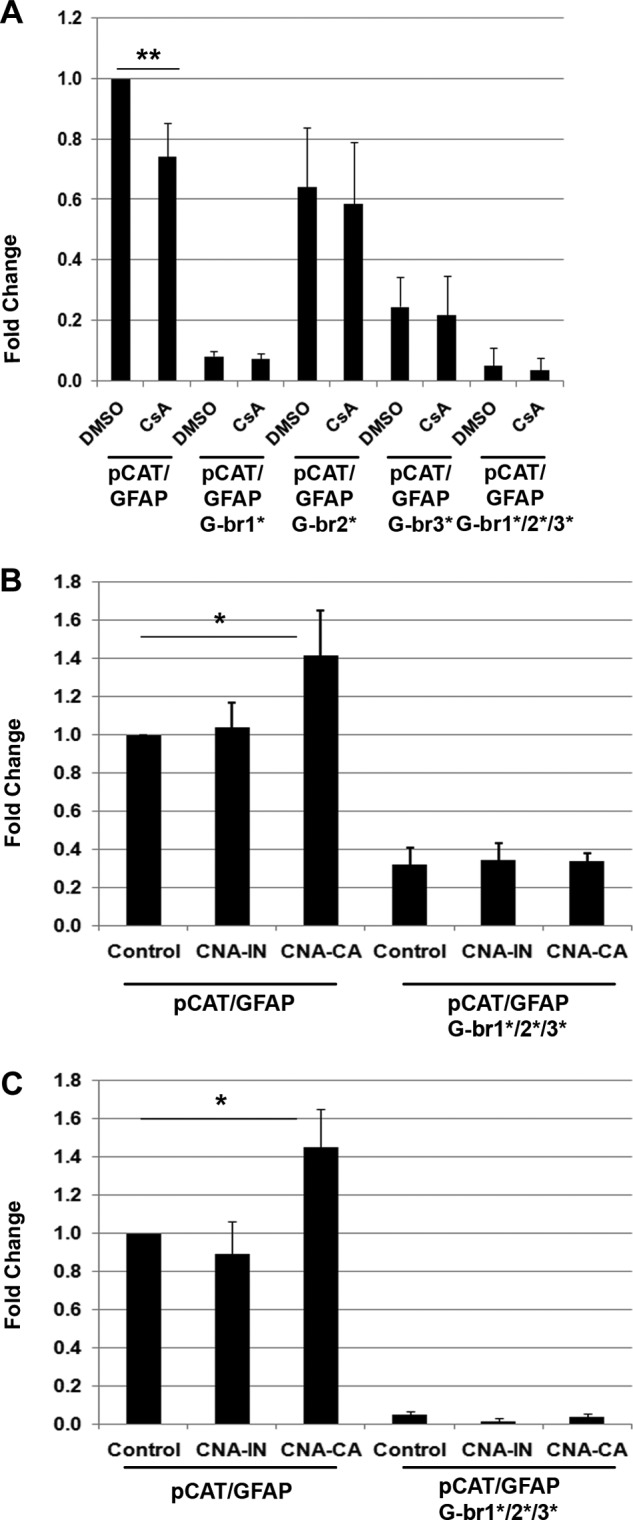
Calcineurin modulates NFI-dependent promoter activity. A, U251 cells were transfected with pCAT/GFAP, pCAT/GFAP G-br1*, pCAT/GFAP G-br2*, pCAT/GFAP G-br3*, or pCAT/GFAP G-br1*/2*/3* (NFI sites mutated) and treated with 1 μm CsA or DMSO (control) for 24 h. Acetylated [14C]chloramphenicol was measured in counts/min from equal aliquots of cell lysates using a scintillation counter. The fold increases in CAT activity are relative to U251 treated with DMSO and transfected with pCAT/GFAP. The results are an average of four (pCAT/GFAP G-br1*, pCAT/GFAP G-br2*, pCAT/GFAP G-br3*, and pCAT/GFAP G-br1*/2*/3* constructs) to six (pCAT/GFAP construct) independent experiments with standard deviation indicated by error bars. B and C, U251 (B) and U87 (C) cells were co-transfected with pCAT/GFAP or pCAT/GFAP G-br1*/2*/3* and HA-DDXI (control), catalytically inactive (CNA-IN), or constitutively active (CNA-CA) HA-tagged calcineurin. The fold increases in CAT activity are relative to U251 pCAT/GFAP as follows: HA-DDXI (control) (B) or U87 pCAT/GFAP HA-DDX1 (control) (C). The results are an average of four independent experiments with standard deviation indicated by error bars. Statistical significance was determined using unpaired t test. *, denotes p < 0.01; **, denotes p < 0.001.
To determine whether increasing calcineurin activity modifies NFI-dependent promoter activity, U251 and U87 cells were co-transfected with the following: (i) pCAT/GFAP or pCAT/GFAP G-br1*/2*/3* and (ii) control (HA-tagged DDX1; used as transfection control), HA-tagged CNA-IN (catalytically inactive CNA), or HA-tagged CNA-CA (constitutively active CNA) construct. Expression of constitutively active CNA resulted in a 1.41-fold increase in CAT activity in U251 with pCAT/GFAP (p < 0.01) (Fig. 2B) and a 1.45-fold increase in CAT activity in U87 cells compared with expression of catalytically inactive CNA (p < 0.01) (Fig. 2C). There was no difference in CAT activity between control and CNA-IN or upon co-transfection of control, CNA-IN, or CNA-CA along with pCAT/GFAP G-br1*/2*/3* in either U251 or U87 cells (Fig. 2, B and C).
The alteration in NFI phosphorylation resulting from inhibition of calcineurin suggests that calcineurin is at least one of the phosphatases responsible for regulating the phosphorylation state of NFI. To address whether calcineurin exists in the same complex as NFI, we carried out co-immunoprecipitations. Using an anti-CNA antibody, we were able to immunoprecipitate a very large fraction of CNA, completely depleting CNA from the supernatant (Fig. 3). A small amount of NFI was co-immunoprecipitated with CNA. No NFI was detected in the IgG control lane. We were unable to co-immunoprecipitate CNA with anti-NFI antibody. These results suggest that there may be a weak interaction between CNA and NFI and/or a small subset of NFI resides in the same cellular complex as calcineurin.
FIGURE 3.
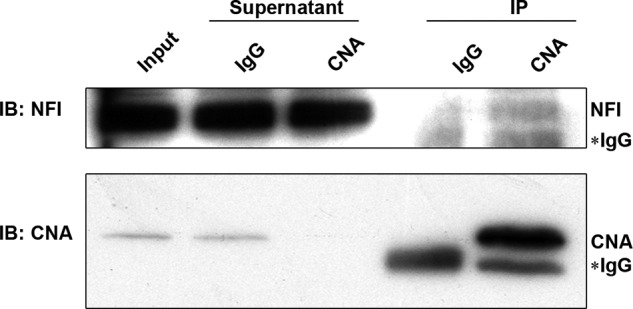
Co-immunoprecipitation of CNA and NFI. Whole cell extracts from U251 cells were incubated with either anti-CNA antibody or purified mouse IgG. The immunocomplexes were subjected to gel electrophoresis, transferred to a PVDF membrane, and immunostained with anti-NFI antibody (Tanese) and anti-CNA antibody. Five percent of the supernatant from each immunoprecipitation was loaded in indicated lanes. IP, immunoprecipitation; IB, immunoblotting.
CNA Expression in MG Cell Lines
Our results indicate a role for CNA in regulating NFI dephosphorylation. We therefore examined CNA expression in a panel of 10 MG cell lines, five that express B-FABP/GFAP and have hypophosphorylated NFI (M016, M049, M103, U251, and U373) and five that do not express B-FABP/GFAP and have hyperphosphorylated NFI (A172, CLA, M021, T98, and U87) (17). Western blot analysis of whole cell extracts revealed significant variation in expression of CNA (60 kDa) with the highest levels in A172, CLA, M021, and T98, and lowest levels in M103, U251, and U373 (Fig. 4A). Intriguingly, we also observed a faster migrating band of ∼57 kDa present exclusively in the five B-FABP/GFAP+ve cell lines with hypophosphorylated NFIs. This 57-kDa form of CNA has previously been reported to be a cleaved form of CNA with ∼2-fold increased activity compared with uncleaved CNA (50). The observed correlation between the 57-kDa form of CNA and NFI hypophosphorylation suggests that the cleaved form of CNA may regulate NFI dephosphorylation in MG cells.
FIGURE 4.
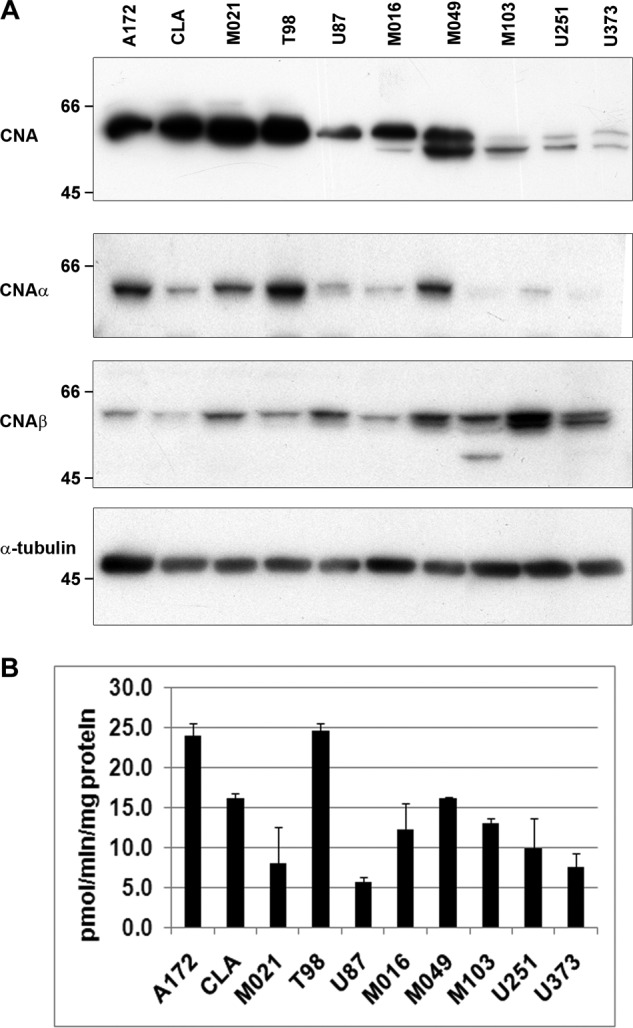
CNA expression in MG cell lines. A, whole cell extracts from MG cell lines were electrophoresed, transferred to PVDF membranes, and probed with anti-CNA, anti-CNAα, anti-CNAβ, and anti-α-tubulin antibodies. B, calcineurin activity (pmol/min/mg protein) in whole cell lysates was measured using γ-32P-RII peptide substrate. The results are an average of three replicates with standard deviation indicated by error bars.
Our antibody to CNA recognizes both the α and β CNA isoforms. A third CNA isoform is testis-specific (51, 52). To identify the CNA isoform(s) cleaved in B-FABP/GFAP+ve MG cells, we immunostained our panel of 10 MG cell lines with antibodies specific to CNAα and CNAβ. Overall, CNAα was expressed at higher levels in B-FABP/GFAP-ve cell lines, with only the higher migrating band observed (Fig. 4A). Immunostaining with anti-CNAβ antibody revealed two bands in B-FABP/GFAP+ve MG cell lines (Fig. 4A). These results suggest that CNAβ, but not CNAα, is being cleaved and activated in B-FABP/GFAP+ve cells.
Specific calcineurin activity in whole cell lysates was measured by dephosphorylation of 32P-RII peptide, a peptide selectively and efficiently dephosphorylated by calcineurin in vitro (53, 54). As with CNA expression, calcineurin activity varied widely among MG cell lines (Fig. 4B). The highest activity was detected in A172 and T98 cells, releasing 23.9 and 24.5 pmol/min/mg protein of 32P from the RII peptide. U87 cells had the lowest level of calcineurin activity, 5.7 pmol/min/mg protein. The remaining cell lines had activities ranging from 7.9 pmol/min/mg protein (M021) to 16.0 pmol/min/mg protein (CLA). Interestingly, although the expression of CNA was lowest in M103, U251, and U373, calcineurin activity in the cell lines using this assay was comparable with CLA and M021, which had very high levels of CNA.
Calpastatin Modulates NFI-dependent Promoter Activity
CNA has been reported to be cleaved to a 57-kDa form by the protease calpain (50, 55). As calpastatin is an endogenous inhibitor of calpain activity (56), we co-transfected U251 and U87 cells with a calpastatin expression construct along with pCAT/GFAP to determine whether inhibition of calpain could alter NFI-dependent promoter activity. In U251 cells, calpastatin significantly decreased CAT activity, to 67% of the control levels obtained with empty vector (p < 0.05) (Fig. 5A). U87 cells, which do not have the 57-kDa cleaved form of CNA, showed no change in CAT activity. We also prepared nuclear and cytoplasmic extracts from U251 and U87 cells transfected with calpastatin. Expression of calpastatin did not alter CNAβ expression or cleavage in the cytoplasm of U251 or U87 cells; however, there was a clear decrease in the amount of cleaved CNAβ in the nucleus of U251 cells treated with calpastatin (Fig. 5B). No CNAβ was detected in the nucleus of U87 cells.
FIGURE 5.
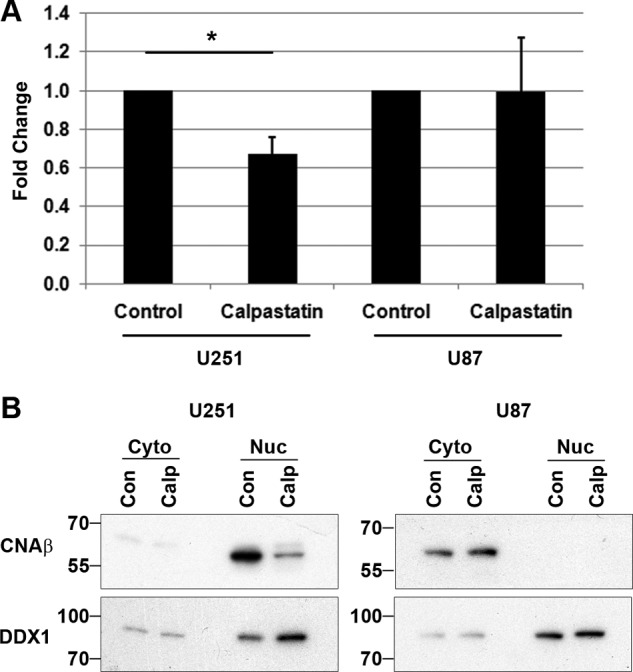
Calpastatin alters NFI-dependent promoter activity. A, U251 and U87 cells were co-transfected with pCAT/GFAP and control (empty vector) or calpastatin expression construct. Acetylated [14C]chloramphenicol was measured from equal aliquots of cell lysates using a scintillation counter. The fold increases in CAT activity are relative to control (empty vector). The results are an average of four independent experiments with standard deviation indicated by error bars. Statistical significance was determined using unpaired t test. *, denotes p < 0.01. B, U251 and U87 cells were transfected with control (Con; empty vector), or calpastatin (Calp), cytoplasmic (Cyto) ,and nuclear (Nuc) fractions were prepared. Extracts were electrophoresed, transferred to PVDF membranes, and probed with anti-CNAβ and anti-DDX1 (loading control) antibodies.
Ionomycin Alters NFI Phosphorylation and NFI-dependent Promoter Activity
As shown previously in Fig. 1D, treatment of U87 cells with the calcium ionophore ionomycin resulted in increased dephosphorylation of NFI. We therefore examined NFI-dependent promoter activity in U251 and U87 cells following treatment with ionomycin to see if changes in NFI phosphorylation might result in altered NFI activity. U251 and U87 cells were transfected with pCAT/GFAP, followed by treatment with ionomycin or DMSO (control) for 24 h. CAT activity was not altered in U251 cells where ionomycin had little effect on NFI phosphorylation (Figs. 1C, 3rd lane, and 6A). However, in U87 cells, treatment with ionomycin increased CAT activity 1.8-fold (p < 0.001) (Fig. 6A).
FIGURE 6.

Ionomycin alters NFI-dependent promoter activity and calcineurin localization. A, U251 and U87 cells were transfected with pCAT/GFAP and treated with 1 μm ionomycin (Iono) or DMSO (control) for 24 h. Acetylated [14C]chloramphenicol was measured in counts/min from equal aliquots of cell lysates using a scintillation counter. The fold increases in CAT activity are relative to the DMSO control. The results are an average of four independent experiments with standard deviation indicated by error bars. B, subcellular localization of calcineurin in U251 and U87 cells treated with DMSO (control) or 1 μm ionomycin (Iono) was analyzed by immunofluorescence using anti-CNA primary antibody followed by Alexa 488-conjugated secondary antibody. DNA was stained with 4′6-diamidino-2-phenylindole (DAPI). Bar, 10 μm. C, percentage of cells with predominantly cytoplasmic staining versus cells with nuclear and cytoplasmic staining for CNA in U87 cells treated with DMSO (control) or 1 μm ionomycin for 1 h. This analysis was carried out >100 cells for each parameter. Briefly, 10 separate fields with ∼10–15 cells per field were randomly selected for each parameter. Line scans through the cytoplasm and nucleus of each cell were used to assess relative signal in the nucleus and cytoplasm. Statistical significance was determined using unpaired t test. **, denotes p < 0.001. D, U251 and U87 cells were treated with DMSO (control) or 1 μm ionomycin (Iono) for 1 h and harvested, and cytoplasmic (Cyto) and nuclear (Nuc) extracts were prepared. Extracts were electrophoresed, transferred to PVDF membranes, and immunostained with anti-CNAβ and anti-DDX1 (loading control) antibodies.
CNA Localization in MG Cell Lines and Astrocytoma Tumors
The differences in CNA expression and activity observed in U251 and U87 cells led us to examine CNA subcellular localization in these cells. In U251 control (DMSO-treated) cells, CNA was concentrated in the nucleus, with dense areas of perinuclear staining. CNA staining in the cytoplasm was much weaker than in the nucleus (Fig. 6B). Interestingly, this pattern was reversed in U87 cells, with CNA primarily found in the cytoplasm. Upon addition of 1 μm ionomycin for 1 h, CNA translocated to the nucleus in U87 cells but had no effect on U251 cells. We then quantitated the subcellular localization of CNA in the cytoplasm and nucleus of U87 cells. As indicated in Fig. 6C, CNA was primarily found in the cytoplasm of 99% of U87 control cells, with only 1% of untreated cells showing a predominantly nuclear pattern. Upon exposure to ionomycin, localization of CNA to the nucleus was observed in 88% of U87 cells. We verified these changes in subcellular localization by nuclear and cytoplasmic fractionation. Cleaved CNAβ was detected in the nucleus of U251 cells treated with DMSO and ionomycin, with uncleaved CNAβ detected only in the cytoplasm (Fig. 6D). In U87 cells, CNAβ was not detected in the nucleus of DMSO-treated cells; however, following treatment with ionomycin, cleaved CNAβ was clearly detected in the nucleus.
Next, we examined CNA expression in brain and MG tumor tissue. CNA was not detected in normal human brain (frontal lobe) based on immunohistochemical analysis (Fig. 7A). Immunostaining of grades I, II, and IV astrocytoma tumor tissues revealed increased expression of CNA in high grade (grade IV) astrocytomas (Fig. 7, E–H) compared with grade I and II tumors (Fig. 7, B and C). Expression of CNA was primarily restricted to the cytoplasm of a small subset of cells in grade I tumors (Fig. 7B). The staining pattern in grade II astrocytomas was similar to that of grade I astrocytomas with CNA expression primarily found in the cytoplasm of a small number of cells. In grade IV tumors, we observed increased expression of CNA in areas of tumor infiltration. Fig. 7, D–F, shows progressively higher levels of CNA staining in regions of low infiltration to high infiltration. In some areas (Fig. 7E), CNA staining was primarily diffuse and cytoplasmic, whereas in other areas (Fig. 7F), CNA staining was primarily perinuclear, with a small subset of cells showing nuclear staining (Fig. 7F, inset). Of note, strong staining was detected in pseudopalisading cells surrounding necrotic areas (Fig. 7, G and H). Hypercellular pseudopalisades are commonly observed in high grade astrocytomas and are formed by actively migrating cells (57).
FIGURE 7.

Immunohistochemical analysis of CNA in human astrocytoma tumors. Tissue sections were immunostained with anti-CNA antibody and counterstained with hematoxylin. A, normal human brain. B, grade I astrocytoma. Arrows indicate positive cells (patient 983). C, grade II astrocytoma (patient 470). Arrow indicates area of positive staining. D, grade IV astrocytoma, no visible tumor cells (patient 1046). E, grade IV astrocytoma, tumor center (patient 1046). F, grade IV astrocytoma, heavy tumor infiltration. Inset, arrow indicates perinuclear staining; arrowhead indicates nuclear staining (patient 335). G and H, grade IV astrocytoma, heavy tumor infiltration (patient 1046).
DISCUSSION
The NFI family of transcription factors is a key regulator of neural cell differentiation, playing an essential role in gliogenesis in the brain and spinal cord (22, 23). In the spinal cord, induction of NFIA and NFIB expression coincides with the start of gliogenesis, and knockdown of NFIA results in loss of glial cell progenitors in the chick embryo (22). Following gliogenesis, NFIA and NFIB promote astrocyte differentiation. In the brain, NFIA expression confers astrocytic potential on neural precursor cells downstream of Notch-induced signaling and Sox9 (23, 24). In MG, NFI regulates expression of B-FABP and GFAP, normally expressed in radial glial cells and in differentiated astrocytes, respectively (16). Importantly, NFI is differentially phosphorylated in MG cells, with hypophosphorylated NFI found in B-FABP/GFAP+ve MG cell lines and hyperphosphorylated NFI found in B-FABP/GFAP-ve MG cell lines (17). These observations agree with previous experiments demonstrating that increased phosphorylation of NFIs results in decreased transactivation of NFI-dependent promoters (58). Importantly, NFI activity appears to be regulated by a phosphatase activity that is specific to B-FABP/GFAP+ve (NFI-hypophosphorylated) MG cells rather than a kinase activity that is specific to B-FABP/GFAP-ve (NFI-hyperphosphorylated) MG cells (17).
Here, we identify the phosphatase calcineurin as an important regulator of NFI dephosphorylation and activity in MG cells. Our combined approaches, including CNA overexpression and inhibition, co-immunoprecipitation with NFI, ChIP analysis of NFI-dependent B-FABP and GFAP promoters, Western blot analysis of CNA, and reporter gene assays, point to a direct link between calcineurin and dephosphorylation of NFI in MG cells. Calcineurin is a serine/threonine phosphatase that is highly expressed in neurons (48, 59). Calcineurin has also been detected in C6 glioma cells and cultured astrocytes (60, 61). In astrocytes, calcineurin plays an important role in regulating the inflammatory response (62), and calcineurin expression increases in astrocytes but not neurons in a mouse aging model (63). Calcineurin has also been shown to increase NFIC transactivation (41), providing a direct link between calcineurin and the NFI family.
The distinguishing characteristics of calcineurin in NFI-hypophosphorylated versus NFI-hyperphosphorylated MG cell lines are neither its expression levels nor its activity as measured using the RII peptide in vitro assay, but rather the following: (i) the presence of a cleaved 57-kDa CNA fragment specifically associated with NFI-hypophosphorylated MG cells, and (ii) the nuclear localization of CNA in NFI-hypophosphorylated MG cells. A 57-kDa cleaved product of CNA has been previously reported in cortical neurons following glutamate exposure and in the brains of Alzheimer disease patients (50, 64). Of note, the 57-kDa form of calcineurin identified in the brains of Alzheimer disease patients shows a 2-fold increase in phosphatase activity compared with full-length CNA (50). There are two CNA isoforms expressed in brain, CNAα and CNAβ (65). In this study, we demonstrate that the CNA isoform that is cleaved in NFI-hypophosphorylated MG cells is CNAβ. Although both CNA isoforms have been shown to dephosphorylate the same substrates in vitro, each isoform has different substrate preference, and differs in substrate binding affinity and turnover (66). The differential expression of CNAα and CNAβ in different MG cell lines, combined with specific cleavage of CNAβ in NFI-hypophosphorylated MG cell lines, suggests unique roles for these two CNA isoforms in these tumor cells.
It is unclear why the 57-kDa form of CNAβ is more active, as the cleavage site is C-terminal to the CNB binding domain, the calmodulin binding domain, and the autoinhibitory domain (50). Furthermore, despite the cleavage site being located downstream of identified nuclear localization signal and nuclear export signal sequences (67), we found that cleaved CNAβ preferentially localizes to the nucleus, and the uncleaved form preferentially localizes to the cytoplasm. We postulate that removal of the C-terminal 20 amino acids of CNAβ increases both its phosphatase activity as the result of an altered protein structure and its ability to localize to the nucleus.
CNA is cleaved by the Ca2+-dependent cysteine protease calpain, and inhibition of calpain inhibits this cleavage. Our results indicate that inhibition of calpain by calpastatin, a specific endogenous inhibitor of calpain (56), decreases NFI-dependent promoter activity in U251 cells but not in U87 cells. These observations are in agreement with CNA cleavage being calpain-dependent in MG cell lines that express the hypophosphorylated form of NFI. It will be interesting to examine expression and activity of calpain and calpain regulators, including calpastatin, in MG cell lines and tumors, as they may be important upstream regulators of calcineurin, and consequently NFI activity.
An intriguing finding is the different subcellular distribution of CNA in MG cell lines. In U251 cells, CNA localizes almost exclusively to the nucleus, whereas CNA is primarily found in the cytoplasm of U87 cells. When U87 cells are treated with ionomycin, a calcium ionophore, CNAβ, which causes an influx of calcium in the cells, is cleaved and translocates to the nucleus. CNA has been detected in the nucleus of a variety of cell types (68–70) and has been reported to translocate to the nucleus following addition of a calcium ionophore (71). Translocation of CNA to the nucleus of MG cells may be an important regulatory step in NFI dephosphorylation as NFI is normally found in the nucleus (72). In keeping with this idea, suppression of CNA translocation to the nucleus in cardiomyocytes obtained from cases of myocardial hypertrophy has been shown to inhibit CNA signaling (67, 73). Thus, we propose that in B-FABP/GFAP+ve NFI-hypophosphorylated MG cells, where CNA is truncated and nuclear, CNA is active and able to dephosphorylate NFI, whereas in B-FABP/GFAP-ve NFI-hyperphosphorylated MG cells, CNA localization to the cytoplasm prevents interaction with and dephosphorylation of NFI.
Differential cleavage and localization of CNA suggests that there may be differences in calcium signaling in MG cell lines. In glial cells, calcium signaling regulates cell function by controlling cellular homeostasis and releasing gliotransmitters (74). Interestingly, overexpression of calcium-permeable AMPA receptors has been reported in brain tumor initiating cells (75). As AMPA receptors allow calcium influx upon stimulation, increased levels of these receptors in MG cells may contribute to the cleavage and activation of calcineurin. In turn, increased activity of calcineurin may have wide ranging effects beyond NFI phosphorylation because calcineurin has many targets. Further examination of MG cell lines and tumor tissues for changes in calcium signaling, including expression of calcium channels and receptors, may elucidate how both calpain and calcineurin are activated in these cells.
In normal brain, CNA is highly expressed in neurons, with little or no expression in astrocytes (48, 76). Calcineurin is activated in astrocytes by inflammatory signals in a cell context-dependent manner, with activation of calcineurin in resting astrocytes resulting in progression of the inflammatory cascade and resolution of the inflammatory cascade in activated astrocytes (62). In low grade astrocytomas, CNA expression is weak and limited to a small percentage of cells. However, CNA levels are considerably higher in grade IV astrocytomas, especially at sites of tumor infiltration and in pseudopalisading cells surrounding necrotic zones. Pseudopalisades, associated with aggressive tumors and a hallmark of grade IV astrocytomas, are regions of hypercellularity formed by tumor cells actively migrating away from hypoxic areas (57). Our results suggest that CNA is preferentially expressed in the most aggressively growing regions of the tumors. B-FABP, a target of NFI, has also been shown to be associated with sites of tumor infiltration and migration in MG tumors and cell lines (5, 12, 14). Thus, calcineurin may reside at the apex of a regulatory cascade centered on the activation of genes associated with migration/infiltration. As such, calcineurin may represent a key target for the treatment of high grade astrocytomas.
There are a number of NFI phosphorylation states in MG cells, suggesting that NFIs are phosphorylated at multiple sites (17). In fact, even the most hypophosphorylated form of NFI in U251 can be further dephosphorylated by addition of potato acid phosphatase (17). Thus, we propose that NFI phosphorylation effectively serves as a rheostat to control function, with increasing phosphorylation resulting in increased negative charge. The outcome is a gradual disruption of molecular interactions and decreased activity. A graded response to multisite phosphorylation has been characterized previously as a way of finely regulating DNA binding by the transcription factor Ets-1 (77) and p53 binding to CREB-binding protein (78). Alternatively, there may be a threshold of phosphorylation/dephosphorylation that acts analogously to an on/off switch. Such a mechanism has been demonstrated for NFAT1, which requires dephosphorylation of 13 phosphoserines by CNA for its activation (79). A third possibility is that phosphorylation/dephosphorylation of one or a small number of specific residues regulates activity. This is the case for Fox03 in which phosphorylation of Ser-207 by MST1, independent of other phosphorylation events, triggers nuclear localization (80). It remains to be seen which of these mechanisms underlies the control of NFI activity.
Taken together, our data support an important role for calcineurin in the dephosphorylation and activation of NFI-dependent promoter activity in MG cells. A complex regulatory picture emerges, consisting of at least four principal steps as follows: (i) cleavage of full-length calcineurin to the more active 57-kDa form; (ii) translocation of calcineurin to the nucleus; (iii) interaction of calcineurin with NFI resulting in NFI dephosphorylation, and (iv) binding of NFI to (and regulation of) NFI target gene promoters (Fig. 8). Based on CNA's position at the top of this regulatory cascade, it should be possible to regulate the expression of NFI target genes in MG through modulation of CNA activity. One NFI target gene, B-FABP, has already been shown to correlate with poor prognosis and reduced survival in MG tumors (12, 81), as well as increased migration in MG cell lines and infiltration in grade IV astrocytomas (5). Thus, we may be able to alter the migratory properties of MG cells through modulation of NFI phosphorylation by inhibiting CNA activation and/or translocation. Future work will involve examining MG tumors to determine whether the CNA detected in these tissues is activated through calpain and how NFI phosphorylation and expression of NFI target genes are affected by CNA expression in MG tumors.
FIGURE 8.
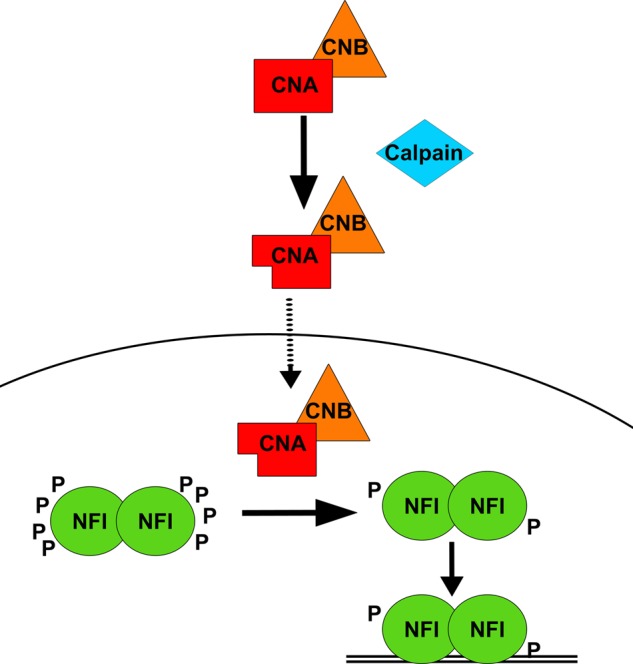
Model of regulation of calcineurin and NFI activity in MG cells. In the cytoplasm, calpain cleaves CNA (bound by regulatory subunit CNB). Cleaved CNA bound to CNB translocates to the nucleus and dephosphorylates NFI. Hypophosphorylated NFI can now interact with NFI consensus binding sites in target gene promoters.
Acknowledgments
We thank Dr. Naoko Tanese for the anti-NFI antibody, Dr. R. Chen for the CNA expression constructs, and Dr. David Jay for the calpastatin construct. We appreciate the expert technical assistance given by Elizabeth Monckton. We are also grateful to Drs. Lei Li and Rong-Zong Liu for reading the manuscript.
This work was supported by a grant from the Alberta Cancer Foundation and a studentship from Alberta Innovates-Health Solutions (to M. B.).
- MG
- malignant gliomas
- GFAP
- glial fibrillary acidic protein
- B-FABP
- brain fatty acid-binding protein
- NFI
- nuclear factor I
- CNA
- calcineurin A
- CNB
- calcineurin B
- CsA
- cyclosporin A
- CAT
- chloramphenicol acetyltransferase
- ChIP
- chromatin immunoprecipitation
- PVDF
- polyvinyl fluoride.
REFERENCES
- 1. Ohgaki H., Kleihues P. (2005) Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J. Neuropathol. Exp. Neurol. 64, 479–489 [DOI] [PubMed] [Google Scholar]
- 2. Mason W. P., Maestro R. D., Eisenstat D., Forsyth P., Fulton D., Laperrière N., Macdonald D., Perry J., Thiessen B., and Canadian GBM Recommendations Committee (2007) Canadian recommendations for the treatment of glioblastoma multiforme. Curr. Oncol. 14, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eng L. F., Rubinstein L. J. (1978) Contribution of immunohistochemistry to diagnostic problems of human cerebral tumors. J. Histochem. Cytochem. 26, 513–522 [DOI] [PubMed] [Google Scholar]
- 4. Sanai N., Alvarez-Buylla A., Berger M. S. (2005) Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353, 811–822 [DOI] [PubMed] [Google Scholar]
- 5. Mita R., Coles J. E., Glubrecht D. D., Sung R., Sun X., Godbout R. (2007) B-FABP-expressing radial glial cells: the malignant glioma cell of origin? Neoplasia 9, 734–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Godbout R., Bisgrove D. A., Shkolny D., Day R. S., 3rd (1998) Correlation of B-FABP and GFAP expression in malignant glioma. Oncogene 16, 1955–1962 [DOI] [PubMed] [Google Scholar]
- 7. Merkle F. T., Tramontin A. D., García-Verdugo J. M., Alvarez-Buylla A. (2004) Radial glia give rise to adult neural stem cells in the subventricular zone. Proc. Natl. Acad. Sci. U.S.A. 101, 17528–17532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kriegstein A., Alvarez-Buylla A. (2009) The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feng L., Hatten M. E., Heintz N. (1994) Brain lipid-binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron 12, 895–908 [DOI] [PubMed] [Google Scholar]
- 10. Kurtz A., Zimmer A., Schnütgen F., Brüning G., Spener F., Müller T. (1994) The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development 120, 2637–2649 [DOI] [PubMed] [Google Scholar]
- 11. Hansen D. V., Lui J. H., Parker P. R., Kriegstein A. R. (2010) Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561 [DOI] [PubMed] [Google Scholar]
- 12. Kaloshi G., Mokhtari K., Carpentier C., Taillibert S., Lejeune J., Marie Y., Delattre J. Y., Godbout R., Sanson M. (2007) FABP7 expression in glioblastomas: relation to prognosis, invasion and EGFR status. J. Neurooncol. 84, 245–248 [DOI] [PubMed] [Google Scholar]
- 13. Liang Y., Diehn M., Watson N., Bollen A. W., Aldape K. D., Nicholas M. K., Lamborn K. R., Berger M. S., Botstein D., Brown P. O., Israel M. A. (2005) Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc. Natl. Acad. Sci. U.S.A. 102, 5814–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Rosa A., Pellegatta S., Rossi M., Tunici P., Magnoni L., Speranza M. C., Malusa F., Miragliotta V., Mori E., Finocchiaro G., Bakker A. (2012) A radial glia gene marker, fatty acid binding protein 7 (FABP7), is involved in proliferation and invasion of glioblastoma cells. PLoS One 7, e52113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mita R., Beaulieu M. J., Field C., Godbout R. (2010) Brain fatty acid-binding protein and ω-3/ω-6 fatty acids: mechanistic insight into malignant glioma cell migration. J. Biol. Chem. 285, 37005–37015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brun M., Coles J. E., Monckton E. A., Glubrecht D. D., Bisgrove D., Godbout R. (2009) Nuclear factor I regulates brain fatty acid-binding protein and glial fibrillary acidic protein gene expression in malignant glioma cell lines. J. Mol. Biol. 391, 282–300 [DOI] [PubMed] [Google Scholar]
- 17. Bisgrove D. A., Monckton E. A., Packer M., Godbout R. (2000) Regulation of brain fatty acid-binding protein expression by differential phosphorylation of nuclear factor I in malignant glioma cell lines. J. Biol. Chem. 275, 30668–30676 [DOI] [PubMed] [Google Scholar]
- 18. Roulet E., Bucher P., Schneider R., Wingender E., Dusserre Y., Werner T., Mermod N. (2000) Experimental analysis and computer prediction of CTF/NFI transcription factor DNA binding sites. J. Mol. Biol. 297, 833–848 [DOI] [PubMed] [Google Scholar]
- 19. Kruse U., Sippel A. E. (1994) Transcription factor nuclear factor I proteins form stable homo- and heterodimers. FEBS Lett. 348, 46–50 [DOI] [PubMed] [Google Scholar]
- 20. Gronostajski R. M. (2000) Roles of the NFI/CTF gene family in transcription and development. Gene 249, 31–45 [DOI] [PubMed] [Google Scholar]
- 21. Amemiya K., Traub R., Durham L., Major E. O. (1992) Adjacent nuclear factor-1 and activator protein binding sites in the enhancer of the neurotropic JC virus. A common characteristic of many brain-specific genes. J. Biol. Chem. 267, 14204–14211 [PubMed] [Google Scholar]
- 22. Deneen B., Ho R., Lukaszewicz A., Hochstim C. J., Gronostajski R. M., Anderson D. J. (2006) The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 52, 953–968 [DOI] [PubMed] [Google Scholar]
- 23. Namihira M., Kohyama J., Semi K., Sanosaka T., Deneen B., Taga T., Nakashima K. (2009) Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev. Cell 16, 245–255 [DOI] [PubMed] [Google Scholar]
- 24. Kang P., Lee H. K., Glasgow S. M., Finley M., Donti T., Gaber Z. B., Graham B. H., Foster A. E., Novitch B. G., Gronostajski R. M., Deneen B. (2012) Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 74, 79–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. das Neves L., Duchala C. S., Tolentino-Silva F., Haxhiu M. A., Colmenares C., Macklin W. B., Campbell C. E., Butz K. G., Gronostajski R. M. (1999) Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc. Natl. Acad. Sci. U.S.A. 96, 11946–11951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steele-Perkins G., Plachez C., Butz K. G., Yang G., Bachurski C. J., Kinsman S. L., Litwack E. D., Richards L. J., Gronostajski R. M. (2005) The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol. Cell. Biol. 25, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shu T., Butz K. G., Plachez C., Gronostajski R. M., Richards L. J. (2003) Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J. Neurosci. 23, 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gründer A., Ebel T. T., Mallo M., Schwarzkopf G., Shimizu T., Sippel A. E., Schrewe H. (2002) Nuclear factor I-B (Nfib) deficient mice have severe lung hypoplasia. Mech. Dev. 112, 69–77 [DOI] [PubMed] [Google Scholar]
- 29. Driller K., Pagenstecher A., Uhl M., Omran H., Berlis A., Gründer A., Sippel A. E. (2007) Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol. Cell. Biol. 27, 3855–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Steele-Perkins G., Butz K. G., Lyons G. E., Zeichner-David M., Kim H. J., Cho M. I., Gronostajski R. M. (2003) Essential role for NFI-C/CTF transcription-replication factor in tooth root development. Mol. Cell. Biol. 23, 1075–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee D. S., Park J. T., Kim H. M., Ko J. S., Son H. H., Gronostajski R. M., Cho M. I., Choung P. H., Park J. C. (2009) Nuclear factor I-C is essential for odontogenic cell proliferation and odontoblast differentiation during tooth root development. J. Biol. Chem. 284, 17293–17303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cebolla B., Vallejo M. (2006) Nuclear factor-I regulates glial fibrillary acidic protein gene expression in astrocytes differentiated from cortical precursor cells. J. Neurochem. 97, 1057–1070 [DOI] [PubMed] [Google Scholar]
- 33. Klee C. B., Crouch T. H., Krinks M. H. (1979) Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc. Natl. Acad. Sci. U.S.A. 76, 6270–6273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klee C. B., Draetta G. F., Hubbard M. J. (1988) Calcineurin. Adv. Enzymol. Relat. Areas Mol. Biol. 61, 149–200 [DOI] [PubMed] [Google Scholar]
- 35. Aramburu J., Rao A., Klee C. B. (2000) Calcineurin: from structure to function. Curr. Top. Cell. Regul. 36, 237–295 [DOI] [PubMed] [Google Scholar]
- 36. Shibasaki F., Hallin U., Uchino H. (2002) Calcineurin as a multifunctional regulator. J. Biochem. 131, 1–15 [DOI] [PubMed] [Google Scholar]
- 37. Yakel J. L. (1997) Calcineurin regulation of synaptic function: from ion channels to transmitter release and gene transcription. Trends Pharmacol. Sci. 18, 124–134 [DOI] [PubMed] [Google Scholar]
- 38. Schwartz N., Schohl A., Ruthazer E. S. (2009) Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron 62, 655–669 [DOI] [PubMed] [Google Scholar]
- 39. Baumgärtel K., Mansuy I. M. (2012) Neural functions of calcineurin in synaptic plasticity and memory. Learn. Mem. 19, 375–384 [DOI] [PubMed] [Google Scholar]
- 40. Ding B., Wang W., Selvakumar T., Xi H. S., Zhu H., Chow C. W., Horton J. D., Gronostajski R. M., Kilpatrick D. L. (2013) Temporal regulation of nuclear factor one occupancy by calcineurin/NFAT governs a voltage-sensitive developmental switch in late maturing neurons. J. Neurosci. 33, 2860–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alevizopoulos A., Dusserre Y., Rüegg U., Mermod N. (1997) Regulation of the transforming growth factor β-responsive transcription factor CTF-1 by calcineurin and calcium/calmodulin-dependent protein kinase IV. J. Biol. Chem. 272, 23597–23605 [DOI] [PubMed] [Google Scholar]
- 42. Chen R., Liu M., Li H., Xue Y., Ramey W. N., He N., Ai N., Luo H., Zhu Y., Zhou N., Zhou Q. (2008) PP2B and PP1α cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 22, 1356–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. García E., Stracher A., Jay D. (2006) Calcineurin dephosphorylates the C-terminal region of filamin in an important regulatory site: a possible mechanism for filamin mobilization and cell signaling. Arch. Biochem. Biophys. 446, 140–150 [DOI] [PubMed] [Google Scholar]
- 44. Pillai S., Dasgupta P., Chellappan S. P. (2009) Chromatin immunoprecipitation assays: analyzing transcription factor binding and histone modifications in vivo. Methods Mol. Biol. 523, 323–339 [DOI] [PubMed] [Google Scholar]
- 45. Andrin C., McDonald D., Attwood K. M., Rodrigue A., Ghosh S., Mirzayans R., Masson J. Y., Dellaire G., Hendzel M. J. (2012) A requirement for polymerized actin in DNA double-strand break repair. Nucleus 3, 384–395 [DOI] [PubMed] [Google Scholar]
- 46. Bléoo S., Sun X., Hendzel M. J., Rowe J. M., Packer M., Godbout R. (2001) Association of human DEAD box protein DDX1 with a cleavage stimulation factor involved in 3′-end processing of pre-MRNA. Mol. Biol. Cell 12, 3046–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fruman D. A., Klee C. B., Bierer B. E., Burakoff S. J. (1992) Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc. Natl. Acad. Sci. U.S.A. 89, 3686–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goto S., Matsukado Y., Mihara Y., Inoue N., Miyamoto E. (1986) The distribution of calcineurin in rat brain by light and electron microscopic immunohistochemistry and enzyme immunoassay. Brain Res. 397, 161–172 [DOI] [PubMed] [Google Scholar]
- 49. Pyrzynska B., Lis A., Mosieniak G., Kaminska B. (2001) Cyclosporin A-sensitive signaling pathway involving calcineurin regulates survival of reactive astrocytes. Neurochem. Int. 38, 409–415 [DOI] [PubMed] [Google Scholar]
- 50. Liu F., Grundke-Iqbal I., Iqbal K., Oda Y., Tomizawa K., Gong C. X. (2005) Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J. Biol. Chem. 280, 37755–37762 [DOI] [PubMed] [Google Scholar]
- 51. Muramatsu T., Kincaid R. L. (1992) Molecular cloning and chromosomal mapping of the human gene for the testis-specific catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin A). Biochem. Biophys. Res. Commun. 188, 265–271 [DOI] [PubMed] [Google Scholar]
- 52. Muramatsu T., Giri P. R., Higuchi S., Kincaid R. L. (1992) Molecular cloning of a calmodulin-dependent phosphatase from murine testis: identification of a developmentally expressed nonneural isoenzyme. Proc. Natl. Acad. Sci. U.S.A. 89, 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Enz A., Shapiro G., Chappuis A., Dattler A. (1994) Nonradioactive assay for protein phosphatase 2B (calcineurin) activity using a partial sequence of the subunit of cAMP-dependent protein kinase as substrate. Anal. Biochem. 216, 147–153 [DOI] [PubMed] [Google Scholar]
- 54. Donella-Deana A., Krinks M. H., Ruzzene M., Klee C., Pinna L. A. (1994) Dephosphorylation of phosphopeptides by calcineurin (protein phosphatase 2B). Eur. J. Biochem. 219, 109–117 [DOI] [PubMed] [Google Scholar]
- 55. Wang M. G., Yi H., Guerini D., Klee C. B., McBride O. W. (1996) Calcineurin A α (PPP3CA), calcineurin A β (PPP3CB) and calcineurin B (PPP3R1) are located on human chromosomes 4, 10q21→q22 and 2p16→p15, respectively. Cytogenet. Cell Genet. 72, 236–241 [DOI] [PubMed] [Google Scholar]
- 56. Goll D. E., Thompson V. F., Li H., Wei W., Cong J. (2003) The calpain system. Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- 57. Brat D. J., Castellano-Sanchez A. A., Hunter S. B., Pecot M., Cohen C., Hammond E. H., Devi S. N., Kaur B., Van Meir E. G. (2004) Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 64, 920–927 [DOI] [PubMed] [Google Scholar]
- 58. Yang B. S., Gilbert J. D., Freytag S. O. (1993) Overexpression of Myc suppresses CCAAT transcription factor/nuclear factor 1-dependent promoters in vivo. Mol. Cell. Biol. 13, 3093–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Steiner J. P., Dawson T. M., Fotuhi M., Glatt C. E., Snowman A. M., Cohen N., Snyder S. H. (1992) High brain densities of the immunophilin FKBP colocalized with calcineurin. Nature 358, 584–587 [DOI] [PubMed] [Google Scholar]
- 60. Matsuda T., Takuma K., Asano S., Kishida Y., Nakamura H., Mori K., Maeda S., Baba A. (1998) Involvement of calcineurin in Ca2+ paradox-like injury of cultured rat astrocytes. J. Neurochem. 70, 2004–2011 [DOI] [PubMed] [Google Scholar]
- 61. Farber L. H., Wilson F. J., Wolff D. J. (1987) Calmodulin-dependent phosphatases of PC12, GH3, and C6 cells: physical, kinetic, and immunochemical properties. J. Neurochem. 49, 404–414 [DOI] [PubMed] [Google Scholar]
- 62. Fernandez A. M., Fernandez S., Carrero P., Garcia-Garcia M., Torres-Aleman I. (2007) Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J. Neurosci. 27, 8745–8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Norris C. M., Kadish I., Blalock E. M., Chen K. C., Thibault V., Porter N. M., Landfield P. W., Kraner S. D. (2005) Calcineurin triggers reactive/inflammatory processes in astrocytes and is up-regulated in aging and Alzheimer's models. J. Neurosci. 25, 4649–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wu H. Y., Tomizawa K., Oda Y., Wei F. Y., Lu Y. F., Matsushita M., Li S. T., Moriwaki A., Matsui H. (2004) Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J. Biol. Chem. 279, 4929–4940 [DOI] [PubMed] [Google Scholar]
- 65. Jiang H., Xiong F., Kong S., Ogawa T., Kobayashi M., Liu J. O. (1997) Distinct tissue and cellular distribution of two major isoforms of calcineurin. Mol. Immunol. 34, 663–669 [DOI] [PubMed] [Google Scholar]
- 66. Kilka S., Erdmann F., Migdoll A., Fischer G., Weiwad M. (2009) The proline-rich N-terminal sequence of calcineurin Aβ determines substrate binding. Biochemistry 48, 1900–1910 [DOI] [PubMed] [Google Scholar]
- 67. Hallhuber M., Burkard N., Wu R., Buch M. H., Engelhardt S., Hein L., Neyses L., Schuh K., Ritter O. (2006) Inhibition of nuclear import of calcineurin prevents myocardial hypertrophy. Circ. Res. 99, 626–635 [DOI] [PubMed] [Google Scholar]
- 68. Santella L., Carafoli E. (1997) Calcium signaling in the cell nucleus. FASEB J. 11, 1091–1109 [PubMed] [Google Scholar]
- 69. Bosser R., Aligué R., Guerini D., Agell N., Carafoli E., Bachs O. (1993) Calmodulin can modulate protein phosphorylation in rat liver cells nuclei. J. Biol. Chem. 268, 15477–15483 [PubMed] [Google Scholar]
- 70. Pujol M. J., Bosser R., Vendrell M., Serratosa J., Bachs O. (1993) Nuclear calmodulin-binding proteins in rat neurons. J. Neurochem. 60, 1422–1428 [DOI] [PubMed] [Google Scholar]
- 71. Shibasaki F., Price E. R., Milan D., McKeon F. (1996) Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature 382, 370–373 [DOI] [PubMed] [Google Scholar]
- 72. Bosher J., Dawson A., Hay R. T. (1992) Nuclear factor I is specifically targeted to discrete subnuclear sites in adenovirus type 2-infected cells. J. Virol. 66, 3140–3150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cyert M. S. (2001) Regulation of nuclear localization during signaling. J. Biol. Chem. 276, 20805–20808 [DOI] [PubMed] [Google Scholar]
- 74. Nedergaard M., Rodríguez J. J., Verkhratsky A. (2010) Glial calcium and diseases of the nervous system. Cell Calcium 47, 140–149 [DOI] [PubMed] [Google Scholar]
- 75. Oh M. C., Kim J. M., Safaee M., Kaur G., Sun M. Z., Kaur R., Celli A., Mauro T. M., Parsa A. T. (2012) Overexpression of calcium-permeable glutamate receptors in glioblastoma derived brain tumor initiating cells. PLoS One 7, e47846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dawson T. M., Steiner J. P., Lyons W. E., Fotuhi M., Blue M., Snyder S. H. (1994) The immunophilins, FK506 binding protein and cyclophilin, are discretely localized in the brain: relationship to calcineurin. Neuroscience 62, 569–580 [DOI] [PubMed] [Google Scholar]
- 77. Pufall M. A., Lee G. M., Nelson M. L., Kang H. S., Velyvis A., Kay L. E., McIntosh L. P., Graves B. J. (2005) Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 309, 142–145 [DOI] [PubMed] [Google Scholar]
- 78. Lee C. W., Ferreon J. C., Ferreon A. C., Arai M., Wright P. E. (2010) Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 107, 19290–19295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Okamura H., Aramburu J., García-Rodríguez C., Viola J. P., Raghavan A., Tahiliani M., Zhang X., Qin J., Hogan P. G., Rao A. (2000) Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 6, 539–550 [DOI] [PubMed] [Google Scholar]
- 80. Lehtinen M. K., Yuan Z., Boag P. R., Yang Y., Villén J., Becker E. B., DiBacco S., de la Iglesia N., Gygi S., Blackwell T. K., Bonni A. (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125, 987–1001 [DOI] [PubMed] [Google Scholar]
- 81. Liang Y., Bollen A. W., Aldape K. D., Gupta N. (2006) Nuclear FABP7 immunoreactivity is preferentially expressed in infiltrative glioma and is associated with poor prognosis in EGFR-overexpressing glioblastoma. BMC Cancer 6, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]